
Abstract
Leptin is the most critical hormone in the homeostatic regulation of energy balance among those so far discovered. Leptin primarily acts on the neurons of the mediobasal part of hypothalamus to regulate food intake, thermogenesis, and the blood glucose level. In the hypothalamic neurons, leptin binding to the long form leptin receptors on the plasma membrane initiates multiple signaling cascades. The signaling pathways known to mediate the actions of leptin include JAK–STAT signaling, PI3K–Akt–FoxO1 signaling, SHP2–ERK signaling, AMPK signaling, and mTOR–S6K signaling. Recent evidence suggests that leptin signaling in hypothalamic neurons is also linked to primary cilia function. On the other hand, signaling molecules/pathways mitigating leptin actions in hypothalamic neurons have been extensively investigated in an effort to treat leptin resistance observed in obesity. These include SOCS3, tyrosine phosphatase PTP1B, and inflammatory signaling pathways such as IKK-NFκB and JNK signaling, and ER stress–mitochondrial signaling. In this review, we discuss leptin signaling pathways in the hypothalamus, with a particular focus on the most recently discovered pathways.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The regulation of food intake and energy expenditure by the central nervous system (CNS) is critical for maintaining whole body energy homeostasis. Among the brain regions, the hypothalamus plays a central role in the control of energy balance. Neurons in the hypothalamic arcuate nucleus (ARC) monitor the body energy state by sensing the blood levels of key metabolic hormones and nutrients. Leptin and leptin receptor signaling are most crucial of the factors identified to date that coordinate the control of food intake and energy expenditure in response to an altered energy state [1]. Leptin primarily acts on the ARC neurons producing anorexigenic proopiomelanocortin (POMC) and orexigenic neuropeptide Y (NPY)/Agouti-related protein (AgRP). Leptin suppresses energy intake and stimulates energy expenditure, leading to a reduction in stored body fat.
In 1950, the first obese mouse (ob/ob, now refer to Lep ob) was reported by the Jackson Laboratory [2]. This mouse showed marked obesity with increased fat mass, massive hyperphagia and mild symptoms of diabetes. After 16 years, Coleman and colleagues introduced another mutant mouse (mutation diabetes; db/db or Lepr db) with marked hyperphagia, obesity, and severe diabetic symptoms such as hyperglycemia, polyuria, and glycouria [3]. As a result of a series of symbolic “parabiosis experiments” by Coleman and colleagues, it was realized that Lepr db mice overproduced a certain circulating factor but were unable to respond to it, whereas the Lep ob mice responded, but could not synthesize this factor [4, 5]. Using a positional cloning technique, Friedman’s group cloned this circulating factor and named it “leptin” [6]. The initial belief that leptin could cure human obesity was thwarted, however, by the fact that most obese individuals are hyperleptinemic with intact leptin receptors and did not lose weight after leptin treatment, so called leptin resistance. It was shown however, similar to a classical endocrine replacement therapy, that the administration of recombinant leptin to both leptin deficient humans and rodents dramatically reduced excessive body weight, hyperphagia, and hyperglycemia [7, 8].
During the past few decades, the leptin research field has dramatically expanded into a wide range of studies that have explored not only the leptin signaling pathways but also the precise site and the function of leptin action and the mechanisms of leptin resistance. Initial findings indicated the CNS as the major action site of leptin action as the brain-specific deletion or reconstitution of leptin receptors demonstrated the sufficiency and requirement of CNS leptin action for the regulation of energy homeostasis [9, 10]. Recent studies have also shown the effects of leptin in diverse sites both in the CNS and in the periphery [11–13]. However, the functional roles of endogenous leptin in peripheral organs have largely remained obscure. In our current review, we seek to provide a comprehensive overview on the various mechanisms through which leptin activates and interacts with intracellular signaling pathways in the hypothalamic neurons.
Leptin and its receptors (Fig. 1)
Leptin is a cytokine-like hormone predominantly produced in the white adipose tissue and secreted into the systemic circulation [6]. Leptin mRNA expression is also found in other tissues including brown adipose tissue, placenta, ovary, skeletal muscle, stomach, pituitary gland, and the CNS [14–21], suggesting the local production of leptin in these organs. Leptin production in various tissues may be suggestive of pleiotropic effects of leptin on various biological functions. In addition to its critical role in appetite regulation and energy balance, leptin modulates immune function, gonadal function, and stress responses [22–24].

Schematic representation of mouse leptin receptor isoforms. The mouse leptin receptor has six isoforms, LepRa–f with a common extracellular domain but diverse intracellular domains. JAK interaction and activation is mediated through the box 1 motif. The long form, LepRb, is the only isoform that contains motifs enabling the leptin-induced activation of the JAK-STAT pathway
Leptin belongs to the long-chain helical cytokine family [25] and exerts its regulatory effects on energy homeostasis through its receptors which have a wide distribution in the body, particularly in the CNS, in both humans and rodents [26]. The leptin receptor (LepR) is a single membrane spanning receptor belonging to the class 1 cytokine receptor family. There are multiple leptin receptor isoforms that are expressed from the primary leptin receptor gene by alternative splicing [27, 28]. The leptin receptor gene was cloned from the murine choroid plexus using tagged proteins to identify cells with leptin binding activity [29]. The identification of the leptin receptor in the brain was a major breakthrough in our understanding of leptin biology as it provided further insights into the mechanism regulating leptin activity and paved the way for further studies on the obese and hyperphagic phenotypes observed in Lep ob and Lepr db mice [25, 27, 30].
The leptin receptor gene contains 17 common exons and several alternatively spliced 3′-exons [31]. To date, six isoforms of the leptin receptor have been identified in rodents (LepRa–f), all possessing the extracellular domains necessary for ligand binding [6, 32] (Fig. 1). LepRs can be categorized into three groups: secreted, short, and long isoforms. The secreted isoforms, such as LepRe, are either products of alternatively spliced messenger RNA species (in the mouse) or proteolytic cleavage products of the membrane bound form (in human) [31, 32]. LepRe is the smallest isoform and contains neither the cytoplasmic nor the transmembrane domains but only the extracellular domains that can bind to circulating leptin [33].
The short form LepRs include LepRa, c, d and f in rodents, whereas LepRb is classified as a long form (Fig. 1). Leptin binds to the short and long form receptors with the same affinity. However, only the long form has the ability to transmit leptin signaling and initiate the necessary intracellular responses [34, 35]. The short and long forms of the leptin receptor are distinguished by their intracellular domain size and the presence of JAK/STAT binding sites. The long form (LepRb) has a large intracellular domain consisting of 304 residues and has several putative JAK and STAT binding sites, whilst the short form (LepRa) comprises a 34 residue-intracellular domain and only one putative JAK binding domain [36–40]. Leptin regulation of JAK/STAT signaling will be further described in a later section. Both the short and long forms contain exons 1–17 of the leptin receptor gene and share extracellular and transmembrane domains as well as the first 29 amino acids of the cytoplasmic domain [41]. In rodents, LepRa, c, and d have different terminal exons, encoding only 3, 5, and 11 amino acids, respectively, whereas the terminal exon of LepRb has 273 amino acids [31]. The shorter isoforms, LepRa, c, d, and f, have truncated cytoplasmic domains with reduced signaling transduction capability. The leptin receptor short isoforms have been identified in multiple species. LepRa–c are widely spread receptors. In contrast, LepRd is found only in mice and LepRf is expressed only in rats. However, the roles of the short form leptin receptors remain to be clearly elucidated.
Despite the normal expression of the short form leptin receptors in Lepr db mice, these mice manifest an obese phenotype, indicating that the short form receptor cannot mediate leptin-induced changes in energy homeostasis [42]. Recent reports have described the phenotype of the LepRa knockout mouse [43, 44]. These mice have slightly lower fasting blood glucose level and improved glucose tolerance with normal body weight and food intake under chow diet-fed conditions. However, when given a high fat diet, they develop mild obesity along with modest leptin resistance and decreased leptin transport into the cerebrospinal fluid. Thus LepRa seems to mediate some of the effects of leptin, but its role in metabolic regulation appears to be limited when compared to LepRb. Further studies are needed to address the importance of the short form leptin receptors in energy homeostasis. It will also be necessary to establish whether there is any interaction between the short form and the long form leptin receptors in mediating the anorectic effects of leptin in the brain.
Signaling pathways that transduce the hypothalamic actions of leptin (Fig. 2)
JAK–STAT signaling
JAK–STAT signaling is a representative signaling pathway through which leptin regulates food intake and energy homeostasis in the hypothalamus. Leptin binds to its receptors and initiates downstream signaling through the sequential phosphorylation of the tyrosine kinase Janus kinase (JAK) and the transcription factor signal transducer and activators of transcription (STAT). The phosphorylation of STAT induces its dimerization and translocation to the nucleus, where it binds DNA and modulates the transcription of genes involved in food intake and energy homeostasis [45].

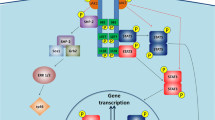
Signaling pathways of leptin and its downstream effectors. Leptin binds to its receptor and activates the JAK–STAT3, PI3K–FoxO1, and ERK pathways. For JAK–STAT activation, activated JAK2 tyrosine kinase induces the phosphorylation of Tyr985 and Tyr1138 of LepRb, which leads to the activation of STAT3/STAT5. Phosphorylated Tyr985 strongly recruits SHP-2 and GRB2 resulting in the activation of ERK signaling. Leptin also activates PI3K by recruiting IRS proteins leading to the inactivation of FoxO1 by sequestering them in the cytoplasm. On the other hand, leptin has been reported to inhibit AMPK activity. AMPK is activated by upstream signaling, including LKB1 and CaMKKβ. In addition, leptin treatment activates mTOR/S6K signaling in the hypothalamus, which phosphorylates Ser491 of AMPK α-subunit and inhibits AMPK activity. AgRP agouti-related protein, FoxO1 forkhead box protein O1, IRS insulin receptor substrate, JAK Janus kinase, PI3K phosphatidylinositol 3-OH kinase, POMC proopiomelanocortin, PTP1B protein-tyrosine phosphatase 1B, SOCS3 suppressor of cytokine signaling 3, STAT signal transducer and activator of transcription
STAT molecules are cytoplasmic proteins activated by numerous factors including cytokines, growth factors, and hormones including leptin. The mammalian STAT family has seven members, STAT1–4, STAT5a, STAT5b and STAT6. Leptin induces the phosphorylation of STAT1, STAT3, STAT5 and STAT6, among which STAT3 and possibly STAT5 mediate leptin-induced anorectic effects [34, 46–50].
STAT3 is widely expressed in the CNS. Mouse models have been employed to explore the role of STAT3 in leptin regulation of energy homeostasis [50]. Neuron-specific STAT3 deletion resulted in hyperphagia, severe obesity, diabetes, and hyperleptinemia in mice [51], traits which were also observed in leptin- and LepRb-deficient mice and classified as hallmarks of leptin resistance [52, 53]. Phosphorylated STAT3 facilitates the leptin-mediated transcriptional regulation of key appetite-regulating neuropeptides such as POMC, AgRP, and NPY. POMC is a precursor peptide which is cleaved by prohormone convertases, giving rise to anorexigenic α-melanocyte stimulating hormone (α-MSH). AGRP and NPY are potent orexigenic peptides synthesized in discrete neurons of the hypothalamic ARC. Leptin increases the transcriptional activity of POMC whereas a dominant-negative form of STAT3 inhibits this activity, indicating that POMC is a major target gene of LepRb–STAT3 signaling [54]. Increased food intake in STAT3 mutant mice can be attributed to the dysregulated transcription of POMC, AgRP and NPY genes [55, 56].
Tyrosine 1138 of LepRb mediates STAT3 activation during leptin action [57] and its substitution for serine (Lepr S1138) specifically disrupts LepRb–STAT3 signaling. Similar to Lepr db mice, mice harboring the Lepr S1138 allele (s/s mice) are hyperphagic and obese from an early age. In these animals, obesity progresses throughout adulthood and is accompanied by elevated plasma leptin levels, implying leptin resistance [50]. Lepr db mice are also infertile, short, and diabetic whereas s/s mice display normal fertility and growth and less severe hyperglycemia. Furthermore, hypothalamic POMC expression is similarly suppressed in both Lepr db and s/s mice, whereas NPY expression is elevated in Lepr db mice but not in s/s mice. Hence, the LepRb–STAT3 signaling pathway is critical for the effects of leptin on melanocortin production and body energy homeostasis, but other signaling pathways may transduce actions of leptin on the hypothalamic NPY, on neuroendocrine functions such as fertility and growth, and on glucose metabolism [50].
Leptin administration increases the transcription of suppressor of cytokine signaling 3 (SOCS3) in the hypothalamus, via tyrosine 1138 of LepRb, whilst SOCS3 is known to inhibit leptin activities. Thus a negative feedback mechanism exists in leptin-induced STAT3 signaling, through the induction of SOCS3 [58]. Leptin phosphorylates and activates STAT3, which then binds to two sites within the SOCS3 promoter to increase the transcription of SOCS3 [59]. Increased SOCS3 expression creates a feedback loop curtailing further activation of leptin–STAT3 signaling [60]. As such, the JAK–STAT3 pathway may be vital for the maintenance of energy homeostasis through the fine tuning of a modulatory loop in the molecular network of leptin action. Enhanced hypothalamic SOCS3 expression under obese conditions has been suggested as a major mechanism limiting leptin signaling and actions, which will be further discussed in the following section.
LRP1 and LRP2 (low-density lipoprotein receptor-related protein-1, 2) are multi-ligand endocytic receptors that belong to the LRP family of proteins [61]. Mice lacking neuronal LRP1 develop leptin resistance and obesity [62]. Moreover, a molecular interaction between LRP1 and leptin–LepRb complex has been demonstrated in the hypothalamus [62], indicating an involvement of LRP1 in hypothalamic leptin signaling. On the other hand, LRP2, also known as megalin/gp330, has been implicated in leptin reuptake in the renal tubules and leptin transport in the choroid plexus [63–65]. A blockade of hypothalamic LRP and endocytosis impairs leptin-induced anorexia and hypothalamic STAT3 phosphorylation [66, 67]. Thus, the LRP-mediated endocytosis of leptin–leptin receptor complex may be required for leptin-activated STAT3 signaling.
