
Abstract
Celastrol, a potent natural triterpene and one of the most promising medicinal molecules, is known to possess a broad range of biological activity. Rotenone, a pesticide and complex I inhibitor, is commonly used to produce experimental models of Parkinson’s disease both in vivo and in vitro. The present study was designed to examine the effects of celastrol on cell injury induced by rotenone in the human dopaminergic cells and to elucidate the possible mechanistic clues in its neuroprotective action. We demonstrate that celastrol protects SH-SY5Y cells from rotenone-induced cellular injury and apoptotic cell death. Celastrol also prevented the increased generation of reactive oxygen species and mitochondrial membrane potential (ΔΨm) loss induced by rotenone. Similarly, celastrol treatment inhibited cytochrome c release, Bax/Bcl-2 ratio changes, and caspase-9/3 activation. Celastrol specifically inhibited rotenone-evoked p38 mitogen-activated protein kinase activation in SH-SY5Y cells. These data suggest that celastrol may serve as a potent agent for prevention of neurotoxin-induced neurodegeneration through multiple mechanisms and thus has therapeutic potential for the treatment of neurodegenerative diseases.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
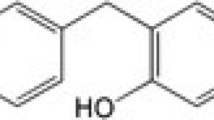
Celastrol (Fig. 1a) is a quinone methide triterpenoid isolated from root bark of Thunder of God Vine (Tripterygium wilfordii Hook F.) of the Celastraceae family. This plant is native to China, Japan and Korea, and has a long history of use in traditional medicine for treating various illnesses [1, 2]. In recent years, celastrol has been emerging as a potent candidate with medicinal prospects for treating inflammatory diseases and cancer [3–5]. It possesses a broad range of biological activity including antioxidant properties. The antioxidant activity of celastrol has been reported to be more potent than alpha-tocopherol [6]. Numerous studies have demonstrated that celasterol can inhibit the inflammatory response in macrophages, microglia and endothelial cells [7–9]. It has been identified as a potential neuroprotective candidate [10, 11]. In a rat model of Alzheimer’s disease (AD), celastrol improved memory and learning in a psychomotor-activity test [7] and reduced β-amyloid via an NF-κB dependent mechanism [12]. Moreover, it was found to prevent MPTP-induced neurotoxicity in mouse [13], induce heat shock proteins in differentiated human and rodent neurons [14], and protect the dopaminergic neurons in DJ-1A model of Drosophila [15]. All these findings suggest that celastrol could play a therapeutic role in neurodegenerative diseases.

Evaluation of cytotoxic effect of celastrol and rotenone by MTT and LDH assays. a The chemical structure of celastrol. Effect of celastrol (b) and rotenone (c) on SH-SY5Y cell viability by MTT assay. Cells were treated with celastrol (1–10 nM) or rotenone (1–100 μM) for 24 h. Cell viability was assessed by MTT reduction assay. Effect of celastrol (d) and rotenone (e) on the leakage of LDH in SH-SY5Y cells. Cells were treated with indicated concentrations of celastrol or rotenone for 24 h and the LDH released in the medium were measured by LDH assay. Values are expressed as percentage of control (vehicle). The data are expressed as mean ± SD of three experiments. *P < 0.05 versus control group
Parkinson’s disease (PD) is the most prevalent neurodegenerative movement disorder. This devastating disease affects 1–2 % of the population over the age of 60 [16] and is expected to impose an increasing socioeconomic burden on societies and a major challenge for biomedicine. PD is characterized clinically by resting tremor, rigidity, bradykinesia, and postural instability and are due to progressive loss of dopaminergic neurons in the substantia nigra and depletion of the neurotransmitter dopamine in the striatum. Although the molecular mechanism associated with the selective degeneration of dopaminergic (DA) neurons are not known completely, oxidative stress [17], and mitochondrial dysfunction [18, 19] have been reported to play a fundamental roles. While the causes of sporadic PD remains unknown, epidemiological studies suggest that environmental factors, particularly pesticide exposure, increase the risk of PD. Rotenone is a common pesticide and naturally occurring plant compound derived from the roots of certain species. Rotenone induces cell destruction by inhibiting mitochondrial complex I and mimics the symptoms of PD, both in vivo and in vitro [20–23]. The rotenone model recapitulates most of the mechanisms that are thought to be relevant in PD pathogenesis and is thus suitable to test neuroprotective strategies [24, 25].
So far, there have been very few reports of effectiveness of celastrol against rotenone-induced PD model. Recently, it has been shown that celastrol protects SH-SY5Y cells from rotenone induced injury through autophagy [26]. In this study, we investigated the effects of celastrol on cell apoptosis/injury in rotenone-treated human dopaminergic cells, and attempted to elucidate the underlying mechanisms by investigating the involvement of mitogen-activated protein kinases (MAPKs) signaling pathways.
Materials and Methods
Materials
Celastrol was purchased from Cayman Chemical (Ann Arbor, MI, USA). Dulbecco’s Modified Eagle Medium (DMEM), Fetal Bovine Serum (FBS), penicillin and streptomycin were obtained from Gibco BRL (Gaithersburg MD, USA). SB203580, Nacety-l-cysteine (NAC), 2′7′-dichlorofluorescein diacetate (DCF-DA), dimethyl sulfoxide (DMSO), 4′,6-diamidino-2-phenylindole (DAPI), Propidium iodide (PI) and Rhodamine 123 were obtained from Sigma-Aldrich (St. Louis, MO, USA). caspase-3, caspase-9, Bcl-2, Bax, cytochrome c, pJNK, JNK, pP38, and p38 antibodies were obtained from SANTA CRUZ (Santa Cruz, CA, USA). Anti-COX-IV antibody was obtained from Abcam (Cambridge, UK). Anti-actin antibody was purchased from Biomeda Crop (Foster City, CA, USA). WEST-ZOL plus was obtained from INTRON Biotech (Seongnam, Korea). Bicinchoninic acid (BCA) protein assay kit was obtained from Pierce (Rockford, IL, USA). Protein inhibitor cocktail was obtained from Calbiochem (Darmstadt, Germany). Cytotoxicity Detection Kit (LDH assay) was purchased from Roche Applied Science (Rotkreuz, Switzerland). DeadEnd™ Fluorometric TUNEL System was purchased from Promega coporation (Madison, WI, USA).
