
Abstract
The hormone leptin is secreted by adipose tissue in proportion to fat mass to signal the repletion of body energy stores to the neuroendocrine system. Leptin acts on neurons in the hypothalamus and elsewhere in the brain to decrease appetite and regulate the activity of the thyroid, adrenal, growth, gonadal, and lactational axes. Conversely, absence of leptin signaling initiates the neuroendocrine starvation response. Leptin mediates these effects by activating the long form (LRb) of its receptor. One LRb signal, STAT3, has recently been shown to play a critical role in the regulation of body weight and some elements of neuroendocrine function (thyroid, adrenal, lactation), although the participation of STAT3 in the gonadal and growth axes is negligible. We discuss these findings in the context of the hypothalamic neuroendocrine system as it is presently understood.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Leptin and leptin action
Leptin production reflects body energy balance
Adipose cells are the primary source of leptin. Leptin production by fat cells in culture is stimulated by glucocorticoids and indicators of acute nutritional influx such as insulin and is inhibited by the counterregulatory hormones and their intracellular signaling mediators [1, 2, 3]. This regulation of leptin production by insulin (feeding→increased insulin→increased leptin) and counterregulatory hormones (fasting→increased counterregulatory hormones→decreased leptin) suggests that leptin serves as an indicator of energy balance, i.e., that increased energy stores yield increased leptin levels. Indeed, circulating leptin levels are strongly correlated with body adiposity and changes in acute nutritional status [4, 5, 6].
The stimulation of leptin production by glucocorticoids appears counterintuitive in this light, however, since circulating glucocorticoids are important mediators of the stress and starvation responses. Indeed, while plentiful data suggest that insulin is a critical mediator of increased leptin production in vivo, the diurnal pattern of leptin levels and numerous other data argue against an important role for circulating glucocorticoids in the stimulation of leptin production [7]. In contrast, adipocyte-produced autocrine- or paracrine-acting glucocorticoids may be involved, since the production of glucocorticoids by adipocytes increases with the accumulation of triglycerides, and mouse models with increased glucocorticoid production specifically in adipocytes have elevated leptin levels [8].
Integrating this information, then, circulating leptin levels reflect adipose mass (perhaps via the production of glucocorticoids, and reflecting long-term energy stores) modified by circulating levels of insulin (reflecting recent food intake) and counterregulatory hormones (reflecting lack of recent food intake). Leptin levels are also regulated by other factors, and are stimulated by infection and cytokines such as leukemia inhibitory factor, tumor necrosis factor, and interleukin-1, which may play a role in infection-induced weight loss [9, 10].
Leptin regulates body energy homeostasis and neuroendocrine responses to fasting
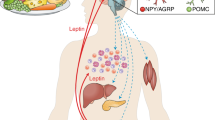
When energy stores are replete, leptin production is high; conversely, fasting and the depletion of fat mass inhibits leptin production [11, 12]. Leptin is thus well designed to communicate body energy status throughout the body. Indeed, low leptin levels enhance appetite and decrease energy utilization by initiating the endocrine starvation response [13]. Adequate leptin levels moderate appetite and permit normal growth and reproduction in addition to activating the thyroid axis and suppressing the production of adrenal corticosteroids (Fig. 1); leptin also activates the sympathetic nervous system [14].

Summary of leptin action. Leptin is secreted by adipocytes as a signal of fat storage. Leptin binds to the long form of the leptin receptor (LRb) in hypothalamic nuclei to increase metabolic rate and to permit the production of hormones required for thyroid function, reproductive function and growth. Leptin also acts to suppress feeding (thus reducing body weight) and also to suppress the production of adrenal corticosteroids (in mice). The ability of leptin to inhibit feeding may be lost in obesity, suggesting leptin resistance
Lack of the leptin signal in mice (and humans) genetically null for leptin (ob/ob mice) or the leptin receptor (LR; db/db mice) results in early-onset obesity secondary to increased feeding and decreased energy utilization [11, 12, 15, 16]. These mouse models also display a phenotype reminiscent of the neuroendocrine starvation response, including hypothyroidism, hypercorticosteronism, decreased growth, and infertility. Indeed, exogenous leptin replacement during food restriction normalizes each of these parameters as well as decreasing appetite [13]. Interestingly, while the evidence from humans supports the idea that leptin regulates appetite as well as thyroid, growth and gonadal function, the adrenal axis does not appear to be regulated by leptin in humans [17].
Leptin receptors and sites of leptin action
The primary LR transcript is alternately spliced to yield a number of isoforms; in the rodent, these are termed LRa–LRf [18, 19, 20]. A related transcript from within the LR genomic DNA produces no receptor, but the transcript (leptin receptor gene-related transcript) may regulate transcription from the leptin receptor promotor [21]. LRa–LRd and LRf contain identical extracellular leptin binding and transmembrane domains as well as the same first 29 intracellular amino acids, and diverge in sequence secondary to alternative splicing of 3′ exons [22]. LRe contains only the coding sequences for extracellular leptin binding domains and is secreted; proteolytic cleavage of the extracellular domains of at least some other isoforms also contributes to circulating LR. These circulating LR isoforms complex with circulating leptin and may regulate leptin availability [23].
LRa–LRd, and LRf, the isoforms containing transmembrane domains, fall into two classes: short, and long. The long form, LRb, is highly conserved among species and possesses an intracellular domain of approximately 300 residues [24, 25]. Of the short forms (LRa, LRc, LRd, LRf) only LRa is conserved across mammalian species, and the intracellular domains contain only 32–40 amino acids in the rodent. While the function of the short LR forms remains unclear, LRb is critical for leptin action. Indeed, the originally described db/db mice lack only LRb (as a consequence of a mutation that results in missplicing of the LRb message) but exhibit a phenotype indistinguishable from that of leptin-deficient ob/ob animals and of db 3J /db 3J mice (which are deficient in all LR isoforms) [11, 24, 25].
