
Abstract
Introduction
Oncogenic Ras-related GTP-binding proteins, referred to as Rabs, are characterized by their intricate interactions with upstream, downstream molecules, and notably, extracellular vesicles (EVs). While the expansive family of Rabs and their associated signaling pathways have been exhaustively dissected, Rab22a emerges as an entity of outstanding interest, owing to its potent influence in many biological processes and its conspicuous correlation with cancer metastasis and migration. A burgeoning interest in the interactions between Rab22a and EVs in the field of oncology underscores the necessity for more in-depth reviews and scholarly discourses.
Methods
We performed a review based on published original and review articles related to Rab22a, tumor, microRNA, exosome, microvesicles, EVs, CD147, lysosome, degradation, endosomal recycling, etc. from PubMed, Web of Science and Google Scholar databases.
Results and conclusions
We summarize the regulatory processes governing the expression of Rab22a and the mutants of Rab22a. Notably, the present understanding of complex interactions between Rab22a and EVs are highlighted, encompassing both the impact of Rab22a on the genesis of EVs and the role of EVs that are affected by Rab22a mutants in propelling tumor advancement. The dynamic interaction between Rab22a and EVs plays a significant role in the progression of tumors, and it can provide novel insights into the pathogenesis of cancers and the development of new therapeutic targets.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Within mammals, the Rab GTPase family includes over 60 members, distinguishing it as the most extensive branch within the Ras superfamily [1]. Rab22a, a pivotal member of the Rab5 subfamily, holds its place alongside Rab21, Rab5, and Rab22b (Rab31). The gene blueprint of Rab22a is etched onto the 20q13.32 region of the chromosome, crafting a protein comprising 194 amino acids. This protein comprises six distinct segments, encompassing one effector region, four regions involved in GTP-binding, and a single disordered region. Rab proteins, when activated, take residence within specific intracellular membranes. For Rab22a, its active form navigates to a diverse array of cellular compartments, inclusive of the Golgi apparatus, the endoplasmic reticulum (ER) membrane, early endosomes, sorting endosomes, non-canonical autophagosomes, and phagosomes [2, 3]. As an oncogene, the expression level of Rab22a presents clinically relevant implications, correlating with distant metastasis-free survival rates and patient mortality [4]. Numerous in vivo and in vitro experiments have shown clearly—that Rab22a regulation significantly influences tumor progression [4], thus highlighting its promising potential as a therapeutic target in oncology. Rab22a is known to contribute to tumor invasion and metastasis [5]; however, experiments that demonstrated the absence of Rab22a expression in cells do not impact primary tumor growth in certain cell lines [4].
Ejected by nearly every cell type and frequently detected in multiple biological fluids, extracellular vesicles (EVs) encapsulate a broad spectrum of membrane-enclosed particles [6, 7]. EVs are typically sorted into three categories: small EVs (sEVs), microvesicles (MVs), and apoptotic bodies, based on their respective sizes [6, 7]. The sEVs, otherwise known as exosomes, possess a diameter spanning from 30 to 100 nm, whereas MVs have a diameter ranging from 100 to 1000 nm, and apoptotic bodies measure between 1000 and 5000 nm [8, 9]. EVs wield a dual influence on tumor progression. Serving as delivery vehicles, EVs can ignite anti-tumor immune responses, thus offering significant therapeutic impacts on tumor progression. Given their ability to stimulate potent tumor-specific immunity, EVs are prime candidates for developing cell-free tumor vaccines. For example, immunostimulatory DNA within sEVs can be recognized by cytoplasmic DNA receptors in activated dendritic cells (DCs), thus triggering a STING-dependent pathway that propels anti-tumor immunity [10]. Furthermore, tumor-associated sEVs serve as a significant alternate source of tumor antigens, offering the potential to construct efficient and personalized cancer vaccines [11]. Conversely, EVs activated by various oncogenic Rab proteins can steer tumor progression in unique ways. For instance, blocking Rab27a and thus controlling EVs can lead to decreased primary tumor growth and lung dissemination of metastatic carcinoma, while it does not impact nonmetastatic carcinoma [12]. Contrarily, Rab31, another Rab protein interacting with EVs, prompts a switch from an invasive to a proliferative phenotype, as evidenced by increased cell proliferation, reduced adhesion and invasion, and a diminished ability to form lung metastases [13].
Interestingly, a significant interplay exists between EVs and Rab22a. The EVs triggered by Rab22a protein assume unique characteristics in tumor progression. Initially, Rab22a participates in the formation of a specialized class of EVs, known as Rab22a-induced EVs (R-EVs). These R-EVs can transmit STING, thus stimulating an anti-tumor response [3]. Second, some types of EVs, such as MVs, induced by Rab22a in tumor progression, only initiate their biogenesis under hypoxic conditions [4]. Third, while these EVs appear not to be involved in primary tumor growth, they actively participate in tumor invasion and metastasis [4]. EVs can also influence Rab22a through various non-coding RNAs. For instance, miR-204 and miR-193b can be enveloped by sEVs, thereby modulating Rab22a gene expression and providing potential avenues for anti-tumor therapy [14, 15].
In this review, we embark on an intellectual journey that spans the breadth and depth of Rab22a’s regulatory landscape. We summarize its changes at transcriptional and post-transcriptional levels and their subsequent contribution to the transformative role Rab22a assumes in cancer development and progression. Furthermore, we explore the multitude of mutations Rab22a can potentially undergo, the implications these alterations may have on its functional characteristics, and their influence on transforming the cancerous phenotype. We delve deeper into the intriguing role of Rab22a, investigating how Rab22a can significantly alter the tumor microenvironment through its interactions with EVs, and how it changes the course of cancer progression. Through these insights, we aspire to augment the scientific understanding of the role of Rab22a in oncogenesis and its potential value as a target for therapeutic interventions in cancers.
Regulation of Rab22a expression
Transcriptional regulation
Hypoxia-inducible factor (HIF) is a dimer composed of constitutively generated subunit β and oxygen-regulated subunit α. Under normoxic conditions, HIF-α is degraded through the ubiquitin-dependent proteasome pathway, effectively hindering subsequent pathways [16]. However, HIF-α becomes stabilized and subsequently translocates to the nucleus in hypoxia [16]. There, it interacts with co-factors such as aryl hydrocarbon receptor nuclear translocator (ARNT), CREB-binding protein (CBP)/p300, and the DNA polymerase II (Pol II) complex, enabling it to bind to hypoxia responsive elements (HREs) and stimulate transcription of target genes [17]. Among the various pathways through which cells respond to low oxygen levels, HIF serves as the principal regulator [18].