Cell Culture and Treatments
The human DA neuronal cell line, SH-SY5Y was obtained from the American Type Culture Collection (Rockville, MD). The cells were cultured in DMEM supplemented with 10 % FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin, and kept at 37 °C in humidified 5 % CO2/95 % air. Media were changed every 2 days. Rotenone and celastrol were dissolved in dimethyl sulphoxide (DMSO) to a final concentration of 0.01 %. To examine possible toxic effects, SH-SY5Y cells were treated with celastrol in a concentration ranging from 1 to 10 nM for 24 h. Similarly, SH-SY5Y cells were treated with rotenone at concentrations ranging from 1 to 100 μM for 24 h. Celastrol at 2.5 nM and 10 μM of rotenone was chosen to evaluate the neuroprotective effects by examining cell viability. To examine the protective effect of celastrol, cells were treated with rotenone in the absence or presence of celastrol for 24 h. Cells were treated with DMSO as a vehicle control. Celastrol was added 90 min prior to treatment with rotenone. In addition, cells were also pre-treated with NAC (1 mM) followed by exposure to rotenone in the absence or presence of celastrol. Moreover, cells were pre-treated for 30 min with SB203580 (p38 inhibitor) and/or NAC then treated with rotenone in the absence or presence of celastrol. For JNK and p38 MAPK Western blot analysis, regular culture medium was replaced with low-serum media 1 h before rotenone treatment to minimize background kinase activity. In a single experiment each treatment was performed in triplicate.
Analysis of Cell Viability
Cell viability was determined by the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. SH-SY5Y cells were seeded in 96-well plates at a density of 1 × 104 cell/well and incubated for 24 h prior to experimental treatments. The cells were then subjected to the treatments of interest. After 24 h incubation, MTT (0.5 mg/mL) was added to each wall. Following an additional 4 h incubation at 37 °C, 100 μL of DMSO was added to dissolve the formazan crystals. The absorbance was then measured at 570 nm using a VERSAmax micro plate reader (Molecular Devices, CA, USA). Wells without cells were used as blanks and were subtracted as background from each sample. Results were expressed as a percentage of control.
Lactate Dehydrogenase (LDH) Release Assay
Cells dying by apoptosis or necrosis released LDH into the supernatant. The amount of LDH in the supernatant was measured with a cytotoxicity detection kit (Roche). Briefly, SH-SY5Y cells (1 × 104 cell/well) were seeded in 96 well plates and then subjected to the indicated treatments. For analysis, 100 μL supernatant was extracted from each well and was placed in separate wells of a new 96-well plate, and 100 μL catalyst solutions was added to each well and incubated at 37 °C for 30 min. Absorbance was measured at 490 nm using a VERSAmax microplate reader (Molecular Devices, Sunnyvale, CA, USA). Total cellular LDH was determined by lysing the cells with 2 % Triton X-100 (high control); the assay medium served as a low control and was subtracted from all absorbance measurements; Cytotoxicity (%) = (exp.value − low control)/(high control − low control) × 100.
Observations of Morphological Changes
SH-SY5Y Cells were seeded in 8-well chamber slide and then treated with rotenone for 24 h after pre-treated with or without celastrol for 90 min. The cells were washed twice with PBS and then fixed in 1 % paraformaldehyde for 15 min. After rinses with PBS, cellular morphology was observed using a phase contrast microscope (Nikon, Eclipse TE 2000-U, Japan) and photographed.
Nuclear Staining for Assessment of Apoptosis
Nuclear morphology was assessed by staining with 4′,6-diamidino-2-phenylindole (DAPI). SH-SY5Y cells (1 × 103 cells/well) were seeded in 8-well chamber slide and then treated with rotenone for 24 h after pre-treated with or without celastrol for 90 min. The cells were washed twice with PBS and then fixed in 1 % paraformaldehyde for 15 min. After 2 rinses with PBS, the cells were stained with DAPI (0.3 μM) for 10 min at 37 °C in dark. Slides were washed twice with PBS and examined under fluorescent microscope (Nikon, Eclipse TE 2000-U, Japan) and photographed.
TUNEL Assay
TUNEL assay was performed using DeadEnd™ Fluorometric TUNEL System (Promega coporation, USA). SH-SY5Y cells were cultured on 8-well chamber slide at a density of 1 × 103 cells/chamber. After treatment with rotenone and celastrol as indicated, cells were washed with PBS and fixed by incubation in 4 % paraformaldehyde for 20 min at 4 °C. The fixed cells were then washed and permeabilized with 0.2 % Triton X-100 in PBS for 5 min. After rinses with PBS, the cells were incubated with terminal deoxynucleotidyl transferase recombinant (rTdT)-catalyzed reaction and nucleotide mixture for 60 min at 37 °C in dark and then immersed in stop/wash buffer for 15 min at room temperature. The cells were then washed with PBS to remove unincorporated fluorescein-12-dUTP. After washing, cells were incubated in 1 μg/mL propidium iodide (PI) solution for 15 min in dark. The cells were observed with fluorescent microscope (Nikon, Eclipse TE 2000-U, Japan) and photographed.