Much of the action of leptin is attributable to effects in the CNS, especially in the basomedial hypothalamus, the site of highest LRb expression [26, 27]. Indeed, transgenic replacement of LRb that is restricted to neurons in db 3J /db 3J animals attenuates the majority of the db/db phenotype [28]. While a saturable transport system has been shown to move leptin across the blood-brain barrier, the sites of highest LRb expression lie within extremely close proximity to the median eminence, and it is likely that leptin reaches these target hypothalamic neurons without requiring transport across the blood-brain barrier [29, 30]. In the hypothalamus leptin acts on neurons that regulate levels of circulating hormones (e.g., thyroid hormone, sex steroids, and growth hormone) [26, 31]. Leptin action on these hypothalamic neurons also regulates the activity of the autonomic nervous system, although direct leptin action on brainstem LRb-expressing neurons likely plays an important role as well [32, 33].
Leptin resistance and obesity
Over one-quarter of adult Americans are obese, and the incidence of obesity continues to rise in industrialized nations. Obesity is a major risk factor for type 2 diabetes, cardiovascular disease, and some forms of cancer [34]. Since administration of leptin to rodents decreases food intake and blocks the diurnal decrease in energy expenditure, resulting in loss of fat mass, leptin was initially hailed as a potential cure for obesity [11, 12, 24, 26, 31]. With the exception of humans with (rare) genetic leptin deficiency [15, 17], however, circulating leptin levels are correlated with body mass index and total body fat mass. Hence obese individuals have elevated circulating leptin levels, but this leptin fails to mediate weight loss, suggesting that most human obesity is a form of leptin resistance. It is not clear whether this leptin resistance is causative or represents feedback inhibition due to elevated circulating leptin in obesity, however. Indeed, although therapy with exogenous leptin does augment weight loss, the effects of leptin are modest under the conditions that have been tested [35].
A number of potential mechanisms have been postulated to underlie leptin resistance, including defects in leptin access into the brain, in LRb signaling, and in pathways/neurons that mediate downstream leptin action. Since leptin can likely access the appetite centers of the basomedial hypothalamus without relying on a specific transport mechanism, we favor the hypothesis that alterations in leptin signaling or in pathways downstream of leptin action in the hypothalamus mediate leptin resistance.
LR signaling
LRb mediates Jak2 activation
LRb belongs to the interleukin-6 receptor family of class 1 cytokine receptors which contain an extracellular ligand-binding domain, a single transmembrane domain, and a cytoplasmic signaling domain [24, 36]. As with other cytokine receptors, LRb does not contain intrinsic enzymatic activity but instead signals via a noncovalently associated tyrosine kinase of the Jak kinase family (Jak2 in the case of LRb) [37, 38, 39]. Unliganded LRb exists as a preformed homodimer; leptin binding alters the conformation of the LRb dimer, enabling transphosphorylation and activation of the intracellular LRb-associated Jak2 molecules [24, 40, 41]. The activated Jak2 molecule then phosphorylates other tyrosine residues within the LRb/Jak2 complex to mediate downstream signaling [42, 43].
Signaling by cytokine receptors requires a proline-rich “box 1” motif critical for Jak kinase interaction and activation; additional less-conserved sequences COOH-terminal to box 1 (sometimes referred to as “box 2”) are also important for Jak kinase interactions and likely function in Jak kinase isoform selectivity [36, 37, 39]. In the case of LRb intracellular residues 31–36 (i.e., immediately downstream of the alternative splice junction following amino acid 29) compose box 2 [39]. Homology between the box 2 regions of LRb and other Jak2-associated cytokine receptors suggests that a loosely conserved E/N-X0–2-E/N-X0–2-L/I motif mediates Jak2 association [39]. This motif is absent from all described short LR isoforms, explaining the inability of these molecules to mediate leptin action in db/db animals [24, 39, 42]. These data suggest that ability of LRa to activate extracellular signal regulated kinase (ERK) signaling in transfected cells likely represents an artifact of Jak2 overexpression [44].
Phosphotyrosine-dependent signaling by LRb
Tyrosine kinase-dependent signaling generally proceeds via the phosphotyrosine-dependent recruitment of signaling proteins that contain specialized phosphotyrosine-binding domains (e.g., Src homology, SH, 2 domains) [45]. Each SH2 domain isoform recognizes phosphotyrosine in a specific amino acid context. Thus, while tyrosine phosphorylation acts as a molecular switch to recruit SH2-containing proteins, each tyrosine phosphorylation site recruits only specific SH2 isoforms since they recognize the surrounding amino acids as well as the phosphotyrosine residue. For instance, the SH2 domain of the latent transcription factor, signal transducer and activator of transcription (STAT) 3, is recruited to phosphotyrosine in the context of a Y (P)XXQ motif [46, 47].
Understanding signaling by the LRb/Jak2 complex thus requires defining the tyrosine phosphorylation sites on LRb and Jak2 and the SH2 proteins that they recruit. There are three conserved residues on the intracellular domain of LRb Tyr985, Tyr1077, and Tyr1138 [24, 43, 42]. Tyr985 and Tyr1138 are phosphorylated upon leptin binding, while Tyr1077 is not phosphorylated and does not contribute to leptin signaling [43].