PI3K–Akt–FoxO1 signaling
The intracerebroventricular (ICV) administration of leptin activates the insulin receptor substrate (IRS)–phosphatidylinositol 3-OH kinase (PI3K) signaling in the mediobasal hypothalamus (MBH) [68]. Involvement of the IRS–PI3K pathway in leptin activity was first delineated from the IRS2-null mouse phenotype, which involved an elevated leptin level, increased food intake and decreased energy expenditure [69]. Subsequently, it was shown that inhibiting the hypothalamic PI3K pathway suppressed the anorexigenic effects of leptin [68], suggesting a crucial role of PI3K in mediating leptin actions. The Sar homology family member SH2B1 mediates leptin-induced PI3K activation. SH2B1 enhances JAK2 activity and recruits IRS proteins to JAK2, which leads to JAK2-mediated IRS phosphorylation and subsequent PI3K activation [70, 71].
In hypothalamic neurons, PI3K integrates insulin and leptin signaling [72]. Leptin directly increases PI3K activity in POMC neurons, but indirectly inhibits it in AGRP neurons. By contrast, insulin activates PI3K signaling in both neuronal cell types [73]. PI3K activation mediates the acute effects of leptin on the neuroelectrical activity of POMC neurons as PI3K inhibitors blocks leptin-induced neuroelectrical changes in POMC neurons [74]. Disruption of PI3K signaling in POMC cells abolishes membrane depolarization and decreases the firing rate in response to leptin [74]. Nevertheless, mice with impaired PI3K signaling in POMC neurons have normal body weights. Thus PI3K signaling in POMC neurons is not critical for the leptin-mediated regulation of energy balance.
PI3K catalyzes the phosphorylation of phosphatidylinositol-4,5-bisphosphate to phosphatidylinositol-3,4,5-trisphosphate (PIP3) whereas tumor suppressor phosphatase and tensin homolog (Pten) promotes the opposite reaction [75]. Activation of PI3K in LepR expressing neurons by ablation of Pten leads to a reduced body fat mass via increased sympathetic outflow to perigonadal white adipose tissue (WAT) resulting in brown adipose tissue-like transdifferentiation of WAT [76]. On the other hand, mice lacking Pten in POMC neurons develop obesity and leptin resistance through the activation of ATP-dependent K+ channels [77]. Thus chronic elevation of PIP3 in POMC neurons may interfere with hypothalamic leptin activity.
The transcription factor forkhead box protein O1 (FoxO1) is a well-known phosphorylation target of PI3K downstream kinase Akt. FoxO1 mediates the anorectic effects of leptin and insulin by regulating the transcription of POMC and AgRP [78–81]. FoxO1 expression is found in the ARC, and ventromedial and dorsomedial hypothalamus, regions which also express leptin receptors. Under nutrition deprivation conditions, FoxO1 translocates from the cytoplasm to the nucleus where it increases the expression of the orexigenic NPY/AgRP genes and suppresses expression of anorexigenic POMC, thereby increasing food intake [78, 82]. Leptin hampers the FoxO1-mediated transcriptional regulation of POMC, NPY and AgRP [78, 82, 83]. Conversely, activated FoxO1 inhibits the ability of leptin to induce anorexia and to stimulate POMC transcription [83]. In line with this, mice with depleted FoxO1 in the POMC neurons are more sensitive to the anorectic effects of leptin [84]. These data suggest that reduced FoxO1 activity via leptin is important for leptin-mediated feeding regulation.
There seems to be a crosstalk between STAT3 and FoxO1 signaling. The overexpression of FoxO1 interrupts STAT3-stimulated POMC promoter activity [85]. FoxO1 does not inhibit leptin-mediated STAT3 phosphorylation nor does it block the translocation of the activated STAT3 to the nucleus. Instead, FoxO1 inhibits the interaction between STAT3 and the SP1 transcription factor, which critically mediates leptin-induced POMC transcriptional activity [85].
SHP2–ERK signaling
The mitogen-activated protein kinases (MAPKs) function to regulate numerous intracellular programs, including cell proliferation and differentiation, by relaying extracellular signals to the cell. The involvement of hypothalamic extracellular signal-regulated kinase (ERK) signaling in energy homeostasis was suggested by ERK activation in the ARC and paraventricular nucleus (PVN) under nutrition deprivation conditions [86] which was reversed after re-feeding [87]. Several studies have demonstrated the role of MAPKs, particularly ERK1/2, in the leptin regulation of energy homeostasis [57, 88, 89]. Leptin activates ERK1/2 but not p38 MARK in the hypothalamic ARC. Furthermore, a pharmacological blockade of hypothalamic ERK1/2 reverses the anorectic and weight-reducing effects of leptin and also inhibits leptin-induced thermogenesis by controlling sympathetic activity on BAT. These findings indicate that the hypothalamic ERK plays an important role in the control of energy balance by leptin [90].
LepRb mediates ERK activation as evidenced by the leptin induced ERK activation in the lean Zucker rat but not in the obese Zucker rat with a defective LepRb [90]. Among the Tyr 985, Tyr 1077 and Tyr 1138 residues of LepRb, Tyr 985 is implicated in leptin activated ERK signaling. The deletion of Tyr 985 inhibits leptin-induced ERK activation by about 70 %, indicating the involvement of another mechanism in ERK activation such as through the leptin receptor short form [57, 91]. ERK1 and 2 are typically activated through the receptor tyrosine kinase upon ligand binding. The activation and autophosphorylation of the tyrosine residue in the intracellular domain of LepR facilitates its interaction with adapter proteins containing the Src homology 2 (SH2) domain [92]. SHP2, a SH2-containing tyrosine-specific protein phosphatase, appears to be a key modulator in leptin–ERK signaling as the phosphatase activity of SHP2 is necessary for leptin-mediated ERK activation [93].
Leptin plays a significant role in the early development of hypothalamic feeding circuits [94]. ARH projections to the PVN, dorsomedial nucleus (DMN), and lateral hypothalamus are markedly reduced in leptin deficient Lep ob mice and restored by leptin treatment during the neonatal period. Both ERK and STAT3 signaling were found to be important for leptin-stimulated formation of hypothalamic neuronal circuits as disrupted ERK and STAT3 signaling impaired ARH projections [95]. Recently, it was reported that ERK signaling in hypothalamic tanycytes is crucial for leptin transport to the MBH [96]. Activation of ERK signaling with epidermal growth factor (EGF) restores decreased leptin transport and leptin sensitivity in the hypothalamus of diet-induced obese animals.
AMPK signaling
Adenosine monophosphate-activated protein kinase (AMPK) is an intracellular energy sensor activated under conditions of metabolic stress causing ATP depletion and an intracellular increase in AMP and ADP levels [97]. Once activated, AMPK restores energy balance by initiating ATP generating catabolic processes such as fatty acid oxidation and suppressing ATP consuming anabolic processes. AMPK is a heterotrimeric serine/threonine kinase consisting of a catalytic α subunit and regulatory β and γ subunits [98, 99]. AMPK activation occurs through the phosphorylation of threonine 172 on the catalytic α subunit by the upstream kinase Peutz–Jegher syndrome tumor-suppressor gene product LKB1 and Ca2+/calmodulin-dependent protein kinase kinase-β (CaMKK β) [100, 101].
AMPK plays a central role in energy homeostasis by integrating hormonal and nutritional signals in both the periphery and the brain. In the hypothalamus, AMPK is activated by fasting but suppressed by refeeding. Leptin inhibits AMPK activity in distinct hypothalamic regions such as the ARC and PVN, hence reducing food intake and body weight [102]. A reduction in AMPK phosphorylation by leptin administration is followed by decreased phosphorylation of the AMP downstream target acetyl-CoA carboxylase (ACC) and elevated malonyl-CoA levels in the hypothalamic neurons [103]. Increased malonyl-CoA reduces mitochondrial fatty acid oxidation by suppressing the carnitine palmitoyl transferase (CPT)-1 activity, thereby increasing the cellular long chain fatty acyl-CoA (LCFA-CoA) level. Increased LCFA-CoA content in the hypothalamus is known to induce anorexia and reduce hepatic glucose production [104]. On the other hand, systemic administration of leptin stimulates skeletal muscle AMPK activity and enhances fatty acid oxidation through both direct and CNS-mediated indirect mechanisms [105]. Hypothalamic AMPK signaling also transduces the effects of leptin on the sympathetic nerve outflows to the kidney, brown and white adipose tissues [106].
Extensive studies have also revealed phosphorylation sites in the α and β subunits that alter the activity of AMPK [107, 108]. Of the two catalytic subunits, α1 and α2, leptin appears to modulate the activity of AMPK by acting on the α2-AMPK subunit [102]. Hypothalamic α2-AMPK activity is important for homeostatic regulation of energy balance. ICV administration of leptin in wild type mice increased the phosphorylation of α2-AMPK at Ser491 in the hypothalamus which decreased α2-AMPK activity. Similarly, ICV injection of leptin had no significant effects on the body weight or food intake of rats with an siRNA silenced α2 AMPK subunit [106]. Mice lacking α2-AMPK in POMC neurons displayed obesity due to reduced energy expenditure and increased food intake whilst an α2-AMPK deletion in AgRP neurons resulted in an age-dependent lean phenotype [109]. Phosphorylation of Ser491 on α2-AMPK by p70 S6 kinase (S6K) has been suggested as the mechanism underlying the leptin-mediated inhibition of AMPK.
mTOR–S6K signaling
The mammalian target of rapamycin (mTOR) is an evolutionally conserved serine–threonine kinase, which senses nutrient (especially amino acids) availability, stimulates protein synthesis, cell growth and proliferation, and inhibits autophagy [110]. Like AMPK, mTOR serves as a cellular fuel sensor [111]. mTOR and its downstream target, S6K1 are expressed in hypothalamic neurons including NPY/AgRP neurons and POMC neurons [112]. Leptin administration acutely increases hypothalamic mTOR activity, determined by increased phospho-S6K1 and phospho-S6 ribosomal protein. Moreover, the coadministration of the mTOR inhibitor rapamycin significantly blocks the anorectic effects of leptin. Mice lacking S6K1 failed to reduce their food intake in response to leptin [113]. Furthermore, S6K activity in the MBH affected hypothalamic leptin sensitivity. Leptin-induced reduction in body weight and food intake is more pronounced in rats expressing constitutively active S6K1 in the MBH whereas it was found to be mitigated in rats expressing dominant negative (DN)-S6K1 [114]. All of these data support an indispensable contribution of mTOR–S6K signaling in the hypothalamic actions of leptin. In addition to feeding regulation, ARC mTOR activation by leptin is important for the sympathetic and cardiovascular effects of leptin [115]. Both rapamycin administration and hypothalamic DN-S6K expression prevent the stimulatory effects of leptin on renal sympathetic nerve outflow and arterial blood pressure. In high-fat-diet-fed animals, leptin is unable to modulate hypothalamic mTOR activity [112], implying the development of leptin resistance in this signaling pathway.
In the hypothalamic neurons, leptin activates mTOR–S6K1 through the activation of PI3K–Akt signaling. As mentioned in the previous section, activated S6K1 in turn phosphorylates α2-AMPK at serine 491 which leads to reduced α2-AMPK activity in the MBH [116]. Thus mTOR–S6K signaling serves as an important pathway upstream of AMPK in the hypothalamic leptin signaling cascades. In contrast to its role in leptin signaling, chronic mTOR activation induced by depleting TSC1 and by aging in POMC neurons causes hyperphagic obesity. Interestingly, rapamycin and hypothalamic S6K1 inactivation successfully treated activated mTOR-induced obesity [117–119]. Thus the metabolic consequences of altered mTOR signaling may differ depending on neuronal cell type and metabolic context.
Cilia-related signaling (Fig. 3)
A cilium is a tiny hair-like organelle projecting from the cell surface. Cilia consist of a 9+2 or 9+0 microtubule-based axonemal complexes, which are assembled on a basal body and covered with a specialized plasma membrane. Most mammalian cells have single immotile (primary) cilia, which have been implicated in various cellular signaling events [120]. Several lines of evidence suggest that leptin signaling is closely tied to the cilia–basal body complex. Obesity is one of the cardinal manifestations of human ciliopathy Bardet–Biedl syndrome (BBS) [121]. Likewise, murine BBS models lacking BBS2, BBS4, and BBS6 proteins display obesity [122]. In these mice, leptin-induced hypothalamic STAT3 phosphorylation, anorexia, and weight loss are severely impaired, even upon calorie restriction to avoid leptin resistance secondary to obesity. These data have suggested that BBS proteins may be involved in hypothalamic leptin signaling and actions. Seven BBS proteins (BBS1, BBS2, BBS4, BBS5, BBS7, BBS8 and BBS9) form a stable complex known as the BBSome [123], which mediates protein/vesicle trafficking to cilia. Interestingly, LepRb specifically interacts with BBS1 [122]. Furthermore, the depletion of BBB1 or BBS2 in retinal pigment epithelial cells disrupts LepRb trafficking to the post-Golgi network (PGN), resulting in aberrant accumulation of LepRb-containing vesicles in the perinuclear area [122]. Therefore, the BBSome might interact with LepRb to mediate LepRb trafficking to the PGN.