Measurement of Intracellular Reactive Oxygen Species (ROS)
Production of ROS was measured using an oxidation sensitive fluorescent probe 2′7′-dichlorofluorescein diacetate (DCF-DA, Sigma-Aldrich, St. Louis, MO, USA) method, based on the ROS-dependent oxidation of DCF-DA to the highly fluorescent compound dichlorofluorescein (DCF). The cells (1 × 104 cell/well) were cultured in 96 well plates and then treated with 10 μM rotenone for 24 h after pre-treated with or without celastrol for 90 min. Medium was removed and cells were washed twice with PBS. After washing, the cells were incubated with DCF-DA (5 μM) for 30 min at 37 °C in the dark. Cellular fluorescence was measured in a fluorescence microplate reader (Spectra Max Gemini EM, Molecular Devices, Sunnyvale, CA, USA) at excitation wavelength 488 nm and emission wavelength 525 nm. Meanwhile, SH-SY5Y cells were cultured in 24-well plates at a density of 1 × 105 cells per well. After similar treatments, DCF-DA incubation, and washing the cells were also monitored by fluorescence microscope (Nikon, Eclipse TE 2000-U, Japan).
Measurement of Mitochondrial Membrane Potential
Mitochondrial membrane potential was determined using the fluorescent dye Rhodamine 123. Briefly, the cells were treated with rotenone for indicated periods after pre-treated with or without celastrol for 90 min. Cells were washed with PBS and incubated with 10 μg/mL Rhodamine 123 for 30 min at 37 °C. The cells were washed and monitored by fluorescent microscope (Nikon, Eclipse TE 2000-U, Japan) and photographed. Moreover, mitochondrial membrane potential was estimated by measuring the uptake of Rhodamine 123. Briefly, mitochondrial fraction was prepared as described below. Thereafter, 20 μg of mitochondrial protein was incubated with solution containing 0.8 μM Rhodamine 123 and 75 mM sucrose in PBS for 30 min at 37 °C and then centrifuged at 10,000×g for 10 min. The resulting supernatant was analyzed for the fluorescence intensity using a Spectra Max Gemini EM fluorometer (Molecular Devices, Sunnyvale, CA, USA) at 490 nm excitation and 515 nm emission. The mitochondrial membrane potential was expressed as fluorescence/μg protein.
Immunoblotting
After treatment, cells were washed once with PBS and then lysed using ice-cold RIPA buffer with protease inhibitor cocktail. Cell lysates were centrifuged at 12,870×g for 25 min, and the protein concentrations were determined by the bicinchoninic acid (BCA) method using bovine serum albumin (BSA) as standard. The proteins were separated by 10 % SDS-PAGE and transferred to polyvinylidine difluoride (PVDF) membrane. The membranes were blocked with 5 % (v/v) nonfat dry milk in Tris-buffered saline with Tween 20 (TBS-T) (10 mM Tris–HCl, 150 mM NaCl, and 0.1 % Tween 20, pH 7.5) and incubated with primary antibodies against Bax, active caspase 3, JNK, pJNK, P38, and pP38, COX-IV (1:1,000 dilution), active caspase 9, Bcl-2 (1:2,000 dilution), or Actin (1:4,000 dilution) for overnight at 4 °C. The membrane was washed in TBS-T and incubated for 2 h at room temperature with horseradish peroxidase (HRP)-conjugated secondary antibody. To reveal the reaction bands, the membrane was reacted with WEST-ZOL (plus) Western blot detection system (Intron Biotechnology, Inc., Korea) and exposed on X-ray film (BioMax MS-1, Eastman Kodak, USA).
Analysis of Cytochrome c in Cytosol and Mitochondria
SH-SY5Y cells were treated with or without rotenone and/or celastrol for indicated periods and harvested with a cell scraper. The collected cells were washed twice in ice-cold PBS and then resuspended in lysis buffer (250 mM sucrose, 50 mM Tris–HCl, 1 mM EGTA, 2.5 mM EDTA, 50 μM Na3VO4, 1 mM DTT, 0.1 mM PMSF, 40 μg/ml aprotinine, 20 μg/ml leupeptine; pH 7.4). After 30 min incubation on ice, cells were homogenized. The homogenates were then centrifuged at 1,100×g for 5 min. The supernatants were transferred to tubes and centrifuged at 12,000×g for 12 min to obtain mitochondrial pellets. The resulting supernatant was then centrifuged at 100,000×g for 60 min to obtain cytosolic fraction. Mitochondrial and cytosolic protein concentrations were determined by BCA assay and subjected to Western blot analysis as mentioned above using cytochrome c primary antibody (1:2,000 dilutions).
Statistical Analysis
The data were expressed as the mean ± SD. Statistical significance was assessed with one-way analysis of variance followed by a post hoc (Bonferroni) test for multiple group comparison. Differences with P value < 0.05 were considered statistically significant.
Results
Celastrol Protects Against Rotenone-Induced Cytotoxicity
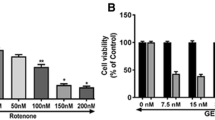
We first evaluated the cytotoxic response of SH-SY5Y cells to celastrol. As shown in Fig. 1b, 1–10 nM celastrol treatment for 24 h did not significantly affect the cell viability of SH-SY5Y cells. Next, we examined the toxic effect of rotenone. As shown in Fig. 1c, rotenone induced a dose dependent cytotoxicity in SH-SY5Y cells. In the presence of 10 μM rotenone, there are only 51.7 % of viable cells as compared to control cells. Similar cytotoxic effects were also observed with the LDH leakage assay. As shown in Fig. 1d, celastrol treatment did not cause significant changes in LDH levels in the culture medium. However, exposure of SH-SY5Y cells to rotenone increased LDH level in the culture medium in a dose-dependent manner (Fig. 1e). Consequently, the treatment of 2.5 nM of celastrol and 10 μM of rotenone for 24 h was chosen in the subsequent experiments.