As an oncogene, Rab22a expression positively correlated with the hypoxic tumor microenvironment through the action of HIF. Supporting this notion, a specific HIF binding sequence, 5′-RCGTG-3′, was identified in the 5′-untranslated region (UTR) of exon 1 of the Rab22a gene, suggesting that Rab22a is indeed a target gene of HIF [4]. In a subsequent study that further substantiates these findings, HIF-knockdown effectively blocked the expression of hypoxia-induced Rab22a mRNA in MDA-435 and MDA-231 breast cancer cell lines exposed to hypoxic conditions [4].
Post-transcriptional regulation
MiR-204
MiR-204 is an intronic miRNA located in the TRPM3 gene [15], and serves as a critical regulator of Rab22a. Typically, miR-204 suppresses Rab22a, contributing to the control of various diseases and participating in numerous physiological processes. As a tumor suppressor, miR-204 has been found to be down-regulated in multiple human cancers, with CpG methylation-induced gene inactivation potentially accounting for this phenomenon [15]. By specifically suppressing Rab22a and other targets, miR-204 effectively inhibits cancer cell proliferation, promotes apoptosis, increases chemosensitivity, and impacts the progression and prognosis of different tumor types [15]. For instance, in gastric cancer, miR-204 represses cancer cell proliferation by silencing the expression of USP47 and Rab22a, both of which have been confirmed targets of miR-204 [19]. UCA1 represents a lncRNA with the ability to sponge miR-204, playing a pivotal role in colorectal cancer tumorigenesis and progression by fostering cell proliferation, impeding apoptosis, and inducing resistance to 5-fluorouracil [20] (Table 1).
MiR-520b
MiR-520b, a tumor suppressor that negatively regulates Rab22a, is negatively correlated with TNM staging [21]. In non-small cell lung cancer (NSCLC) and gallbladder carcinoma, miR-520b has been found to be down-regulated significantly [22]. Moreover, the relationship between down-regulated miR-520b expression and overexpressed Rab22a is also observed in different cancers [21, 22] (Table 1).
MiR-193b
MiR-193b is associated with cancers such as breast cancer (BC) and colon cancer (CC). It can regulate the transcription factor DNAJC13 and inhibit the expression of oncogene Rab22a. Its down-regulation will increase cell proliferation, clonogenicity, migration, and invasion [23]. In CC, overexpression of miR-193b decreases colon cancer cell cycle progression and migration ability significantly. One study showed that increased Rab22a expression by miR-193b inhibited downstream proteins involved in the Ras signaling pathway, including Ras and extracellular signal-associated kinases, which may suppress tumor proliferation and migration [24] (Table 1).
MiR-373
MiR-373 has abnormal methylation patterns in its promoter region. This abnormality tends to result in a marked reduction in miR-373 expression, which, in turn, instigates an elevation in the expression of the target gene, Rab22a. This chain of molecular events potentiates a heightened proliferation capacity within CC cells, thereby underlining the critical role of miR-373 in dictating the dynamics of cellular growth within the context of CC [25] (Table 1). Furthermore, aberrant expression of miR-373, along with other miRNAs, may exert regulatory influence over the activation of the Ras oncogene, particularly during arsenic-induced malignant transformation [26]. These findings spotlight the potential of miR-373 and similar molecular players in modulating oncogenic signaling pathways and thereby present new potential avenues for targeted cancer therapeutics.
Other miRNAs
MiR-151a-3p, miR-211, and miR-300 have additionally been reported to target Rab22a, down-regulating its expression, thereby impacting the invasive and migratory potentials as well as the epithelial–mesenchymal transition process [27, 28] (Table 1).
Mutants of Rab22a oncogene
Rab22a-S19N, Rab22a-Q64L, and other point mutations
Recognized as a mutant variant of the Rab22a protein, Rab22a-S19N is distinguished by a critical modification at the 19th amino acid in the protein sequence, wherein serine (S) is substituted by asparagine (N). This alteration precipitates a diminished affinity of the protein for guanosine triphosphate (GTP), a molecule involved in intracellular energy transfer. Because of its reduced interaction with GTP, this mutation is frequently referred to as a negative mutant [29]. Rab22a-S19N is characteristically localized within the cytosol of cells. Pioneering studies have demonstrated that cells expressing Rab22a-S19N exhibit decreased endocytosis, a fundamental cellular process entailing the internalization of extracellular materials [29].
In a similar vein, Rab22a-Q64L emerges as a specific point mutant of the Rab22a oncogene. This mutation is defined by the substitution of the 64th amino acid in the protein sequence, glutamine (Q), with leucine (L). This genetic alteration prompts a decrease in the endogenous GTPase activity of the protein, resulting in Rab22a-Q64L being a constitutively active mutant [29]. Contrary to both the wild-type Rab22a and the negative Rab22a-S19N mutant, Rab22a-Q64L is uniquely associated with early and late endosomes, lysosomes, and autophagosomes within cells. Notably, cells overexpressing Rab22a-Q64L have been observed to undergo significant morphological transformations of endosomes without the inhibition of bulk endocytosis [29].
Owing to the advent of site-directed mutagenesis, these two mutants, Rab22a-S19N and Rab22a-Q64L, were successfully generated [29]. Thus, they have become indispensable tools in the investigative landscape of Rab22a, aiding in the elucidation of the diverse functions attributed to different forms of Rab22a. Remarkably, akin to the active form of Rab22a, Rab22a-Q64L, several Rab22a mutants, such as D31H, D31Y, P32S, M43I, V84L, S94I, R104Q, and E127Q, have been discovered within the context of human cancers [3]. The utilization of these mutations, while promising, necessitates further refinement and development.
Rab22a-NeoF1
The activation of oncogenes frequently involves translocation and gene rearrangement, with the most well-known example being the Philadelphia chromosome found in chronic myeloid leukemia patients. Typically, the majority of fusion genes originate from exons, similar to how the BCR and ABL genes combined in the Philadelphia chromosome case. Nevertheless, it has been discovered that exon–intron fusion can also generate a functional protein, as demonstrated by one of Rab22a’s derivatives, known as Rab22a-NeoF1 [30].
