
Abstract
Introduction
Multiple sclerosis (MS) is a heterogeneous neurological disorder with multifactorial etiologies characterized by demyelination, axonal degeneration, and oligodendroglial death. It is believed that both genetics and environmental risk factors such as infection are involved in disease etiology. Accumulating evidence indicates that alteration in purinergic system signaling is involved in immunity and inflammation. Adenosine, a key purine nucleoside, has been shown to be produced during metabolic stress, including ischemia, inflammatory condition, and tissue injury.
Methods
Extracellular adenosine directly affects various physiological and pathological processes of MS by stimulating G protein-coupled adenosine receptors A1, A2A, A2B, and A3 on the surface of immune cells. It has been suggested that promotion of adenosinergic system may be an important factor in MS pathophysiology and considered as promising therapeutic target for this disease.
Conclusion
In this review, we will discuss about the immunopathologic effects of adenosine on MS and its animal model, experimental autoimmune encephalomyelitis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Multiple Sclerosis (MS) is the central nervous system (CNS)-related autoimmune disease which affects about 1.3 million people worldwide [1, 2]. The onset of MS occurs among individual’s most productive years and is more common among women than men [3, 4]. MS patients exhibit several clinical symptoms such as chronic pain, depression, ataxia, sensory organ disorders, and overall cognitive disorders [5]. Despite extensive research the precise cause of disease still remains unknown [6]. MS is a chronic neuroinflammatory demyelinating disease which is associated with activation of immune cells including neutrophils, macrophages, and lymphocytes [7]. As the adenosine can control the immune system and inflammation, it seems that adenosine receptors may be considered as novel potent therapeutic targets in MS [8]. Increasing evidence suggests that adenosine can directly control different aspects of the pathophysiology of MS, particularly neural inflammation and demyelination [9]. It has also been reported that concentration of adenosine is decreased in blood of MS patients [10]. In this review, we tried to clarify the role of adenosine and adenosine receptors in the immunopathogenesis and treatment of MS and its animal models.
Multiple sclerosis
Two major hypotheses are considered in etiology of MS, including autoimmunity and oligodendrogliapathy. Autoimmune hypothesis, the most accepted hypothesis, indicates that neuroinflammation is responsible for demyelination and auto-reactive leukocyte-induced disease [11]. A recent theory suggests that MS is an immunological convolution between initial degenerative disorder and aberrant immune response of the host. In both events, infiltration of leukocytes (including T lymphocytes, monocytes, and other immune cells) into the CNS is a main event which is mediated through interaction of integrins expressed on the surface of leukocytes with their ligands [cell adhesion molecules (CAMs)] expressed on the surface of endothelial cells [12]. However, it is accepted that a combination of genetic and environmental factors is involved in disease susceptibility. High frequency of certain genetic variants in MHC-II gene loci among MS patients compared to normal population supports the genetic involvement. HLA DRB1 * 1501 allele shows a high association with MS [1, 8, 13]. Among environmental factors, infection with Epstein-Barr virus has the greatest involvement in susceptibility to disease [1].
Inflammatory events in MS are associated with self-reactive responses against myelin antigens in which both the innate and adaptive immunity play a key role [14, 15]. In the early stages of the disease, antigen-presenting cells (APCs) present myelin-derived antigens to naive T CD4+ cells and prime these cells in the peripheral lymph nodes. Stimulated T CD4+ lymphocytes then cross blood–brain barrier and react with myelin and oligodendroglia antigens in the CNS [16]. Secretion of inflammatory mediators such as proinflammatory cytokines including interleukin (IL)-12 and tumor necrosis factor-α (TNF-α) enhances activation of microglia in the CNS [14]. Subsequently, these factors together with other soluble factors which were secreted from glial cells induce demyelination via oligodendroglia TNF receptor 1, which affects oligodendrocytes and myelin sheaths [1, 11]. Axonal damage commonly is observed with chronicity and progression of the disease, which leads to an important and irreversible neurological disability [17, 18]. Inflammation, demyelination, and neurodegeneration in human cortex are the pyramid of MS as suggested by Fisher et al. [19]. Moreover, they compared various inflammatory and neurodegenerative diseases, using neuropathological and genome-wide microarray analysis, and highlight some of the common pathways leading to MS-specific lesions. These data suggest that plaques of primary demyelination (complete loss of myelin but axonal maintenance) are the main hallmark of cortical damage in cases of secondary progressive MS. These subpial focal lesions with demyelination confirmed by the presence of activated macrophages containing myelin degradation products are further distinguished by profound inflammation in the meninges, activation of microglia, perivascular inflammatory cuffs, and T/B lymphocyte infiltration [19]. In addition to T cells, it is supposed that B cells play an important role in the pathogenesis of MS, possibly as APC [20]. B cells may play an important role in antibody-dependent demyelination through complement activation or antibody-dependent cellular cytotoxicity. Consistently, the histopathologic findings have demonstrated deposition of IgG at the border of MS plaques [1, 21].