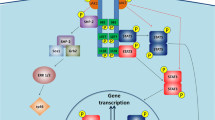
There are thus three primary intracellular signaling pathways that emanate from LRb (Fig. 2): Those originating directly from Jak2 tyrosine phosphorylation sites, from Tyr985 of LRb, and from Tyr1138 of LRb. The phosphorylation of Tyr985 creates a binding site for the COOH-terminal SH2 domain of the tyrosine phosphatase, src homology 2 containing phosphatase (SHP) 2. Recruitment of SHP-2 results in its tyrosine phosphorylation and recruitment of GRB2, the first step in the canonical p21ras→ERK signaling pathway. While Tyr985 thus mediates the majority of ERK stimulation during LRb signaling, a small amount of ERK activity occurs independently of LRb phosphorylation, presumably via tyrosine phosphorylation sites on Jak2 [39, 43, 48].

Intracellular leptin signaling via LRb. Leptin binding to LRb activates the associated Jak2 tyrosine kinase, which in turn phosphorylates Tyr985 and Tyr1138 of the intracellular tail of LRb. Phosphorylated Tyr985 recruits SHP-2, which becomes phosphorylated, recruits GRB2 and activates the ERK signaling pathway. Phosphorylated Tyr1138 binds STAT3, resulting in its tyrosine phosphorylation and subsequent nuclear translocation and transcriptional activation, mediating the translation of several genes including the feedback inhibitor, SOCS3. Multiple signals emanate directly from Jak2, including a minor component of the ERK pathway and a number of other poorly defined pathways. While the Tyr1138→STAT3 pathway is central for melanocortin action and lactation, other signals predominate in the control of NPY-mediated physiology
Phosphorylation of Tyr1138 recruits STAT3 to the LRb/Jak2 complex, resulting in tyrosine phosphorylation and subsequent nuclear translocation of STAT3 to mediate transcriptional regulation [42, 43]. Among other genes, STAT3 mediates the transcription of the SH2 domain containing feedback inhibitor, suppressor of cytokine signaling (SOCS) 3 [43, 49]. SOCS3 binds to Tyr985 of LRb to mediate inhibition of LRb→STAT3 signaling [50]. Jak2 tyrosine phosphorylation during LRb stimulation mediates some signals independently of tyrosine phosphorylation sites on LRb (e.g., a portion of ERK activation) [43]. Unfortunately, most Jak2 tyrosine phosphorylation sites have not been defined, impairing our understanding of the mechanisms by which Jak2-dependent signals are mediated. One Jak2-dependent pathway that is partially understood is the activation of the insulin receptor substrate protein/phosphatidylinositol 3 kinase pathway that is an area of crosstalk with insulin signaling [51, 52].
Leptin regulation of neural networks and neurophysiology
While LRb is expressed at other sites, the highest levels of LRb expression in the body are found in neurons of the nuclei of the basomedial hypothalamus, including the arcuate (ARC), dorsomedial hypothalamic, and ventromedial hypothalamic (VMH) nuclei and possibly in lower levels in the paraventricular nucleus [27]. Chemical or physical ablation of these nuclei results in increased feeding and neuroendocrine abnormalities that are similar to the phenotypes of db/db or ob/ob mice, suggesting that these hypothalamic nuclei that make up the so-called “satiety center” are critical sites of leptin action [26, 53].
Arcuate NPY and POMC neurons are major sites of leptin action
Within these nuclei of the basomedial hypothalamus LRb is expressed at its highest levels in the ARC. Within the ARC, LRb is found in at least two distinct populations of neurons: (a) neurons that coexpress neuropeptide Y (NPY) and agouti-related peptide (AgRP) and (b) neurons that express pro-opiomelanocortin (POMC) [26, 53]. POMC is processed to α-melanocyte stimulating hormone (αMSH) in the LRb/POMC neuron, which mediates a powerful anorectic (appetite-suppressing) signal via activation of melanocortin receptors (MC3R and MC4R). LRb stimulates the expression of POMC and activates the LRb/POMC neuron [53, 54]. AgRP is an antagonist of αMSH signaling, and NPY is itself an orexigenic (appetite-stimulating) hormone that also acts to suppress the central LRb growth and reproductive axes [55, 56, 57, 58]. Leptin acts via LRb to inhibit the NPY/AgRP neurons and to suppress expression of these neuropeptides. Thus leptin→LRb signaling stimulates the production of anorectic neuropeptides and suppresses levels of orexigenic peptides. Conversely, when leptin action is decreased or deficient (e.g., starvation, ob/ob, and db/db mice), appetite is stimulated via the suppression of anorectic neuropeptides (e.g., POMC) and by increased expression of orexigenic peptides (e.g., NPY and AgRP) [26, 53]. Other distinct populations of LRb-expressing neurons may also be found in the ARC [59].
Neuroendocrine control in the hypothalamus
The regulation of endocrine function begins in the hypothalamus, where neurons that synthesize releasing factors (e.g., thyrotropin-releasing hormone, TRH) secrete these factors in to the specialized portal circulation that carries these factors to the anterior portion of the pituitary gland. Within the pituitary the hypothalamic-releasing factors influence the synthesis of stimulating factors (e.g., thyrotropin) and their elaboration into the general circulation, where they regulate the activity of endocrine organs and the production of hormones (e.g., thyroid hormone) by these organs.
Much of the central control of thyroid and adrenal function occurs in the parvocellular neurons of the paraventricular hypothalamus (PVH) [60]. TRH neurons within the VMH release TRH to stimulate the secretion of thyrotropin in the pituitary and promote thyroid hormone production in the thyroid gland. Similarly, corticotropin-releasing hormone (CRH) neurons in the VMH elaborate CRH/corticotropin-releasing factor to promote the elaboration of corticotropin in the pituitary and the production of glucocorticoids from the adrenal cortex. Leptin acts to stimulate the thyroid axis by increasing the production and release of TRH. In mice, leptin also decreases circulating levels of corticotropin and corticosteroids, and these leptin effects are correlated with the inhibition of PVH CRH production by leptin [13].