Rab22a-NeoF1 is comprised of two exons from the Rab22a gene and a fragment of an inverted intron from the DOK5 gene. Consequently, the 1–38 amino acids of the fusion protein are derived from Rab22a, while the remaining amino acids are produced by the DOK5 intron [30]. In the absence of the inhibitory effect of the remaining Rab22a amino acids (Rab22a39-186), Lys7 of Rab22a-NeoF1 can directly interact with specific effectors that are not involved in the wild-type Rab22a binding spectrum. As a result, the oncogene becomes active and exerts carcinogenic functions, which will be further discussed in the subsequent sections [30]. Correspondingly, fusion proteins from various malignancies, such as Rab22a1-38-MYO9B from breast cancer and Rab22a1-38-TEF from lung cancer, share a similar mechanism with Rab22a-NeoF1 in osteosarcoma [30]. Rab22a-NeoF1 has demonstrated its capacity to activate oncogenic functions, making it a subject of significant interest. Further, it has been recognized as a critical hallmark in osteosarcoma, thereby underscoring its clinical relevance and the importance of its thorough investigation [30]. Moreover, the close connection between Rab22a-NeoF1 and EVs adds another layer of complexity and potential biological significance to this intriguing mutation. Therefore, in the ensuing section, we will pivot our focus towards this mutant to reveal more about its role in disease biology.
The interplay of Rab22a, its mutants, and EVs in the tumor: a comprehensive overview
Rab22a in EV biogenesis and tumor progression
Rab22a in rapid recycling and sEV biogenesis
Rab22a orchestrates both conventional and rapid intercellular recycling, wherein cargoes transported via the conventional recycling pathway, such as MHC-I and CD1a, play a significant role in immune response and antigen presentation [31]. Interestingly, exocytic vesicles in this process are frequently directed to early endosomal antigen (EEA)-positive early endosomes. Subsequently, sorting endosomes emerge from early endosomes and either undergo recycling as recycling endosomes or experience degradation through interactions with lysosomes [32,33,34]. While this conventional recycling pathway and its immunological functions have been previously addressed [35], our focus will be on the rapid recycling mechanism and certain tumor-related molecules in the pathway.
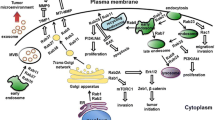
Research has elucidated that certain cargo molecules demonstrate distinct dynamics, characterized by accelerated recycling speed, diminished degradation, and extended surface longevity, a departure from their counterparts [36]. This led to the validation of a unique recycling process, henceforth referred to as the “rapid recycling pathway” [37]. In this pathway, specific cargoes such as CD44, CD98, and CD147 are promptly redirected to the plasma membrane following internalization, bypassing the EEA-positive early endosome and recycling by tubular recycling endosomes (Fig. 1). Throughout this procedure, Rab22a plays a pivotal role in the efficient sorting of payloads into tubular endosomes [37]. It is noteworthy that this intricate process is orchestrated by the BLOC-1-Rab22a-KIF13A-BLOC-2 complex [38,39,40]. Consequently, Rab22a depletion has been observed to curtail the plasma membrane expression of tumor-related molecules via the rapid recycling pathway, as evidenced in lung cancer and hepatocellular carcinoma cases [37, 41]. This revelation underscores the rapid recycling pathway as a key mechanism through which Rab22a manifests its oncogenic traits.

Rab22a in extracellular vesicle biogenesis and tumor progression. The figure illuminates the multifaceted manners in which Rab22a shapes oncogenesis through alterations in extracellular pathways. a HG-CD147 molecules undergo endocytosis, with the majority rapidly recycling via the recycling tubule. This leads to an elevation in HG-CD147 expression on the plasma membrane, subsequently triggering the production of MMPs, and consequentially, alterations in the tumor microenvironment. b MCT-1 and CD147 increased in tumor cells on the plasma membrane, and with the facilitation of Rab22a, they might be endocytosed into the early endosome. Concurrently, HG-CD147 from the trans-Golgi network, previously matured from LG-CD147 in the cis-Golgi, is secreted as secretory vesicles. The transport of CD147 from Golgi to EE might be regulated by Rab22a. Then EE matures into MVE. As a result of inward budding of MVE, MCT-1, and CD147 are incorporated into the membrane of sEVs. Their overexpression significantly augments sEVs release and enhances the adhesive force of these vesicles. c Rab22a interacts with PI4K2A to generate PI4P, which in turn attracts the Atg12-Atg5-Atg16L1 complex. This interaction enables the attachment of LC3-II to the membrane derived from the ER, thereby initiating the creation of a Rab22a-guided non-canonical autophagosome. This autophagosome subsequently merges with an early endosome to form Rafeesome, which releases the R-EV. Notably, the R-EV carries the STING protein, which is instrumental in instigating an anti-tumor response. d Rab22a potentially packages cargo proteins whose expression is stimulated by hypoxia (RhoA, etc.) in an HIF-dependent manner. Subsequently, Rab22a proteins colocalize with budding MVs, markedly increasing MVs release and fostering the production of MVs that stimulate focal adhesion formation and ECM invasion. Color-coded pathways represent different processes: green signifies the MV release pathway, grey denotes the non-canonical autophagy pathway, red illustrates the MVE pathway, and purple designates the rapid recycling pathway. Dotted lines are used to depict mechanisms that are currently not well defined. HG-CD147 highly glycosylated clusters of differentiation 147, MMPs matrix metalloproteinases, MCT-1 monocarboxylate transporter-1, LG-CD147 lowly glycosylated clusters of differentiation 147, EE early endosome, MVE multivesicular endosome, sEVs small extracellular vesicles, ER endoplasmic reticulum, STING stimulator of interferon genes, PI4P phosphatidylinositol 4-phosphate, PI4K2A phosphatidylinositol-4 kinase type 2alpha, Atg autophagy-related, LC3 microtubule-associated protein light chain 3, R-EV Rab22a-induced extracellular vesicle, GDI guanine nucleotide dissociation inhibitor, RhoA Ras homolog gene family member A, MVs microvesicles, ECM extracellular matrix
Regarded as a key constituent of the rapid recycling pathway, CD147 is hypothesized to foster the malignant traits observable in cancer cells, such as the acceleration of proliferation and the inhibition of apoptosis. It has been scientifically validated that the overexpression of Rab22a amplifies CD147 membrane expression via the rapid recycling mechanism [37]. Paradoxically, it is noteworthy that in one empirical study, the expression of CD147 escalates rather than diminishes upon the down-regulation of Rab22a [2]. Moreover, it has been postulated that only the highly glycosylated form of CD147 can promote the production of other oncogenic molecules like matrix metalloproteases (MMP), thereby indicating the indispensable role of CD147 glycosylation in its functionality [2, 42]. Rab22a, which is typically localized on the Golgi apparatus and the ER membrane [9], might play a pivotal role in the glycosylation of CD147. This assertion is supported by the concurrence of Rab22a depletion and a decrease in highly glycosylated CD147 in the ER and Golgi fractions [2]. However, to test the hypothesis, additional direct tests and proof are required.