MS is divided into four different subtypes based on clinical manifestation, including (i) relapsing-remitting MS (RRMS), (ii) primary-progressive MS (PPMS), (iii) secondary-progressive MS (SPMS), and (iv) progressive-relapsing MS (PRMS) [6]. Approximately 85 % of patients exhibit RRMS characterized with recurrent and unpredictable episodes of neurological deficits such as loss of vision and paralysis followed by a recovery period [22]. About 10 % of the patients show PPMS phenotype in which symptoms are persistent and sometimes small temporary improvement occurs. SPMS begins as RR form characterized by minor improvement followed by attacks and worsening of the disease. PRMS is the rarest disease course (approximately 5 % of the patients), in which disability from onset of disease is progressive [6]. The overall mean duration of the disease is 25–30 years. Benign course of disease is observed in one-third of the patients, who remain fully functional and show partial disability only 15 years after the onset of disease. On the other hand, malignant MS rapidly progresses, which leads to significant neurological impairment and even death [8]. During the past 20 years, many attempts have been made for treatment of MS, although none of the existing drugs can efficiently control the neurodegeneration process. Moreover, these drugs are often associated with several side effects [23]. Therefore, further investigations are required to design the new therapeutic approaches for treatment of MS [4, 24].
Adenosine
Adenosine is an intermediate metabolite and building blocks of nucleic acids and a part of common biological energy, i.e. adenosine 5′-triphosphate (ATP) [25]. This metabolite was first identified by Drury et al. as a physiological regulator of coronary vascular tone [26]. Adenosine directly affects a number of synaptic processes involved in signaling pathways and plays an important role in regulation of several neurotransmitters in the CNS [27]. Newby is proposed a hypothesis for adenosine function according to which adenosine is referred to as a retaliatory metabolite, as a consequence of its ability to mediate an autoregulatory loop. According to this hypothesis, it is assumed that adenosine is released in response to a wide variety of noxious stimuli and stressors, which induce a self-regulatory cycle. This function protects the organ from the damage following the initiating stressful stimuli [27, 28]. Adenosine protects the organs through several mechanisms. Adenosine reduces energy requirement of the tissues, increases the supply of oxygen and nitrogen through vasodilation, and regulates uncontrolled inflammation or immune activation against foreign agents [29]. The baseline level of adenosine has trivial effects on the immune response; however, it becomes high enough to have considerable immunomodulatory effects in ischemic and inflammatory conditions [29]. The genetic defect in the adenosine deaminase (the enzymes involved in metabolism of adenosine) is associated with severe combined immune deficiency disease (SCID) which demonstrates the immunomodulatory effects of adenosine [30].
Adenosine constitutively found at low concentrations in the extracellular space [25]. Under physiological conditions extracellular adenosine concentration in the tissues is less than 1 μM; however, following metabolic stress its concentration increases up to 100 μM. It seems that the inflammatory conditions such as ischemic lesions increase extracellular adenosine concentrations to suppress immune responses [31]. Several biological processes affect extracellular concentration of adenosine, including formation of adenosine through intracellular adenosine reservoirs, extracellular adenosine production and adenosine transport outside the cell [32]. Moreover, since the majority of cells possess adenosine transporters, extracellular concentration of the adenosine is limited to basic physiological conditions [33].
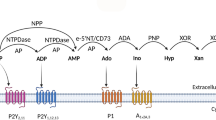
Adenosine bioavailability depends on its production, secretion, cellular uptake, and enzymatic degradation. Pathophysiological conditions such as hypoxia and intracellular stress promote ATP dephosphorylation to adenosine by 5′ nucleotidase enzyme, which is accompanied by suppression of adenosine kinase enzyme leading to increased intracellular adenosine [8]. Overexpression of intracellular adenosine leads to adenosine transportation to the extracellular space by nucleoside transporters [8, 34]. Extracellular catabolism of ATP, ADP, and AMP by ectonucleotidases including CD39 (nucleoside triphosphate diphosphohydrolase [NTPDase]) and CD73 (5′-ectonucleotidase) can also increase the extracellular concentration of adenosine [35, 36]. These enzymes exhibit high expression, particularly in lymphocytes and endothelial cells and can be overexpressed in response to chronic hypoxia [37]. Two other enzymes including adenosine deaminase (ADA) and adenosine kinase (AK) are also involved in regulation of extracellular adenosine. It seems that ADA is a main enzyme in metabolism of purines and irreversibly catalyzes deamination of adenosine and deoxyadenosine to inosine and doxyinosine, respectively [28]. Thus, ADA is involved in removal of adenosine from extracellular area. AK is an intracellular enzyme, which catalyzes rapid phosphorylation of adenosine to adenosine monophosphate (AMP). Since the cellular uptake of adenosine from extracellular spaces is controlled by concentration gradient, AK indirectly regulates extracellular adenosine uptake by controlling the intracellular concentration of adenosine [30, 38, 39]. After formation, adenosine starts cellular function through specific receptors on the cell surface.
Adenosine receptors
Extracellular purines such as adenosine, adenosine diphosphate (ADP), and ATP mediate a variety of biological effects by G protein-coupling receptors, called purinergic receptors [25]. Two families of purinergic receptors are defined based on pharmacological and molecular cloning characteristics including p1 and p2 receptors [38]. While the p1 receptor binds to adenosine, p2 receptors identify a wide range of natural ligands such as ATP, ADP, uridine-5′-triphosphate(UTP), and uridine diphosphate (UDP) [40]. According to IUPHAK nomenclature, p1 receptors are also called adenosine receptors (ARs). These receptors belong to the super family of seven transmembrane receptors bound to G protein divided into subcategories of A1, A2A, A2B, and A3. Except A3R which exhibits significant variation between species, other ARs are highly conserved during evolution (i.e. 80–95 % sequence homology) [25, 41]. A1R is the most abundant AR [42], which is widely expressed throughout the body with highest expression in the brain, particularly in excitatory nerve endings [25, 43]. These receptors have high affinity for adenosine analogs such as L-N6-phenyl isopropyl adenosine (L-PIA) [44]. Ligation of A1R inhibits the activity of adenylyl cyclase, activates potassium channels, inhibits voltage-dependent calcium channels, and increases intracellular calcium and Inositol trisphosphate (IP3) by inducing activation of phospholipase Cγ [45].