Association of cilia signaling and leptin signaling. Primary cilia are associated with leptin signaling in hypothalamic neurons. BBS proteins form a protein complex called BBSome, which is involved in the trafficking of vesicles laden with ciliary proteins from the Golgi to cilia–basal bodies. The BBSome, especially BBS1, may mediate the trafficking of LepRb to the cellular surface or periciliary area as the deletion of BBS1 causes the aberrant distribution of LepRb in the perinuclear area. Another ciliary gene, RPGRIP1L, interacts with LepRb and mediates LepRb trafficking to the periciliary area. RPGRIP1L expression is regulated by CUX1, which binds to the first intron of FTO gene and stimulates the transcription of RPGRIP1L and FTO. BBS Bardet–Biedl syndrome, CUX1 cut-like homeobox 1, FTO fat mass and obesity associated, RPGRIP1L retinitis pigmentosa GTPase regulator-interacting protein-1 like
Another ciliary gene, retinitis pigmentosa GTPase regulator-interacting protein-1 (RPGRIP1L) has been shown to coordinate leptin signaling in hypothalamic neurons [124, 125]. RPGRIP1L-haploinsufficient mice exhibit increased fat mass and food intake and reduced energy expenditure [125]. These mice show reduced anorectic responses and hypothalamic STAT3 activation by exogenous leptin, even before the development of obesity. Like BBS proteins, RPGRIP1L interacts with LepRb and mediates its trafficking to the periciliary area. In LepRb-expressing normal fibroblasts, LepRb localizes to the vicinity of cilium upon leptin treatment [125]. In line with this, LepRb colocalizes with the PGN markers TGN46 and ARF4 near the base of cilium in the hypothalamic arcuate nuclei of mice following leptin treatment. LepRb fails however to locate around the base of the cilium in fibroblasts with an RPGRIP1L mutation and in RPGRIP1L-haploinsufficient mice. Thus, RPGRIP1L-mediated LepRb trafficking to the TGN and periciliary area may be critical in hypothalamic leptin signaling. Interestingly, the expression of RPGRIP1L was found to be regulated by the cut-like homeobox 1 (CUX1), which binds to the first intron of the fat mass and obesity associated (FTO) gene and stimulates the transcription of RPGRIP1L and FTO. Common single nucleotide polymorphisms (SNPs) in the FTO gene are significantly associated with the fat mass in humans [126, 127] and have been shown to disrupt CUX1-stimulated RPGRIP1L and FTO transcription. Hence, common variants in the FTO gene may be attributable to adiposity through the impairment of leptin receptor trafficking to the cilia in hypothalamic neurons.
Loss of cilia in neurons, especially in POMC neurons, by depleting the intraflagellar transport protein 88 homolog (IFT88) gene leads to hyperphagic obesity and hyperleptinemia [128]. Likewise, the intrahypothalamic injection of small inhibitory RNAs targeting kinesin-2 subunit KIF3A and IFT88 significantly reduces the effects of exogenous leptin on food intake and hypothalamic STAT3 phosphorylation [129]. Mice lacking the type 3 adenylyl cyclase (AC3), used in the immunostaining of neuronal cilia, were found to be obese and hyperleptinemic [130]. All of these data suggest that neuronal cilia and ciliary proteins may be required for leptin signal transduction and/or the maintenance of an energy balance. In contrast, a recent study has reported that IFT88- and BBS4-knockout mice normally respond to leptin under preobese conditions [131], suggesting a dispensable role of the cilia in hypothalamic leptin signaling. Further studies are needed to clarify how cilia influence leptin signaling processes.
On the other hand, leptin regulates the cilia lengths in hypothalamic neurons. The average lengths of hypothalamic cilia are shorter in Lep ob and Lepr db mice compared to normal mice [129]. In line with this, a 7 day-leptin treatment of Lep ob mice rescued the short cilia phenotype. The control of hypothalamic neuron cilia lengths by leptin was found to be mediated through PTEN–GSK3β signaling which involves ciliary gene transcription and actin cytoskeleton rearrangement [132]. Thus it is plausible that leptin modulates its own signaling by promoting cilia growth in hypothalamic neurons.
Signaling pathways that inhibit the actions of leptin (Fig. 4)
The discovery of leptin generated considerable attention and expectations for its putative use as a therapeutic agent against human obesity. However, obese individuals were found to be refractory to leptin therapy in clinical trials [133, 134]. In fact, obese individuals generally exhibit elevated circulating leptin concentrations in proportion to their increased body fat mass [135]. Thus, an understanding of why an increased leptin level under obese conditions does not yield the expected effects is essential for any future strategies to combat human obesity in this way.

Signaling molecules which negatively regulate leptin signaling. Several signaling pathways are activated in the hypothalamus of DIO mice and attenuate signaling cascades downstream from the leptin receptors via interactions with JAK2, STAT3, IRS2, and PI3K. IKKβ IκB kinase-β, NFκB nuclear factor-κB, PKC-θ protein kinase-θ, PPARγ peroxisome proliferator activated transcript-γ, PTP1B protein tyrosine phosphatase 1B, SOCS3 suppressor of cytokine signaling 3
The number of publications per year citing ‘leptin resistance’ has been growing steadily since this hormone was discovered [136]. In clinical settings, the term “leptin resistance” can be defined literally as the inability of exogenous leptin to promote desired outcomes including reduced appetite and body weight. In contrast, in many other situations, the meaning of this term is considered to be hyperleptinemia accompanied by obesity, which reflects expanded adipose tissue [136].
Leptin resistance emanates from reduced leptin transport into the brain or defects in LepRb signaling cascades in the hypothalamic neurons. Leptin-induced activation of hypothalamic neurons, including ARC, ventromedial hypothalamus (VMH), and lateral hypothalamic areas, can be traced by immunohistochemical analysis of phosphorylated STAT3 [137]. Upon administration of a pharmacological dose of leptin, diet-induced obesity (DIO) rodents show a limited amplitude of leptin signaling in the hypothalamus, as evidenced by reduced STAT3 phosphorylation compared with lean animals [138]. Putative negative regulators of leptin receptor signaling are described below.
SOCS3
In the hypothalamic neurons, activated STAT3 in response to leptin signaling induces SOCS3 expression, which in turn yields a negative feedback loop by binding to the LepRb–JAK2 complex and thereby attenuating leptin signaling [139]. Since hypothalamic SOCS3 expression was found to be elevated in leptin-resistant obese yellow (A y/a) mice, an increased SOCS3 level was the first proposed mechanism of leptin resistance [139]. A role of SOCS as a negative regulator of leptin signaling was confirmed by the fact that obesity and leptin resistance upon high-fat diet (HFD) feeding are less severe in SOCS3 heterozygous knockout mice as well as in mice with neuronal SOCS3 deletion [140–142]. Consistently, increased SOCS3 expression in POMC neurons led to impaired STAT3 signaling, leptin resistance and obesity. However, SOCS3 overexpression in LepR-expressing neurons did not cause obesity, suggesting that the impacts of SOCS3 signaling on energy balance may be neuron type-specific [143].
ARC SOCS3 expression increases from 1 week after HFD feeding, ahead of the establishment of DIO [53, 144, 145]. During the development of DIO, SOCS3 activation in AgRP neurons precedes that in POMC- and other hypothalamic neurons [146]. From these findings, it appears that the HFD-induced increase of SOCS3 may have a distinct temporal and spatial pattern and AgRP neurons may be the predominant early responders to subtle changes in leptin signaling upon short-term HFD feeding [146].
A recent paper has shown that the central and peripheral administration of high-dose leptin receptor antagonist induces comparable changes in food intake, body weight, and hypothalamic POMC and SOCS3 mRNA levels in lean and obese mice [53, 144, 145]. Thus endogenous leptin signaling may not be much reduced in obese mice on a HFD, which challenges the previous concept of leptin resistance under obese conditions. Instead, elevated hypothalamic SOCS3 expression in DIO mice may limit additional activation of LepRb signaling by exogenous leptin [147].
PTP1B and other tyrosine phosphatases
Protein tyrosine phosphatase-1B (PTP1B) is a cytoplasmic enzyme predominantly localized in the endoplasmic reticulum (ER) [148]. PTP1B is expressed in hypothalamic areas such as the ARC, VMH, and dorsomedial hypothalamus (DMH) [149]. Like SOCS3, PTP1B inhibits LepRb signaling by targeting JAK2. PTP1B binds to and dephosphorylates JAK2, thereby attenuating STAT3 tyrosine phosphorylation in the hypothalamus [149–151]. PTP1B also negatively regulates hypothalamic leptin signaling via α2-AMPK [152]. Consistent with these mechanisms, mice with a mutation or depletion of PTP1B in hypothalamic neurons are leaner, more sensitive to leptin and resistant to DIO and display improved glucose homeostasis [138, 152–156], supporting an antagonistic effect of PTP1B in hypothalamic leptin signaling. The impact of PTP1B on energy balance appears to rely on hypothalamic LepR signaling as hypothalamic PTP1B depletion leads to reduced adiposity, an effect that is abolished by the concomitant hypothalamic deletion of LepR [157].
Previous studies have revealed increased PTP1B expression in the MBH of rodent DIO models [156, 158, 159] although one study has reported an unaltered hypothalamic PTP1B level during HFD feeding [144]. Aging is also related to elevated MBH PTP1B expression [160]. Hyperleptinemia may contribute to an obesity-associated increase in hypothalamic PTP1B as chronic leptin treatment significantly increases the hypothalamic PTP1B mRNA level [155, 159].
Several molecules are involved in hypothalamic PTP1B signaling. Coexpression of PTP1B with SH2B1 blocks the inhibitory effect of PTP1B on leptin-induced STAT3 phosphorylation [71]. LIM domain Only 4 (LMO4), a transcription cofactor involved in STAT3 activation in VMH neurons [161, 162], downregulates PTP1B activity by augmenting the PTP1B inactive form via oxidation [163]. There have been trials to treat obesity and leptin resistance by inhibiting PTP1B. Trodusquemine, a spermine metabolite of cholesterol, and its polyaminosteroid derivative claramine, cause a loss of fat mass in DIO mice by inhibiting PTP1B [164, 165].
Other PTPs may also participate in leptin receptor signaling. Tyrosine phosphatase ε (RPTPε) dephosphorylates JAK2 and thus downregulates leptin receptor signaling in the hypothalamus. Leptin induces the phosphorylation and activation of RPTPε at its C-terminal Y695 residue, which constitutes a negative feedback pathway of leptin signaling. Increased RPTPε activity is found in the hypothalamus of obese, leptin-resistant mice [166]. Moreover, mice lacking RPTPε are leptin-sensitive and protected from obesity induced by HFD, aging, and ovariectomy [166]. Another PTP variant, T cell PTP (TCPTP), is elevated in the hypothalamus of DIO mice where it negatively modulates leptin-induced STAT3 signaling. Genetic ablation or pharmacologic suppression of neuronal TCPTP enhances leptin sensitivity [167]. Other PTPs such as SHP2 or PTEN may also have regulatory roles in leptin receptor signaling. Further studies are needed to elucidate the activities of these PTPs in LepRb signaling [168].
IKK–NFκB signaling
De Souza et al. reported for the first time that a long-term high fat diet increases the hypothalamic expression of proinflammatory cytokines such as interleukin-1 (IL-1), IL-6 and tumor necrosis factor-α (TNFα) and activates the inflammatory signaling pathways c-Jun N-terminal kinase (JNK) and nuclear factor-κB (NF-κB) in the rat hypothalamus. These authors suggested this phenomenon as a mechanism of hypothalamic insulin resistance [169]. Thereafter, the cumulative evidence has indicated that activated proinflammatory signaling in the hypothalamus is an important mechanism underlying HFD-induced leptin resistance. Notably, only 1 day on a HFD induced hypothalamic inflammation in rodents [170]. The key mediators of inflammation/innate immunity, IκB kinase-β (IKKβ) and NFκB, are highly expressed in MBH neurons [171]. Activation of hypothalamic IKKβ–NFκB signaling is observed upon exposure to 1 day of HFD or following a single ICV administration of glucose or fatty acid [170–172]. Thus, HFD-induced hypothalamic inflammation may be an early event in the development of overnutrition-induced leptin resistance.
Artificial activation of hypothalamic IKKβ–NFκB signaling by viral mediated IKKβ overexpression in the MBH increases food intake and promotes weight gain along with impaired leptin signaling. Conversely, the neuronal knockout of IKKβ or the virus-mediated deletion of IKKβ in MBH attenuates the development of DIO [171, 173, 174]. These results demonstrate a causative role of IKKβ–NFκB signaling in leptin resistance in DIO mice. Interestingly, the metabolic impacts resulting from activated IKKβ–NFκB signaling in POMC and AgRP neurons appear to be opposing. Inactivation of IKKβ–NFκB signaling in the AgRP neurons protects against obesity, suggesting the obesity promoting effect of NFκB signaling in AgRP neurons [171]. However, a recent study has demonstrated that activation of IKKβ signaling in AgRP neurons fails to cause obesity and leptin resistance but instead disrupts insulin signaling in AgRP neurons and impairs systemic glucose homeostasis [175]. In contrast, NFκB signaling in POMC neurons is activated by leptin and mediates its anorexigenic effects [176]. NFκB binding sites within the POMC promoter are hypermethylated in mice with HFD-induced obesity, which limits the ability of leptin to increase POMC expression [177].
In the hypothalamus, activation of IKKβ–NFκB signaling induced by overnutrition leads to increased expression of proinflammatory cytokines such as TNFα, IL-1β, and IL-6 [169, 178, 179]. In support of a role of hypothalamic proinflammatory cytokines in overnutrition induced leptin resistance, the lack of TNF receptor-1 or central infusion of TNFα neutralizing antibody was found to mitigate leptin resistance under HFD conditions [180, 181]. Consistently, ICV coinjection of TNFα partially blocks the anorexigenic effect of leptin through the inhibition of PI3K–Akt–FoxO1 signaling [182]. On the other hand, central infusion of recombinant IL-6 increases hypothalamic STAT3 phosphorylation. Furthermore, IL-6 treatment of DIO mice suppresses hyperphagia and inhibits hypothalamic IKKβ activation [183], suggesting beneficial metabolic effects of IL-6. Interestingly, injection of leptin increases the IL-1β level in the hypothalamus of normal Sprague–Dawley rats [184]. A central injection of IL-1 receptor antagonist inhibits the satiety effect caused by central or peripheral leptin injection and abolishes the leptin-induced increase in body temperature. Mice lacking IL-1 receptor show no reduction in food intake in response to leptin, suggesting that IL-1β acts as an important downstream mediator of hypothalamic leptin actions [184]. Thus, proinflammatory cytokines may have differential roles in hypothalamic leptin signaling [170, 185, 186].