Simultaneously, it is plausible that CD147 assumes a crucial role as an intermediary molecule in modulating the Rab22a-induced promotion of sEV in tumors. Empirical research has provided evidence that the expression of Rab22a can indeed stimulate the biogenesis of sEVs [14], with plenty of experiments proposing a potential involvement of CD147. In one particular tumor study, it was observed that the upregulated expression of CD147 can bind with monocarboxylate transporter-1 (MCT-1), ultimately producing increased numbers of sEVs with greater adhesion [43] (Fig. 1). In a separate investigation, overexpressed CD147 was found to associate with CD44 standard splice isoform (CD44s), facilitating the formation of complexes that trigger the activator of transcription 3 (STAT3) signaling pathway, thereby promoting the release of sEVs in cancer [44, 45]. Ultimately, these sEVs are involved in the tumor progression process [43,44,45]. Despite these promising findings, the underlying hypothesis still necessitates further validation. Consequently, future research should focus on conducting additional direct tests and furnishing compelling proof to substantiate these preliminary observations.
Rab22a in non-canonical autophagy and R-EV biogenesis
While the conventional perspective considers autophagy primarily as a lysosomal degradation pathway, an accumulating body of evidence suggests the existence of a variant form, termed secretory or non-canonical autophagy [5]. In this context, Rab22a has been reported to play a pivotal role in facilitating this process.
In the initiation phase, Rab22a forms a complex with PI4K2A, culminating in the production of PI4P. This molecule subsequently serves to attract the Atg12-Atg5-Atg16L1 complex. Following this interaction, the complex induces the anchoring of LC3-II on the membrane derived from the endoplasmic reticulum, leading to the genesis of the Rab22a-mediated non-canonical autophagosome. In the concluding stage of this process, the newly formed non-canonical autophagosome merges with the early endosome, engendering a structure resembling a multivesicular endosome (MVE). This complex structure named Rafeesome subsequently releases a unique EV, which is both ER-derived and modulated by Rab22a, and has been named the Rab22a-induced extracellular vesicle (R-EV) [3] (Fig. 1). This highlights the essential and specific role Rab22a plays in the orchestration of non-canonical autophagy and extracellular vesicle biogenesis.
STING, an intrinsic anti-cancer protein responsible for modulating innate immune responses, has been identified as one of the crucial cargoes incorporated by LC3-II within the R-EV [3]. Upon uptake of these STING-enriched R-EVs by tumor-associated macrophages, the STING protein housed within the R-EV orchestrates the activation of the interferon (IFN) pathway in the recipient cells situated within the tumor microenvironment, thereby amplifying anti-tumor immunity. In a seemingly paradoxical turn of events, it has been noted that the Rab22a oncogene, traditionally associated with cancer pathogenesis, might paradoxically act to enhance anti-tumor immunity [3]. This intriguing proposition finds further support in clinical observations, which have demonstrated that patients exhibiting elevated Rab22a expression tend to present with a more favorable prognosis in the context of nasopharyngeal cancer [3]. This nuanced role of Rab22a underscores the complexity and dynamism of the cancer–immunity interface.
Rab22a in microvesicles biogenesis
The interactions between MVs and Rab22a have been established (Fig. 1). Specifically, the diminution of Rab22a expression within breast cancer cells evidently reduces the production of MVs, a phenomenon which is predominantly observed within a hypoxic environment [4]. This phenomenon has been further confirmed by experimental evidence showing that Rab22a knockdown leads to a marked reduction in MVs formation [4]. Conversely, in conditions of normoxia, the expression of MVs remains unaltered, suggesting the potential involvement of another Rab protein in the biogenesis of MVs, aside from Rab22a [4].
Nevertheless, the precise molecular mechanisms through which Rab22a orchestrates MVs development remain an active area of investigation. In hypoxic environments, Rab22a may have other potential effects in addition to the determinative effect of stimulating MVs. Empirical evidence has revealed a statistically significant variation in the protein composition of MVs between the control and Rab22a overexpression groups [4]. Moreover, MVs originating from hypoxic cells with elevated Rab22a expression seem to contribute to the emergence of more aggressive cancer subtypes, augmenting the invasive potential of cancer cells both in vivo and in vitro. This evidence underscores the possibility that Rab22a may selectively incorporate proteins induced under hypoxic conditions into MVs [4]. Such findings offer a tantalizing glimpse into the complex role of Rab22a in the context of cancer pathogenesis and progression.
Rab22a in vesicle degradation and secretion
One of the fundamental roles of Rab22a is to prevent degradation. Indeed, the fusion of subcellular vesicles with lysosomes necessitates active Rab7 binding to its effector, RILP [46]. Consequently, by interfering with Rab5–Rab7 conversion, Rab22a can effectively prevent vesicles from degradation [47]. This observation may elucidate why Rab22a-coated subcellular vesicles tend to develop a secretory or recycling phenotype, thereby preventing destruction (Fig. 1). For instance, it might provide insight into why Rab22a-induced fast recycling pathways circumvent degradation [36]. In a related context, Mycobacterium TB, a pathogen, can influence phagolysosome development by recruiting Rab22a to its phagosome [47]. Similarly, non-canonical autophagy displays a comparable phenomenon, hindering the degradation of an MVE-like structure enveloped in Rab22a [3].