A2R is found at pre and post synaptic nerve terminals, mast cells, and airway smooth muscle [25]. High levels of A2Rs are found in the striatum of the brain, immune cells, spleen, thymus, and leukocytes [46]. A2Rs are subdivided into two subgroups of A2A and A2B. A2A has high affinity for adenosine while A2B has low affinity [25]. Ligation of A2AR stimulates cAMP-protein kinase A (PKA) pathway [6, 45]. A2ARs play a key role in modulation of inflammatory events in peripheral tissues [47]. It has been shown that activation of A2AR in mice inhibits tissue injury caused by inflammation, which represents anti-inflammatory effect of these receptors [48]. Evaluation of tissue damage caused by T cell, macrophages, and cytokines in A2AR-deficient mice led to significant increase in tissue damage and increased proinflammatory cytokines such as, TNF-α, IFN-γ, and IL-12 [48–50]. These data indicate that signaling of A2ARs plays an important role as anti-inflammatory pathway. Thus, these receptors may be considered as an important pharmacological target for the treatment of a wide range of immunoinflammatory diseases [51].
A2BRs are highly expressed in bladder, gastrointestinal tract, and lung [52]. A2BR is inactive under physiological conditions and is activated by high concentrations of extracellular adenosine during hypoxia, ischemia, and inflammation [52].
A3R is mainly expressed in the kidney, lung, heart, brain cortex, and immune cells such as mast cell, eosinophil, and neutrophil [44]. A3Rs show significant differences in pharmacology, distribution, and function among the various species. In mice, A3R signaling is associated with degranulation of mast cells [53]. Ligation of A3R inhibits adenylate cyclase, stimulates phospholipase C and B, and induces calcium withdrawal and release of intracellular reservoirs [25].
ARs act as metabolic sensors in the tissues [51] and can be targeted for therapeutic purposes. Many drugs used in clinics (including methotrexate and dipyridamole) exert their effects in part through altering the concentration of extracellular adenosine. Accordingly, many AR agonists and antagonists are being developed for clinical investigation [54–56]. Furthermore, adenosine can modulate immune function through inhibitory effects on lymphocytes, neutrophils, monocytes/macrophages, and dendritic cells [57]. Thus, modulation of neuroinflamation through ARs offers a new mechanism, which provides new therapeutic opportunities for MS and other demyelinating diseases (Table 1) [58].
Effects of adenosine on the immune function
Neutrophils
Adenosine is an important modulator of neutrophil function [8, 59]. It has been shown that the frequency, function, and survival of neutrophils are increased in MS patients [60]. Neutrophils may be involved in chronic neuroinflammatory process leading to demyelination of neurons [61, 62]. Neutrophils express CD39 and CD73 molecules and release adenosine after activation during inflammation [38]. Neutrophils also express all four subtypes of ARs and their expression pattern can be affected by inflammatory mediators [38]. The effect of adenosine on neutrophil migration and homing is controversial. Adenosine stimulates neutrophils binding to vascular endothelium in sub-micromolar concentrations in part through A1R [38]. A1R enhances the expression of p-selectin on endothelial cells and Mac1 on neutrophils [38]. Controversially, signaling of A2ARs (A2A and A2B) on endothelial cells inhibits the binding of neutrophils to these cells at micromolar concentrations of adenosine [38]. It has also been suggested that A3R may also inhibit binding of neutrophils to endothelial cells [38]. Ligation of A2AR on neutrophils can potently suppress production of several inflammatory mediators such as TNF-α, CCL3, CCL4, CCL20, CXCL2, LTB4, and platelet activating factor (PAF) [38]. It is reported that administration of A2AR agonist CGS21680 to the striatum inhibits neutrophil accumulation and reduced cell death [25]. Moreover, the A2AR agonist ATL193 could also inhibit neutrophil oxidative activity through a cAMP- and PKA-dependent mechanisms [25, 37]. Thus, the progress of inflammation increases extracellular levels of adenosine as a negative feedback to inhibit over-recruitment and over-activation of harmful neutrophils [38].
Monocytes/macrophages
All four types of ARs express on both monocytes and macrophages; however, their expression changes during inflammation [63]. Adenosine modulates adhesion of monocytes during inflammation in part through downregulation of adhesion molecules on endothelial cells [38]. Ligation of A2AR inhibits the binding of monocytes to endothelial cells via NF-κB-dependent downregulation of E-selectin [41]. Adenosine can also inhibit the expression of VCAM-1 on the surface of stimulated endothelial cells by A2A and A2B receptors [38]. Moreover, adenosine inhibits the differentiation of monocytes into macrophages. Adenosine can also dramatically suppress production of active nitrogen and oxygen intermediates by monocyte/macrophages [41]. It seems that all ARs are involved in downregulation of TNF-α in macrophages. Treatment of LPS-stimulated human monocytes with AR agonists including NECA, R-PIA, CGS 21680, and 2 -CADO led to downregulation of TNF-α [24, 41, 63–65]. Hasko et al. using A2AR knockout mice showed that adenosine is involved in the decreasing IL-12 production which was associated with upregulation of cAMP levels in mice [41]. Moreover, it is reported that in human monocytes A3R is also involved in downregulating of IL-12 [66].