To determine the protective effects of celastrol against rotenone-induced loss of cell viability, SH-SY5Y cells were pre-treated with 2.5 nM celastrol for 90 min, followed by 10 μM rotenone for 24 h. As illustrated in Fig. 2A, rotenone substantially decreased cell viability to 51.7 %, while celastrol prevented cells from rotenone-induced injury, restoring cell survival to 85.4 %. To further investigate the protective effect of celastrol, the release of LDH was measured. As shown in Fig. 2b, release of LDH was increased to 91.2 % after exposure to 10 μM rotenone. However, celastrol pre-treated cells showed decreased release of LDH (67.41 %) compared with rotenone-exposed cell group, indicating that celastrol rescued the viability of cells against the neurotoxicity induced by rotenone. The protective effect of celastrol was further confirmed by microscopic evaluation of the cells. Microscopic analysis indicated that celastrol profoundly inhibited rotenone-induced round shape phenotype and monolayer detachment, typical features of apoptosis occurrences (Fig. 2c). These data suggest that celastrol may protect SH-SY5Y cells against rotenone induced cell cytotoxicity.

Celastrol protects SH-SY5Y cells against rotenone-induced cytotoxicity. a Cells were exposed for 90 min with 2.5 nM celastrol and then treated with 10 μM rotenone. After 24 h viable cells were identified by the MTT assay (a) and LDH assay (b). SH-SY5Y cells were treated with 2.5 nM celastrol for 90 min followed by rotenone (10 μM) treatment for 24 h, and then morphology visualized directly by phase-contrast microscopy (c). The data are represented as mean ± SD of three independent experiments. *P < 0.05 versus vehicle (control) group. **P < 0.05, compared to rotenone-treated group
Celastrol Suppresses Rotenone Induced Oxidative Stress
There is considerable evidence that ROS are involved in the pathogenesis of various neurodegenerative disorders including PD. Rotenone has long been known to produce ROS by inhibiting mitochondrial electron transfer chain at the level of complex I. We therefore investigated whether celastrol could attenuate rotenone-induced intracellular ROS production in SH-SY5Y cells using fluorescent dye DCF-DA. As shown in Fig. 3, exposure of SH-SY5Y cells to rotenone (10 μM, for 24 h) caused a significant increase in intracellular ROS levels. However, pretreatment with celastrol (2.5 nM) markedly attenuated rotenone induced ROS production.

Celastrol reduces rotenone-induced ROS formation. SH-SY5Y cells were treated with 2.5 nM celastrol for 90 min then treated with vehicle or rotenone (10 μM) for 24 h. Intracellular ROS accumulation was measured using the fluorescence probe DCF-DA. a SH-SY5Y cells were incubated with DCF-DA and accumulation of intracellular ROS was monitored by fluorescent microscope (Nikon, Eclipse TE 2000-U, Japan). b The fluorescence intensity was determined using a Spectra Max Gemini EM fluorometer (Molecular Devices, Sunnyvale, CA, USA) at 485 nm excitation and 535 nm emission. Arrowhead indicates DCF-positive cells. Values are presented as mean ± SD of three experiments. *P < 0.05 versus vehicle (control) group. **P < 0.05, compared to rotenone treated group
Celastrol Prevents Rotenone-Induced Mitochondrial Dysfunction
As the major source of intracellular ROS production and target of rotenone, mitochondria play the crucial role in apoptosis. We first evaluated mitochondrial membrane potential (ΔΨm), one of the important events related to apoptosis. To investigate this event, SH-SY5Y cells were incubated with 10 μM rotenone for 24, and then the dynamics of the rotenone induced loss of ΔΨm was monitored by Rodamine-123 using confocal microscope. The loss of ΔΨm was visualized as indicated by the reduction of Rodamine-123 fluorescence intensity (Fig. 4a). Conversely, incubation with 2.5 nM celastrol significantly protected mitochondria from ΔΨm loss induced by rotenone (Fig. 4a (d)). Mitochondrial membrane potential was also assessed by measuring the uptake of Rhodamine-123 after incubating mitochondrial fraction with rotenone in the presence or absence of celastrol using spectrofluorometer. The amount of dye remaining in the supernatant was inversely proportional to the membrane potential. As shown in Fig. 4b, there was a significant increase in Rodamine-123 fluorescence due to a lack of uptake by the mitochondrial fraction incubated with rotenone, suggesting the loss in mitochondrial membrane potential. However, celastrol inhibited the increase in Rhodamine-123 fluorescence caused by rotenone. Next, we examined mitochondrial and cytosolic levels of cytochrome c by Western blot. Treatment with rotenone induced the release of cytochrome c into the cytosol and a decline in the mitochondrial cytochrome c content. Celastrol, which itself had no significant effect, blocked this displacement (Fig. 4c). These results suggest that celastrol may provide antiapoptotic effects by preserving the mitochondrial function.