Rab22a-NeoF1 in EV biogenesis and tumor progression
A recent study found a new fusion protein, Rab22a-NeoF1, in patients with lung metastases from osteosarcoma. Unlike the wild-type protein, Rab22a-NeoF1 is secreted into sEVs and influences cell signaling when absorbed by recipient cells [48]. This finding challenges the belief that Rab22a mainly controls tumor behavior by managing EV biogenesis, expanding our understanding of Rab22a’s interplay with EVs. We will explore how Rab22a-NeoF1 potentially enhances metastasis, creating pre-metastatic niches, and the latest research on the underlying mechanisms (Fig. 2).

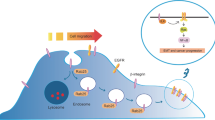
Rab22a-NeoF1 in extracellular vesicle biogenesis and tumor progression. The multifaceted impacts exerted by Rab22a-NeoF1 in the context of cancer progression, directly within Rab22a-NeoF1-positive cells and indirectly via sEVs in Rab22a-NeoF1-negative cells. a For Rab22a-NeoF1-positive osteosarcoma cells, an intriguing interplay is depicted wherein Rab22a-NeoF1 showcases a high affinity for SmgGDS-607, thus fostering the release of pre-prenylated GTP-RhoA. Crucially, this interaction is contingent upon the acetylation of the Lys7 residue of Rab22a-NeoF1 by P300/CBP. The figure underscores the critical role of two negatively charged regions within SmgGDS-607, indispensable for its binding to RhoA and Rab22a-NeoF1’s two positively charged residues (Arg4 and acetylated Lys7). In the subsequent progression, the formation of a Rab22a-NeoF1-SmgGDS-607-RhoA trimeric complex is illustrated, which bolsters the GEF activity of SmgGDS and orchestrates the departure of RhoA from SmgGDS-607. Following this, the figure illustrated the prenylation of GTP-RhoA by cytoplasmic GGTase-I, post-prenylation modifications, and its migration towards the plasma membrane. The final portrayal centers around the activation of RhoA, which consequently instigates cancer cell migration and metastasis. Simultaneously, Rab22a, along with its binding partner, are encapsulated into MVE and subsequently released as sEVs. When these sEVs are taken by Rab22a-NeoF1-negative cancer cells, they incite RhoA activation via PYK2, thereby driving migration and metastasis of Rab22a-NeoF1-negative cancer cells. b When these sEVs are taken up by macrophages, PYK2 is instrumental in orchestrating the transition to the M2 macrophage-polarized phenotype in TAMs. This is achieved through the sustained activation of STAT3 signaling, thereby proving pivotal in the formation of a PMN. RhoA Ras homolog gene family member A, K7 Lysine at position 7, GGTase geranylgeranyltransferase-I, PYK2 proline-rich tyrosine kinase 2, MVE multivesicular endosome, STAT3 signal transducer and activator of transcription 3, M2-TAM M2-type tumor-associated macrophages, PMN pre-metastatic niches, GEF guanine nucleotide exchange factor
Signal pathway in Rab22a-NeoF1-positive cells
In Rab22a-NeoF1-positive cells, Rab22a-NeoF1 exhibits novel protein–protein interaction with its changed spatial structure compared with Rab22a. In Rab22a-NeoF1 fusion protein, the positively charged residue of Rab22a1-10, namely Lys7, is surface-exposed and indispensable to bind with small G protein guanine dissociation stimulator (SmgGDS) via electrostatic force [30]. However, in the entire length of Rab22a, the residue Lys7 is blocked by main-chain oxygen atoms of Met73, Tyr74, and Arg76 [49], showing little affinity to SmgGDS [30].
Given the protein–protein interaction between Rab22a-NeoF1 and SmgGDS is important for oncogenic function, we will give brief information on the SmgGDS. First, SmgGDS incorporates two splice variants. The longer form which contains 607 amino acid residues was named SmgGDS-607 [50]; the shorter form of SmgGDS, which is made up of 558 amino acid residues, is called SmgGDS-558 [45]. However, in osteosarcoma cell lines, compared with SmgGDS-558, the expression of SmgGDS-607 is primarily higher [30]. This phenomenon is in line with the results that cells containing a higher 607:558 ratio proliferate and migrate rapidly [51]. Second, as prenylation of the C-terminus of the Ras and Rho family is necessary for them to anchor at the membrane, which is also called compartmentalization, SmgGDS serves as an emerging master regulator of this process [4, 52]. SmgGDS-607 binds with newly synthesized hydrophilic Ras and Rho family members, which is pre-prenylated [50]. SmgGDS-607 may retain this small GTPase until the arrival of releasing signals, like GDP/GTP exchange, serving as a valve that controls the flow into the prenylation pathway [18, 50]. Third, SmgGDS-607, which does not have a typical guanine exchange factor (GEF) catalytic domain and is entirely composed of armadillo-repeat motifs (ARMs), is a specific non-canonical GEF for only RhoA and RhoC, not for other small GTPases [16, 53]. However, the regular activity of GEF is relatively low and can be further facilitated by releasing signals [52]. Rab22a-NeoF1 exhibits a high affinity to SmgGDS-607 and facilitates the release of pre-prenylated GTP-RhoA. Nevertheless, only when P300/CBP acetylates the Lys7 residue of Rab22a-NeoF1 can it bind with SmgGDS-607 [54]. There are two negatively charged regions of SmgGDS-607 (E254K/D256K), which are crucial for its association with RhoA [16, 54]. Interestingly, these regions are also required for binding with two positively charged residues of Rab22a-NeoF1, namely Arg4 and acetylated Lys7 [30]. Rab22a-NeoF1 binds SmgGDS-607 when RhoA is bound to form a Rab22a-NeoF1-SmgGDS-607-RhoA trimeric complex, thereby speeding up GEF activity of SmgGDS and also sequence RhoA out of SmgGDS-607 [30]. Once pre-prenylated GTP-RhoA is released, it can be prenylated by the cytoplasmic geranylgeranyltransferase-I (GGTase-I), undergo further post-prenylation, and ultimately move directly towards the plasma membrane [52]. RhoA activation can promote migration and metastases of cancer cells [48] (Fig. 2).
Signal pathways in Rab22a-NeoF1-negative cells
Remarkably, only a minuscule fraction of osteosarcoma cells test positive for Rab22a-NeoF1 in Rab22a-NeoF1-positive osteosarcoma [30]. It appears that these cells wield their influence over other negative recipient cells, including osteosarcoma cells and macrophages, thereby catalyzing metastasis and the genesis of pre-metastatic niches (PMN) in the lung [48].