Ligation of A3R suppressed LPS-stimulated INF-γ production in murine macrophages [38]. Adenosine enhances release of anti-inflammatory cytokine IL-10 by macrophages [37]. Macrophages from A2AR-deficient mice showed also impaired IL-10 production [37]. Treating human and murine monocytes with CGS21680, AR agonist, enhances LPS-induced IL-10 production [37, 41]. In addition, Koscso et al. showed that A2BR activation stimulates IL-10 production by murine microglia cells [67]. Recently, it has been reported that adenosine can also stimulate activation of M2 macrophages in part through A2A and A2B receptors [67]. It is shown that A1, A2A, and A3 receptors are expressed on microglia and are involved in regulating neuroinflammatory responses [68–70].
Lymphocytes
T Lymphocytes
Lymphocytemalfunction is observed in patients with ADA-SCID syndrome, which may be in part due to accumulation of intracellular and extracellular adenosine [38]. This indicates that adenosine is an important signaling molecule in lymphocytes function [38].
T cells express A3, A2B, and A2A receptors. A2ARs are the main receptors expressed on peripheral T lymphocytes, so modulation of A2AR may have a significant effect on T cell responses [38]. Extracellular adenosine at micromolar concentrations attenuates proliferation and function of T cells through A2ARs [38]. In addition, adenosine can inhibit IL-2 production via A2BR activation. It is stated that NF-κB signaling pathway may be stimulated by A2BR signaling in activated T lymphocyte [38, 51]. Ligation of A2AR decreases the expression of co-stimulatory molecule CD40L and increases the expression of inhibitory molecules cytotoxic T-lymphocyte antigen (CTLA)-4 and programmed cell death (PD)-1 on T cells [71]. A2AR signaling can also inhibit IFN-γ secretion by T cells [72]. Adenosine suppresses the expression of lymphocyte peyer’s patch adhesion molecule (LPAM)-1 and intercellular adhesion molecules (ICAM)-1 on lymphocytes [73]. Therefore, extracellular adenosine may inhibit the migration of lymphocytes to inflammatory area [74, 75]. Moreover, A2BR blocking inhibits Th17 cell differentiation through preventing IL-6 production from APCs [76].
Activation of ARs regulates immunosuppressive effects of Regulatory T (Treg) cells [77]. Treg cells can specifically differentiate from T cells in the presence of high levels of CD39 and CD73. The adenosine produced by these enzymes suppresses T cell function by binding to A2AR on target cells. In addition, A2AR activation induces Treg formation through FoxP3 overexpression [78, 79].
B lymphocyte
Adenosine activation inhibits NF-κB signaling pathway in mouse B cells. This is due to activation of ARs along with increase in cAMP and PKA activation leading to inhibition of Iκβ phosphorylation. In addition, several purine nucleosides are adenosine analogs and 2′ doxycycline adenosine like tubercidin (tub) can inhibit the proliferative response of B cells [38].
Adenosine and adenosine receptors in MS
It is suggested that adenosine and ARs are involved in modulation of neuroinflammation in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE), an animal model for the central nervous system (CNS) inflammatory disease, MS (Table 2) [80]. CD73, a cell surface enzyme that catalyzes the breakdown of AMP to adenosine, is expressed in brain endothelial cells in choroid plexus epithelium and modulates lymphocytes immune surveillance in the blood and cerebrospinal fluid (CSF) [81]. Due to the substantial immunosuppressive and anti-inflammatory properties of adenosine, it is presumed that CD73-deficient mice would develop severe EAE. In contrast to this finding, Mills et al. demonstrated that CD73-deficient mice show preserved T cell responses and are resistant to EAE induction [81]. Consistent with these findings, It was shown that CD4 T cells from CD73-deficient mice secreted higher levels of the pro-inflammatory cytokines IL-1β and IL-17 after MOG stimulation than wild-type (WT) mice and were able to induce EAE when cd73 −/− T cells adoptively transferred into tcr −/− recipients. So, resistance to EAE induction that observed in CD73-deficient mice was not induced by a deficiency in T cell responsiveness. Consequently, in CNS lymphocyte infiltration during EAE, CD73 and adenosine play more profound role than modulation of neuroinflammation [81]. In addition, it was observed that infiltration of lymphocytes into CNS in CD73-deficient mice were fewer than WT mice. And also, in order to determining of the expression of CD73 in the CNS, that help to lymphocyte migration, immunohistochemistry results showed that expression level of in the choroid plexus epithelium was high, while brain endothelial cells not expressed CD73 [81].
Dysfunction of ARs in the CNS shows that signaling of these receptors is contributed to development of MS in humans and EAE in animal models [82]. It has been suggested that extracellular adenosine plays a critical and complex role in the severity and development of EAE. It is likely the expression of adhesion molecules at the choroid plexus is modulated by adenosine signaling. Previous studies showed that overexpression of the adhesion molecules such as VCAM-1, ICAM-1, and MadCAM-1 at the choroid plexus is correlated with progression of EAE [83]. Therefore, it is concluded that, CD73 and AR signaling are essential for proper entry of lymphocytes into the CNS to induce EAE [82].