The elaboration of growth hormone (GH) by the pituitary gland is regulated by numerous hypothalamic inputs, including the GH-releasing hormone GHRH that is secreted into the tubuloinfundibular system by ARC GHRH neurons to stimulate GH secretion, and somatostatin, which is secreted by ARC and periventricular neurons to block GH production. Leptin increases GHRH synthesis and secretion from the hypothalamus and inhibits somatostatin production and release [61, 62, 63]. The hypothalamic control of the release of pituitary gonadotropins is mediated in large part by the pulsatile secretion of gonadotropin-releasing hormone (GnRH) in the median preoptic area of the hypothalamus. Leptin increases the amplitude and frequency of GnRH pulses and the production of pituitary gonadotropins [64].
LRb→STAT3 Signaling in the regulation of physiology
LRb signaling via STAT3 mediates a subset of leptin actions
We have directly addressed the contribution of the LRb→STAT3 signaling pathway to the control of mammalian physiology by studying homologously targeted “knock-in” mice in which LRb is replaced by a mutant molecule (LRbS1138) that contains a substitution mutation of Tyr1138 (the STAT3 binding site) [58]. While LRbS1138 fails to mediate activation of STAT3 during leptin signaling, this mutant regulates all other LRb signaling pathways normally. Use of the “knock-in” approach ensured that the expression pattern and levels of LRbS1138 mirror that of wild-type LRb. As in the case of db/db animals, mice homozygous for LRbS1138 (s/s) display hyperphagia and decreased energy expenditure, resulting in massive early-onset obesity that is associated with increased serum leptin levels. The high circulating leptin levels in s/s animals are not only correlated with increased adipose mass in these mice but also indicate resistance to the energy homeostatic effects of leptin. Furthermore, as with db/db mice, s/s animals display elevated glucocorticoid and decreased thyroid levels (S.H. Bates and M.G. Myers Jr., unpublished observations). Although the role that LRb→STAT3 signaling plays in the leptin-mediated activation of the sympathetic nervous system is not yet known, phosphatidylinositol 3 kinase signaling is required for this leptin action, as it is for feeding [14, 51].
Important differences exist between the phenotypes of s/s mice (missing only the LRb→STAT3 signal) and db/db mice (devoid of all leptin signals), however [58]. While db/db animals are infertile and demonstrate decreased linear growth, s/s mice retain relatively normal gonad function and actually demonstrate increased linear growth compared to wild-type animals. Hence LRb→STAT3 signaling is central to the regulation of energy balance and the neuroendocrine thyroid and adrenal axes but is dispensable for regulation of the hypothalamic control of the gonadal and growth axes.
Interestingly, although s/s animals are fertile, the offspring of s/s females die within 48 h after birth without milk in their stomachs, although it is possible to foster them to wild-type females. Similarly, leptin-deficient ob/ob females rendered fertile by leptin treatment fail to lactate if leptin is withdrawn at the onset of pregnancy, but lactate normally if leptin therapy is continued, suggesting that leptin provides a permissive signal to the control of lactation [65, 66]. To investigate the hormonal basis for this defect we assayed prolactin levels in wild-type and s/s females immediately postpartum and found that circulating prolactin was undetectable in the s/s mothers (Fig. 3). Thus, although LRb→STAT3 signaling is not required for fertility, it is required in a pathway that mediates the elaboration of prolactin and lactation.

Gonad function and lactation in s/s animals. Left panel Ovaries from 20-week-old female s/s and db/db mice (n=5 per genotype) were fixed and stained with hematoxylin and eosin and scored for the number of ovulated follicles (corpora lutea) in each ovary (mean ±SEM). Ovaries from s/s mice revealed histological evidence of ovulation and had normal number of corpora lutea, while db/db females had atrophic reproductive organs (data not shown) and displayed no evidence of ovulation. Right panel Since s/s females were unable to suckle their progeny, serum prolactin was assessed in female mice within 24 h postpartum (Biotrak assay, Amersham). Circulating prolactin levels were virtually undetectable in s/s mice compared to wild-type, indicating that the LRb-STAT3 signal is required for leptin control of prolactin secretion and lactation, although not for gonadal function
LRb→STAT3 signaling and the regulation of ARC neuropeptides
Analysis of hypothalamic neuropeptide expression reveals that, as with db/db mice, s/s mice have decreased POMC and increased AgRP mRNA levels in the hypothalamus [58]. In contrast, while db/db animals display dramatic induction of hypothalamic NPY mRNA, levels of NPY message are near normal in s/s animals. These data suggest that LRb→STAT3 signaling is a critical regulator of hypothalamic melanocortin action, and that dysregulated melanocortin signaling (as opposed to alterations in NPY) accounts for the obesity of s/s animals. Additionally, non-STAT3 LRb signals are critical regulators of NPY expression in the LRb/NPY neuron.
An integrated model for the control of hypothalamic neuroendocrine function by leptin
Our analysis of s/s animals that display severe impairment of the melanocortin system with relative preservation of NPY regulation suggests a model in which melanocortin signaling is dominant signal in the control of feeding, although the slightly decreased feeding in s/s than in db/db animals suggests a role for other pathways, such as NPY (Fig. 4). Indeed, the feeding and weight gain phenotype of s/s animals closely mirrors that of ob/ob animals genetically devoid of NPY (ob/ob, Npy --/--) [57].