Several molecules engaged in proinflammatory signaling pathway have been implicated in hypothalamic inflammation and leptin resistance. First, the Toll-like receptors (TLRs) are membrane-bound pattern recognition receptors, which play an important role in innate immune defense [187]. Saturated fatty acids serve as a naturally occurring ligand for TLR4, which mediates the fatty acid-induced activation of IKKβ-NFκB signaling [188]. Hypothalamic TLR4 expression and its downstream signaling activity are increased by both the acute ICV infusion of saturated fatty acids and chronic HFD feeding [178, 179]. Furthermore, inhibition and a loss-of-function mutation of TLR4 protects mice from DIO and reduces hypothalamic inflammation [179, 181]. These results demonstrate a critical role of TLR4 in diet-induced hypothalamic inflammation. As hypothalamic TLR4 expression is mostly found in activated microglia [179], interactions between microglia and hypothalamic neurons may be crucial for the progression of hypothalamic inflammation. Second, the myeloid-differentiation primary-response gene 88 (MyD88) is an essential signaling adaptor molecule that couples TLR4 to inflammatory signaling cascades [189]. Similarly to TLR4, hypothalamic MyD88 expression is stimulated by the ICV administration of saturated fatty acids [190]. Mice lacking MyD88 in the CNS are more sensitive to leptin upon acute and chronic exposure to fatty acids, indicating a role of MyD88 in the development of central leptin resistance [190]. Third, IKKε is a downstream molecule of NFκB signaling. Inhibition of IKKε activity reduces leptin resistance by restoring JAK2–STAT3 and IRS–PI3K–Akt signaling in the hypothalamus of HFD-fed mice [191].
Interestingly, endotoxin lipopolysaccharides (LPS) and leptin share a common signaling pathway in the hypothalamus. Similar to leptin, a single intraperitoneal injection of LPS increases hypothalamic STAT3 phosphorylation and POMC expression but decreases hypothalamic AMPK activity, thereby inducing a hypophagic response [192, 193]. Moreover, exposure to LPS acutely provokes an excitatory tone in most POMC neurons and an inhibitory tone in AgRP/NPY neurons. Blockade of the TLR4 receptor in microglia reduces the inhibitory effect of LPS on AgRP/NPY neurons, indicating an indirect and inhibitory regulation by LPS in these cells [194]. Repeated injection of LPS leads to a reduced response to LPS, implying the development of LPS resistance. Moreover, leptin-resistant DIO rats show a blunted hypophagic response to LPS whilst mice receiving a repeated LPS injection show reduced leptin sensitivity [195]. These findings indicate that the mechanisms underlying hypothalamic resistance to endotoxin largely overlap with those of leptin resistance.
ER stress–mitochondrial signaling
The rough endoplasmic reticulum is the cellular organelle responsible for the correct folding and sorting of eukaryotic proteins undergoing maturation [196]. A chronic failure of this maturation causes the accumulation of degradation-resistant misfolded proteins, resulting in ER stress. The adaptive mechanism that can resolve ER stress is termed the unfolded protein response (UPR) [197]. ER stress was initially identified as a central feature of insulin resistance in peripheral tissues [198], and later suggested as a possible link between overnutrition and hypothalamic leptin resistance [171]. The expression level of UPR signaling molecules such as PKR-like ER kinase (PERK), inositol-requiring-1 (IRE1), spliced form of X-box-binding protein-1 (XBP-1s), glucose-regulated/binding immunoglobulin protein-78 (GPR78) and C/EBP homology protein (CHOP) are increased in the hypothalamus of DIO models [171, 179, 183, 199, 200]. Hypothalamic UPR signaling is induced by the short-term ICV infusion of fatty acids [179]. In addition to DIO, ER stress is related to genetic human obesity caused by melanocortin-4 receptor (MC4R) gene variants. Obesity-associated MC4R variants are retained in the ER and cause ER stress, which results in impaired MC4R signaling and obesity. Pharmacological chaperone 4-phenyl butyric acid promotes the location of MC4R to the cell surface and subsequent MC4R signaling [201, 202].
Evidence for the contribution of ER stress to central leptin resistance comes from the finding that the prior ICV injection of the ER stress inducer thapsigargin inhibits the activation of hypothalamic STAT3 and Akt by leptin and insulin [200]. Consistently, ER stress inducers suppress the leptin-induced phosphorylation of STAT3 in the ARC of hypothalamic slice preparations [203]. In addition, the central administration of chemical chaperones or ER stress inhibitors improves leptin sensitivity and alleviates weight gain in the DIO animal models [171, 199, 200].
HFD-induced hypothalamic ER stress may be caused by ectopic accumulation of lipids in the hypothalamic neurons (termed ‘lipotoxicity’) [204]. HFD induces the accumulation of palmitoyl- and stearoyl-CoA in the rat hypothalamus [172]. Treatment of hypothalamic neurons with palmitate causes ER stress which leads to reduced MC4R protein expression and a profound loss of MC4R signaling in response to anorexigenic melanocortin α-MSH [205]. As the hypothalamic melanocortin system is an important mediator of leptin actions, ER stress-induced impairment of hypothalamic melanocortin signaling may underlie the reduced hypothalamic response to leptin in animals on a palmitate-rich HFD.
Several mechanisms are suggested to bridge HFD-induced ER stress to impaired leptin receptor signaling. ER stress may lead to the activation of IKKβ–NFκB signaling in the hypothalamus [171]. Overnutrition has been shown to activate IKKβ–NFκB signaling in the hypothalamus through ER stress responses [171]. Conversely, induction of hypothalamic ER stress by HFD is abrogated by the inhibition of NFκB signaling [206], which implies a positive feedback loop between NFκB signaling and ER stress [171]. Peroxisomes are generated from the domain in the ER under increased metabolic pressure [207]. The peroxisome proliferator-activated receptor-γ (PPARγ) is a nuclear receptor that senses the flux of increased lipids and regulates the transcription of genes involved in glucose and lipid metabolism [208]. PPARγ agonist stimulates the formation of peroxisomes in hypothalamic neurons [209]. Hypothalamic activation of PPARγ causes hyperphagia and weight gain. By contrast, a blockade of hypothalamic PPARγ activity leads to a negative energy balance and improved leptin resistance in HFD-fed rats [209]. In POMC neurons, PPARγ reduces the level of reactive oxygen species (ROS), that are a critical signal that stimulates POMC neuronal activity, via peroxisome proliferation and thus results in reduced neuronal activity [210, 211].
It was recently suggested that disruption of the mitochondrial-ER interaction may be a key element in leptin resistance in POMC neurons. Mitochondrial-ER contacts are decreased in the anorexigenic POMC neurons of DIO mice [212]. Furthermore, deletion of PPARγ in POMC neurons enhances mitochondrial-ER interactions and increases leptin sensitivity in these neurons [211]. Mitofusin-2 is a mitochondrial transmembrane GTPase protein that mediates mitochondrial-ER contacts. Mice lacking mitofusin-2 in POMC neurons display a loss of these interactions, defective POMC processing, ER stress-induced leptin resistance, hyperphagia, reduced energy expenditure, and obesity. In these mice, the pharmacological reduction of hypothalamic ER stress rescues these metabolic defects. In contrast, the ablation of mitofusin-2 and mitofusin-1 in AgRP neurons disrupts mitochondrial fusion without inducing ER stress and alleviates HFD-induced obesity [213]. These findings indicate the importance of mitochondrial dynamics in AgRP neurons during the establishment of DIO.
On the other hand, certain UPS signaling pathways may have a protective role against overnutrition-induced leptin resistance. X-box binding protein 1 (XBP1) is an UPR transcription factor under the control of the IRE1 pathway and is processed to an active form by splicing upon ER stress [214]. The neuron-specific deletion of XBP1 increases the susceptibility to leptin resistance [199] whereas constitutive expression of XBP1 in POMC neurons improves leptin sensitivity by suppressing PTP1B and SOCS3 [203]. These results demonstrate the beneficial metabolic effects of XBP1 under conditions of overnutrition.
Miscellaneous
Several other signaling molecules have been shown to regulate leptin sensitivity in the hypothalamus. The JNK-signaling pathway plays a crucial role in the development of obesity and insulin resistance [215]. JNK1 disrupts insulin signaling via the serine phosphorylation of IRS-1 in peripheral organs [215]. JNK1 expression is elevated in the hypothalamus of DIO mice, which may result from lipotoxic stress and low grade inflammation [216, 217]. The neuronal ablation of JNK1 in mice prevents HFD-induced obesity and increases energy expenditure and insulin sensitivity, indicating a deleterious impact of neuronal JNK1 on leptin signaling and energy balance. These effects have been associated with the activation of a hypothalamo-pituitary-thyroid axis [216]. In line with this, the acute ICV injection of JNK inhibitor into Lep ob mice rescues impaired hypothalamic insulin signaling and glucose intolerance [217]. Recently, it was also demonstrated that AgRP neuron-specific JNK activation increases the spontaneous firing of AgRP neurons and causes leptin resistance, hyperphagia and obesity [175].
A member of a novel class of PKC serine–threonine kinases, protein kinase-θ (PKC-θ), is activated by lipid metabolite diacylglycerol (DAG) and inhibits insulin-induced IRS1-tyrosine phosphorylation and PI3K activation in peripheral tissues [218]. PKC-θ is expressed in discrete neuronal populations including the ARC NPY/AgRP neurons and the neurons of the hypothalamic DMH [219]. The systemic and central administration of palmitic acid increases the hypothalamic DAG level and promotes the membrane localization of PKC-θ in the hypothalamus, a marker of PKC-θ activation and of dampened hypothalamic insulin and leptin signaling. The knockdown of PKC-θ in hypothalamic ARC attenuates DIO and improves HFD-induced glucose intolerance. These results have suggested that the activation of hypothalamic PKC-θ may mediate the deleterious effects of a HFD on hypothalamic insulin and leptin signaling.
Elevation of the adenosine 3′, 5′-monophosphate (cAMP) level impairs leptin-activated STAT3 and S6K signaling cascades via the induction of SOCS3 and PTP1B in organotypic hypothalamic slices [220]. This effect is mediated by Epac, a cAMP-regulated guanine nucleotide exchange factor for the small G protein Rap1 [221]. Hypothalamic Epac activation blunts the leptin-induced depolarization of hypothalamic POMC neurons and attenuates the anorectic responses to exogenous leptin. As hypothalamic Rap1 activity is enhanced in the hypothalamus of DIO mice, cAMP–Epac–Rap1 signaling may contribute to HFD-induced leptin resistance.
Closing remarks
In the last two decades since the discovery of leptin, numerous studies have demonstrated how this hormone functions in the hypothalamic neurons to maintain energy homeostasis. To explore signaling pathways that transduce the effects leptin in hypothalamic neurons, different research groups have used animal models lacking critical signaling molecules in these cells. The results obtained using this approach should be carefully interpreted however as deletion of essential signaling components in hypothalamic neurons may lead to an energy imbalance phenotype without affecting leptin signaling. It is also possible that abnormal metabolic phenotypes in genetically engineered animal models are the result of developmental defects in the neuronal circuits regulating energy balance.
Meanwhile, as obese humans and mice exhibit elevated levels of leptin in the systemic circulation and reduced responses to exogenous leptin, the molecular mechanisms underlying leptin resistance in hypothalamic neurons have been a major area of obesity research. To date, multiple negative regulatory pathways of leptin receptor signaling in the hypothalamus have been identified and are reviewed herein. However, recent studies using a leptin receptor antagonist have reported that endogenous leptin receptor signaling in the hypothalamus in DIO mice is comparable to lean mice, arguing against the concept of ‘leptin resistance’ in DIO [88, 145]. Future studies will be required to address whether DIO mice have a reduced ability to respond to endogenous leptin and to reveal why obesity develops in subjects or animals with fully functioning leptin signaling pathways.
Several issues in hypothalamic leptin signaling pathways also need to be addressed in future studies. There is no doubt that LepRb is a primary receptor responsible for the metabolic and neuro-hormonal effects of leptin. However, it is possible that other receptors (for example, LRP proteins) or signaling molecules on the cellular surface may interact with leptin to evoke downstream signaling cascades that amplify the classical signaling cascades mediated through LepRb. Approaches to the discovery of novel receptors of leptin may help to develop therapeutic strategies to modulate leptin signaling. Recent evidence links classical leptin signaling to cilia signaling. Future studies are needed to elucidate where and how these signaling pathways are connected. Although POMC and AgRP neurons in the hypothalamic ARC are known as a major target of leptin, the deletion of LepRb in other neurons such as GABAnergic neurons and neuronal nitric oxide synthase (NOS1)-expressing neurons recapitulates the obese phenotype seen in LepRb-null mice [222, 223]. Moreover, recent studies have shown that leptin acts on astrocytes to indirectly regulate hypothalamic neuronal activity [224, 225]. Identifying new cellular targets for hypothalamic leptin functions may greatly contribute to our understanding of the functional networks between hypothalamic neurons, or between neurons and glial cells or cells of the innate immune system within the hypothalamus, that control the energy balance.