Celastrol prevents rotenone-induced mitochondrial membrane potential loss and cytochrome c release in SH-SY5Y cells. Cells were treated with celastrol (2.5 nM) for 90 min followed by 10 μM rotenone for 24 h. a Treated cells were incubated with Rhodamine 123 and the mitochondrial membrane potential was observed by fluorescent microscope (Nikon, Eclipse TE 2000-U, Japan). b Assessment of mitochondrial membrane potential by Rhodamine uptake assay. Mitochondrial fractions from cells treated with rotenone in the presence and absence of celastrol were incubated with Rhodamine 123. Following centrifugation, the supernatant was analyzed for Rhodamine-123 using using a Spectra Max Gemini EM fluorometer (Molecular Devices, Sunnyvale, CA, USA) at 490 nm excitation and 515 nm emission. Higher the fluorescence lower is the mitochondrial membrane potential, expressed as Rhodamine-123 fluorescence/μg protein. When membrane potential decreased by rotenone, Rhodamine-123 was released, resulting in an increase in Rhodamine-123 fluorescence. This effect was prevented when rotenone-treated cells were incubated with celastrol. c immunoblot of cytochrome c on mitochondria and cytosol fraction. Mitochondrial and cytosolic fractions were subjected to immunoblot with anti-cytochrome c antibody. Intensity of each band was estimated by densitometric analysis. Results are expressed as mean ± SD of three independent experiments. *P < 0.05 versus vehicle (control) group. **P < 0.05, compared to rotenone-treated group
Celastrol Attenuates Rotenone-Induced Apoptosis
The effects of celastrol on rotenone-induced apoptosis in SH-SY5Y cells were examined by staining cells with the DNA dye DAPI to visualize nuclear morphology or with PI staining and TUNEL assay. Representative photomicrographs are shown in Fig. 5. Nuclear staining with DAPI demonstrated that control SH-SY5Y cells had normal regular and oval shaped nuclei (Fig. 5a). The nuclear morphology of cells exposed to celastrol alone was intact and similar to that of untreated control cells (Fig. 5b). In contrast, treatment with rotenone induced condensed and fragmented nuclei, hallmarks of morphological changes associated with apoptosis (Fig. 5c). SH-SY5Y cells with fragmented or condensed nuclei were also positively labeled with PI and TUNEL after rotenone treatment (Fig. 5g, j). Pretreatment of celastrol significantly inhibited the rotenone-induced apoptosis (Fig. 5d, h, l). These results indicate that celastrol has an anti-apoptotic effect in SH-SY5Y cells.

Celastrol suppresses rotenone-induced cell injury. SH-SY5Y cells were pre-treated with 2.5 nM celastrol for 90 min then treated with vehicle or rotenone (10 μM) for 24 h. Cell apoptosis was detected by DAPI (a–d), PI (e–h), and TUNEL (i–l) staining. Arrowheads indicate apoptotic cells. Each image is representative of three experiments. Pictures were taken using a fluorescent microscope (Nikon, Eclipse TE 2000-U, Japan) and photographed at ×100 magnification
Celastrol Alters Rotenone-Induced Changes of Bcl-2 and Bax Levels in SH-SY5Y Cells
Antiapoptotic protein Bcl-2 maintains the integrity of the mitochondrial membranes to prevent cytochrome c release, whereas proapoptotic Bax disturb the integrity of these membranes and triggers cytochrome c release. The expression ratio of Bcl-2 to Bax is useful index to evaluate whether a cell has undergone apoptosis. Therefore, we examined whether celastrol affects the expression of proapoptotic and antiapoptotic proteins in SH-SY5Y cells. As shown in Fig. 6a, rotenone treatment increased the expression levels of proapoptotic Bax, whereas it decreased expression levels of antiapoptotic Bcl-2. This resulted in a high Bax to Bcl-2 ratio. However, pretreatment with celastrol prevented the increase of the ratio of Bax to Bcl-2 in the rotenone treated cells.

Celastrol alters rotenone induced changes in the expression levels of apoptotic proteins in SH-SY5Y cells. Cells were treated with 2.5 nM celastrol for 90 min then treated with vehicle or rotenone (10 μM) for 24 h. Expression of Bcl-2, and Bax (a) and caspase-9, and caspase-3 (b) were assessed by immunoblots and intensity of each band was estimated by densitometric analysis. Actin was used as an internal loading control. Results are expressed as mean ± SD of three independent experiments. *P < 0.05 versus vehicle (control) group. **P < 0.05, compared to rotenone treated group
Celastrol Counteracts Rotenone Induced Caspase-3, and Caspase-9 Activation
Indeed, caspase-3 and caspase-9 have been recognized to play a critical role in apoptosis, we examined whether celastrol affects their expression in rotenone-treated cells by Western blot analysis. As shown in Fig. 6b, treatment with rotenone increased the expression of caspase-3 and caspase-9 by 2.3-fold, and 3.13-fold respectively compared with that of the control group. In contrast, celastrol significantly inhibited caspase-3 and caspase-9 cleavage induced by rotenone.
Celastrol Suppresses Rotenone-Induced p38 MAPK Phosphorylation
Because rotenone stimulates the activities of JNK and p38 MAPK in human dopaminergic cells [27], we investigated the effects of celastrol on the activation of JNK, and p38 MAPK in SH-SY5Y cells treated with rotenone. As shown in Fig. 7a, b, rotenone activated both JNK and p38 MAPK. Pretreatment with celastrol markedly reduced the rotenone induced p38 MAPK activation. However, phosphorylation of JNK was not affected by celastrol treatment.

Effects of celastrol on rotenone-induced JNK and p38 MAPK activation as well as the effect of pharmacological interventions on p38 MAPK activity and on cell viability of SH-SY5Y cells exposed to rotenone. a Celastrol did not suppress rotenone-induced JNK activation. b Celastrol attenuated rotenone-induced p38 MAPK phosphorylation. Cells were pre-treated with 2.5 nM celastrol for 90 min followed by 10 μM rotenone. Cell extracts were prepared and analyzed by immunoblotting using antibodies against phospho-and total-form of JNK and p38 MAPK and intensity of each band was estimated by densitometric analysis. c NAC and celastrol attenuated rotenone-induced p38 MAPK phosphorylation. Cells were pre-treated with NAC (1 mM) before exposure to rotenone in the presence or absence of celastrol. Cell lysates were subjected to immunoblot using antibodies against phospho-and total-form p38 MAPK and intensity of each band was estimated by densitometric analysis. d p38 MAPK mediated rotenone-induced cell death in SH-SY5Y cells. Cells were pre-treated with p38 inhibitor SB203580 or NAC and subsequently treated with rotenone in the absence or presence of celastrol and the cell viabilities were assessed by MTT assay. Results are expressed as mean ± SD of three independent experiments. *P < 0.05 versus vehicle (control) group. **P < 0.05, compared to rotenone treated group
We further investigated whether ROS was involved in the activation of p38 MAPK in response to rotenone. We found that rotenone-induced p38 activation was inhibited by pre-treatment with an antioxidant NAC and this effect was more pronounced when the cells were co-treated with celastrol and NAC (Fig. 7c). This finding suggests that rotenone-induced p38 activation could be associated with ROS generation. To examine the role p38 MAPK in rotenone induced cell death, we evaluated the effect of p38 inhibitor SB203580 on the viability of SH-SY5Y cells exposed to rotenone. As shown in Fig. 7d, cells pretreated with SB203580 considerably attenuated rotenone induced cell loss. In addition, pretreatment with SB203580 enhanced the cytoprotective effects of celastrol on rotenone-induced cell death. The protective effect of celastrol against rotenone was also found to be similar with NAC. These data indicated that ROS production was involved in p38 MAPK activation and p38 plays important role in rotenone-induced cell death.