Studies suggested that AR activation exhibits a bidirectional effect in neuroinflammation and brain injury [82]. It is demonstrated that the expression of A1R macrophages in both the brain and blood cells is reduced in MS patients, which proposes its role in macrophage activation and the CNS inflammation [58]. A1R regulates severity of EAE-related neurobehavioral and neuropathological outcomes such as demyelination and axonal damage. Activation of A1R also reduces inflammatory response in the CNS [58]. Furthermore the function of adenosine on mononuclear cells varies between the MS patients and control groups. In RRMS patients, A1R has a prominent role in regulating levels of proinflammatory cytokines such as TNF-α and IL-6. It was shown that stimulation of A1R in peripheral blood mononuclear cells (PBMC) from control groups, but not RRMS patients, markedly inhibited TNF-α level. In contrast, IL-6 production was reduced in RRMS patients but not control groups following stimulation of A1R [84, 85]. This observation is important, since the pathogenesis of MS is thought to involve a cascade of events in which activated microglia and macrophages release mediators that cause myelin and white matter damage. Moreover, the ability of adenosine analogues to inhibit TNF-α production by stimulated mononuclear cells was attenuated by the A1R antagonist DPCPX [58]. As well, in the other study demonstrated that the A1R is downregulated on microglia during MOG-EAE, which it suggests that transcriptional control or transcript degradation of A1R gene was disturbed during the CNS inflammation [86]. Interestingly, glucocorticoids, commonly used in the treatment of MS, also increase A1R expression. In vivo study of A1R null mice showed that expression of mRNA MMP12 (macrophage metalloelastase) was increased. Of interest, expression of mRNA MMP12 also increased in the brains of MS patients. MMP12 is considered a neurotoxic mediator since it causes chemokine cleavage and converts it to a highly neurotoxic molecule [86].
A1R null mice also show more severe forms of progressive/relapsing EAE compared to wild-type mice [86]. One of the most important early determinants of MS and EAE onset is the infiltration and activation of lymphocytes, while A1R expression is diminished. Because of this severity of EAE at the onset of disease did not differ between A1R null and wild-type animals. However, A1R knockdown is seemingly related to induction of proinflammatory and inhibition of anti-inflammatory molecules during EAE in the A1R null mouse and might contribute to development of demyelination and axonal injury [86]. It has been reported chronic caffeine treatment attenuates EAE pathological symptoms, reduces cell infiltration, demyelination and inflammatory cytokines. It is recommended that caffeine’s protective function in EAE is probably due to overexpression of A1R and downregulation of IFN-γ mRNA [87]. Likewise, in vitro caffeine treatment of human monocytoid cells also enhanced A1R expression and decreased proinflammatory cytokine production [86].
In contrast, receptor-specific A2AR antagonist, ScH58261, makes mice resistant to EAE by restricting the entry of lymphocytes into the CNS during EAE development [81]. Moreover, lymphocyte infiltration into the CNS wAS noticeably diminished in mice treated with an A2AR antagonist or that lack A2AR expression in their CNS compared to control groups [88]. It was proposed that that the activation of adenosine receptors causes oligodendroglial death. Of interest, the protective roles of A2AR blockade may also prevent oligodendrocyte damage, since its antagonism reduces activation of JNK/MAPK signaling in oligodendrocytes [89] (Fig. 1). Furthermore, other studies have identified that extracellular adenosine signaling plays an important role in controlling cell migration in a cell- and tissue-specific manner. For example, it was reported that extracellular adenosine signaling stimulates migration of immature oligodendrocytes, dendritic cells, bronchial epithelial cells, and endothelial cells. In contrast it can restrain chemotaxis in eosinophils, mast cells, and microglia/monocytes [88].

An A2AR- antagonist (SCH58261) suppresses inflammatory process and prevents oligodendrocyte damage through suppressing of JNK/MAPK pathway in oligodendrocyte. JNK c-Jun N-terminal kinases, MAPK Mitogen-activated protein kinases
Interestingly, A2AR knockout mice showed increased chemokine production and immune cell infiltration into spinal cord compared to wild-type mice. It was found that severe EAE in the A2AR knockout mice is related to pro-inflammatory nature of A2AR-deficient immune cells and thereby A2AR signaling is important in limiting inflammation and tissue injury [88]. In addition to the findings described above, it is also observed that A2AAR−/− lymphocytes are more proliferative and produced more IFN-ε than did their wild-type counterparts. These protective effects have likely attributed A2AR ability to inhibit the production of the proinflammatory cytokines IL-12, INF-γ, IL-6, and TNF-α [88]. Therefore, A2AR is capable to affect EAE progression from several different pathways including magnitude of the inflammatory response via expression on lymphocytes and efficient lymphocyte entry/infiltration into the CNS via expression at the choroid plexus [88]. In another study the efficacy of intrathecal A2AR agonist, ATL313, was assessed in a model of robust neuroinflammation, RR-EAE. It was shown that A2AR agonist has anti-inflammatory effect on macrophages and microglia with reduced markers of activation in microglia and monocyte-lineage cells (CD68 and OX-42) [90]. Furthermore, the A2AR agonist treatment was effective in alleviating motor symptoms associated with reduced neuroinflammation [90].
Wei et al. have recently showed that A2BR expression increased in peripheral blood leukocytes of MS patients and in peripheral lymphoid tissues of EAE mice [76]. Moreover, A2BR-specific antagonists, CVT-6883 and MRS-1754, diminish the clinical signs of EAE and protect the CNS from immune damage [76].