References
Schwartz MW, Woods SC, Porte D Jr, Seeley RJ, Baskin DG (2000) Central nervous system control of food intake. Nature 404:661–671
Ingalls AM, Dickie MM, Snell GD (1950) Obese, a new mutation in the house mouse. J Hered 41:317–318
Hummel KP, Dickie MM, Coleman DL (1966) Diabetes, a new mutation in the mouse. Science 153:1127–1128
Coleman DL (1973) Effects of parabiosis of obese with diabetes and normal mice. Diabetologia 9:294–298
Coleman DL, Hummel KP (1969) Effects of parabiosis of normal with genetically diabetic mice. Am J Physiol 217:1298–1304
Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM (1994) Positional cloning of the mouse obese gene and its human homologue. Nature 372:425–432
Farooqi IS, Jebb SA, Langmack G, Lawrence E, Cheetham CH, Prentice AM, Hughes IA, McCamish MA, O’Rahilly S (1999) Effects of recombinant leptin therapy in a child with congenital leptin deficiency. N Engl J Med 341:879–884
Harris RB, Zhou J, Redmann SM Jr, Smagin GN, Smith SR, Rodgers E, Zachwieja JJ (1998) A leptin dose–response study in obese (ob/ob) and lean (+/?) mice. Endocrinology 139:8–19
Cohen P, Zhao C, Cai X, Montez JM, Rohani SC, Feinstein P, Mombaerts P, Friedman JM (2001) Selective deletion of leptin receptor in neurons leads to obesity. J Clin Invest 108:1113–1121
de Luca C, Kowalski TJ, Zhang Y, Elmquist JK, Lee C, Kilimann MW, Ludwig T, Liu SM, Chua SC Jr (2005) Complete rescue of obesity, diabetes, and infertility in db/db mice by neuron-specific LEPR-B transgenes. J Clin Invest 115:3484–3493
Tadokoro S, Ide S, Tokuyama R, Umeki H, Tatehara S, Kataoka S, Satomura K (2015) Leptin promotes wound healing in the skin. PLoS One 10:e0121242
Huynh FK, Levi J, Denroche HC, Gray SL, Voshol PJ, Neumann UH, Speck M, Chua SC, Covey SD, Kieffer TJ (2010) Disruption of hepatic leptin signaling protects mice from age- and diet-related glucose intolerance. Diabetes 59:3032–3040
Marroqui L, Gonzalez A, Neco P, Caballero-Garrido E, Vieira E, Ripoll C, Nadal A, Quesada I (2012) Role of leptin in the pancreatic beta-cell: effects and signaling pathways. J Mol Endocrinol 49:R9–17
Hoggard N, Mercer JG, Rayner DV, Moar K, Trayhurn P, Williams LM (1997) Localization of leptin receptor mRNA splice variants in murine peripheral tissues by RT-PCR and in situ hybridization. Biochem Biophys Res Commun 232:383–387
Bado A, Levasseur S, Attoub S, Kermorgant S, Laigneau JP, Bortoluzzi MN, Moizo L, Lehy T, Guerre-Millo M, Le Marchand-Brustel Y, Lewin MJ (1998) The stomach is a source of leptin. Nature 394:790–793
Wang J, Liu R, Hawkins M, Barzilai N, Rossetti L (1998) A nutrient-sensing pathway regulates leptin gene expression in muscle and fat. Nature 393:684–688
Dessolin S, Schalling M, Champigny O, Lonnqvist F, Ailhaud G, Dani C, Ricquier D (1997) Leptin gene is expressed in rat brown adipose tissue at birth. FASEB J 11:382–387
Cioffi JA, Van Blerkom J, Antczak M, Shafer A, Wittmer S, Snodgrass HR (1997) The expression of leptin and its receptors in pre-ovulatory human follicles. Mol Hum Reprod 3:467–472
Masuzaki H, Ogawa Y, Sagawa N, Hosoda K, Matsumoto T, Mise H, Nishimura H, Yoshimasa Y, Tanaka I, Mori T, Nakao K (1997) Nonadipose tissue production of leptin: leptin as a novel placenta-derived hormone in humans. Nat Med 3:1029–1033
Jin L, Burguera BG, Couce ME, Scheithauer BW, Lamsan J, Eberhardt NL, Kulig E, Lloyd RV (1999) Leptin and leptin receptor expression in normal and neoplastic human pituitary: evidence of a regulatory role for leptin on pituitary cell proliferation. J Clin Endocrinol Metab 84:2903–2911
Wilkinson M, Brown R, Imran SA, Ur E (2007) Adipokine gene expression in brain and pituitary gland. Neuroendocrinology 86:191–209
Roubos EW, Dahmen M, Kozicz T, Xu L (2012) Leptin and the hypothalamo-pituitary-adrenal stress axis. Gen Comp Endocrinol 177:28–36
Matarese G, Moschos S, Mantzoros CS (2005) Leptin in immunology. J Immunol 174:3137–3142
Chan JL, Mantzoros CS (2001) Leptin and the hypothalamic-pituitary regulation of the gonadotropin-gonadal axis. Pituitary 4:87–92
Houseknecht KL, Portocarrero CP (1998) Leptin and its receptors: regulators of whole-body energy homeostasis. Domest Anim Endocrinol 15:457–475
Gotoda T, Manning BS, Goldstone AP, Imrie H, Evans AL, Strosberg AD, McKeigue PM, Scott J, Aitman TJ (1997) Leptin receptor gene variation and obesity: lack of association in a white British male population. Hum Mol Genet 6:869–876
Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J, Devos R, Richards GJ, Campfield LA, Clark FT, Deeds J, Muir C, Sanker S, Moriarty A, Moore KJ, Smutko JS, Mays GG, Wool EA, Monroe CA, Tepper RI (1995) Identification and expression cloning of a leptin receptor, OB-R. Cell 83:1263–1271
Tartaglia LA (1997) The leptin receptor. J Biol Chem 272:6093–6096
Devos R, Richards JG, Campfield LA, Tartaglia LA, Guisez Y, van der Heyden J, Travernier J, Plaetinck G, Burn P (1996) OB protein binds specifically to the choroid plexus of mice and rats. Proc Natl Acad Sci USA 93:5668–5673
Friedman JM (1998) Leptin, leptin receptors, and the control of body weight. Nutr Rev 56:S38–S46
Munzberg H, Bjornholm M, Bates SH, Myers MG Jr (2005) Leptin receptor action and mechanisms of leptin resistance. Cell Mol Life Sci 62:642–652
Bates SH, Myers MG (2004) The role of leptin → STAT3 signaling in neuroendocrine function: an integrative perspective. J Mol Med (Berl) 82:12–20
Maamra M, Bidlingmaier M, Postel-Vinay MC, Wu Z, Strasburger CJ, Ross RJ (2001) Generation of human soluble leptin receptor by proteolytic cleavage of membrane-anchored receptors. Endocrinology 142:4389–4393
Baumann H, Morella KK, White DW, Dembski M, Bailon PS, Kim HK, Lai CF, Tartaglia LA (1996) The full-length leptin receptor has signaling capabilities of interleukin 6-type cytokine receptors. Proc Natl Acad Sci USA 93:8374–8378
Rosenblum CI, Tota M, Cully D, Smith T, Collum R, Qureshi S, Hess JF, Phillips MS, Hey PJ, Vongs A, Fong TM, Xu L, Chen HY, Smith RG, Schindler C, Van der Ploeg LH (1996) Functional STAT 1 and 3 signaling by the leptin receptor (OB-R); reduced expression of the rat fatty leptin receptor in transfected cells. Endocrinology 137:5178–5181
Stahl N, Yancopoulos GD (1993) The alphas, betas, and kinases of cytokine receptor complexes. Cell 74:587–590
Schindler C, Darnell JE Jr (1995) Transcriptional responses to polypeptide ligands: the JAK-STAT pathway. Annu Rev Biochem 64:621–651
Ihle JN (1996) STATs: signal transducers and activators of transcription. Cell 84:331–334
Kelesidis T, Kelesidis I, Chou S, Mantzoros CS (2010) Narrative review: the role of leptin in human physiology: emerging clinical applications. Ann Intern Med 152:93–100
Fong TM, Huang RR, Tota MR, Mao C, Smith T, Varnerin J, Karpitskiy VV, Krause JE, Van der Ploeg LH (1998) Localization of leptin binding domain in the leptin receptor. Mol Pharmacol 53:234–240
Leinninger GM, Myers MG Jr (2008) LRb signals act within a distributed network of leptin-responsive neurones to mediate leptin action. Acta Physiol (Oxf) 192:49–59
Murakami T, Yamashita T, Iida M, Kuwajima M, Shima K (1997) A short form of leptin receptor performs signal transduction. Biochem Biophys Res Commun 231:26–29
Li Z, Ceccarini G, Eisenstein M, Tan K, Friedman JM (2013) Phenotypic effects of an induced mutation of the ObRa isoform of the leptin receptor. Mol Metab 2:364–375
Dam J, Jockers R (2013) Hunting for the functions of short leptin receptor isoforms. Mol Metab 2:327–328
Akira S (1999) Functional roles of STAT family proteins: lessons from knockout mice. Stem Cells 17:138–146
Ghilardi N, Ziegler S, Wiestner A, Stoffel R, Heim MH, Skoda RC (1996) Defective STAT signaling by the leptin receptor in diabetic mice. Proc Natl Acad Sci USA 93:6231–6235
Gong Y, Ishida-Takahashi R, Villanueva EC, Fingar DC, Munzberg H, Myers MG Jr (2007) The long form of the leptin receptor regulates STAT5 and ribosomal protein S6 via alternate mechanisms. J Biol Chem 282:31019–31027
Vaisse C, Halaas JL, Horvath CM, Darnell JE Jr, Stoffel M, Friedman JM (1996) Leptin activation of Stat3 in the hypothalamus of wild-type and ob/ob mice but not db/db mice. Nat Genet 14:95–97
Kim KW, Zhao L, Donato J Jr, Kohno D, Xu Y, Elias CF, Lee C, Parker KL, Elmquist JK (2011) Steroidogenic factor 1 directs programs regulating diet-induced thermogenesis and leptin action in the ventral medial hypothalamic nucleus. Proc Natl Acad Sci USA 108:10673–10678
Bates SH, Stearns WH, Dundon TA, Schubert M, Tso AWK, Wang YP, Banks AS, Lavery HJ, Haq AK, Maratos-Flier E, Neel BG, Schwartz MW, Myers MG (2003) STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature 421:856–859
Gao Q, Wolfgang MJ, Neschen S, Morino K, Horvath TL, Shulman GI, Fu XY (2004) Disruption of neural signal transducer and activator of transcription 3 causes obesity, diabetes, infertility, and thermal dysregulation. Proc Natl Acad Sci USA 101:4661–4666
Scarpace PJ, Zhang Y (2009) Leptin resistance: a prediposing factor for diet-induced obesity. Am J Physiol Regul Integr Comp Physiol 296:R493–R500
Enriori PJ, Evans AE, Sinnayah P, Jobst EE, Tonelli-Lemos L, Billes SK, Glavas MM, Grayson BE, Perello M, Nillni EA, Grove KL, Cowley MA (2007) Diet-induced obesity causes severe but reversible leptin resistance in arcuate melanocortin neurons. Cell Metab 5:181–194
Munzberg H, Huo L, Nillni EA, Hollenberg AN, Bjorbaek C (2003) Role of signal transducer and activator of transcription 3 in regulation of hypothalamic proopiomelanocortin gene expression by leptin. Endocrinology 144:2121–2131
Varela L, Horvath TL (2012) Leptin and insulin pathways in POMC and AgRP neurons that modulate energy balance and glucose homeostasis. EMBO Rep 13:1079–1086
Joly-Amado A, Denis RG, Castel J, Lacombe A, Cansell C, Rouch C, Kassis N, Dairou J, Cani PD, Ventura-Clapier R, Prola A, Flamment M, Foufelle F, Magnan C, Luquet S (2012) Hypothalamic AgRP-neurons control peripheral substrate utilization and nutrient partitioning. EMBO J 31:4276–4288
Banks AS, Davis SM, Bates SH, Myers MG Jr (2000) Activation of downstream signals by the long form of the leptin receptor. J Biol Chem 275:14563–14572
Starr R, Willson TA, Viney EM, Murray LJ, Rayner JR, Jenkins BJ, Gonda TJ, Alexander WS, Metcalf D, Nicola NA, Hilton DJ (1997) A family of cytokine-inducible inhibitors of signalling. Nature 387:917–921
Auernhammer CJ, Bousquet C, Melmed S (1999) Autoregulation of pituitary corticotroph SOCS-3 expression: characterization of the murine SOCS-3 promoter. Proc Natl Acad Sci USA 96:6964–6969
Bjorbaek C, El-Haschimi K, Frantz JD, Flier JS (1999) The role of SOCS-3 in leptin signaling and leptin resistance. J Biol Chem 274:30059–30065
May P, Woldt E, Matz RL, Boucher P (2007) The LDL receptor-related protein (LRP) family: an old family of proteins with new physiological functions. Ann Med 39:219–228
Liu Q, Zhang J, Zerbinatti C, Zhan Y, Kolber BJ, Herz J, Muglia LJ, Bu G (2011) Lipoprotein receptor LRP1 regulates leptin signaling and energy homeostasis in the adult central nervous system. PLoS Biol 9:e1000575
Ceccarini G, Flavell RR, Butelman ER, Synan M, Willnow TE, Bar-Dagan M, Goldsmith SJ, Kreek MJ, Kothari P, Vallabhajosula S, Muir TW, Friedman JM (2009) PET imaging of leptin biodistribution and metabolism in rodents and primates. Cell Metab 10:148–159
Hama H, Saito A, Takeda T, Tanuma A, Xie Y, Sato K, Kazama JJ, Gejyo F (2004) Evidence indicating that renal tubular metabolism of leptin is mediated by megalin but not by the leptin receptors. Endocrinology 145:3935–3940
Dietrich MO, Spuch C, Antequera D, Rodal I, de Yebenes JG, Molina JA, Bermejo F, Carro E (2008) Megalin mediates the transport of leptin across the blood–CSF barrier. Neurobiol Aging 29:902–912