A model of LRb signaling in the control of hypothalamic neuropeptides and physiology. In the LRb/POMC coexpressing arcuate neuron leptin increases POMC production via STAT3, generating an anorectic signal via αMSH and the melanocortin receptors MC3R and MC4R. In the LRb/NPY/AgRP neuron, leptin inhibits AgRP production in part via the LRb-STAT3 pathway, disinhibiting melanocortin signaling. Melanocortins powerfully stimulate anorexia and energy expenditure. Melanocortin action likely regulates TRH expression in the PVH. Leptin acts to inhibit NPY expression independently of STAT3 signaling, perhaps via the insulin receptor substrate protein→phosphatidylinositol 3′ kinase pathway. Elevated NPY levels (suppressed by leptin) contribute a component of energy balance via the regulation of feeding and energy expenditure but strongly inhibit the function of the reproductive and growth axes. Although likely to be regulated by LRb→STAT3 signaling in mice, the details by which CRH expression in the PVH is regulated by this signal have not been established. Red pathways Regulation by STAT3; blue pathways independent of STAT3; arrowheads positive effectors; bars inhibition
In the growth axis arcuate NPY blocks GHRH and stimulates somatostatin, effectively inhibiting GH elaboration from the hypothalamus [67] The suppression of ARC NPY action by leptin may mediate the permissive effect of leptin on this axis [63, 68]. Similarly, much of the leptin effect on the gonadal axis may be mediated via blockade of NPY action by leptin, as NPY provides a powerful block on GnRH secretion [68, 69]. The phenotype of s/s animals is consistent with the proposed role for NPY in suppressing the hypothalamic growth and gonadal axes; thus the increased NPY signaling in ob/ob and db/db mice may only modestly increase feeding, but may be responsible primarily for infertility and growth retardation in these mouse models. Indeed the phenotype of ob/ob, Npy −/− animals displays important similarities with the s/s phenotype [70]. Both display restoration of the hypothalamic/gonadal axis and increased linear growth with only modestly attenuated obesity compared to ob/ob and db/db mice.
Similarly, numerous data suggest that melanocortin action is dispensable for reproduction and growth. Comparison to the dramatic effects observed with manipulation of the NPY system, pharmacological, or genetic manipulation of hypothalamic melanocortin action does not appreciably alter fertility or its hypothalamic control [68, 71]. Furthermore, acute blockade of the melanocortin system does not alter growth or the hypothalamic control of the growth axis but chronic attenuation of melanocortin function (as in MC4R−/− and s/s mice) actually increases the activity of the growth axis, presumably by increasing adiposity (and thus leptin levels) and exaggerating the (STAT3-independent) inhibition of ARC NPY neurons by leptin.
The mechanisms by which leptin regulates the thyroid and adrenal axes have been more difficult to dissect. LRb is expressed at very low levels within the PVH (where TRH and CRH neurons are found), suggesting that much of this regulation is indirect [29, 31]. Indeed, while some data suggest a direct role for leptin on TRH neurons, many data point to a critical role for αMSH and NPY projections from the ARC in the regulation of TRH [17, 72, 73]. Our unpublished observations suggest that thyroid function is similarly suppressed in s/s and db/db animals, which is consistent with the notion that melanocortin action is a critical mechanism by which leptin controls thyroid function.
The mechanism by which leptin regulates the CRH neurons of the PVH is unclear; although both melanocortin agonists and NPY may modulate CRH levels and activity, no arcuate NPY or POMC projections have yet been mapped onto these neurons. Furthermore, the PVH CRH neuron may be regulated divergently by leptin in different species, as leptin increases CRH expression in the PVH in the rat [74, 75]. Recall, as well, that the adrenal axis does not appear to be altered in humans that lack leptin [17].
Perhaps the least understood of all of the neuroendocrine systems regulated by leptin is lactation, however. Since lactation is blocked in s/s animals, NPY is not likely to be the hypothalamic regulator of PRL secretion. Furthermore, it is clear that melanocortin action is dispensable for lactation, as animals that overexpress the agouti melanocortin antagonist or that lack melanocortin receptors are fertile and suckle their young normally [71, 76]. Identifying the leptin-regulated neuron that controls prolactin secretion and lactation will clearly be an important step in understanding this process.
Summary
Models of disrupted leptin signaling have provided important tools in the investigation of leptin physiology, it is clear however, that there are many issues that remain to be explored regarding leptin signals in the control of physiology and especially in s/s mice. The regulation of a number of neuropeptides has yet to be explored in s/s animals (including CART and GALP, among others). Also the development of the LRb-expressing neurons and the ability of leptin to control membrane potential in these animals need to be investigated. The physiological roles of most other intracellular LRb-mediated signals have yet to be investigated, as well.