Discussion
PD is the prototypical movement disorder described almost 200 years ago, but despite decades of intensive study, the etiology of this disease remains poorly understood. However, our knowledge of the pathogenesis of PD has advanced considerably in recent years. Many studies have shown that mitochondrial dysfunction, oxidative stress, and cellular death are common factors in many neurodegenerative diseases including PD [28]. Among these factors, oxidative stress acts as a unifying factor in the pathogenesis of PD [29]. In fact, a common denominator in the pathogenesis of PD by rotenone is the involvement of oxidative stress mediated apoptotic processes [23, 30] and intervention in rotenone induced oxidative stress has been suggested to be critical for survival of DA neurons [31, 32]. Therefore, inhibition of oxidative damage may offer potential therapeutic benefit for oxidative stress related neurodegenerative diseases. In the present study, we provide evidence that celastrol, which can easily penetrate biological membranes [12], is able to protect dopaminergic neurons against rotenone-induced injury via regulation of p38 MAPK pathway.
Mitochondria, the “powerhouse of the cell” are highly dynamic organelles which play an important role in regulating the life and death of neuronal cells. Defects in mitochondrial function have been implicated in dopaminergic cell degeneration in PD [33]. In PD, the major mitochondrial defect is associated with complex I activity [34]. Rotenone acts as a mitochondrial complex I inhibitor. Inhibition of mitochondrial respiratory chain complex I has several damaging consequences. One of the expected consequences of complex I inhibition is an increased production of ROS. Increased ROS can damage virtually all biological macromolecules [35]. Oxidative damage is thought to be a key mechanism of mitochondrial toxicity in the rotenone-induced dopaminergic neuronal cell death [36]. Consistent with previous findings [26], we observed that celastrol significantly attenuated the ROS production in SH-SY5Y cells caused by rotenone, indicating its potent ability to scavenge free radicals and protect the dopaminegic cells against rotenone-induced injury.
Because mitochondria are primary target of rotenone and site where ROS is produced, they are likely to play a central role in neuronal cell death. The inhibition of complex I by rotenone is accompanied with excess ROS formation, which may induce loss of ΔΨm and release of cytochrome c. Similar to the findings by Deng et al. [26], we found that celastrol prevented rotenone induced ΔΨm loss and cytosolic accumulation of cytochrome c, suggesting that neuroprotective effects of celastrol are mediated in part by the preservation of mitochondrial function. Mitochondrial membrane permeabilization is considered as the point-of-no-return in the apoptotic pathway [35]. Permeabilization of mitochondrial membrane is regulated by proteins of the Bcl-2 family [37]. The anti-apoptotic factor Bcl-2, residing in the outer mitochondrial membrane, inhibits cytochrome c release [38]. The pro-apoptotic factor Bax resides in the cytosol and translocates from cytoplasm to the mitochondria following apoptotic signaling, where it promotes the permeabilization of the mitochondrial membrane. Increased mitochondrial permeability results in the release of cytochrome c from the mitochondria [39]. Released cytochrome c triggers activation of caspase-9 which in turn activates caspase-3, and activated caspase-3 induces cell death. In this study, we found that Bax/Bcl-2 protein ratio is increased following a treatment with rotenone, but it decreases with the administration of celastrol prior to rotenone. Moreover, we found that celastrol attenuated the effects of rotenone on activation of caspase-9, and caspase-3. These results suggest that celastrol protects the dopaminergic cells, at least in part, through inhibition of mitochondrial-dependent apoptotic pathway.
We further investigated the involvement of MAPKs signaling pathways underlying the protective effects of celastrol on rotenone-induced cell injury. We found that celastrol inhibited rotenone-induced activation of p38 MAPK in SH-SY5Y cells. JNK and p38 MAPK are potent effectors of neuronal apoptosis [40]. Persistent activation of these two kinases has been suggested to mediate neuronal death in PD [41]. Although, studies have shown that JNK is also involved in neuronal cell death, celastrol did not reduce its phosphorylation in rotenone induced activation. It has been shown that p38 MAPK mediate pro-apoptotic protein Bax induced mitochondrial membrane permeabilization [42] and the death signal mediated by this kinase is initiated at least by ROS [43]. To confirm the involvement of ROS in activation of p38 MAPK, we evaluated the effects of NAC, a potent ROS scavenger and celastrol on rotenone induced p38 MAPK activation. We observed that incubation of cells with NAC and celastrol prevented the rotenone-induced activation of p38 MAPK. We further examined the role of p38 MAPK in rotenone induced cell death using p38 inhibitor SB203580 by MTT assay. We found that blockage of p38 MAPK with SB203580 attenuated the rotenone induced cell death and the protective effect was found to be more pronounced when the cells were incubated with SB203580 and celastrol. These results suggested a relationship between the production of ROS by rotenone and initiation of p38 MAPK activation. Moreover, studies have shown that the p38 MAPK pathways play an important role in regulation of autophagy. Recently, it has been suggested that autophagic pathway is involved in celastrol neuroprotection [26]. These findings indicate that the protective effect of celastrol on rotenone induced cell injury was regulated via p38 MAPK dependent pathway.