Gil SY, Youn BS, Byun K, Huang H, Namkoong C, Jang PG, Lee JY, Jo YH, Kang GM, Kim HK, Shin MS, Pietrzik CU, Lee B, Kim YB, Kim MS (2013) Clusterin and LRP2 are critical components of the hypothalamic feeding regulatory pathway. Nat Commun 4:1862
Byun K, Gil SY, Namkoong C, Youn BS, Huang H, Shin MS, Kang GM, Kim HK, Lee B, Kim YB, Kim MS (2014) Clusterin/ApoJ enhances central leptin signaling through Lrp2-mediated endocytosis. EMBO Rep 15:801–808
Niswender KD, Morton GJ, Stearns WH, Rhodes CJ, Myers MG Jr, Schwartz MW (2001) Intracellular signalling. Key enzyme in leptin-induced anorexia. Nature 413:794–795
Withers DJ, Gutierrez JS, Towery H, Burks DJ, Ren JM, Previs S, Zhang Y, Bernal D, Pons S, Shulman GI, Bonner-Weir S, White MF (1998) Disruption of IRS-2 causes type 2 diabetes in mice. Nature 391:900–904
Duan C, Li M, Rui L (2004) SH2-B promotes insulin receptor substrate 1 (IRS1)- and IRS2-mediated activation of the phosphatidylinositol 3-kinase pathway in response to leptin. J Biol Chem 279:43684–43691
Ren D, Li M, Duan C, Rui L (2005) Identification of SH2-B as a key regulator of leptin sensitivity, energy balance, and body weight in mice. Cell Metab 2:95–104
Niswender KD, Baskin DG, Schwartz MW (2004) Insulin and its evolving partnership with leptin in the hypothalamic control of energy homeostasis. Trends Endocrinol Metab 15:362–369
Xu AW, Kaelin CB, Takeda K, Akira S, Schwartz MW, Barsh GS (2005) PI3K integrates the action of insulin and leptin on hypothalamic neurons. J Clin Invest 115:951–958
Hill JW, Williams KW, Ye C, Luo J, Balthasar N, Coppari R, Cowley MA, Cantley LC, Lowell BB, Elmquist JK (2008) Acute effects of leptin require PI3K signaling in hypothalamic proopiomelanocortin neurons in mice. J Clin Invest 118:1796–1805
Backer JM, Myers MG Jr, Shoelson SE, Chin DJ, Sun XJ, Miralpeix M, Hu P, Margolis B, Skolnik EY, Schlessinger J et al (1992) Phosphatidylinositol 3′-kinase is activated by association with IRS-1 during insulin stimulation. EMBO J 11:3469–3479
Plum L, Rother E, Munzberg H, Wunderlich FT, Morgan DA, Hampel B, Shanabrough M, Janoschek R, Konner AC, Alber J, Suzuki A, Krone W, Horvath TL, Rahmouni K, Bruning JC (2007) Enhanced leptin-stimulated Pi3k activation in the CNS promotes white adipose tissue transdifferentiation. Cell Metab 6:431–445
Plum L, Ma X, Hampel B, Balthasar N, Coppari R, Munzberg H, Shanabrough M, Burdakov D, Rother E, Janoschek R, Alber J, Belgardt BF, Koch L, Seibler J, Schwenk F, Fekete C, Suzuki A, Mak TW, Krone W, Horvath TL, Ashcroft FM, Bruning JC (2006) Enhanced PIP3 signaling in POMC neurons causes KATP channel activation and leads to diet-sensitive obesity. J Clin Invest 116:1886–1901
Kitamura T, Feng Y, Kitamura YI, Chua SC Jr, Xu AW, Barsh GS, Rossetti L, Accili D (2006) Forkhead protein FoxO1 mediates Agrp-dependent effects of leptin on food intake. Nat Med 12:534–540
Baskin DG, Schwartz MW, Seeley RJ, Woods SC, Porte D Jr, Breininger JF, Jonak Z, Schaefer J, Krouse M, Burghardt C, Campfield LA, Burn P, Kochan JP (1999) Leptin receptor long-form splice-variant protein expression in neuron cell bodies of the brain and co-localization with neuropeptide Y mRNA in the arcuate nucleus. J Histochem Cytochem 47:353–362
Elmquist JK, Bjorbaek C, Ahima RS, Flier JS, Saper CB (1998) Distributions of leptin receptor mRNA isoforms in the rat brain. J Comp Neurol 395:535–547
Kim KW, Donato J Jr, Berglund ED, Choi YH, Kohno D, Elias CF, Depinho RA, Elmquist JK (2012) FOXO1 in the ventromedial hypothalamus regulates energy balance. J Clin Invest 122:2578–2589
Huang H, Kong D, Byun KH, Ye C, Koda S, Lee DH, Oh BC, Lee SW, Lee B, Zabolotny JM, Kim MS, Bjorbaek C, Lowell BB, Kim YB (2012) Rho-kinase regulates energy balance by targeting hypothalamic leptin receptor signaling. Nat Neurosci 15:1391–1398
Kim MS, Pak YK, Jang PG, Namkoong C, Choi YS, Won JC, Kim KS, Kim SW, Kim HS, Park JY, Kim YB, Lee KU (2006) Role of hypothalamic Foxo1 in the regulation of food intake and energy homeostasis. Nat Neurosci 9:901–906
Plum L, Lin HV, Dutia R, Tanaka J, Aizawa KS, Matsumoto M, Kim AJ, Cawley NX, Paik JH, Loh YP, DePinho RA, Wardlaw SL, Accili D (2009) The obesity susceptibility gene Cpe links FoxO1 signaling in hypothalamic pro-opiomelanocortin neurons with regulation of food intake. Nat Med 15:1195–1201
Yang G, Lim CY, Li C, Xiao X, Radda GK, Li C, Cao X, Han W (2009) FoxO1 inhibits leptin regulation of pro-opiomelanocortin promoter activity by blocking STAT3 interaction with specificity protein 1. J Biol Chem 284:3719–3727
Morikawa Y, Ueyama E, Senba E (2004) Fasting-induced activation of mitogen-activated protein kinases (ERK/p38) in the mouse hypothalamus. J Neuroendocrinol 16:105–112
Ueyama E, Morikawa Y, Yasuda T, Senba E (2004) Attenuation of fasting-induced phosphorylation of mitogen-activated protein kinases (ERK/p38) in the mouse hypothalamus in response to refeeding. Neurosci Lett 371:40–44
Myers MG Jr (2004) Leptin receptor signaling and the regulation of mammalian physiology. Recent Prog Horm Res 59:287–304
Takahashi Y, Okimura Y, Mizuno I, Iida K, Takahashi T, Kaji H, Abe H, Chihara K (1997) Leptin induces mitogen-activated protein kinase-dependent proliferation of C3H10T1/2 cells. J Biol Chem 272:12897–12900
Rahmouni K, Sigmund CD, Haynes WG, Mark AL (2009) Hypothalamic ERK mediates the anorectic and thermogenic sympathetic effects of leptin. Diabetes 58:536–542
Bjorbaek C, Uotani S, da Silva B, Flier JS (1997) Divergent signaling capacities of the long and short isoforms of the leptin receptor. J Biol Chem 272:32686–32695
Schlessinger J (2000) Cell signaling by receptor tyrosine kinases. Cell 103:211–225
Bjorbaek C, Buchholz RM, Davis SM, Bates SH, Pierroz DD, Gu H, Neel BG, Myers MG Jr, Flier JS (2001) Divergent roles of SHP-2 in ERK activation by leptin receptors. J Biol Chem 276:4747–4755
Bouret SG, Draper SJ, Simerly RB (2004) Trophic action of leptin on hypothalamic neurons that regulate feeding. Science 304:108–110
Bouret SG, Bates SH, Chen S, Myers MG Jr, Simerly RB (2012) Distinct roles for specific leptin receptor signals in the development of hypothalamic feeding circuits. J Neurosci 32:1244–1252
Balland E, Dam J, Langlet F, Caron E, Steculorum S, Messina A, Rasika S, Falluel-Morel A, Anouar Y, Dehouck B, Trinquet E, Jockers R, Bouret SG, Prevot V (2014) Hypothalamic tanycytes are an ERK-gated conduit for leptin into the brain. Cell Metab 19:293–301
Hardie DG, Ross FA, Hawley SA (2012) AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 13:251–262
Sanders MJ, Ali ZS, Hegarty BD, Heath R, Snowden MA, Carling D (2007) Defining the mechanism of activation of AMP-activated protein kinase by the small molecule A-769662, a member of the thienopyridone family. J Biol Chem 282:32539–32548
Mihaylova MM, Shaw RJ (2011) The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol 13:1016–1023
Hawley SA, Boudeau J, Reid JL, Mustard KJ, Udd L, Makela TP, Alessi DR, Hardie DG (2003) Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J Biol 2:28
Racioppi L, Means AR (2012) Calcium/calmodulin-dependent protein kinase kinase 2: roles in signaling and pathophysiology. J Biol Chem 287:31658–31665
Minokoshi Y, Alquier T, Furukawa N, Kim YB, Lee A, Xue B, Mu J, Foufelle F, Ferre P, Birnbaum MJ, Stuck BJ, Kahn BB (2004) AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature 428:569–574
Andersson U, Filipsson K, Abbott CR, Woods A, Smith K, Bloom SR, Carling D, Small CJ (2004) AMP-activated protein kinase plays a role in the control of food intake. J Biol Chem 279:12005–12008
Obici S, Feng Z, Arduini A, Conti R, Rossetti L (2003) Inhibition of hypothalamic carnitine palmitoyltransferase-1 decreases food intake and glucose production. Nat Med 9:756–761
Minokoshi Y, Kim YB, Peroni OD, Fryer LG, Muller C, Carling D, Kahn BB (2002) Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 415:339–343
Tanida M, Yamamoto N, Shibamoto T, Rahmouni K (2013) Involvement of hypothalamic AMP-activated protein kinase in leptin-induced sympathetic nerve activation. PLoS One 8:e56660
Djouder N, Tuerk RD, Suter M, Salvioni P, Thali RF, Scholz R, Vaahtomeri K, Auchli Y, Rechsteiner H, Brunisholz RA, Viollet B, Makela TP, Wallimann T, Neumann D, Krek W (2010) PKA phosphorylates and inactivates AMPKalpha to promote efficient lipolysis. EMBO J 29:469–481
Hurley RL, Barre LK, Wood SD, Anderson KA, Kemp BE, Means AR, Witters LA (2006) Regulation of AMP-activated protein kinase by multisite phosphorylation in response to agents that elevate cellular cAMP. J Biol Chem 281:36662–36672
Claret M, Smith MA, Batterham RL, Selman C, Choudhury AI, Fryer LG, Clements M, Al-Qassab H, Heffron H, Xu AW, Speakman JR, Barsh GS, Viollet B, Vaulont S, Ashford ML, Carling D, Withers DJ (2007) AMPK is essential for energy homeostasis regulation and glucose sensing by POMC and AgRP neurons. J Clin Invest 117:2325–2336
Jacinto E, Hall MN (2003) Tor signalling in bugs, brain and brawn. Nat Rev Mol Cell Biol 4:117–126
Dennis PB, Jaeschke A, Saitoh M, Fowler B, Kozma SC, Thomas G (2001) Mammalian TOR: a homeostatic ATP sensor. Science 294:1102–1105
Cota D, Proulx K, Smith KA, Kozma SC, Thomas G, Woods SC, Seeley RJ (2006) Hypothalamic mTOR signaling regulates food intake. Science 312:927–930
Cota D, Matter EK, Woods SC, Seeley RJ (2008) The role of hypothalamic mammalian target of rapamycin complex 1 signaling in diet-induced obesity. J Neurosci 28:7202–7208
Blouet C, Ono H, Schwartz GJ (2008) Mediobasal hypothalamic p70 S6 kinase 1 modulates the control of energy homeostasis. Cell Metab 8:459–467
Harlan SM, Guo DF, Morgan DA, Fernandes-Santos C, Rahmouni K (2013) Hypothalamic mTORC1 signaling controls sympathetic nerve activity and arterial pressure and mediates leptin effects. Cell Metab 17:599–606
Dagon Y, Hur E, Zheng B, Wellenstein K, Cantley LC, Kahn BB (2012) p70S6 kinase phosphorylates AMPK on serine 491 to mediate leptin’s effect on food intake. Cell Metab 16:104–112
Kim JG, Horvath TL (2012) mTOR signaling fades POMC neurons during aging. Neuron 75:356–357
Yang SB, Tien AC, Boddupalli G, Xu AW, Jan YN, Jan LY (2012) Rapamycin ameliorates age-dependent obesity associated with increased mTOR signaling in hypothalamic POMC neurons. Neuron 75:425–436
Mori H, Inoki K, Munzberg H, Opland D, Faouzi M, Villanueva EC, Ikenoue T, Kwiatkowski D, MacDougald OA, Myers MG Jr, Guan KL (2009) Critical role for hypothalamic mTOR activity in energy balance. Cell Metab 9:362–374
Marshall WF, Nonaka S (2006) Cilia: tuning into the cell’s antenna. Curr Biol 16:R604–R614
Badano JL, Mitsuma N, Beales PL, Katsanis N (2006) The ciliopathies: an emerging class of human genetic disorders. Annu Rev Genomics Hum Genet 7:125–148
Seo S, Guo DF, Bugge K, Morgan DA, Rahmouni K, Sheffield VC (2009) Requirement of Bardet–Biedl syndrome proteins for leptin receptor signaling. Hum Mol Genet 18:1323–1331
Nachury MV, Loktev AV, Zhang Q, Westlake CJ, Peranen J, Merdes A, Slusarski DC, Scheller RH, Bazan JF, Sheffield VC, Jackson PK (2007) A core complex of BBS proteins cooperates with the GTPase Rab8 to promote ciliary membrane biogenesis. Cell 129:1201–1213
Stratigopoulos G, LeDuc CA, Cremona ML, Chung WK, Leibel RL (2011) Cut-like homeobox 1 (CUX1) regulates expression of the fat mass and obesity-associated and retinitis pigmentosa GTPase regulator-interacting protein-1-like (RPGRIP1L) genes and coordinates leptin receptor signaling. J Biol Chem 286:2155–2170