Abbreviations
- AgRP :
-
Agouti-related peptide
- ARC :
-
Arcuate nucleus
- CRH :
-
Corticotropin-releasing hormone
- ERK :
-
Extracellular signal regulated kinase
- GH :
-
Growth hormone
- GHRH :
-
Growth hormone releasing hormone
- GnRH :
-
Gonadotropin-releasing hormone
- LR :
-
Leptin receptor
- MCR :
-
Melanocortin receptor
- MSH :
-
Melanocyte stimulating hormone
- NPY :
-
Neuropeptide Y
- POMC :
-
Pro-opiomelanocortin
- PVH :
-
Paraventricular hypothalamus
- SH :
-
Src homology
- SHP :
-
Src homology 2 containing phosphatase
- SOCS :
-
Suppressor of cytokine signaling
- STAT :
-
Signal transducer and activator of transcription
- TRH :
-
Thyrotropin-releasing hormone
- VMH :
-
Ventromedial hypothalamus
References
MacDougald OA, Hwang CS, Fan H, Lane MD (1995) Regulated expression of the obese gene product (leptin) in white adipose tissue and 3T3–L1 adipocytes. Proc Natl Acad Sci U S A 92:9034–9037
Rentsch J, Chiesi M (1996) Regulation of ob gene mRNA levels in cultured adipocytes. FEBS Lett 379:55–59
Slieker LJ, Sloop KW, Surface PL, Kriauciunas A, LaQuier F, Manetta J, Bue-Valleskey J, Stephens TW (1996) Regulation of expression of ob mRNA and protein by glucocorticoids and cAMP. J Biol Chem 271:5301–5304
Maffei M, Halaas J, Ravussin E, Pratley RE, Lee GH, Zhang Y, Fei H, Kim S, Lallone R, Ranganathan S, Kern PA, Friedman JM (1995) Leptin levels in human and rodent: measurement of plasma leptin and ob RNA in obese and weight-reduced subjects. Nat Med 1:1155–1161
Frederich RC, Hamann A, Anderson S, Lollmann B, Lowell BB, Flier JS (1995) Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nat Med 1:1311–1314
Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, Ohannesian JP, Marco CC, McKee LJ, Bauer TL, Caro JF (1996) Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med 334:292–295
Licinio J, Mantzoros C, Negrao AB, Cizza G, Wong ML, Bongiorno PB, Chrousos GP, Karp B, Allen C, Flier JS, Gold PW (1997) Human leptin levels are pulsatile and inversely related to pituitary-adrenal function. Nat Med 3:575–579
Bornstein SR, Uhlmann K, Haidan A, Ehrhart-Bornstein M, Scherbaum WA (1997) Evidence for a novel peripheral action of leptin as a metabolic signal to the adrenal gland: leptin inhibits cortisol release directly. Diabetes 46:1235–1238
Grunfeld C, Zhao C, Fuller J, Pollack A, Moser A, Friedman J, Feingold KR (1996) Endotoxin and cytokines induce expression of leptin, the ob gene product, in hamsters. J Clin Invest 97:2152–2157
Sarraf P, Frederich RC, Turner EM, Ma G, Jaskowiak NT, Rivet DJ, III, Flier JS, Lowell BB, Fraker DL, Alexander HR (1997) Multiple cytokines and acute inflammation raise mouse leptin levels: potential role in inflammatory anorexia. J Exp Med 185:171–175
Friedman JM, Halaas JL (1998) Leptin and the regulation of body weight in mammals. Nature 395:763–770
Elmquist JK, Maratos-Flier E, Saper CB, Flier JS (1998) Unraveling the central nervous system pathways underlying responses to leptin. Nature Neuroscience 1:445–449
Ahima RS, Prabakaran D, Mantzoros CS, Qu D, Lowell BB, Maratos-Flier E, Flier JS (1996) Role of leptin in the neuroendocrine response to fasting. Nature 382:250–252
Rahmouni K, Haynes WG, Morgan DA, Mark AL (2003) Intracellular mechanisms involved in leptin regulation of sympathetic outflow. Hypertension 41:763–767
Montague CT, Farooqi IS, Whitehead JP, Soos MS, Rau H, Wareham NJ, Sewter CP, Digby JE, Mohammed SN, Hurst JA, Cheetham CH, Early AR, Barnett AH, Prins JB, O’Rahilly S (1997) Congenital leptin deficiency is associated with severe early onset obesity in humans. Nature 387:903–908
Clement K, Vaisse C, Lahlou N, Cabrol S, Pelloux V, Cassuto D, Gourmelen M, Dina C, Chambaz J, Lacorte JM, Basdevant A, Bougneres P, leBouc Y, Froguel P, Guy-Grand B (1998) A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature 392:398–401
Farooqi IS, Matarese G, Lord GM, Keogh JM, Lawrence E, Agwu C, Sanna V, Jebb SA, Perna F, Fontana S, Lechler RI, Depaoli AM, O’Rahilly S (2002) Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J Clin Invest 110:1093–1103
Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J, Devos R, Richards GJ, Campfield LA, Clark FT, Deeds J, Muir C, Sanker S, Moriarty A, Moore KJ, Smutko JS, Mays GG, Woolf EA, Monroe CA, Tepper RI (1995) Identification and expression cloning of a leptin receptor, OB-R. Cell 83:1263–1271
Lee GH, Proenca R, Montez JM, Carroll KM, Darvishzadeh JG, Lee JI, Friedman JM (1996) Abnormal splicing of the leptin receptor in diabetic mice. Nature 379:632–635
Wang MY, Zhou YT, Newgard CB, Unger RH (1996) A novel leptin receptor isoform in rat. Growth Regul 392:87–90
Mercer JG, Moar KM, Hoggard N, Strosberg AD, Froguel P, Bailleul B (2000) B219/OB-R 5’-UTR and leptin receptor gene-related protein gene expression in mouse brain and placenta: tissue-specific leptin receptor promoter activity. J Neuroendocrinol 12:649–655
Chua SC Jr, Chung WK, Wu-Peng XS, Zhang Y, Liu SM, Tartaglia LA, Leibel RL (1996) Phenotypes of mouse diabetes and rat fatty due to mutations in the OB (leptin) receptor. Science 271:994–996