Indeed, oncogenic signals can radiate from Rab22a-NeoF1-positive cells to Rab22a-NeoF1-negative tumor cells. This suggests that the signals, originating from Rab22a-NeoF1-positive cells, with Rab22a-NeoF1 and proline-rich tyrosine kinase 2 (PYK2), serve as a binding partner [48]. Subsequently, this engagement induces intercellular communication mediated by sEV. This implies that Rab22a-NeoF1 and PYK2 can be secreted into the lumen of intraluminal vesicles (ILVs). Interestingly, contrary to wild-type Rab22a, Rab22a-NeoF1, due to changes in spatial structure, can be secreted as cargo rather than anchoring on the membrane (Fig. 2).
Upon being uptake by Rab22a-NeoF1 negative cancer cells, the excreted sEVs induce RhoA activation via PYK2, thereby promoting migration and metastases of cancer cells [48]. Simultaneously, when taken up by macrophages, PYK2 plays a critical role in enabling the transition to the M2-macrophage-polarized phenotype of tumor-associated macrophages (TAMs) through the continual activation of STAT3 signaling [48, 55,56,57]. Given that the M2 subtype is tumor-supportive and immunosuppressive, they subsequently curtail the median survival time of Rab22a-NeoF1-positive osteosarcoma patients [30] (Fig. 2).
sEV-derived Rab22a-NeoF1 also orchestrates the recruitment of bone marrow-derived macrophages (BMDCs), which are instrumental in the formation of PMN [48]. This phenomenon is further confirmed when BMDCs are preliminarily depleted by liposomes, resulting in a significant decrease in lung metastasis when treated with small EV containing Rab22a-NeoF1, underscoring the vital role of BMDCs in the pulmonary metastasis of osteosarcoma [48] (Fig. 2).
Conclusion and perspective
It is indicated that the Rab22a and its derivatives have a marked influence in facilitating tumor invasion and metastasis [27, 48]. A notable correlation has been established between the abnormal expression of Rab22a and an unfavorable prognosis across a broad spectrum of tumor types. In a curious turn of events, despite a substantial volume of literature focusing on the correlation between elevated Rab22a levels and tumor behavior, a conspicuous absence of further research regarding the underlying mechanisms or plausible hypotheses persists. This lack of thorough investigation into potential pathways leaves a gap in our understanding. Given the established role of Rab22a as a small GTPase orchestrating vesicle behavior and the phenomenon that it is implicated in the long-distance effects exhibited by tumors, particularly their propensity for invasion and metastasis [27]. These lend credence to the notion that EVs may represent a pivotal mechanism in the function of Rab22a. Intriguingly, our review has identified three specific EVs: sEVs, MVs, and R-EVs-under the regulatory purview of Rab22a [3, 4, 14]. While sEVs and MVs have been reported to have several biogenesis pathways, R-EVs are uniquely biogenesis via Rab22a, an exciting finding indicating the unique function of Rab22a in tumor behavior [3]. There, we notice that the profound link between Rab22a and tumor behavior mediated by EVs, Rab22a holds significant promise as a therapeutic target in tumor therapy.
Furthermore, the correlation between Rab22a expression levels and prognosis may exhibit a counterintuitive pattern in certain types of cancer, as exemplified in the case of nasopharyngeal cancer [3]. This intriguing deviation from the norm could potentially be attributed to the unique characteristics of the cancer type in question or to the influence of specific chemotherapeutic agents used. Regardless of the underlying cause, this unexpected observation presents a tantalizing possibility of inverting the oncogenic function of Rab22a. Such a finding could hold significant implications for future therapeutic strategies in oncology. Despite the above strides made in understanding, there is a pressing need for more precise and comprehensive research to unravel the intricate mechanisms of extracellular vesicles in the context of Rab22a and tumor development. The outcome of such investigations would facilitate the development of tailored therapeutics that could disrupt tumor behavior, thus ushering in an exciting era in cancer treatment.
Availability of data and materials
Not applicable.
Abbreviations
- Rabs:
-
Ras-related GTP-binding proteins
- EVs:
-
Extracellular vesicles
- ER:
-
Endoplasmic reticulum
- sEVs:
-
Small extracellular vesicles
- MVs:
-
Microvesicles
- DCs:
-
Dendritic cells
- STING:
-
Stimulator of interferon genes
- R-EVs:
-
Rab22a-induced extracellular vesicles
- HIF:
-
Hypoxia-inducible factor
- ARNT:
-
Aryl hydrocarbon receptor nuclear translocator
- CBP:
-
CREB-binding protein
- Pol II:
-
DNA polymerase II
- HREs:
-
Hypoxia responsive elements
- UTR:
-
Untranslated region
- TNM:
-
Tumor node metastasis
- NSCLC:
-
Non-small cell lung cancer
- BC:
-
Breast cancer
- CC:
-
Colon cancer
- GTP:
-
Guanosine triphosphate
- EEA:
-
Early endosomal antigen
- MMP:
-
Matrix metalloproteinases
- MCT-1:
-
Monocarboxylate transporter-1
- CD44s:
-
CD44 standard splice isoform
- STAT3:
-
Activator of transcription 3
- MVE:
-
Multivesicular endosome
- SmgGDS:
-
Small G protein guanine dissociation stimulator
- ARMs:
-
Armadillo-repeat motifs
- GEF:
-
Guanine exchange factor
- GGTase-I:
-
Geranylgeranyltransferase-I
- PMN:
-
Pre-metastatic niches
- PYK2:
-
Proline-rich tyrosine kinase 2
- ILVs:
-
Intraluminal vesicles
- TAMs:
-
Tumor-associated macrophages
- BMDCs:
-
Bone marrow-derived macrophages
References
Hutagalung AH, Novick PJ. Role of Rab GTPases in membrane traffic and cell physiology. Physiol Rev. 2011;91(1):119–49.
Qi S, Su L, Li J, Zhao P, Zhang Q, Niu X, et al. YIPF2 is a novel Rab-GDF that enhances HCC malignant phenotypes by facilitating CD147 endocytic recycle. Cell Death Dis. 2019;10(6):462.
Gao Y, Zheng X, Chang B, Lin Y, Huang X, Wang W, et al. Intercellular transfer of activated STING triggered by RAB22A-mediated non-canonical autophagy promotes antitumor immunity. Cell Res. 2022;32(12):1086–104.
Wang T, Gilkes DM, Takano N, Xiang L, Luo W, Bishop CJ, et al. Hypoxia-inducible factors and RAB22A mediate formation of microvesicles that stimulate breast cancer invasion and metastasis. Proc Natl Acad Sci U S A. 2014;111(31):E3234–42.
Ao X, Zou L, Wu Y. Regulation of autophagy by the Rab GTPase network. Cell Death Differ. 2014;21(3):348–58.
van Niel G, D’Angelo G, Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat Rev Mol Cell Biol. 2018;19(4):213–28.
Wu P, Zhang B, Ocansey DKW, Xu W, Qian H. Extracellular vesicles: a bright star of nanomedicine. Biomaterials. 2021;269: 120467.
Huang T, Song C, Zheng L, Xia L, Li Y, Zhou Y. The roles of extracellular vesicles in gastric cancer development, microenvironment, anti-cancer drug resistance, and therapy. Mol Cancer. 2019;18(1):62.
Bai Y, Huang W, Ma L-T, Jiang J-L, Chen Z-N. Importance of N-glycosylation on CD147 for its biological functions. Int J Mol Sci. 2014;15(4):6356–77.
Xunian Z, Kalluri R. Biology and therapeutic potential of mesenchymal stem cell-derived exosomes. Cancer Sci. 2020;111(9):3100–10.
Alia Moosavian S, Hashemi M, Etemad L, Daneshmand S, Salmasi Z. Melanoma-derived exosomes: versatile extracellular vesicles for diagnosis, metastasis, immune modulation, and treatment of melanoma. Int Immunopharmacol. 2022;113(Pt A): 109320.
Bobrie A, Krumeich S, Reyal F, Recchi C, Moita LF, Seabra MC, et al. Rab27a supports exosome-dependent and -independent mechanisms that modify the tumor microenvironment and can promote tumor progression. Cancer Res. 2012;72(19):4920–30.
Grismayer B, Sölch S, Seubert B, Kirchner T, Schäfer S, Baretton G, et al. Rab31 expression levels modulate tumor-relevant characteristics of breast cancer cells. Mol Cancer. 2012;11:62.
Sun L, He M, Xu N, Xu D-H, Ben-David Y, Yang Z-Y, et al. Regulation of RAB22A by mir-193b inhibits breast cancer growth and metastasis mediated by exosomes. Int J Oncol. 2018;53(6):2705–14.
Yin Y, Zhang B, Wang W, Fei B, Quan C, Zhang J, et al. miR-204-5p inhibits proliferation and invasion and enhances chemotherapeutic sensitivity of colorectal cancer cells by downregulating RAB22A. Clin Cancer Res. 2014;20(23):6187–99.
Huang LE, Gu J, Schau M, Bunn HF. Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci U S A. 1998;95(14):7987–92.
Yamashita K, Discher DJ, Hu J, Bishopric NH, Webster KA. Molecular regulation of the endothelin-1 gene by hypoxia. Contributions of hypoxia-inducible factor-1, activator protein-1, GATA-2, AND p300/CBP. J Biol Chem. 2001;276(16):12645–53.
Semenza GL. HIF-1 and mechanisms of hypoxia sensing. Curr Opin Cell Biol. 2001;13(2):167–71.
Zhang B, Yin Y, Hu Y, Zhang J, Bian Z, Song M, et al. MicroRNA-204-5p inhibits gastric cancer cell proliferation by downregulating USP47 and RAB22A. Med Oncol. 2015;32(1):331.
Bian Z, Jin L, Zhang J, Yin Y, Quan C, Hu Y, et al. LncRNA-UCA1 enhances cell proliferation and 5-fluorouracil resistance in colorectal cancer by inhibiting miR-204-5p. Sci Rep. 2016;6:23892.
Zhang L, Yu S. Role of miR-520b in non-small cell lung cancer. Exp Ther Med. 2018;16(5):3987–95.
Zhou J, Gao F, Zhang H, Xing M, Xu Z, Zhang R. MiR-520b inhibits proliferation, migration and invasion in gallbladder carcinoma by targeting RAB22A. Arch Med Sci. 2021;17(2):481–91.
Yang Z, He M, Wang K, Sun G, Tang L, Xu Z. Tumor suppressive microRNA-193b promotes breast cancer progression via targeting DNAJC13 and RAB22A. Int J Clin Exp Pathol. 2014;7(11):7563–70.
Fang Z, Li C, Li S. MicroRNA-193b acts as a tumor suppressor in colon cancer progression via targeting RAB22A. Exp Ther Med. 2019;17(5):3921–8.
Tanaka T, Arai M, Wu S, Kanda T, Miyauchi H, Imazeki F, et al. Epigenetic silencing of microRNA-373 plays an important role in regulating cell proliferation in colon cancer. Oncol Rep. 2011;26(5):1329–35.
Ngalame NNO, Tokar EJ, Person RJ, Xu Y, Waalkes MP. Aberrant microRNA expression likely controls RAS oncogene activation during malignant transformation of human prostate epithelial and stem cells by arsenic. Toxicol Sci. 2014;138(2):268–77.
Zheng S, Jiang F, Ge D, Tang J, Chen H, Yang J, et al. LncRNA SNHG3/miRNA-151a-3p/RAB22A axis regulates invasion and migration of osteosarcoma. Biomed Pharmacother. 2019;112:108695.
Yu H, Yang W. MiR-211 is epigenetically regulated by DNMT1 mediated methylation and inhibits EMT of melanoma cells by targeting RAB22A. Biochem Biophys Res Commun. 2016;476(4):400–5.
Mesa R, Salomón C, Roggero M, Stahl PD, Mayorga LS. Rab22a affects the morphology and function of the endocytic pathway. J Cell Sci. 2001;114(Pt 22):4041–9.
Liao D, Zhong L, Yin J, Zeng C, Wang X, Huang X, et al. Chromosomal translocation-derived aberrant Rab22a drives metastasis of osteosarcoma. Nat Cell Biol. 2020;22(7):868–81.
Barral DC, Cavallari M, McCormick PJ, Garg S, Magee AI, Bonifacino JS, et al. CD1a and MHC class I follow a similar endocytic recycling pathway. Traffic. 2008;9(9):1446–57.