In vivo studies have shown that administration of celastrol in low nanomolar doses provided excellent neuroprotection in a Drosophila model of PD [15]. Celastrol at nanomolar doses (7 μg/kgxd), improved learning and memory in a mouse AD model, and higher doses (3 mg/kgxd) of celastrol have been used in the MPTP mice model of PD without any observable side effects [8, 13, 44]. In vitro studies have been shown that celastrol induced heat shock protein 70 (Hsp70) via activation of heat shock transcription factor 1 in undifferentiated neuroblastoma cells at 3 μM [45], a dosage that Chow and Brown [14] found to be detrimental to viability of SH-SY5Y cells. It has been reported that the minimal doses of celastrol that could induce the various Hsps in both differentiated and undifferentiated SH-SY5Y cells were 0.25 and 0.5 μM respectively with the threshold of 1 μM in undifferentiated state and 0.75 μM in differentiated state at which SH-SY5Y cell viability was found to affect [14]. In fact, celastrol at lower doses might be effective in neuroprotection against toxic insults on either in vivo or in vitro. Recently, Deng et al. [26] have been reported that celastrol at 0.5 μM enhanced cell viability by 28.99 %, and decreased cell apoptosis by 54.38 %, in rotenone treated SH-SY5Y cells. In this study, we found that celastrol at 2.5 nM protected SH-SY5Y cells against rotenone-induced injury. Although the protection was modest at 2.5 nM, based on the above studies, it could be possible that celastrol at higher concentration (0.25–0.75 μM in differentiated state; 0.5–1 μM in undifferentiated state) could provide better protection in rotenone treated SH-SY5Y cells.
In conclusion, the present observations identify a potential neuroprotective role of celastrol against neurotoxicity induced by rotenone. The protective effect of celastrol appears to be associated with its capacity to suppress oxidative stress, inhibit apoptotic features and maintain the ΔΨm stability through the inhibition of p38 MAPK activation. Although, more study is needed to further explore the neuroprotective actions of celastrol, our results indicate that celastrol may have potential therapeutic value in the treatment of neurodegenerative diseases including PD.
References
Ahn DK (1998) Illustrated book of Korean medicinal herbs. Kyo-Hak Publishing, Seoul
Brinker AM, Ma J, Lipsky PE, Raskin I (2007) Medicinal chemistry and pharmacology of genus Tripterygium (Celastraceae). Phytochemistry 68:732–766
Salminen A, Lehtonen M, Paimela T, Kaarniranta K (2010) Celastrol: molecular targets of Thunder God Vine. Biochem Biophys Res Commun 394:439–442
Celastrol MoritaT (2010) a new therapeutic potential of traditional chinese medicine. Am J Hypertens 23:821
Lee JH, Koo TH, Yoon H, Jung HS, Jin HZ, Lee K et al (2006) Inhibition of NF-kappa B activation through targeting I kappa B kinase by celastrol, a quinone methide triterpenoid. Biochem Pharmacol 72:1311–1321
Sassa H, Takaishi Y, Terada H (1990) The triterpene celastrol as a very potent inhibitor of lipid peroxidation in mitochondria. Biochem Biophys Res Commun 172:890–897
Kannaiyan R, Shanmugam MK, Sethi G (2011) Molecular targets of celastrol derived from Thunder of God Vine: potential role in the treatment of inflammatory disorders and cancer. Cancer Lett 303:9–20
Allison AC, Cacabelos R, Lombardi VR, Alvarez XA, Vigo C (2001) Celastrol, a potent antioxidant and anti-inflammatory drug, as a possible treatment for Alzheimer’s disease. Prog Neuropsychopharmacol Biol Psychiatr 25:1341–1357
Kim DH, Shin EK, Kim YH, Lee BW, Jun JG, Park JH et al (2009) Suppression of inflammatory responses by celastrol, a quinone methide triterpenoid isolated from Celastrus regelii. Eur J Clin Invest 39:819–827
Abbott A (2002) Neurologists strike gold in drug screen effort. Nature 417:109
Heemskerk J, Tobin AJ, Bain LJ (2002) Teaching old drugs new tricks. Meeting of the neurodegeneration drug screening consortium, 7–8 April 2002, Washington, DC, USA. Trends Neurosci 25:494–506
Paris D, Ganey NJ, Laporte V, Patel NS, Beaulieu-Abdelahad D, Bachmeier C et al (2010) Reduction of beta-amyloid pathology by celastrol in a transgenic mouse model of Alzheimer’s disease. J Neuroinflammation 7:17
Cleren C, Calingasan NY, Chen J, Beal MF (2005) Celastrol protects against MPTP- and 3-nitropropionic acid–induced neurotoxicity. J Neurochem 94:995–1004
Chow AM, Brown IR (2007) Induction of heat shock proteins in differentiated human and rodent neurons by celastrol. Cell Stress Chaperones 12:237–244
Faust K, Gehrke S, Yang Y, Yang L, Beal MF, Lu B (2009) Neuroprotective effects of compounds with antioxidant and anti-inflammatory properties in a Drosophila model of Parkinson’s disease. BMC Neurosci. doi:10.1186/1471-2202-10-109
Thomas B, Beal MF (2007) Parkinson’s disease. Hum Mol Genet 16(2):183–194
Jenner P (2003) Oxidative stress in Parkinson’s disease. Ann Neurol 53:S26–S38
Greenamyre JT, Sherer TB, Betarbet R, Panov AV (2001) Complex I and Parkinson’s disease. IUBMB Life 52:135–141