Ge H, Huang L, Pourbahrami T, Li C (2002) Generation of soluble leptin receptor by ectodomain shedding of membrane-spanning receptors in vitro and in vivo. J Biol Chem 277:45898–45903
Tartaglia LA (1997) The leptin receptor. J Biol Chem 272:6093–6096
Chua SC Jr, Koutras IK, Han L, Liu SM, Kay J, Young SJ, Chung WK, Leibel RL (1997) Fine structure of the murine leptin receptor gene: splice site suppression is required to form two alternatively spliced transcripts. Genomics 45:264–270
Elmquist JK, Elias CF, Saper CB (1999) From lesions to leptin: hypothalamic control of food intake and body weight. Neuron 22:221–232
Elmquist JK, Bjorbaek C, Ahima RS, Flier JS, Saper CB (1998) Distributions of leptin receptor mRNA isoforms in the rat brain. J Comp Neurol 395:535–547
Kowalski TJ, Liu SM, Leibel RL, Chua SC Jr (2001) Transgenic complementation of leptin-receptor deficiency. I. Rescue of the obesity/diabetes phenotype of LEPR-null mice expressing a LEPR-B transgene. Diabetes 50:425–435
Bjorbaek C, Elmquist JK, Michl P, Ahima RS, van Bueren A, McCall AL, Flier JS (1998) Expression of leptin receptor isoforms in rat brain microvessels. Endocrinology 139:3485–3491
Banks WA, Kastin AJ, Huang W, Jaspan JB, Maness LM (1996) Leptin enters the brain by a saturable system independent of insulin. Peptides 17:305–311
Inui A (1999) Feeding and body-weight regulation by hypothalamic neuropeptides-mediation of the actions of leptin. Trends Neurosci 22:62–67
Grill HJ, Schwartz MW, Kaplan JM, Foxhall JS, Breininger J, Baskin DG (2002) Evidence that the caudal brainstem is a target for the inhibitory effect of leptin on food intake. Endocrinology 143:239–246
Elmquist JK, Ahima RS, Maratos-Flier E, Flier JS, Saper CB (1997) Leptin activates neurons in ventrobasal hypothalamus and brainstem. Endocrinology 138:839–842
Roth J (1998) Diabetes and obesity. Diabetes Metab Rev 13:1–2
Gura T (1999) Obesity research. Leptin not impressive in clinical trial. Science 286:881–882
Taga T, Kishimoto T (1997) gp130 and the interleukin-6 family of cytokines. Annu Rev Immunol 15:797–819
Ihle J N, Kerr IM (1995) Jaks and Stats in signaling by the cytokine receptor superfamily. Trends Genet 11:69–74
Taniguchi T (1995) Cytokine signaling through nonreceptor protein tyrosine kinases. Science 268:251–255
Kloek C, Haq AK, Dunn SL, Lavery HJ, Banks AS, Myers MG Jr (2002) Regulation of Jak kinases by intracellular leptin receptor sequences. J Biol Chem 277:41547–41555
Devos R, Guisez Y, Van der Heyden J, White DW, Kalai M, Fountoulakis M, Plaetinck G (1997) Ligand-independent dimerization of the extracellular domain of the leptin receptor and determination of the stoichiometry of leptin binding. J Biol Chem 272:18304–18310
Couturier C, Jockers R (2003) Activation of the leptin receptor by a ligand-induced conformational change of constitutive receptor dimers. J Biol Chem 278:26604–26611
White DW, Kuropatwinski KK, Devos R, Baumann H, Tartaglia LA (1997) Leptin receptor (OB-R) signaling. J Biol Chem 272:4065–4071
Banks AS, Davis SM, Bates SH, Myers MG Jr (2000) Activation of downstream signals by the long form of the leptin receptor. J Biol Chem 275:14563–14572
Bjorbaek C, Uotani S, da Silva B, Flier JS (1997) Divergent signaling capacities of the long and short isoforms of the leptin receptor. J Biol Chem 272:32686–32695
Koch CA, Anderson DJ, Moran MF, Ellis CA, Pawson T (1991) SH2 and SH3 domains: elements that control interactions of cytoplasmic signaling proteins. Science 252:668–674
Songyang Z, Shoelson SE, Chaudhuri M, Gish GD, Pawson T, Haser WG, King F, Roberts T, Ratnofsky S, Lechleider RJ, Neel BG, Birge RB, Fajardo JE, Chou MM, Hanafusa H, Schaffhausen B, Cantley LC (1993) SH2 domains recognize specific phosphopeptide sequences. Cell 72:767–778
Haan S, Hemmann U, Hassiepen U, Schaper F, Schneider-Mergener J, Wollmer A, Heinrich PC, Grotzinger J (1999) Characterization and binding specificity of the monomeric STAT3-SH2 domain. J Biol Chem 274:1342–1348
Bjorbaek C, Buchholz RM, Davis SM, Bates SH, Pierroz DD, Gu H, Neel BG, Myers MG Jr, Flier JS (2001) Divergent roles of SHP-2 in ERK activation by leptin receptors. J Biol Chem 276:4747–4755
Bjorbaek C, Elmquist JK, Frantz JD, Shoelson SE, Flier JS (1998) Identification of SOCS-3 as a potential mediator of central leptin resistance. Mol Cell 1:619–625
Bjorbak C, Lavery HJ, Bates SH, Olson RK, Davis SM, Flier JS, Myers MG Jr (2000) SOCS3 mediates feedback inhibition of the leptin receptor via Tyr985. J Biol Chem 275:40649–40657
Niswender KD, Morton GJ, Stearns WH, Rhodes CJ, Myers MG Jr, Schwartz MW (2001) Intracellular signalling. Key enzyme in leptin-induced anorexia. Nature 413:794–795
Niswender KD, Morrison CD, Clegg DJ, Olson R, Baskin DG, Myers MG Jr, Seeley RJ, Schwartz MW (2003) Insulin activation of phosphatidylinositol 3-kinase in the hypothalamic arcuate nucleus: a key mediator of insulin-induced anorexia. Diabetes 52:227–231