Stratigopoulos G, Martin Carli JF, O’Day DR, Wang L, Leduc CA, Lanzano P, Chung WK, Rosenbaum M, Egli D, Doherty DA, Leibel RL (2014) Hypomorphism for RPGRIP1L, a ciliary gene vicinal to the FTO locus, causes increased adiposity in mice. Cell Metab 19:767–779
Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, Perry JR, Elliott KS, Lango H, Rayner NW, Shields B, Harries LW, Barrett JC, Ellard S, Groves CJ, Knight B, Patch AM, Ness AR, Ebrahim S, Lawlor DA, Ring SM, Ben-Shlomo Y, Jarvelin MR, Sovio U, Bennett AJ, Melzer D, Ferrucci L, Loos RJ, Barroso I, Wareham NJ, Karpe F, Owen KR, Cardon LR, Walker M, Hitman GA, Palmer CN, Doney AS, Morris AD, Smith GD, Hattersley AT, McCarthy MI (2007) A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 316:889–894
Meyre D, Delplanque J, Chevre JC, Lecoeur C, Lobbens S, Gallina S, Durand E, Vatin V, Degraeve F, Proenca C, Gaget S, Korner A, Kovacs P, Kiess W, Tichet J, Marre M, Hartikainen AL, Horber F, Potoczna N, Hercberg S, Levy-Marchal C, Pattou F, Heude B, Tauber M, McCarthy MI, Blakemore AI, Montpetit A, Polychronakos C, Weill J, Coin LJ, Asher J, Elliott P, Jarvelin MR, Visvikis-Siest S, Balkau B, Sladek R, Balding D, Walley A, Dina C, Froguel P (2009) Genome-wide association study for early-onset and morbid adult obesity identifies three new risk loci in European populations. Nat Genet 41:157–159
Davenport JR, Watts AJ, Roper VC, Croyle MJ, van Groen T, Wyss JM, Nagy TR, Kesterson RA, Yoder BK (2007) Disruption of intraflagellar transport in adult mice leads to obesity and slow-onset cystic kidney disease. Curr Biol 17:1586–1594
Han YM, Kang GM, Byun K, Ko HW, Kim J, Shin MS, Kim HK, Gil SY, Yu JH, Lee B, Kim MS (2014) Leptin-promoted cilia assembly is critical for normal energy balance. J Clin Invest 124:2193–2197
Wang Z, Li V, Chan GC, Phan T, Nudelman AS, Xia Z, Storm DR (2009) Adult type 3 adenylyl cyclase-deficient mice are obese. PLoS One 4:e6979
Berbari NF, Pasek RC, Malarkey EB, Yazdi SM, McNair AD, Lewis WR, Nagy TR, Kesterson RA, Yoder BK (2013) Leptin resistance is a secondary consequence of the obesity in ciliopathy mutant mice. Proc Natl Acad Sci USA 110:7796–7801
Kang GM, Han YM, Ko HW, Kim J, Oh BC, Kwon I, Kim MS (2015) Leptin Elongates Hypothalamic Neuronal Cilia via Transcriptional Regulation and Actin Destabilization. J Biol Chem 290:18146–18155
Heymsfield SB, Greenberg AS, Fujioka K, Dixon RM, Kushner R, Hunt T, Lubina JA, Patane J, Self B, Hunt P, McCamish M (1999) Recombinant leptin for weight loss in obese and lean adults: a randomized, controlled, dose-escalation trial. JAMA 282:1568–1575
Hukshorn CJ, Saris WH, Westerterp-Plantenga MS, Farid AR, Smith FJ, Campfield LA (2000) Weekly subcutaneous pegylated recombinant native human leptin (PEG-OB) administration in obese men. J Clin Endocrinol Metab 85:4003–4009
Frederich RC, Hamann A, Anderson S, Lollmann B, Lowell BB, Flier JS (1995) Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nat Med 1:1311–1314
Myers MG Jr, Heymsfield SB, Haft C, Kahn BB, Laughlin M, Leibel RL, Tschop MH, Yanovski JA (2012) Challenges and opportunities of defining clinical leptin resistance. Cell Metab 15:150–156
Hubschle T, Thom E, Watson A, Roth J, Klaus S, Meyerhof W (2001) Leptin-induced nuclear translocation of STAT3 immunoreactivity in hypothalamic nuclei involved in body weight regulation. J Neurosci 21:2413–2424
El-Haschimi K, Pierroz DD, Hileman SM, Bjorbaek C, Flier JS (2000) Two defects contribute to hypothalamic leptin resistance in mice with diet-induced obesity. J Clin Invest 105:1827–1832
Bjorbaek C, Elmquist JK, Frantz JD, Shoelson SE, Flier JS (1998) Identification of SOCS-3 as a potential mediator of central leptin resistance. Mol Cell 1:619–625
Howard JK, Cave BJ, Oksanen LJ, Tzameli I, Bjorbaek C, Flier JS (2004) Enhanced leptin sensitivity and attenuation of diet-induced obesity in mice with haploinsufficiency of Socs3. Nat Med 10:734–738
Mori H, Hanada R, Hanada T, Aki D, Mashima R, Nishinakamura H, Torisu T, Chien KR, Yasukawa H, Yoshimura A (2004) Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nat Med 10:739–743
Kievit P, Howard JK, Badman MK, Balthasar N, Coppari R, Mori H, Lee CE, Elmquist JK, Yoshimura A, Flier JS (2006) Enhanced leptin sensitivity and improved glucose homeostasis in mice lacking suppressor of cytokine signaling-3 in POMC-expressing cells. Cell Metab 4:123–132
Reed AS, Unger EK, Olofsson LE, Piper ML, Myers MG Jr, Xu AW (2010) Functional role of suppressor of cytokine signaling 3 upregulation in hypothalamic leptin resistance and long-term energy homeostasis. Diabetes 59:894–906
Munzberg H, Flier JS, Bjorbaek C (2004) Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology 145:4880–4889
Ottaway N, Mahbod P, Rivero B, Norman LA, Gertler A, D’Alessio DA, Perez-Tilve D (2015) Diet-induced obese mice retain endogenous leptin action. Cell Metab 21:877–882
Olofsson LE, Unger EK, Cheung CC, Xu AW (2013) Modulation of AgRP-neuronal function by SOCS3 as an initiating event in diet-induced hypothalamic leptin resistance. Proc Natl Acad Sci USA 110:E697–E706
Myers MG Jr (2015) Leptin keeps working, even in obesity. Cell Metab 21:791–792
Frangioni JV, Beahm PH, Shifrin V, Jost CA, Neel BG (1992) The nontransmembrane tyrosine phosphatase PTP-1B localizes to the endoplasmic reticulum via its 35 amino acid C-terminal sequence. Cell 68:545–560
Zabolotny JM, Bence-Hanulec KK, Stricker-Krongrad A, Haj F, Wang Y, Minokoshi Y, Kim YB, Elmquist JK, Tartaglia LA, Kahn BB, Neel BG (2002) PTP1B regulates leptin signal transduction in vivo. Dev Cell 2:489–495
Kaszubska W, Falls HD, Schaefer VG, Haasch D, Frost L, Hessler P, Kroeger PE, White DW, Jirousek MR, Trevillyan JM (2002) Protein tyrosine phosphatase 1B negatively regulates leptin signaling in a hypothalamic cell line. Mol Cell Endocrinol 195:109–118
Myers MP, Andersen JN, Cheng A, Tremblay ML, Horvath CM, Parisien JP, Salmeen A, Barford D, Tonks NK (2001) TYK2 and JAK2 are substrates of protein-tyrosine phosphatase 1B. J Biol Chem 276:47771–47774
Xue B, Pulinilkunnil T, Murano I, Bence KK, He H, Minokoshi Y, Asakura K, Lee A, Haj F, Furukawa N, Catalano KJ, Delibegovic M, Balschi JA, Cinti S, Neel BG, Kahn BB (2009) Neuronal protein tyrosine phosphatase 1B deficiency results in inhibition of hypothalamic AMPK and isoform-specific activation of AMPK in peripheral tissues. Mol Cell Biol 29:4563–4573
Klaman LD, Boss O, Peroni OD, Kim JK, Martino JL, Zabolotny JM, Moghal N, Lubkin M, Kim YB, Sharpe AH, Stricker-Krongrad A, Shulman GI, Neel BG, Kahn BB (2000) Increased energy expenditure, decreased adiposity, and tissue-specific insulin sensitivity in protein-tyrosine phosphatase 1B-deficient mice. Mol Cell Biol 20:5479–5489
Bence KK, Delibegovic M, Xue B, Gorgun CZ, Hotamisligil GS, Neel BG, Kahn BB (2006) Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat Med 12:917–924
Cheng A, Uetani N, Simoncic PD, Chaubey VP, Lee-Loy A, McGlade CJ, Kennedy BP, Tremblay ML (2002) Attenuation of leptin action and regulation of obesity by protein tyrosine phosphatase 1B. Dev Cell 2:497–503
Picardi PK, Calegari VC, Prada PO, Moraes JC, Araujo E, Marcondes MC, Ueno M, Carvalheira JB, Velloso LA, Saad MJ (2008) Reduction of hypothalamic protein tyrosine phosphatase improves insulin and leptin resistance in diet-induced obese rats. Endocrinology 149:3870–3880
Tsou RC, Rak KS, Zimmer DJ, Bence KK (2014) Improved metabolic phenotype of hypothalamic PTP1B-deficiency is dependent upon the leptin receptor. Mol Metab 3:301–312
Zabolotny JM, Kim YB, Welsh LA, Kershaw EE, Neel BG, Kahn BB (2008) Protein-tyrosine phosphatase 1B expression is induced by inflammation in vivo. J Biol Chem 283:14230–14241
White CL, Whittington A, Barnes MJ, Wang Z, Bray GA, Morrison CD (2009) HF diets increase hypothalamic PTP1B and induce leptin resistance through both leptin-dependent and -independent mechanisms. Am J Physiol Endocrinol Metab 296:E291–E299
Morrison CD, White CL, Wang Z, Lee SY, Lawrence DS, Cefalu WT, Zhang ZY, Gettys TW (2007) Increased hypothalamic protein tyrosine phosphatase 1B contributes to leptin resistance with age. Endocrinology 148:433–440
Chen HH, Schock SC, Xu J, Safarpour F, Thompson CS, Stewart AF (2007) Extracellular ATP-dependent upregulation of the transcription cofactor LMO4 promotes neuron survival from hypoxia. Exp Cell Res 313:3106–3116
Zhou X, Gomez-Smith M, Qin Z, Duquette PM, Cardenas-Blanco A, Rai PS, Harper ME, Tsai EC, Anisman H, Chen HH (2012) Ablation of LMO4 in glutamatergic neurons impairs leptin control of fat metabolism. Cell Mol Life Sci 69:819–828
Pandey NR, Zhou X, Qin Z, Zaman T, Gomez-Smith M, Keyhanian K, Anisman H, Brunel JM, Stewart AF, Chen HH (2013) The LIM domain only 4 protein is a metabolic responsive inhibitor of protein tyrosine phosphatase 1B that controls hypothalamic leptin signaling. J Neurosci 33:12647–12655
Lantz KA, Hart SG, Planey SL, Roitman MF, Ruiz-White IA, Wolfe HR, McLane MP (2010) Inhibition of PTP1B by trodusquemine (MSI-1436) causes fat-specific weight loss in diet-induced obese mice. Obesity (Silver Spring) 18:1516–1523
Qin Z, Pandey NR, Zhou X, Stewart CA, Hari A, Huang H, Stewart AF, Brunel JM, Chen HH (2015) Functional properties of Claramine: a novel PTP1B inhibitor and insulin-mimetic compound. Biochem Biophys Res Commun 458:21–27
Rousso-Noori L, Knobler H, Levy-Apter E, Kuperman Y, Neufeld-Cohen A, Keshet Y, Akepati VR, Klinghoffer RA, Chen A, Elson A (2011) Protein tyrosine phosphatase epsilon affects body weight by downregulating leptin signaling in a phosphorylation-dependent manner. Cell Metab 13:562–572
Loh K, Fukushima A, Zhang X, Galic S, Briggs D, Enriori PJ, Simonds S, Wiede F, Reichenbach A, Hauser C, Sims NA, Bence KK, Zhang S, Zhang ZY, Kahn BB, Neel BG, Andrews ZB, Cowley MA, Tiganis T (2011) Elevated hypothalamic TCPTP in obesity contributes to cellular leptin resistance. Cell Metab 14:684–699
St-Pierre J, Tremblay ML (2012) Modulation of leptin resistance by protein tyrosine phosphatases. Cell Metab 15:292–297
De Souza CT, Araujo EP, Bordin S, Ashimine R, Zollner RL, Boschero AC, Saad MJ, Velloso LA (2005) Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology 146:4192–4199
Thaler JP, Yi CX, Schur EA, Guyenet SJ, Hwang BH, Dietrich MO, Zhao X, Sarruf DA, Izgur V, Maravilla KR, Nguyen HT, Fischer JD, Matsen ME, Wisse BE, Morton GJ, Horvath TL, Baskin DG, Tschop MH, Schwartz MW (2012) Obesity is associated with hypothalamic injury in rodents and humans. J Clin Invest 122:153–162
Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D (2008) Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell 135:61–73
Posey KA, Clegg DJ, Printz RL, Byun J, Morton GJ, Vivekanandan-Giri A, Pennathur S, Baskin DG, Heinecke JW, Woods SC, Schwartz MW, Niswender KD (2009) Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am J Physiol Endocrinol Metab 296:E1003–E1012
Meng Q, Cai D (2011) Defective hypothalamic autophagy directs the central pathogenesis of obesity via the IkappaB kinase beta (IKKbeta)/NF-kappaB pathway. J Biol Chem 286:32324–32332
Benzler J, Ganjam GK, Pretz D, Oelkrug R, Koch CE, Legler K, Stohr S, Culmsee C, Williams LM, Tups A (2015) Central inhibition of IKKbeta/NF-kappaB signaling attenuates high-fat diet-induced obesity and glucose intolerance. Diabetes 64:2015–2027
Tsaousidou E, Paeger L, Belgardt BF, Pal M, Wunderlich CM, Bronneke H, Collienne U, Hampel B, Wunderlich FT, Schmidt-Supprian M, Kloppenburg P, Bruning JC (2014) Distinct roles for JNK and IKK activation in agouti-related peptide neurons in the development of obesity and insulin resistance. Cell Rep 9:1495–1506