Schapira AH (2004) Disease modification in Parkinson’s disease. Lancet Neurol 3:362–368
Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT (2000) Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci 3:1301–1306
Alam M, Schmidt WJ (2002) Rotenone destroys dopaminergic neurons and induces parkinsonian symptoms in rats. Behav Brain Res 136:317–324
Greenamyre JT, Betarbet R, Sherer TB (2003) The rotenone model of Parkinson’s disease: genes, environment and mitochondria. Parkinsonism Relat Disord Suppl 2:S59–S64
Sherer TB, Richardson JR, Testa CM, Seo BB, Panov AV, Yagi T et al (2007) Mechanism of toxicity of pesticides acting at complex I: relevance to environmental etiologies of Parkinson’s disease. J Neurochem 100:1469–1479
Sherer TB, Betarbet R, Testa CM, Seo BB, Richardson JR, Kim JH et al (2003) Mechanism of toxicity in rotenone models of Parkinson’s disease. J Neurosci 23:10756–10764
Cannon JR, Tapias V, Na HM, Honick AS, Drolet RE, Greenamyre JT (2009) A highly reproducible rotenone model of Parkinson’s disease. Neurobiol Dis 34:279–290
Deng YN, Shi J, Liu J, Qu QM (2013) Celastrol protects human neuroblastoma SH-SY5Y cells from rotenone-induced injury through induction of autophagy. Neurochem Int pii:S0197-0186(13)00128-9. doi:10.1016/j.neuint.2013.04.005 [Epub ahead of print]
Newhouse K, Hsuan SL, Chang SH, Cai B, Wang Y, Xia Z (2004) Rotenone induced apoptosis is mediated by p38 and JNK MAP kinases in human dopaminergic SH-SY5Y cells. Toxicol Sci 79:137–146
Lin MT, Beal MF (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443:787–795
Cavalli A, Bolognesi ML, Minarini A, Rosini M, Tumiatti V, Recanatini M et al (2008) Multi-target-directed ligands to combat neurodegenerative diseases. J Med Chem 51:347–372
Betarbet R, Canet-Aviles RM, Sherer TB, Mastroberardino PG, McLendon C, Kim JH et al (2006) Intersecting pathways to neurodegeneration in Parkinson’s disease: effects of the pesticide rotenone on DJ-1, alpha-synuclein, and the ubiquitin-proteasome system. Neurobiol Dis 22:404–420
Kim SJ, Kim JS, Cho HS, Lee HJ, Kim SY, Kim S et al (2006) Carnosol a component of rosemary (Rosmarinus officinalis L.) protects nigral dopaminergic neuronal cells. NeuroReport 17:1729–1733
Sapkota K, Kim S, Park SE, Kim SJ (2011) Detoxified extract of Rhus verniciflua stokes inhibits rotenone-induced apoptosis in human dopaminergic cells, SH-SY5Y. Cell Mol Neurobiol 31:213–223
Jenner P, Olanow CW (1996) Oxidative stress and the pathogenesis of Parkinson’s disease. Neurology 47:161–170
Schapira AH (1999) Science, medicine, and the future: Parkinson’s disease. BMJ 318:311–314
Perier C, Vila M (2012) Mitochondrial biology and Parkinson’s disease. Cold Spring Harb Perspect Med 2(2):a009332
Testa CM, Sherer TB, Greenamyre JT (2005) Rotenone induces oxidative stress and dopaminergic neuron damage in organotypic substantia nigra cultures. Brain Res Mol Brain Res 134:109–118
Vila M, Przedborski S (2003) Targeting programmed cell death in neurodegenerative diseases. Nat Rev Neurosci 4:365–375
Borner C (2003) The Bcl-2 protein family: sensors and checkpoints for life-or-death decisions. Mol Immunol 39:615–647
Chinnaiyan AM, Orth K, O’Rourke K, Duan H, Poirier GG, Dixit VM (1996) Molecular ordering of the cell death pathway-Bcl-2 and Bcl-x (L) function upstream of ced-3-like apoptotic proteases. J Biol Chem 271:4573–4576
Mielke K, Herdegen T (2000) JNK and p38 stresskinases–degenerative effectors of signal-transduction-cascades in the nervous system. Prog Neurobiol 61:45–60
Kim EK, Choi EJ (2010) Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta 1802:396–405
Chipuk JE, Green DR (2008) How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol 18:157–164
Choi WS, Eom DS, Han BS, Kim WK, Han BH, Choi EJ et al (2004) Phosphorylation of p38 MAPK induced by oxidative stress is linked to activation of both caspase-8- and -9-mediated apoptotic pathways in dopaminergic neurons. J Biol Chem 279:20451–20460
Pinna GF, Fiorucci M, Reimund JM, Taquet N, Arondel Y, Muller CD (2004) Celastrol inhibits pro-inflammatory cytokine secretion in Crohn’s disease biopsies. Biochem Biophys Res Commun 322:778–786
Westerheide SD, Bosman JD, Mbadugha BN, Kawahara TL, Matsumoto G, Kim S et al (2004) Celastrols as inducers of the heat shock response and cytoprotection. J Biol Chem 279:56053–56060
Acknowledgments
This study was supported by research funds from Chosun University, 2012.
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Additional information
Bong-Suk Choi and Hyool Kim have contributed equally to this study.
Rights and permissions
About this article
Cite this article
Choi, BS., Kim, H., Lee, H.J. et al. Celastrol from ‘Thunder God Vine’ Protects SH-SY5Y Cells Through the Preservation of Mitochondrial Function and Inhibition of p38 MAPK in a Rotenone Model of Parkinson’s Disease. Neurochem Res 39, 84–96 (2014). https://doi.org/10.1007/s11064-013-1193-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-013-1193-y