Conclusion
Adenosine, a molecule with immunosuppressive activity, is an important compensatory mechanism to reduce inflammation and the inappropriate immune response in MS. On the other hand, neuroinflammation modulation through ARs offers a new mechanism, which provides new therapeutic opportunities for MS and other demyelinating diseases. Nevertheless, high frequency of ARs in various tissues and their contrasting protective effects necessitate preclinical studies prior to approving their use in the treatment of MS.
References
Selter RC, Hemmer B. Update on immunopathogenesis and immunotherapy in multiple sclerosis. Immunotargets Ther. 2013;2:21–30.
Gharibi T, et al. Immunomodulatory characteristics of mesenchymal stem cells and their role in the treatment of multiple sclerosis. Cell Immunol. 2015;293(2):113–21.
Alonso A, Hernán MA. Temporal trends in the incidence of multiple sclerosis a systematic review. Neurology. 2008;71(2):129–35.
Damal K, Stoker E, Foley JF. Optimizing therapeutics in the management of patients with multiple sclerosis: a review of drug efficacy, dosing, and mechanisms of action. Biol Targets Ther. 2013;7:247.
Mirshafiey A, Jadidi-Niaragh F. Prostaglandins in pathogenesis and treatment of multiple sclerosis. Immunopharmacol Immunotoxicol. 2010;32(4):543–54.
Mulakayala N, et al. Synthesis of novel therapeutic agents for the treatment of multiple sclerosis: a brief overview. Eur J Med Chem. 2013;60:170–86.
Jadidi-Niaragh F, Mirshafiey A. Histamine and histamine receptors in pathogenesis and treatment of multiple sclerosis. Neuropharmacology. 2010;59(3):180–9.
Masino S, Boison D. Adenosine: a key link between metabolism and brain activity. New York: Springer; 2012.
Sperlágh B, Illes P. Purinergic modulation of microglial cell activation. Purinergic Signal. 2007;3(1–2):117–27.
Mayne M, et al. Dysregulation of adenosine A1 receptor–mediated cytokine expression in peripheral blood mononuclear cells from multiple sclerosis patients. Ann Neurol. 1999;45(5):633–9.
Nakahara J, et al. Current concepts in multiple sclerosis: autoimmunity versus oligodendrogliopathy. Clin Rev Allergy Immunol. 2012;42(1):26–34.
Ciccarelli O, et al. Pathogenesis of multiple sclerosis: insights from molecular and metabolic imaging. Lancet Neurol. 2014;13(8):807–22.
Jadidi-Niaragh F, Mirshafiey A. Th17 cell, the new player of neuroinflammatory process in multiple sclerosis. Scand J Immunol. 2011;74(1):1–13.
Mirshafiey A, et al. The significance of matrix metalloproteinases in the immunopathogenesis and treatment of multiple sclerosis. Sultan Qaboos Univ Med J. 2014;14(1):e13.
Mirshafiey A, Jadidi-Niaragh F. Immunopharmacological role of the leukotriene receptor antagonists and inhibitors of leukotrienes generating enzymes in multiple sclerosis. Immunopharmacol Immunotoxicol. 2010;32(2):219–27.
Jadidi-Niaragh F, Mirshafiey A. Regulatory T-cell as orchestra leader in immunosuppression process of multiple sclerosis. Immunopharmacol Immunotoxicol. 2011;33(3):545–67.
Lassmann H. Pathology and disease mechanisms in different stages of multiple sclerosis. J Neurol Sci. 2013;333(1):1–4.
D’Aversa TG, et al. Myelin basic protein induces inflammatory mediators from primary human endothelial cells and blood–brain barrier disruption: implications for the pathogenesis of multiple sclerosis. Neuropathol Appl Neurobiol. 2013;39(3):270–83.
Fischer M, et al. Research highlights inflammation, demyelination and neurodegeneration: risky buddies in multiple sclerosis. CNS Neurol Disord Drug Targets. 2014;13(1):1.
Hussain RZ, et al. Immune surveillance of the central nervous system in multiple sclerosis—Relevance for therapy and experimental models. J Neuroimmunol. 2014;276(1):9–17.
Neuhaus O, Archelos JJ, Hartung H-P. Immunomodulation in multiple sclerosis: from immunosuppression to neuroprotection. Trends Pharmacol Sci. 2003;24(3):131–8.
Aktas O, Kieseier B, Hartung H-P. Neuroprotection, regeneration and immunomodulation: broadening the therapeutic repertoire in multiple sclerosis. Trends Neurosci. 2010;33(3):140–52.
Jadidi-Niaragh F, Mirshafiey A. Therapeutic approach to multiple sclerosis by novel oral drugs. Recent Pat Inflamm Allergy Drug Discov. 2011;5(1):66–80.
Vincenzi F, et al. Multiple sclerosis lymphocytes upregulate A2A adenosine receptors that are antiinflammatory when stimulated. Eur J Immunol. 2013;43(8):2206–16.
Sachdeva S, Gupta M. Adenosine and its receptors as therapeutic targets: an overview. Saudi Pharm J. 2013;21(3):245–53.
Cacciari B, et al. A2B adenosine receptor antagonists: recent developments. Mini Rev Med Chem. 2005;5(12):1053–60.
Newby AC. Adenosine and the concept of ‘retaliatory metabolites’. Trends Biochem Sci. 1984;9(2):42–4.
Desrosiers MD, et al. Adenosine deamination sustains dendritic cell activation in inflammation. J Immunol. 2007;179(3):1884–92.
Gessi S, et al. A2A adenosine receptors in human peripheral blood cells. Br J Pharmacol. 2000;129(1):2–11.
