
Abstract
Growing human population demands a doubling of food production by 2050 necessitating significant increase in agricultural production, a challenge that is exacerbated by the changing climatic conditions. To tackle this, new lines of crops are continuously being identified, varieties bred, and transgenic technology developed, made possible by the deployment of modern molecular tools. Finger millet (Eleusine coracana (L.) Gaertn.) is a nutritious millet, assuming high significance as a rich source of good quality protein, micro-nutrients like calcium and iron and fiber, apart from being drought-hardy. This crop has spectacular adaptive abilities, and the grain has superior keeping quality. The plethora of micronutrients consisting of an abundance of calcium and moderately high contents of iron, manganese, and phosphorus that are present in this grain in comparison to other cereals makes it a promising crop for nutritional studies. The crop’s status as an “orphan millet” has been remodelled by the recent attempts on genome sequencing and various transcriptomics studies in the crop making the time ripe for harnessing this information for genomic and functional genomic applications. The nutritional supremacy and extreme adaptability make the crop an interesting candidate for nutri-genomics and climate resilience studies. Modern genetic tools will aid in unravelling the untapped gene pool of the crop which might contribute towards agronomic and nutritional improvement of other crops. This chapter discusses the advancements in the areas of molecular marker technology, gene mapping, NGS based genomics, and modern breeding tools and genetic transformation in finger millet and how these tools are accelerating crop improvement.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
14.1 Introduction
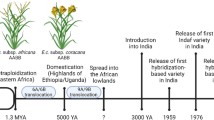
Finger millet (Eleusine coracana (L.) Gaertn.) is a nutritious millet that offers good quality protein, micro-nutrients like calcium, manganese, and iron as well as dietary fiber and antioxidants, apart from being drought-hardy. The crop has recently assumed high significance as a promising nutri-cereal for the future even in the policy level. Finger millet or ragi (Eleusine coracana) is an allotetrapoloid species (2n = 4x = 36) belonging to the genus Eleusine grown primarily in semi-arid regions of India, