
Abstract
Objective and design
Multiple sclerosis (MS) is a debilitating autoimmune disease involving immune dysregulation of the pathogenic T helper 17 (Th17) versus protective T regulatory (Treg) cell subsets, besides other cellular aberrations. Studies on the mechanisms underlying these changes have unraveled the involvement of mitogen-activated protein kinase (MAPK) pathway in the disease process. We describe here a gene expression- and bioinformatics-based study showing that celastrol, a natural triterpenoid, acting via MAPK pathway regulates the downstream genes encoding serum/glucocorticoid regulated kinase 1 (SGK1), which plays a vital role in Th17/Treg differentiation, and brain-derived neurotrophic factor (BDNF), which is a neurotrophic factor, thereby offering protection against experimental autoimmune encephalomyelitis (EAE) in mice.
Methods
We first tested the gene expression profile of splenocytes of EAE mice in response to the disease-related antigen, myelin oligodendrocyte glycoprotein (MOG), and then examined the effect of celastrol on that profile.
Results
Interestingly, celastrol reversed the expression of many MOG-induced genes involved in inflammation and immune pathology. The MAPK pathway involving p38MAPK and ERK was identified as one of the mediators of celastrol action. It involved suppression of SGK1 but upregulation of BDNF, which then contributed to protection against EAE.
Conclusion
Our results not only provide novel insights into disease pathogenesis, but also offer promising therapeutic targets for MS.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Multiple sclerosis (MS) is a chronic debilitating autoimmune disease involving inflammation and damage of the central nervous system (CNS) [1,2,3,4]. MS affects about 400,000 people in the U.S. alone and over 2 million people globally [2, 3]. Typically, MS affects people between the ages of 20 and 50 years, and women are affected more than men. The breakdown of immune tolerance to the antigenic components of the myelin sheath leads to the induction of a pathogenic T helper 17 (Th17) response as well as suppression of the T regulatory (Treg) response, which is required for the maintenance of tolerance to self-antigens and inhibition of excessive immune response. This results in an imbalance of Th17 and Treg cells [5,6,7,8]. In addition, B cells, CD8+ T cells, macrophages, and microglia also play an important role in the immune pathology of this disease [4].
The activation of T cells under a defined cytokine milieu results in the differentiation into the Th17 or Treg phenotype [9, 10]. Furthermore, significant advances have been made over the past decade in defining the pathogenic effector functions of Th17 cells and the immunoregulatory functions of Treg cells [11,12,13,14]. These advances have set the stage for developing novel therapeutic approaches that target specific mediators of Th17/Treg balance and help reset this balance to reinforce immune homeostasis as well as control the progression of autoimmunity [8, 15,16,17]. One of the cellular signaling pathway influencing Th17 differentiation is the p38 mitogen-activated protein kinase (p38 MAPK) pathway, which in turn targets serum/glucocorticoid regulated kinase 1 (SGK1) [18, 19]. Using complementary approaches, different investigators have unraveled the role of p38 MAPK in the pathogenesis of MS [8, 18,19,20,21] and its animal model, experimental autoimmune encephalomyelitis (EAE) [19, 22,23,24,25]. In addition, this pathway has also been invoked in oxidative stress [26, 27].
Therefore, it is anticipated that small molecules with anti-oxidant activity might also be of use in the treatment of MS/EAE. We previously identified celastrol, a small molecule compound derived from a Chinese medicinal herb, Celastrus aculeatus, as a potent immunomodulatory agent for rat adjuvant arthritis, a model for human rheumatoid arthritis (RA) [28,29,30,31]. Specifically, we showed that celastrol modulated the antigen-mediated cellular and humoral responses, including the Th17/Treg balance in arthritic rats. Subsequently, celastrol was shown by other investigators to inhibit EAE [32,33,34]. Two of these studies that were performed in the myelin-basic protein (MBP)-induced EAE in SD rats [32, 34] revealed the role of immune deviation from Th1 to Th2, of reduced expression of NF-kB and nitrite levels [32], and of anti-inflammatory and anti-apoptotic effects [34] in the protective effect of celastrol treatment. The third study based on the myelin oligodendrocyte glycoprotein (MOG)-induced EAE in C57BL/6 mice showed that celastrol inhibited the Th17 response [33]. These studies have laid the foundation for more detailed examination of the molecular basis of the regulation of the Th17/Treg balance as well as neuroprotection offered by celastrol in EAE, which remain to be fully explored.
In this study, using the MOG-induced EAE model, we first performed a comprehensive gene expression analysis of 77 genes of immune cells (splenocytes) of the diseased mice to gain an insight into the mediators/pathways that are most affected by the disease process. Then we examined the effect of celastrol on that gene expression profile. Thereafter, we selected 20 genes for further analysis by ingenuity pathway analysis (IPA) in an attempt to identify their upstream regulators. These analyses revealed that celastrol modulated the MAPK (p38, ERK)–SGK1 pathway as well as induced the expression of brain-derived neurotrophic factor (BDNF), and suggested the role of SGK1 in regulating the Th17/Treg balance and that of T cell-derived BDNF as a neurotrophic factor, leading to inhibition of the progression of EAE. To the best of our knowledge, this is the first report to inter-connect the key components (MAPK, SGK1, BDNF, and the Th17/Treg balance) that both play a vital role in EAE pathogenesis as well as serve as targets of celastrol action, leading to protection against EAE.
Materials and methods
Induction and evaluation of experimental autoimmune encephalomyelitis (EAE)
All animal experiments performed in this study were done according to the guidelines of the Institutional Animal Care and Use Committee (IACUC) of UMB. EAE was induced in 8 weeks old, male C57BL/6 mice as described elsewhere [35]. Briefly, mice were immunized subcutaneously with MOG35–55 peptide (150 µg) in complete Freund’s adjuvant (CFA) followed by concomitant intraperitoneal (i.p.) administration of pertussis toxin (200 ng) on day 0 and day 2. Following immunization, these mice were observed regularly for at least 25 days. The signs of EAE typically developed on about day 10 after immunization. The severity of EAE was graded on a scale of 0–5 as described [36] : (1 = partial or total flaccid paralysis of the tail; 2 = hind limb weakness/disrupted righting reflex; 3 = flaccid paralysis in one hind limb; 4 = flaccid paralysis in both hind limbs; and 5 = moribund/dead.)
Treatment of EAE mice with celastrol
A stock solution of celastrol was prepared in dimethyl sulfoxide (DMSO; Sigma) at a concentration of 10 mg/300 µl and stored at − 20 °C in small aliquots. The experimental group of EAE mice received celastrol (20 µg per mouse) diluted in 200 µl of PBS, which was injected i.p. every 2 days starting from the onset of the disease and continued uninterrupted until the end of the observation period. The control group of EAE mice received equal amount of the vehicle (DMSO in PBS). All these mice were then observed and graded regularly for the signs of EAE. The clinical scores of EAE were recorded, and the representative spinal cords were processed for histological evaluation following staining with hematoxylin and eosin.
Preparation of splenocytes and their re-stimulation with MOG in the presence or absence of celastrol
EAE mice were euthanized between day 15 and 20 after MOG-immunization, which correspond to day 5–10 of onset of disease, and their spleens were harvested. A single cell suspension of splenocytes was prepared. These cells were treated with ammonium chloride potassium (ACK) buffer to remove red blood cells followed by three washings with Hanks' balanced salt solution (HBSS). These splenocytes in culture were then restimulated with MOG peptide (MOG35–55) (25 µg/ml) in the presence or absence of celastrol (100 nM) [31] for different time points, specifically 3 h, 6 h, 12 h and 24 h. This concentration of celastrol is not toxic for cells. The culture conditions consisted of 6 × 106 cells/well in a 6-well culture plate in Dulbecco’s modified eagle medium (DMEM) containing 10% fetal bovine serum (FBS) supplemented with 2 mM l-glutamine, 100 units/ml penicillin G sodium, and 100 µg/ml of streptomycin sulfate in an atmosphere of 95% air and 5% CO2. Cells cultured without the addition of MOG or celastrol were referred to as ‘Medium control’.
Measurement of mRNA expression using qRT-PCR
Total RNA was isolated from splenocytes cultured as described above and then cDNA was prepared from RNA using iScript cDNA synthesis kit. The resulting cDNA was amplified in an Applied Biosystems StepOne instrument using SYBR Green PCR Master Mix and appropriate primers. The sequences of the forward and reverse primers for the detection of mRNAs were selected from the Primer Depot [National Institutes of Health (NIH)] and the corresponding primers were then obtained from Sigma-Aldrich. The mRNA levels of specific genes were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) or hypoxanthine–guanine phosphoribosyltransferase (HPRT) mRNA levels, and the relative gene expression levels were determined. The results were expressed either as ‘Fold change’ or ‘Relative message’. The optimal time point of expression of individual genes was used for further characterization.
T cell isolation and T helper cell differentiation
Using magnetic-activated cell sorting (MACS), naïve CD4+ T cells and natural Treg (nTreg) were isolated from the splenocytes of 5–6-week-old C57BL/6 mice employing ‘Naïve CD4+ T cell isolation kit’ and ‘nTreg isolation kit’ (both from Miltenyi Biotech), respectively. For the T cell differentiation assay, 96-well plates were pre-coated with anti-CD3 antibodies (2.5 µg/ml). For Th17 cell differentiation, 1 × 105 naïve T cells per well were cultured for 4 days in DMEM-containing soluble anti-CD28 (2.5 µg/ml), TGFβ1 (2.5 ng/ml), IL-6 (25 ng/ml), IL-1β (10 ng/ml), IL-23 (50 ng/ml), anti-IFNγ (1 µg/ml), anti-IL-4 (1 µg/ml), and 10% FBS in the presence of absence of celastrol (100 nM). For Treg differentiation, 1 × 105 naïve T cells per well were cultured for 4 days in DMEM-containing soluble anti-CD28 (2.5 µg/ml), TGFβ1 (2.5 ng/ml), anti-IL-6 (1 µg/ml), anti-IFNγ (1 µg/ml), anti-IL-4 (1 µg/ml), and 10% FBS in the presence of absence of celastrol (100 nM). For nTreg stimulation, 1 × 105 cells per well were cultured for 48 h in DMEM-containing soluble anti-CD28 (2.5 µg/ml), IL-2 (20 ng/ml) and 10% FBS. Naïve T cells cultured in the presence of anti-CD3 and anti-CD28 served as Th0 controls. The mRNA expression in these T cells was determined as described above for splenocytes.
Flow cytometry analysis of the T cells
The T cells were treated for 10 min with 10% normal mouse serum to block non-specific Fc-binding sites. Thereafter, the cells were surface-stained with anti-mouse CD3-FITC and anti-mouse CD4-APC. Following that, the cells were fixed and permeabilized using the BD fixation/permeabilization kit and stained with anti-mouse RORγt-PerCP eFluor 710 and/or anti-mouse FOXP3-eFlour 450. For IL-17 expression analysis, cells were stimulated for 4–5 h with 50 ng/mL phorbol myristate acetate (PMA) and 2 µM ionomycin in the presence of Brefeldin A. This was followed by surface staining and intracellular staining, the latter with anti-mouse IL-17A-eFluor 450. Flow cytometric data were acquired on LSRII (BD Bioscience) with gating set on unstained/isotype controls, and data analysis was performed with FCS Express 6 software.
Ingenuity pathway analysis (IPA)
Information about 20 genes (including 15 downregulated and 5 upregulated), whose expression was altered by celastrol in response to MOG re-stimulation, was uploaded onto the Ingenuity Pathway Analysis (IPA) software (Qiagen, Inc.). A ‘Core functional analysis’ was performed to predict the effects of these genes on ‘Diseases and biological functions’ and ‘Canonical pathways’. Also identified were the upstream regulators that are most significantly associated with the regulation of these genes based on Ingenuity Knowledge Base in the IPA. ‘Fisher’s exact test’ was used to calculate the p value and presented as the negative logarithm of the p value (− log (p value)) for the association between the genes and the ‘Disease and function’/‘Canonical pathway’. The predicted activation state (activated or inhibited) of the canonical pathways or upstream regulators was determined by a ‘z score’ algorithm; it compared the gene expression dataset with the expected canonical pathway patterns or the upstream regulators (http://ingenuity.force.com/ipa). For canonical pathways, the ratio was determined by the number of genes in the uploaded dataset that mapped to the pathway divided by the total number of genes that mapped to the canonical pathway. A mechanistic network, indicating the relationships between the genes in the dataset with upstream regulator (p38 MAPK) as well as a network of the celastrol-regulated genes associated with EAE pathology, were also constructed by IPA.
SGK1 enzyme inhibition assay
This assay was performed using SGK1 kinase assay kit (Promega). Briefly, the enzyme (SGK1) was incubated with the substrate [Adenosine triphosphate (ATP)] along with either celastrol (test product) or Staurosporine (a known inhibitor of SGK1) in the kinase buffer. Following incubation, the adenosine diphosphate (ADP) formed by the enzyme activity was determined by adding ADP-Glo™ reagent and kinase detection reagent, followed by recording of luminescence.
Statistical analysis
The difference in gene expression comparing diseased (EAE) and celastrol-treated EAE mice for a given gene was assessed by Student’s ‘t’ test and p values below 0.05 were considered to be statistically significant. In regard to the disease scores, Wilcoxon rank sum test was performed and p value of less than 0.05 was considered significant.
Results
Celastrol suppresses EAE in mice and regulates antigen (MOG)-induced gene expression in immune cells (splenocytes)
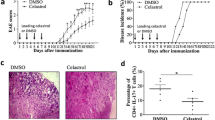
EAE was induced in C57BL/6 mice by immunization with MOG. Beginning from the onset of the disease, the experimental group of EAE mice was treated with celastrol intraperitoneal (i.p.) on alternate days and then continued for 25 days. The control EAE mice received the vehicle i.p. The celastrol-treated mice showed significant reduction in the clinical scores of EAE compared with the control group (Fig. 1a). The suppression of EAE by celastrol was further validated by histological examination of the spinal cord using hematoxylin and eosin staining. The mononuclear cell infiltration into the spinal cord of celastrol-treated mice was markedly decreased in contrast to the control mice (Fig. 1b).

Celastrol suppresses EAE and regulates antigen-induced gene expression in splenocytes. a The clinical scores of EAE mice treated with celastrol or the vehicle on alternate days starting from the day of onset of the disease are shown. The graph represents mean ± SEM (n = 5–6 mice per group). (*p < 0.05 by Wilcoxon rank sum test). b H&E-stained spinal cord sections of representative mice from celastrol-treated and control group on day 25 after MOG-immunization are shown. The arrows indicate the site of mononuclear cell infiltration. c–g qRT-PCR analysis of mRNA expression in splenocytes from EAE mice. The optimal difference in mRNA expression in cells after re-stimulation with MOG35–55 in the presence of celastrol versus MOG35-55 alone is presented as a ratio. Graphs show a representative profile of gene expression
We then examined the relative gene expression in splenocytes of untreated EAE mice and also assessed the effect of celastrol on 77 genes that are involved in the pathogenesis of EAE (Fig. 1c–g, Suppl. Figure 1). These genes included those encoding cytokines, chemokines, cytokine/chemokine receptors, transcription factors and other mediators of inflammation. As expected, exposure to MOG upregulated the expression of genes for most of the pro-inflammatory mediators of EAE, but downregulated the expression of some of the anti-inflammatory mediators. Noticeably, celastrol largely reversed the MOG-induced gene expression (Fig. 1c–g). Significant inhibition of the expression of pro-inflammatory cytokines (e.g., TNFα, IFNγ, IL-1β and IL-17A), pro-inflammatory chemokines or their receptors (e.g., CCL2 and CCR5), T cell-related transcription factors (e.g., RUNX1) and other inflammatory mediators (e.g., SGK1 and NOS2) was observed in cells treated with MOG in the presence of celastrol versus MOG alone. On the contrary, some of the other mediators (e.g., IL6ST and CCR3) showed significant increase in expression. Further, we observed a good trend towards the inhibition of TGFβ, IL-12β, IL-12α, CCL1, CXCL9 and TBX21, but upregulation of IL-11, HEYL and BDNF following celastrol treatment, although these changes were not statistically significant. However, in regard to BDNF, subsequent qRT-PCR testing confirmed its significant upregulation following celastrol treatment, as described below.
IPA analysis reveals critical disease-related pathways and upstream regulator of genes
‘Core analysis’ of the ingenuity pathway analysis (IPA) was performed on the top 20 celastrol-regulated genes, which were selected on the basis of the results described above (Fig. 1c–g), to determine the major ‘Diseases and function’ (Fig. 2) and ‘Canonical pathways’ (Fig. 3) affected by celastrol, as well as the upstream regulators of the celastrol-modulated gene expression (Table 1 and Fig. S2). Interestingly, ‘Cell-mediated immune response’ was the top ‘Diseases and function’ associated with celastrol-regulated genes, whereas, ‘T helper cell differentiation’ was the top canonical pathway affected by celastrol. Among these pathways, the high mobility group box 1 (HMGB1) signaling pathway is predicted to be significantly inhibited (with a negative z score), whereas the Th2 pathway is predicted to be activated (with a positive z score) (the assessment of ‘z score’ is described under “Ingenuity pathway analysis (IPA)”). Also, the neuroinflammation signaling pathway is predicted to be inhibited.

‘Diseases and biological functions’ identified using ingenuity pathway analysis (IPA) of celastrol-regulated genes. Top 15 ‘Disease and function’ of the 20 genes, whose expression is altered by celastrol treatment as identified by the IPA. ‘Threshold levels’ are based on significance (p < 0.05)

‘Canonical pathways’ identified using Ingenuity Pathway Analysis (IPA) of celastrol-regulated genes. Top 15 ‘Canonical pathways’ of the 20 genes, whose expression is altered by celastrol treatment as identified by the IPA. ‘Threshold levels’ are based on significance (p < 0.05). A negative z score indicates that the pathway is inhibited, whereas a positive z score indicates that the pathway is activated. Gray bars represent ‘no activity pattern available’. The ratio (orange dots connected by an orange line) indicates the ratio of genes from the dataset that map to the pathway divided by the total number of genes that map to the same pathway
In regard to the upstream regulators of celastrol-modulated genes, a subset of transcription factors and kinases that have a significant p value, a reported activation z score, and a predicted activation status of the upstream regulator is shown in Table 1. Of the top 10 upstream regulators identified, p38 MAPK matched the directional relationship with 13 of 20 target genes, with 65% match. Likewise, IKBKB (inhibitor of nuclear factor kappa-B kinase subunit beta) matched 9 target genes (45% match) and RELA (nuclear factor NF-kappa-B P65) matched 8 targets genes (40% match). The remaining upstream regulators had a directional match of less than 8 target genes (less than 40% match). Based on this analysis, p38 MAPK was the optimal choice for further analysis. Therefore, we then examined in more detail the network of p38 MAPK upstream regulator, which included 13 direction-matched target genes and 3 target genes with no specified direction (RUNX1, CXCL9 and CCR5) together with other participating molecules (Fig. S2). Interestingly, RELA and IKBKB [as part of nuclear factor kappa B subunit 1(NFKB1)] mentioned above are associated with the p38 MAPK network. Furthermore, ERK is also involved in the regulation of some of the genes shown to be regulated by p38 MAPK but it is shown below p38MAPK in IPA, indicating a more predominant role of p38 MAPK in regulating various genes examined. Taken together, our results suggest that p38 MAPK is one of the key upstream regulators of celastrol-regulated genes.
We generated another network using ‘Molecular activity prediction’ tool on IPA involving 20 celastrol-regulated genes and the 3 upstream regulators to visualize the effect of those molecules on the pathology of EAE (Fig. S3). Interestingly, celastrol-induced gene regulation together with predicted inhibition of their upstream regulators was found to be associated with the suppression of EAE.
Celastrol inhibits SGK1 expression but induces BDNF expression in T cells under Th17-differentiating conditions
As shown in Figs. 2 and 3, ‘Cell-mediated immune response’ and ‘T helper cell differentiation’ were the top ‘Diseases and function’ and canonical pathway, respectively, affected by celastrol. As in some other autoimmune conditions, the Th17/Treg balance is a critical regulator of EAE. Therefore, we tested the expression of two of the celastrol-modulated genes, SGK1 and BDNF, in Th17 and natural Treg (nTreg) cells (Fig. 4a). SGK1 is a recently identified regulator of Th17/Treg balance [37], whereas BDNF is known to regulate neuronal development and function [38, 39]. Our pilot IPA analysis showed that BDNF is a stabilizer of FOXP3 expression, implying the role of BDNF in Treg function. Interestingly, these two genes (SGK1 and BDNF) were found to be differentially expressed in Th17 and nTreg cells (Fig. 4a). The expression of SGK1 was significantly increased in Th17, but decreased in nTreg compared to Th0 cells. In contrast, the expression of BDNF was significantly decreased in Th17, but increased in nTreg.

Celastrol regulates the expression of SGK1 and BDNF in T cells. a RNA isolated from Th17 cells, natural Treg (nTreg), as well as Th0 cells was analyzed by qRT-PCR for mRNA expression. b Naïve T cells were cultured under Th17-differentiation conditions for 4 days in the presence or absence of celastrol. The expression of SGK1, BDNF, and IL-17 mRNA was measured using qRT-PCR. c Th17/Treg differentiation was performed in the presence or absence of celastrol and then the cells analyzed for various cellular markers (RORγt, FOXP3, or CD25) by flow cytometry. Bar graphs represent the relative levels of RORγt and FOXP3 in cells under the indicated culture conditions
We also compared the expression of SGK1 and BDNF in the T cells cultured under Th17-differentiation conditions in the absence or presence of celastrol (Fig. 4b). Interestingly, celastrol significantly inhibited SGK1, but induced BDNF in these cells. Furthermore, the pattern of expression of these two genes matched the results of celastrol-modulated gene expression shown in Fig. 1.
Finally, we tested the effect of celastrol on Th17 differentiation and induction of Treg in vitro. We observed that celastrol inhibited Th17, but facilitated Treg differentiation (Fig. 4c). Our results suggest that in Th17 cells, celastrol treatment inhibited RORγt, but did not have much effect on FOXP3. However, in Treg cells, celastrol markedly increased FOXP3 expression. On the basis of our other results (Fig. 4a, b), we suggest that the effect of celastrol on the differentiation of Th17 and Treg is mediated in part via the inhibition of SKG1. This suggestion was further examined as described below.
Celastrol regulates expression of SGK1 and BDNF in T cells via MAPK (p38, ERK) pathway
We described above results indicating that p38 MAPK and ERK serve as mediators of celastrol action (Table 1 and Fig. S2). Furthermore, p38 MAPK and ERK are upstreams of the two genes (SGK1 and BDNF) mentioned above. Accordingly, we hypothesized that celastrol might regulates SGK1 and BDNF via inhibiting p38 MAPK. To directly examine this proposition, we determined the expression of SGK1 and BDNF in the T cells in response to celastrol (test compound), using SB203580, a known p38 MAPK inhibitor as a positive control (Fig. 5). The T cells activated with anti-CD3/anti-CD28 in the presence of IL-6 showed an increase of SGK1, but reduction of BDNF expression (Fig. 5a). Interestingly, this pattern of expression of SGK1 and BNDF was reversed in the presence of either celastrol or p38 MAPK inhibitor (Fig. 5a). Importantly, the overall effect of celastrol was comparable to that of p38 MAPK inhibitor, although the latter was slightly more effective than celastrol under the test conditions. These results suggest that the expression of SGK1 and BDNF is controlled by p38 MAPK. Furthermore, we tested whether celastrol can directly inhibit the enzyme activity of SGK1. Also tested in parallel was staurosporine, a known SGKI inhibitor. Our results (Fig. 5b) showed that celastrol did not inhibit the activity of SGK1 enzyme, whereas staurosporine was quite effective in inhibiting SGK1 activity. Taken together, these results show that celastrol regulates SGK1 indirectly via MAPK pathway.

Celastrol regulates SGK1 and BDNF via p38 MAPK. a Naïve T cells were stimulated with anti-CD3 and anti-CD28 combination in the presence of IL-6 and/or indicated concentration of celastrol (test compound) or SB203580 (a known p38 MAPK inhibitor; positive control). RNA isolated from these cells was analyzed by qRT-PCR for mRNA expression. b SGK1 inhibition assay involving pre-incubation of SGK1 and celastrol/staurosporine followed by incubation with an enzyme substrate. The enzyme activity was determined by measuring luminescence intensity
Discussion
Several studies, including ours have reported diverse targets of celastrol, including NFkB, certain cell signaling molecules, and anti-oxidant and anti-inflammatory mediators [32,33,34, 40,41,42,43,44,45,46], which in turn contribute to protection against EAE. However, the search continues for newer targets of celastrol and to further understand its mechanism of action in EAE and other autoimmune diseases. In this context, using the EAE model, we examined the antigen (MOG)-induced gene expression profile in splenocytes of the diseased mice, and also evaluated the changes induced by celastrol. As expected, several pro-inflammatory mediators were induced, along with induction of a few anti-inflammatory mediators, albeit to a lesser extent, in splenocytes of the diseased mice. This latter effect might represent the host immune system’s effort to combat the pro-inflammatory response induced by MOG. Importantly, celastrol reversed the expression of many genes that were upregulated by MOG, but it also induced the expression of certain other genes. These changes in gene expression highlight the potential mediators and pathways that are influenced by celastrol. This is further elaborated by our extensive bioinformatics analyses described above. The present study is the first comprehensive testing of 77 genes, providing a broader landscape of molecular changes occurring during EAE development. The ‘Upstream regulator’ analysis of celastrol-modulated genes suggested p38 MAPK and ERK as targets of celastrol. Furthermore, IPA analysis suggested a prominent effect of celastrol on ‘Cell-mediated immune response’ and ‘T helper cell differentiation’, which then directed us to examine the Th17/Treg differentiation in more detail. A couple of earlier reports showing the inhibitory activity of celastrol against p38 MAPK [47, 48] support our results on celastrol targets, although EAE was not tested in those two studies. Furthermore, the significance of p38MAPK in EAE is demonstrated by a few studies mentioned above, but celastrol was not tested in those studies. For example, in a study using genetically manipulated mice, it was shown that regulation of the p38 MAPK pathway in CD4 T cells alone was sufficient to modulate EAE severity [25]. Furthermore, p38 MAPK inhibition was shown to suppress the expression of IL-17 in CD4 T cells. Another study examined the relationship between p38 activity and IL-17 mRNA expression, showing that p38 positively regulated the transcription of the IL17 gene, and that an orally administered p38a inhibitor reduced the severity of EAE [24]. Our results are also supported by another study showing the inhibitory effect of celastrol against EAE [33]. We further provide here a mechanism of inhibition of Th17 via MAPK-SGK1 axis. Recently, SGK1 is reported to be a critical regulator for the reciprocal control of RORγt+ Th17 cell and FOXP3+ Treg cells [19, 37, 49, 50]. The inhibition of SGK1 was shown to suppress Th17, but facilitate Treg differentiation [37]. Interestingly, our IPA-based analysis also revealed that SGK1 expression is controlled by p38 MAPK. Previous studies have suggested the p38 MAPK-mediated regulation of SGK1 [51,52,53], although celastrol was not tested in those studies. Therefore, we hypothesized that celastrol-regulated Th17/Treg through inhibition of the p38 MAPK–SGK1 axis. This conclusion is supported by our results showing that SGK1 is highly expressed in Th17 cells, but its expression is inhibited following treatment with celastrol. The inhibition of p38 MAPK directly by celastrol is reported by few other investigators [47, 48] but those studies were not based on EAE. Furthermore, the activation of SGK1 by cytokines such as IL-23 [50] provides another link with Th17 response, and we suggest that celastrol might mediate its effect in part via inhibiting pro-inflammatory cytokines. Taken together, our results suggest that the activation of the p38 MAPK–SGK1 axis can dysregulate the Th17/Treg balance in favor of Th17, and thereby, inhibition of this axis can reset this T subset balance, leading to the control of EAE. Furthermore, p38-MAPK-SGK1 axis has been implicated in oxidative stress [26]. Celastrol is known to have anti-oxidant activity [40, 41, 45], and thereby, the p38-MAPK-SGK1 pathway can contribute to the EAE-protective effect of celastrol in two ways, via anti-oxidant effect and restoring the Th17/Treg balance. Another notable target of p38 MAPK revealed in our IPA analysis was BDNF, whose expression was enhanced. This was in contrast to the effect of p38 MAPK on SGK1, whose expression was inhibited. The role of BDNF as a neurotrophic factor is well known [54, 55]. In addition, it has been shown that BDNF also can be expressed in the T cells, and that the T cells infiltrating the demyelinating lesions in MS patients secrete BDNF locally [54,55,56]. This local BDNF secretion is believed to contribute to the recovery process following neuroinflammation in the course of MS. Whether BDNF has any direct effect on the programming of T cell differentiation into Th17 and Treg remains to be examined. Our results of T cell subset analysis show that nTreg express much higher levels of BDNF than Th17 cells. In addition, celastrol enhances the expression of BDNF in T cells under Th17-differentiating conditions. Interestingly, Fingolimod, a drug used for the treatment of MS, has also been shown to increases BDNF expression [57]. Thus, new drugs or natural products that enhance BDNF expression can be further examined for the treatment of MS. In this study, we focused our attention on Th17 and Treg cells. However, it is well known that Th1 cells are also critical in EAE pathogenesis [58, 59]. In fact, our data show marked downregulation of IFN-γ and Tbx21 (Fig. 1). While IFN-γ itself may not be necessary for EAE, T-bet and IFN-γ-expressing Th1 cells are pathogenic in EAE and many other autoimmune diseases. Further, as mentioned above, p38 signaling in DCs and macrophages is also critical for EAE. Therefore, celastrol’s effect on suppressing EAE not only involves Th17 cells, but also Th1, DCs, and macrophages. Finally, the IPA analysis also suggested an association between certain intermediate mediators including members of the NF-kB complex and ERK 1/2 in the regulation of celastrol-modulated genes. In this regard, celastrol has previously shown by others [32, 60] and us [28, 31] in different experimental systems to inhibit NF-kB and ERK 1/2. Based on these observations, we suggest that celastrol might also modulate the expression of some of the genes tested in this study by inhibiting one or both of these two mediators besides inhibiting p38 MAPK. Furthermore, our bioinformatics analysis showed celastrol targeting both p38 MAPK and ERK, which are components of MAPK pathway, with a predominant role of p38 MAPK in hierarchy. Our study on celastrol has further expanded the mechanistic aspects of modulation of EAE by traditional Chinese medicine, as also exemplified by comprehensive recent studies using the testing of Pien Tze Huang (PZH) [61] and Astragaloside IV from Astragalus membranaceus [62] in the EAE model. In summary, our results highlight novel pathways targeted by celastrol in mediating its protective effect against EAE. These involve MAPK (p38 MAPK, ERK), SGK1, and BDNF. These results have advanced our understanding of the disease process as well as offered novel therapeutic targets for MS.
Conclusion
Our previous studies [28, 29, 31] and those by other investigators [32, 33] have revealed the immunomodulatory activity of celastrol in animal models of RA and MS. This disease-suppressing activity of celastrol is partly attributed to the regulation of the Th17/Treg balance [28]. However, the mechanisms by which celastrol alters Th17/Treg balance are not yet fully defined. The results of this study based on the EAE model suggest that celastrol’s effects are mediated via MAPK and SGK1 to reset the Th17/Treg balance, and to enhance BDNF production in T cells, leading to protection against EAE.
Abbreviations
- AP-1:
-
Activator protein 1
- BDNF:
-
Brain-derived neurotrophic factor
- CEBPB:
-
CCAAT enhancer binding protein β1
- ERK1/2:
-
Extracellular signal-regulated kinase ½
- GSK3B:
-
Glycogen synthase kinase 3 beta
- IKBKB:
-
Inhibitor of Nuclear factor kappa B kinase subunit beta
- IRF8:
-
Interferon regulatory factor 8
- MAPK:
-
Mitogen-activated protein kinase
- MOG:
-
Myelin oligodendrocyte glycoprotein
- NFKB1:
-
Nuclear factor kappa B subunit 1
- NFKBIA:
-
NFKB inhibitor alpha
- NR3C1:
-
Nuclear receptor subfamily 3, group C, member 1/glucocorticoid receptor
- PKC:
-
Protein kinase C
- RELA:
-
Rel-like domain-containing proteins
- SGK1:
-
Serum/glucocorticoid regulated kinase 1
- SP1:
-
Specificity protein 1
- STAT4:
-
Signal transducer and activator of transcription 4
- TLR:
-
Toll-like receptor
References
Bhise V, Dhib-Jalbut S. Further understanding of the immunopathology of multiple sclerosis: impact on future treatments. Expert Rev Clin Immunol. 2016;12(10):1069–89.
Zwibel HL, Smrtka J. Improving quality of life in multiple sclerosis: an unmet need. Am J Manag Care. 2011;17(Suppl 5 Improving):139–45.
Dilokthornsakul P, Valuck RJ, Nair KV, Corboy JR, Allen RR, Campbell JD. Multiple sclerosis prevalence in the United States commercially insured population. Neurology. 2016;86(11):1014–21.
Yadav SK, Mindur JE, Ito K, Dhib-Jalbut S. Advances in the immunopathogenesis of multiple sclerosis. Curr Opin Neurol. 2015;28(3):206–19.
Bettelli E, Korn T, Oukka M, Kuchroo VK. Induction and effector functions of T(H)17 cells. Nature. 2008;453(7198):1051–7.
Holmoy T. The immunology of multiple sclerosis: disease mechanisms and therapeutic targets. Minerva Med. 2008;99(2):119–40.
Zozulya AL, Wiendl H. The role of regulatory T cells in multiple sclerosis. Nat Clin Pract Neurol. 2008;4(7):384–98.
Di Mitri D, Sambucci M, Loiarro M, De Bardi M, Volpe E, Cencioni MT, Gasperini C, Centonze D, Sette C, Akbar AN, et al. The p38 mitogen-activated protein kinase cascade modulates T helper type 17 differentiation and functionality in multiple sclerosis. Immunology. 2015;146(2):251–63.
Lochner M, Wang Z, Sparwasser T. The special relationship in the development and function of T helper 17 and regulatory T cells. Prog Mol Biol Transl Sci. 2015;136:99–129.
Wang C, Collins M, Kuchroo VK. Effector T cell differentiation: are master regulators of effector T cells still the masters? Curr Opin Immunol. 2015;37:6–10.
Miossec P, Korn T, Kuchroo VK. Interleukin-17 and type 17 helper T cells. N Engl J Med. 2009;361(9):888–98.
Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Ann Rev Immunol. 2009;27:485–517.
Shevach EM. Biological functions of regulatory T cells. Adv Immunol. 2011;112:137–76.
Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: mechanisms of differentiation and function. Ann Rev Immunol. 2012;30:531–64.
Fasching P, Stradner M, Graninger W, Dejaco C, Fessler J. Therapeutic potential of targeting the Th17/Treg axis in autoimmune disorders. Molecules 2017;22(1):134.
Fessler J, Felber A, Duftner C, Dejaco C. Therapeutic potential of regulatory T cells in autoimmune disorders. BioDrugs. 2013;27(4):281–91.
Mai J, Wang H, Yang XF. Th 17 cells interplay with Foxp3+ Tregs in regulation of inflammation and autoimmunity. Front Biosci (Landmark Ed). 2010;15:986–1006.
Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, Muller DN, Hafler DA. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature. 2013;496(7446):518–22.
Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, Kishi Y, Regev A, Kuchroo VK. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature. 2013;496(7446):513–7.
Krementsov DN, Thornton TM, Teuscher C, Rincon M. The emerging role of p38 mitogenactivated protein kinase in multiple sclerosis and its models. Mol Cell Biol. 2013;33(19):3728–34.
Canto E, Isobe N, Didonna A, Hauser SL, Oksenberg JR. Aberrant STAT phosphorylation signaling in peripheral blood mononuclear cells from multiple sclerosis patients. J Neuroinflammation. 2018;15(1):72.
Huang G, Wang Y, Vogel P, Chi H. Control of IL-17 receptor signaling and tissue inflammation by the p38alpha-MKP-1 signaling axis in a mouse model of multiple sclerosis. Sci Signal. 2015;8(366):ra24.
Krementsov DN, Noubade R, Dragon JA, Otsu K, Rincon M, Teuscher C. Sex-specific control of central nervous system autoimmunity by p38 mitogen-activated protein kinase signaling in myeloid cells. Ann Neurol. 2014;75(1):50–66.
Namiki K, Matsunaga H, Yoshioka K, Tanaka K, Murata K, Ishida J, Sakairi A, Kim J, Tokuhara N, Shibakawa N, et al. Mechanism for p38alpha-mediated experimental autoimmune encephalomyelitis. J Biol Chem. 2012;287(29):24228–38.
Noubade R, Krementsov DN, Del Rio R, Thornton T, Nagaleekar V, Saligrama N, Spitzack A, Spach K, Sabio G, Davis RJ, et al. Activation of p38 MAPK in CD4 T cells controls IL-17 production and autoimmune encephalomyelitis. Blood. 2011;118(12):3290–300.
Wang L, Li B, Quan MY, Li L, Chen Y, Tan GJ, Zhang J, Liu XP, Guo L. Mechanism of oxidative stress p38MAPK-SGK1 signaling axis in experimental autoimmune encephalomyelitis (EAE). Oncotarget. 2017;8(26):42808–16.
Kim EK, Choi EJ. Pathological roles of MAPK signaling pathways in human diseases. Biochimica et biophysica acta. 2010;1802(4):396–405.
Astry B, Venkatesha SH, Laurence A, Christensen-Quick A, Garzino-Demo A, Frieman MB, O’Shea JJ, Moudgil KD. Celastrol, a Chinese herbal compound, controls autoimmune inflammation by altering the balance of pathogenic and regulatory T cells in the target organ. Clin Immunol. 2015;157(2):228–38.
Nanjundaiah SM, Venkatesha SH, Yu H, Tong L, Stains JP, Moudgil KD. Celastrus and its bioactive celastrol protect against bone damage in autoimmune arthritis by modulating osteoimmune cross-talk. J Biol Chem. 2012;287(26):22216–26.
Venkatesha SH, Dudics S, Astry B, Moudgil KD. Control of autoimmune inflammation by celastrol, a natural triterpenoid. Pathogens Dis 2016;74(6):ftw059.
Venkatesha SH, Yu H, Rajaiah R, Tong L, Moudgil KD. Celastrus-derived celastrol suppresses autoimmune arthritis by modulating antigen-induced cellular and humoral effector responses. J Biol Chem. 2011;286(17):15138–46.
Abdin AA, Hasby EA. Modulatory effect of celastrol on Th1/Th2 cytokines profile, TLR2 and CD3 + T-lymphocyte expression in a relapsing-remitting model of multiple sclerosis in rats. Eur J Pharmacol. 2014;742:102–12.
Wang Y, Cao L, Xu LM, Cao FF, Peng B, Zhang X, Shen YF, Uzan G, Zhang DH. Celastrol ameliorates EAE induction by suppressing pathogenic T cell responses in the peripheral and central nervous systems. J Neuroimmune Pharmacol. 2015;10(3):506–16.
Yang H, Liu C, Jiang J, Wang Y, Zhang X. Celastrol attenuates multiple sclerosis and optic neuritis in an experimental autoimmune encephalomyelitis model. Front Pharmacol. 2017;8:44.
Bittner S, Afzali AM, Wiendl H, Meuth SG. Myelin oligodendrocyte glycoprotein (MOG35-55) induced experimental autoimmune encephalomyelitis (EAE) in C57BL/6 mice. J Vis Exp. 2014;86:e51275.
Kwong B, Rua R, Gao Y, Flickinger J Jr, Wang Y, Kruhlak MJ, Zhu J, Vivier E, McGavern DB, Lazarevic V. T-bet-dependent NKp46(+) innate lymphoid cells regulate the onset of TH17-induced neuroinflammation. Nat Immunol. 2017;18(10):1117–27.
Wu C, Chen Z, Xiao S, Thalhamer T, Madi A, Han T, Kuchroo V. SGK1 governs the reciprocal development of Th17 and regulatory T cells. Cell Rep. 2018;22(3):653–65.
Bathina S, Das UN. Brain-derived neurotrophic factor and its clinical implications. Arch Med Sci. 2015;11(6):1164–78.
Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. 2001;24:677–736.
Venkatesha SH, Moudgil KD. Celastrol and its role in controlling chronic diseases. Adv Exp Med Biol. 2016;928:267–89.
Kannaiyan R, Shanmugam MK, Sethi G. Molecular targets of celastrol derived from Thunder of God Vine: potential role in the treatment of inflammatory disorders and cancer. Cancer Lett. 2011;303(1):9–20.
Salminen A, Lehtonen M, Paimela T, Kaarniranta K. Celastrol: molecular targets of thunder god vine. Biochem Biophys Res Commun. 2010;394(3):439–42.
Faust K, Gehrke S, Yang Y, Yang L, Beal MF, Lu B. Neuroprotective effects of compounds with antioxidant and anti-inflammatory properties in a Drosophila model of Parkinson’s disease. BMC Neurosci. 2009;10:109.
Gu L, Bai W, Li S, Zhang Y, Han Y, Gu Y, Meng G, Xie L, Wang J, Xiao Y, et al. Celastrol prevents atherosclerosis via inhibiting LOX-1 and oxidative stress. PLoS One. 2013;8(6):e65477.
Cascao R, Fonseca JE, Moita LF. Celastrol: a spectrum of treatment opportunities in chronic diseases. Front Med (Lausanne). 2017;4:69.
Gupta SC, Kim JH, Prasad S, Aggarwal BB. Regulation of survival, proliferation, invasion, angiogenesis, and metastasis of tumor cells through modulation of inflammatory pathways by nutraceuticals. Cancer Metastasis Rev. 2010;29(3):405–34.
Choi BS, Kim H, Lee HJ, Sapkota K, Park SE, Kim S, Kim SJ. Celastrol from ‘Thunder God Vine’ protects SH-SY5Y cells through the preservation of mitochondrial function and inhibition of p38 MAPK in a rotenone model of Parkinson’s disease. Neurochem Res. 2014;39(1):84–96.
Luo D, Guo Y, Cheng Y, Zhao J, Wang Y, Rong J. Natural product celastrol suppressed macrophage M1 polarization against inflammation in diet-induced obese mice via regulating Nrf2/HO-1, MAP kinase and NF-kappaB pathways. Aging (Albany NY). 2017;9(10):2069–82.
Hernandez AL, Kitz A, Wu C, Lowther DE, Rodriguez DM, Vudattu N, Deng S, Herold KC, Kuchroo VK, Kleinewietfeld M, et al. Sodium chloride inhibits the suppressive function of FOXP3+ regulatory T cells. J Clin Invest. 2015;125(11):4212–22.
van der Meer JW, Netea MG. A salty taste to autoimmunity. The New England journal of medicine. 2013;368(26):2520–1.
BelAiba RS, Djordjevic T, Bonello S, Artunc F, Lang F, Hess J, Gorlach A. The serum- and glucocorticoid-inducible kinase Sgk-1 is involved in pulmonary vascular remodeling: role in redox-sensitive regulation of tissue factor by thrombin. Circ Res. 2006;98(6):828–36.
Pastore D, Della-Morte D, Coppola A, Capuani B, Lombardo MF, Pacifici F, Ferrelli F, Arriga R, Mammi C, Federici M, et al. SGK-1 protects kidney cells against apoptosis induced by ceramide and TNF-alpha. Cell Death Dis. 2015;6:e1890.
Bell LM, Leong ML, Kim B, Wang E, Park J, Hemmings BA, Firestone GL. Hyperosmotic stress stimulates promoter activity and regulates cellular utilization of the serum- and glucocorticoid-inducible protein kinase (Sgk) by a p38 MAPK-dependent pathway. J Biol Chem. 2000;275(33):25262–72.
De Santi L, Annunziata P, Sessa E, Bramanti P. Brain-derived neurotrophic factor and TrkB receptor in experimental autoimmune encephalomyelitis and multiple sclerosis. J Neurol Sci. 2009;287(1–2):17–26.
Stadelmann C, Kerschensteiner M, Misgeld T, Bruck W, Hohlfeld R, Lassmann H. BDNF and gp145trkB in multiple sclerosis brain lesions: neuroprotective interactions between immune and neuronal cells? Brain. 2002;125(Pt 1):75–85.
Linker R, Gold R, Luhder F. Function of neurotrophic factors beyond the nervous system: inflammation and autoimmune demyelination. Crit Rev Immunol. 2009;29(1):43–68.
Smith PA, Schmid C, Zurbruegg S, Jivkov M, Doelemeyer A, Theil D, Dubost V, Beckmann N. Fingolimod inhibits brain atrophy and promotes brain-derived neurotrophic factor in an animal model of multiple sclerosis. J Neuroimmunol. 2018;318:103–13.
Murphy AC, Lalor SJ, Lynch MA, Mills KH. Infiltration of Th1 and Th17 cells and activation of microglia in the CNS during the course of experimental autoimmune encephalomyelitis. Brain Behav Immun. 2010;24(4):641–51.
Stromnes IM, Cerretti LM, Liggitt D, Harris RA, Goverman JM. Differential regulation of central nervous system autoimmunity by T(H)1 and T(H)17 cells. Nat Med. 2008;14(3):337–42.
Veerappan K, Natarajan S, Ethiraj P, Vetrivel U, Samuel S. Inhibition of IKKbeta by celastrol and its analogues - an in silico and in vitro approach. Pharm Biol. 2017;55(1):368–73.
Qiu X, Luo H, Liu X, Guo Q, Zheng K. Fan D1, Shen J. Lu C1, He X, Zhang G, Lu A. Therapeutic Potential of Pien Tze Huang on Experimental AutoimmuneEncephalomyelitis Rat. J Immunol Res. 2018;2018:2952471
Yang L, Xing F, Han X, Li Q, Wu H, Shi H, Wang Z, Huang F, Wu X. Astragaloside IV regulates differentiation and induces apoptosis of activated CD4+ T cells in the pathogenesis of experimental autoimmune encephalomyelitis. Toxicol Appl Pharmacol. 2018;362:105–115.
Acknowledgements
This work was supported in part by Grants (1R21NS082918 and R01 AT 004321) from the National Institutes of Health, Bethesda, MD and in part by VA Merit Review Award # 5 I01 BX002424 (to KDM) from the United States (U.S.) Department of Veterans Affairs [Biomedical Laboratory Research and Development Service]. We thank Jason Lees (USUHS, Bethesda) and Bodhraj Acharya (UMB) for helpful advice and discussion regarding the MOG-EAE model. We thank Rakeshchandra Reddy Meka and Steven Dudics for help with some experiments. We also thank Carol Fowler and Tom Bowen for help with the VA Research Facilities. This material is the result of work supported in part with resources and the use of facilities at the VA Maryland Health Care System, Baltimore, Maryland.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest. The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
Additional information
Responsible Editor: John Di Battista.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Venkatesha, S.H., Moudgil, K.D. Celastrol suppresses experimental autoimmune encephalomyelitis via MAPK/SGK1-regulated mediators of autoimmune pathology. Inflamm. Res. 68, 285–296 (2019). https://doi.org/10.1007/s00011-019-01219-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-019-01219-x