Cristalli G, et al. Adenosine deaminase: functional implications and different classes of inhibitors. Med Res Rev. 2001;21(2):105–28.
Haskó G, Cronstein BN. Adenosine: an endogenous regulator of innate immunity. Trends Immunol. 2004;25(1):33–9.
King AE, et al. Nucleoside transporters: from scavengers to novel therapeutic targets. Trends Pharmacol Sci. 2006;27(8):416–25.
Fredholm BB. Adenosine receptors as drug targets. Exp Cell Res. 2010;316(8):1284–8.
Deussen A. Metabolic flux rates of adenosine in the heart. Naunyn Schmiedeberg’s Arch Pharmacol. 2000;362(4–5):351–63.
Thompson LF, et al. Crucial role for ecto-5′-nucleotidase (CD73) in vascular leakage during hypoxia. J Exp Med. 2004;200(11):1395–405.
Eltzschig HK, et al. Endogenous adenosine produced during hypoxia attenuates neutrophil accumulation: coordination by extracellular nucleotide metabolism. Blood. 2004;104(13):3986–92.
Milne GR, Palmer TM. Anti-inflammatory and immunosuppressive effects of the A 2A adenosine receptor. Sci World J. 2011;11:320–39.
Bours M, et al. Adenosine 5′-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol Ther. 2006;112(2):358–404.
Moriwaki Y, Yamamoto T, Higashino K. Enzymes involved in purine metabolism-a review of histochemical localization and functional implications. Histol Histopathol. 1999;14(4):1321–40.
Matsumoto T, Tostes RC, Webb RC. Alterations in vasoconstrictor responses to the endothelium-derived contracting factor uridine adenosine tetraphosphate are region specific in DOCA-salt hypertensive rats. Pharmacol Res. 2012;65(1):81–90.
Haskó G, et al. Shaping of monocyte and macrophage function by adenosine receptors. Pharmacol Ther. 2007;113(2):264–75.
Fredholm BB, et al. Structure and function of adenosine receptors and their genes. Naunyn Schmiedeberg’s Arch Pharmacol. 2000;362(4–5):364–74.
Daly JW, Padgett WL. Agonist activity of 2-and 5′-substituted adenosine analogs and their N 6-cycloalkyl derivatives at A1− and A2−adenosine receptors coupled to adenylate cyclase. Biochem Pharmacol. 1992;43(5):1089–93.
Livingston M, Heaney L, Ennis M. Adenosine, inflammation and asthma–a review. Inflamm Res. 2004;53(5):171–8.
Chen J-F, Eltzschig HK, Fredholm BB. Adenosine receptors as drug targets—what are the challenges? Nat Rev Drug Discov. 2013;12(4):265–86.
Fredholm BB, et al. International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors—an update. Pharmacol Rev. 2011;63(1):1–34.
Carlsson J, et al. Structure-based discovery of A2A adenosine receptor ligands. J Med Chem. 2010;53(9):3748–55.
Sitkovsky MV. Use of the A 2A adenosine receptor as a physiological immunosuppressor and to engineer inflammation in vivo. Biochem Pharmacol. 2003;65(4):493–501.
Cronstein BN. Adenosine, an endogenous anti-inflammatory agent. J Appl Physiol. 1994;76(1):5–13.
Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature. 2001;414(6866):916–20.
Gessi S, et al. Adenosine and lymphocyte regulation. Purinergic Signal. 2007;3(1–2):109–16.
Ryzhov S, et al. Host A2B Adenosine Receptors Promote Carcinoma Growth. Neoplasia. 2008;10(9):987–95.
Zhong H, et al. Activation of murine lung mast cells by the adenosine A3 receptor. J Immunol. 2003;171(1):338–45.
Haskó G, et al. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov. 2008;7(9):759–70.
Müller CE, Jacobson KA. Recent developments in adenosine receptor ligands and their potential as novel drugs. Biochi Biophys Acta (BBA) Biomembr. 2011;1808(5):1290–308.
Jacobson KA, Gao Z-G. Adenosine receptors as therapeutic targets. Nat Rev Drug Discov. 2006;5(3):247–64.
Mills J, et al. Never underestimate the power of adenosine in multiple sclerosis. CNS Neurol Disorders Drug Targets. 2013;12(7):1.
Johnston JB, et al. Diminished adenosine A1 receptor expression on macrophages in brain and blood of patients with multiple sclerosis. Ann Neurol. 2001;49(5):650–8.
Koedel U, et al. Apoptosis is essential for neutrophil functional shutdown and determines tissue damage in experimental pneumococcal meningitis. PLoS Pathog. 2009;5(5):e1000461.
Naegele M, et al. Neutrophils in multiple sclerosis are characterized by a primed phenotype. J Neuroimmunol. 2012;242(1):60–71.
Mossberg N, et al. Oxygen radical production in leukocytes and disease severity in multiple sclerosis. J Neuroimmunol. 2009;213(1):131–4.
Hernández-Pedro NY et al. Initial immunopathogenesis of multiple sclerosis: innate immune response. Clin Dev Immunol. 2013;2013:413465.
Csóka B, et al. A2A adenosine receptors and C/EBPβ are crucially required for IL-10 production by macrophages exposed to Escherichia coli. Blood. 2007;110(7):2685–95.
Tsutsui S, et al. Glucocorticoids regulate innate immunity in a model of multiple sclerosis: reciprocal interactions between the A1 adenosine receptor and β-arrestin-1 in monocytoid cells. FASEB J. 2008;22(3):786–96.