Schwartz MW, Woods SC, Porte D Jr, Seeley RJ, Baskin DG (2000) Central nervous system control of food intake. Nature 404:661–671
Cowley MA, Smart JL, Rubinstein M, Cerdan MG, Diano S, Horvath TL, Cone RD, Low MJ (2001) Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 411:480–484
Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD (1997) Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature 385:165–168
Seeley RJ, Yagaloff KA, Fisher SL, Burn P, Thiele TE, van Dijk G, Baskin DG, Schwartz MW (1997) Melanocortin receptors in leptin effects. Nature 390:349
Erickson JC, Hollopeter G, Palmiter RD (1996) Attenuation of the obesity syndrome of ob/ob mice by the loss of neuropeptide Y. Science 274:1704–1707
Bates SH, Stearns WH, Schubert M, Tso AWK, Wang Y, Banks AS, Dundon TA, Lavery HJ, Haq AK, Maratos-Flier E, Neel BG, Schwartz MW, Myers MG Jr (2003) STAT3 signaling is required for leptin regulation of energy balance but not reproduction. Nature 421:856–859
Jureus A, Cunningham MJ, McClain ME, Clifton DK, Steiner RA (2000) Galanin-like peptide (GALP) is a target for regulation by leptin in the hypothalamus of the rat. Endocrinology 141:2703–2706
Hisano S, Fukui Y, Chikamori-Aoyama M, Aizawa T, Shibasaki T (1993) Reciprocal synaptic relations between CRF-immunoreactive- and TRH-immunoreactive neurons in the paraventricular nucleus of the rat hypothalamus. Brain Res 620:343–346
Burton KA, Kabigting EB, Clifton DK, Steiner RA (1992) Growth hormone receptor messenger ribonucleic acid distribution in the adult male rat brain and its colocalization in hypothalamic somatostatin neurons. Endocrinology 131:958–963
LaPaglia N, Steiner J, Kirsteins L, Emanuele M, Emanuele N (1998) Leptin alters the response of the growth hormone releasing factor-growth hormone-insulin-like growth factor-I axis to fasting. J Endocrinol 159:79–83
Tannenbaum GS, Gurd W, Lapointe M (1998) Leptin is a potent stimulator of spontaneous pulsatile growth hormone (GH) secretion and the GH response to GH-releasing hormone. Endocrinology 139:3871–3875
Yu WH, Kimura M, Walczewska A, Karanth S, McCann SM (1997) Role of leptin in hypothalamic-pituitary function. Proc Natl Acad Sci U S A 94:1023–1028
Mounzih K, Qiu J, Ewart-Toland A, Chehab FF (1998) Leptin is not necessary for gestation and parturition but regulates maternal nutrition via a leptin resistance state. Endocrinology 139:5259–5262
Malik NM, Carter ND, Murray JF, Scaramuzzi RJ, Wilson CA, Stock MJ (2001) Leptin requirement for conception, implantation, and gestation in the mouse. Endocrinology 142:5198–5202
Chan YY, Clifton DK, Steiner RA (1996) Role of NPY neurones in GH-dependent feedback signalling to the brain. Horm Res 45 [Suppl 1]:12–14
Pinkney JH, Goodrick SJ, Katz J, Johnson AB, Lightman SL, Coppack SW, Mohamed-Ali V (1998) Leptin and the pituitary-thyroid axis: a comparative study in lean, obese, hypothyroid and hyperthyroid subjects. Clin Endocrinol (Oxf) 49:583–588
Smith MS, Grove KL (2002) Integration of the regulation of reproductive function and energy balance: lactation as a model. Front Neuroendocrinol 23:225–256
Erickson JC, Hollopeter G, Palmiter RD (1996) Attenuation of the obesity syndrome of ob/ob mice by the loss of neuropeptide Y. Science 274:1704–1707
Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, Smith FJ, Campfield LA, Burn P, Lee F (1997) Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell 88:131–141
Legradi G, Emerson CH, Ahima RS, Flier JS, Lechan RM (1997) Leptin prevents fasting-induced suppression of prothyrotropin-releasing hormone messenger ribonucleic acid in neurons of the hypothalamic paraventricular nucleus. Endocrinology 138:2569–2576
Harris M, Aschkenasi C, Elias CF, Chandrankunnel A, Nillni EA, Bjoorbaek C, Elmquist JK, Flier JS, Hollenberg AN (2001) Transcriptional regulation of the thyrotropin-releasing hormone gene by leptin and melanocortin signaling. J Clin Invest 107:111–120
Mantzoros CS, Rosen HN, Greenspan SL, Flier JS, Moses AC (1997) Short-term hyperthyroidism has no effect on leptin levels in man. J Clin Endocrinol Metab 82:497–499
Schwartz MW, Baskin DG, Bukowski TR, Kuijper JL, Foster D, Lasser G, Prunkard DE, Porte D Jr, Woods SC, Seeley RJ, Weigle DS (1996) Specificity of leptin action on elevated blood glucose levels and hypothalamic neuropeptide Y gene expression in ob/ob mice. Diabetes 45:531–535
Butler AA, Cone RD (2002) The melanocortin receptors: lessons from knockout models. Neuropeptides 36:77–84
Acknowledgements
This research was supported by NIH DK56731 and DK 57768 and grants from the American Diabetes Association (to M.G.M.) and an American Diabetes Association/European Association for the Study of Diabetes Transatlantic Fellowship (to S.H.B.). We thank Michael Schwartz, M.D. for helpful discussions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bates, S.H., Myers, M.G. The role of leptin→STAT3 signaling in neuroendocrine function: an integrative perspective. J Mol Med 82, 12–20 (2004). https://doi.org/10.1007/s00109-003-0494-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-003-0494-z