Radhakrishna H, Donaldson JG. ADP-ribosylation factor 6 regulates a novel plasma membrane recycling pathway. J Cell Biol. 1997;139(1):49–61.
Naslavsky N, Weigert R, Donaldson JG. Convergence of non-clathrin- and clathrin-derived endosomes involves Arf6 inactivation and changes in phosphoinositides. Mol Biol Cell. 2003;14(2):417–31.
Naslavsky N, Weigert R, Donaldson JG. Characterization of a nonclathrin endocytic pathway: membrane cargo and lipid requirements. Mol Biol Cell. 2004;15(8):3542–52.
Mayorga LS, Cebrian I. Rab22a: A novel regulator of immune functions. Mol Immunol. 2019;113:87–92.
Eyster CA, Cole NB, Petersen S, Viswanathan K, Früh K, Donaldson JG. MARCH ubiquitin ligases alter the itinerary of clathrin-independent cargo from recycling to degradation. Mol Biol Cell. 2011;22(17):3218–30.
Maldonado-Báez L, Cole NB, Krämer H, Donaldson JG. Microtubule-dependent endosomal sorting of clathrin-independent cargo by Hook1. J Cell Biol. 2013;201(2):233–47.
Shakya S, Sharma P, Bhatt AM, Jani RA, Delevoye C, Setty SR. Rab22A recruits BLOC-1 and BLOC-2 to promote the biogenesis of recycling endosomes. EMBO Rep. 2018;19(12):e45918.
Thankachan JM, Setty SRG. KIF13A-A Key regulator of recycling endosome dynamics. Front Cell Dev Biol. 2022;10: 877532.
Kong L, Huang S, Bao Y, Chen Y, Hua C, Gao S. Crucial roles of Rab22a in endosomal cargo recycling. Traffic. 2023. https://doi.org/10.1111/tra.12907.
Zhou Y, Wu B, Li J-H, Nan G, Jiang J-L, Chen Z-N. Rab22a enhances CD147 recycling and is required for lung cancer cell migration and invasion. Exp Cell Res. 2017;357(1):9–16.
Tang W, Chang SB, Hemler ME. Links between CD147 function, glycosylation, and caveolin-1. Mol Biol Cell. 2004;15(9):4043–50.
Thakur A, Qiu G, Xu C, Han X, Yang T, Ng SP, et al. Label-free sensing of exosomal MCT1 and CD147 for tracking metabolic reprogramming and malignant progression in glioma. Sci Adv. 2020;6(26):eaaz6119.
Li L, Tang W, Wu X, Karnak D, Meng X, Thompson R, et al. HAb18G/CD147 promotes pSTAT3-mediated pancreatic cancer development via CD44s. Clin Cancer Res. 2013;19(24):6703–15.
Dorayappan KDP, Wanner R, Wallbillich JJ, Saini U, Zingarelli R, Suarez AA, et al. Hypoxia-induced exosomes contribute to a more aggressive and chemoresistant ovarian cancer phenotype: a novel mechanism linking STAT3/Rab proteins. Oncogene. 2018;37(28):3806–21.
Jordens I, Fernandez-Borja M, Marsman M, Dusseljee S, Janssen L, Calafat J, et al. The Rab7 effector protein RILP controls lysosomal transport by inducing the recruitment of dynein-dynactin motors. Curr Biol. 2001;11(21):1680–5.
Roberts EA, Chua J, Kyei GB, Deretic V. Higher order Rab programming in phagolysosome biogenesis. J Cell Biol. 2006;174(7):923–9.
Zhong L, Liao D, Li J, Liu W, Wang J, Zeng C, et al. Rab22a-NeoF1 fusion protein promotes osteosarcoma lung metastasis through its secretion into exosomes. Signal Transduct Target Ther. 2021;6(1):59.
Brahimi-Horn MC, Chiche J, Pouysségur J. Hypoxia and cancer. J Mol Med (Berl). 2007;85(12):1301–7.
Gillies RJ, Gatenby RA. Hypoxia and adaptive landscapes in the evolution of carcinogenesis. Cancer Metastasis Rev. 2007;26(2):311–7.
King HW, Michael MZ, Gleadle JM. Hypoxic enhancement of exosome release by breast cancer cells. BMC Cancer. 2012;12:421.
Hsu YL, Hung JY, Chang WA, Lin YS, Pan YC, Tsai PH, et al. Hypoxic lung cancer-secreted exosomal miR-23a increased angiogenesis and vascular permeability by targeting prolyl hydroxylase and tight junction protein ZO-1. Oncogene. 2017;36(34):4929–42.
Kaelin WG. Proline hydroxylation and gene expression. Annu Rev Biochem. 2005;74:115–28.
Kumar A, Deep G. Hypoxia in tumor microenvironment regulates exosome biogenesis: molecular mechanisms and translational opportunities. Cancer Lett. 2020;479:23–30.
Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S, et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med. 2005;11(12):1314–21.
Yan D, Wang HW, Bowman RL, Joyce JA. STAT3 and STAT6 signaling pathways synergize to promote cathepsin secretion from macrophages via IRE1alpha activation. Cell Rep. 2016;16(11):2914–27.
Wortzel I, Dror S, Kenific CM, Lyden D. Exosome-mediated metastasis: communication from a distance. Dev Cell. 2019;49(3):347–60.
Acknowledgements
We thank all the subjects who participated in this study.
Funding
This work was supported by the Science and Technology Plan Project of Wenzhou (Grant Nos. Y20220389, Y20220045), Zhejiang Province Natural Science Foundation (Grant No. LTGY23H100001), and National Natural Science Foundation of China (Grant No. 81901660).
Author information
Authors and Affiliations
Contributions
Writing, review, and editing, SHH, YXB; writing the first draft, SHH, YXB; final editing and proof reading, LJK, SG, CYH. All the authors have read and agreed to the published version of the article.
Corresponding authors
Ethics declarations
Conflict of interests
The authors declare no competing interests.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable as no patient’s or individual data have been used.
Additional information
Responsible Editor: John Di Battista.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Huang, S., Bao, Y., Kong, L. et al. Insights into the complex interactions between Rab22a and extracellular vesicles in cancers. Inflamm. Res. 73, 99–110 (2024). https://doi.org/10.1007/s00011-023-01821-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-023-01821-0