Jang PG, Namkoong C, Kang GM, Hur MW, Kim SW, Kim GH, Kang Y, Jeon MJ, Kim EH, Lee MS, Karin M, Baik JH, Park JY, Lee KU, Kim YB, Kim MS (2010) NF-kappaB activation in hypothalamic pro-opiomelanocortin neurons is essential in illness- and leptin-induced anorexia. J Biol Chem 285:9706–9715
Shi X, Wang X, Li Q, Su M, Chew E, Wong ET, Lacza Z, Radda GK, Tergaonkar V, Han W (2013) Nuclear factor kappaB (NF-kappaB) suppresses food intake and energy expenditure in mice by directly activating the Pomc promoter. Diabetologia 56:925–936
Wang X, Ge A, Cheng M, Guo F, Zhao M, Zhou X, Liu L, Yang N (2012) Increased hypothalamic inflammation associated with the susceptibility to obesity in rats exposed to high-fat diet. Exp Diabetes Res 2012:847246
Milanski M, Degasperi G, Coope A, Morari J, Denis R, Cintra DE, Tsukumo DM, Anhe G, Amaral ME, Takahashi HK, Curi R, Oliveira HC, Carvalheira JB, Bordin S, Saad MJ, Velloso LA (2009) Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: implications for the pathogenesis of obesity. J Neurosci 29:359–370
Romanatto T, Roman EA, Arruda AP, Denis RG, Solon C, Milanski M, Moraes JC, Bonfleur ML, Degasperi GR, Picardi PK, Hirabara S, Boschero AC, Curi R, Velloso LA (2009) Deletion of tumor necrosis factor-alpha receptor 1 (TNFR1) protects against diet-induced obesity by means of increased thermogenesis. J Biol Chem 284:36213–36222
Milanski M, Arruda AP, Coope A, Ignacio-Souza LM, Nunez CE, Roman EA, Romanatto T, Pascoal LB, Caricilli AM, Torsoni MA, Prada PO, Saad MJ, Velloso LA (2012) Inhibition of hypothalamic inflammation reverses diet-induced insulin resistance in the liver. Diabetes 61:1455–1462
Romanatto T, Cesquini M, Amaral ME, Roman EA, Moraes JC, Torsoni MA, Cruz-Neto AP, Velloso LA (2007) TNF-alpha acts in the hypothalamus inhibiting food intake and increasing the respiratory quotient–effects on leptin and insulin signaling pathways. Peptides 28:1050–1058
Ropelle ER, Flores MB, Cintra DE, Rocha GZ, Pauli JR, Morari J, de Souza CT, Moraes JC, Prada PO, Guadagnini D, Marin RM, Oliveira AG, Augusto TM, Carvalho HF, Velloso LA, Saad MJ, Carvalheira JB (2010) IL-6 and IL-10 anti-inflammatory activity links exercise to hypothalamic insulin and leptin sensitivity through IKKbeta and ER stress inhibition. PLoS Biol 8:e1000465
Luheshi GN, Gardner JD, Rushforth DA, Loudon AS, Rothwell NJ (1999) Leptin actions on food intake and body temperature are mediated by IL-1. Proc Natl Acad Sci USA 96:7047–7052
Cintra DE, Ropelle ER, Moraes JC, Pauli JR, Morari J, Souza CT, Grimaldi R, Stahl M, Carvalheira JB, Saad MJ, Velloso LA (2012) Unsaturated fatty acids revert diet-induced hypothalamic inflammation in obesity. PLoS One 7:e30571
Oh IS, Thaler JP, Ogimoto K, Wisse BE, Morton GJ, Schwartz MW (2010) Central administration of interleukin-4 exacerbates hypothalamic inflammation and weight gain during high-fat feeding. Am J Physiol Endocrinol Metab 299:E47–E53
Takeda K, Kaisho T, Akira S (2003) Toll-like receptors. Annu Rev Immunol 21:335–376
Suganami T, Tanimoto-Koyama K, Nishida J, Itoh M, Yuan X, Mizuarai S, Kotani H, Yamaoka S, Miyake K, Aoe S, Kamei Y, Ogawa Y (2007) Role of the Toll-like receptor 4/NF-kappaB pathway in saturated fatty acid-induced inflammatory changes in the interaction between adipocytes and macrophages. Arterioscler Thromb Vasc Biol 27:84–91
Kawai T, Adachi O, Ogawa T, Takeda K, Akira S (1999) Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity 11:115–122
Kleinridders A, Schenten D, Konner AC, Belgardt BF, Mauer J, Okamura T, Wunderlich FT, Medzhitov R, Bruning JC (2009) MyD88 signaling in the CNS is required for development of fatty acid-induced leptin resistance and diet-induced obesity. Cell Metab 10:249–259
Weissmann L, Quaresma PG, Santos AC, de Matos AH, Pascoal VD, Zanotto TM, Castro G, Guadagnini D, da Silva JM, Velloso LA, Bittencourt JC, Lopes-Cendes I, Saad MJ, Prada PO (2014) IKKepsilon is key to induction of insulin resistance in the hypothalamus, and its inhibition reverses obesity. Diabetes 63:3334–3345
Borges Bde C, Rorato RC, Uchoa ET, Marangon PB, Elias CF, Antunes-Rodrigues J, Elias LL (2015) Protein tyrosine phosphatase-1B contributes to LPS-induced leptin resistance in male rats. Am J Physiol Endocrinol Metab 308:E40–E50
Sachot C, Poole S, Luheshi GN (2004) Circulating leptin mediates lipopolysaccharide-induced anorexia and fever in rats. J Physiol 561:263–272
Reis WL, Yi CX, Gao Y, Tschop MH, Stern JE (2015) Brain innate immunity regulates hypothalamic arcuate neuronal activity and feeding behavior. Endocrinology 156:1303–1315
Borges BC, Rorato R, Avraham Y, da Silva LE, Castro M, Vorobiav L, Berry E, Antunes-Rodrigues J, Elias LL (2011) Leptin resistance and desensitization of hypophagia during prolonged inflammatory challenge. Am J Physiol Endocrinol Metab 300:E858–E869
Marciniak SJ, Ron D (2006) Endoplasmic reticulum stress signaling in disease. Physiol Rev 86:1133–1149
Ron D, Walter P (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol 8:519–529
Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS (2004) Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 306:457–461
Ozcan L, Ergin AS, Lu A, Chung J, Sarkar S, Nie D, Myers MG Jr, Ozcan U (2009) Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab 9:35–51
Won JC, Jang PG, Namkoong C, Koh EH, Kim SK, Park JY, Lee KU, Kim MS (2009) Central administration of an endoplasmic reticulum stress inducer inhibits the anorexigenic effects of leptin and insulin. Obesity (Silver Spring) 17:1861–1865
Alfieri A, Pasanisi F, Salzano S, Esposito L, Martone D, Tafuri D, Daniele A, Contaldo F, Sacchetti L, Zagari A, Buono P (2010) Functional analysis of melanocortin-4-receptor mutants identified in severely obese subjects living in Southern Italy. Gene 457:35–41
Granell S, Mohammad S, Ramanagoudr-Bhojappa R, Baldini G (2010) Obesity-linked variants of melanocortin-4 receptor are misfolded in the endoplasmic reticulum and can be rescued to the cell surface by a chemical chaperone. Mol Endocrinol 24:1805–1821
Williams KW, Liu T, Kong X, Fukuda M, Deng Y, Berglund ED, Deng Z, Gao Y, Liu T, Sohn JW, Jia L, Fujikawa T, Kohno D, Scott MM, Lee S, Lee CE, Sun K, Chang Y, Scherer PE, Elmquist JK (2014) Xbp1s in Pomc neurons connects ER stress with energy balance and glucose homeostasis. Cell Metab 20:471–482
Ramirez S, Claret M (2015) Hypothalamic ER stress: a bridge between leptin resistance and obesity. FEBS Lett 589:1678–1687
Cragle FK, Baldini G (2014) Mild lipid stress induces profound loss of MC4R protein abundance and function. Mol Endocrinol 28:357–367
Purkayastha S, Zhang H, Zhang G, Ahmed Z, Wang Y, Cai D (2011) Neural dysregulation of peripheral insulin action and blood pressure by brain endoplasmic reticulum stress. Proc Natl Acad Sci USA 108:2939–2944
Hoepfner D, Schildknegt D, Braakman I, Philippsen P, Tabak HF (2005) Contribution of the endoplasmic reticulum to peroxisome formation. Cell 122:85–95
Semple RK, Chatterjee VK, O’Rahilly S (2006) PPAR gamma and human metabolic disease. J Clin Invest 116:581–589
Ryan KK, Li B, Grayson BE, Matter EK, Woods SC, Seeley RJ (2011) A role for central nervous system PPAR-gamma in the regulation of energy balance. Nat Med 17:623–626
Diano S, Liu ZW, Jeong JK, Dietrich MO, Ruan HB, Kim E, Suyama S, Kelly K, Gyengesi E, Arbiser JL, Belsham DD, Sarruf DA, Schwartz MW, Bennett AM, Shanabrough M, Mobbs CV, Yang X, Gao XB, Horvath TL (2011) Peroxisome proliferation-associated control of reactive oxygen species sets melanocortin tone and feeding in diet-induced obesity. Nat Med 17:1121–1127
Long L, Toda C, Jeong JK, Horvath TL, Diano S (2014) PPARgamma ablation sensitizes proopiomelanocortin neurons to leptin during high-fat feeding. J Clin Invest 124:4017–4027
Schneeberger M, Dietrich MO, Sebastian D, Imbernon M, Castano C, Garcia A, Esteban Y, Gonzalez-Franquesa A, Rodriguez IC, Bortolozzi A, Garcia-Roves PM, Gomis R, Nogueiras R, Horvath TL, Zorzano A, Claret M (2013) Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell 155:172–187
Dietrich MO, Liu ZW, Horvath TL (2013) Mitochondrial dynamics controlled by mitofusins regulate Agrp neuronal activity and diet-induced obesity. Cell 155:188–199
Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, Clark SG, Ron D (2002) IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415:92–96
Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS (2002) A central role for JNK in obesity and insulin resistance. Nature 420:333–336
Belgardt BF, Mauer J, Wunderlich FT, Ernst MB, Pal M, Spohn G, Bronneke HS, Brodesser S, Hampel B, Schauss AC, Bruning JC (2010) Hypothalamic and pituitary c-Jun N-terminal kinase 1 signaling coordinately regulates glucose metabolism. Proc Natl Acad Sci USA 107:6028–6033
Benzler J, Ganjam GK, Legler K, Stohr S, Kruger M, Steger J, Tups A (2013) Acute inhibition of central c-Jun N-terminal kinase restores hypothalamic insulin signalling and alleviates glucose intolerance in diabetic mice. J Neuroendocrinol 25:446–454
Kim JK, Fillmore JJ, Sunshine MJ, Albrecht B, Higashimori T, Kim DW, Liu ZX, Soos TJ, Cline GW, O’Brien WR, Littman DR, Shulman GI (2004) PKC-theta knockout mice are protected from fat-induced insulin resistance. J Clin Invest 114:823–827
Benoit SC, Kemp CJ, Elias CF, Abplanalp W, Herman JP, Migrenne S, Lefevre AL, Cruciani-Guglielmacci C, Magnan C, Yu F, Niswender K, Irani BG, Holland WL, Clegg DJ (2009) Palmitic acid mediates hypothalamic insulin resistance by altering PKC-theta subcellular localization in rodents. J Clin Invest 119:2577–2589
Fukuda M, Williams KW, Gautron L, Elmquist JK (2011) Induction of leptin resistance by activation of cAMP-Epac signaling. Cell Metab 13:331–339
de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL (1998) Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 396:474–477
Leshan RL, Greenwald-Yarnell M, Patterson CM, Gonzalez IE, Myers MG Jr (2012) Leptin action through hypothalamic nitric oxide synthase-1-expressing neurons controls energy balance. Nat Med 18:820–823
Vong L, Ye C, Yang Z, Choi B, Chua S Jr, Lowell BB (2011) Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron 71:142–154
Fuente-Martin E, Garcia-Caceres C, Granado M, de Ceballos ML, Sanchez-Garrido MA, Sarman B, Liu ZW, Dietrich MO, Tena-Sempere M, Argente-Arizon P, Diaz F, Argente J, Horvath TL, Chowen JA (2012) Leptin regulates glutamate and glucose transporters in hypothalamic astrocytes. J Clin Invest 122:3900–3913
Kim JG, Suyama S, Koch M, Jin S, Argente-Arizon P, Argente J, Liu ZW, Zimmer MR, Jeong JK, Szigeti-Buck K, Gao Y, Garcia-Caceres C, Yi CX, Salmaso N, Vaccarino FM, Chowen J, Diano S, Dietrich MO, Tschop MH, Horvath TL (2014) Leptin signaling in astrocytes regulates hypothalamic neuronal circuits and feeding. Nat Neurosci 17:908–910
Acknowledgments
The authors thank Ann W. Kinyua (Wonju College of Medicine, Yonsei University) for critical reading of this manuscript and Dong Ju Yang (Wonju College of Medicine, Yonsei University) for providing the illustrations. This project was supported by Grants from the National Research Foundation (NRF-2013R1A1A1007693 for K.W.K. and 2013M3C7A1056024 for M.S.K.) and the Asan Institute for Life Sciences (Grant No. 13-326) for M.S.K.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Additional information
O. Kwon and K. W. Kim contributed equally to this work.
Rights and permissions
About this article

Cite this article
Kwon, O., Kim, K.W. & Kim, MS. Leptin signalling pathways in hypothalamic neurons. Cell. Mol. Life Sci. 73, 1457–1477 (2016). https://doi.org/10.1007/s00018-016-2133-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-016-2133-1