Prabhakar U, et al. Inhibition of LPS-induced TNFα production in human monocytes by adenosine (A2) receptor selective agonists. Int J Immunopharmacol. 1995;17(3):221–4.
La Sala A, Gadina M, Kelsall BL. Gi-protein-dependent inhibition of IL-12 production is mediated by activation of the phosphatidylinositol 3-kinase-protein 3 kinase B/Akt pathway and JNK. J Immunol. 2005;175(5):2994–9.
Koscsó B, et al. Adenosine augments IL-10 production by microglial cells through an A2B adenosine receptor-mediated process. J Immunol. 2012;188(1):445–53.
Saijo K, Crotti A, Glass CK. Regulation of microglia activation and deactivation by nuclear receptors. Glia. 2013;61(1):104–11.
Bi W, et al. Rifampicin inhibits microglial inflammation and improves neuron survival against inflammation. Brain Res. 2011;1395:12–20.
Koning N, et al. Downregulation of macrophage inhibitory molecules in multiple sclerosis lesions. Ann Neurol. 2007;62(5):504–14.
Sevigny CP, et al. Activation of adenosine 2A receptors attenuates allograft rejection and alloantigen recognition. J Immunol. 2007;178(7):4240–9.
Lappas CM, Rieger JM, Linden J. A2A adenosine receptor induction inhibits IFN-γ production in murine CD4 + T cells. J Immunol. 2005;174(2):1073–80.
Johnston A, et al. The anti-inflammatory action of methotrexate is not mediated by lymphocyte apoptosis, but by the suppression of activation and adhesion molecules. Clin Immunol. 2005;114(2):154–63.
MacKenzie WM, Hoskin DW, Blay J. Adenosine suppresses α4β7 integrin-mediated adhesion of T lymphocytes to colon adenocarcinoma cells. Exp Cell Res. 2002;276(1):90–100.
Yang Z, et al. Infarct-sparing effect of A2A-adenosine receptor activation is due primarily to its action on lymphocytes. Circulation. 2005;111(17):2190–7.
Wei W, et al. Blocking A2B adenosine receptor alleviates pathogenesis of experimental autoimmune encephalomyelitis via inhibition of IL-6 production and Th17 differentiation. J Immunol. 2013;190(1):138–46.
Buc M. Role of regulatory T cells in pathogenesis and biological therapy of multiple sclerosis. Mediators Inflamm. 2013;2013:11. doi:10.1155/2013/963748.
Lowther DE, Hafler DA. Regulatory T cells in the central nervous system. Immunol Rev. 2012;248(1):156–69.
Buckner JH. Mechanisms of impaired regulation by CD4 + CD25 + FOXP3 + regulatory T cells in human autoimmune diseases. Nat Rev Immunol. 2010;10(12):849–59.
Yao SQ, et al. Genetic inactivation of the adenosine A2A receptor exacerbates brain damage in mice with experimental autoimmune encephalomyelitis. J Neurochem. 2012;123(1):100–12.
Mills JH, et al. CD73 is required for efficient entry of lymphocytes into the central nervous system during experimental autoimmune encephalomyelitis. Proc Natl Acad Sci. 2008;105(27):9325–30.
Dai S-S, Zhou Y-G. Adenosine 2A receptor: a crucial neuromodulator with bidirectional effect in neuroinflammation and brain injury. Rev Neurosci. 2011;22(2):231–9.
Engelhardt B. T cell migration into the central nervous system during health and disease: different molecular keys allow access to different central nervous system compartments. Clin Exp Neuroimmunol. 2010;1(2):79–93.
Stone TW, Ceruti S, Abbracchio MP. Adenosine receptors and neurological disease: neuroprotection and neurodegeneration. Handb Exp Pharmacol. 2009;193:535–587.
Chen J-F, et al. Adenosine A 2A receptors and brain injury: broad spectrum of neuroprotection, multifaceted actions and “fine tuning” modulation. Prog Neurobiol. 2007;83(5):310–31.
Tsutsui S, et al. A1 adenosine receptor upregulation and activation attenuates neuroinflammation and demyelination in a model of multiple sclerosis. J Neurosci. 2004;24(6):1521–9.
Chen GQ, et al. Chronic caffeine treatment attenuates experimental autoimmune encephalomyelitis induced by guinea pig spinal cord homogenates in Wistar rats. Brain Res. 2010;1309:116–25.
Mills JH, et al. A2A adenosine receptor signaling in lymphocytes and the central nervous system regulates inflammation during experimental autoimmune encephalomyelitis. J Immunol. 2012;188(11):5713–22.
Melani A, et al. Selective adenosine A2a receptor antagonism reduces JNK activation in oligodendrocytes after cerebral ischaemia. Brain. 2009;132(6):1480–95.
Loram LC, et al. Adenosine 2A receptor agonism: a single intrathecal administration attenuates motor paralysis in experimental autoimmune encephalopathy in rats. Brain Behav Immun. 2015;46:50–4.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible Editor: Yoshiya Tanaka.
Rights and permissions
About this article

Cite this article
Safarzadeh, E., Jadidi-Niaragh, F., Motallebnezhad, M. et al. The role of adenosine and adenosine receptors in the immunopathogenesis of multiple sclerosis. Inflamm. Res. 65, 511–520 (2016). https://doi.org/10.1007/s00011-016-0936-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00011-016-0936-z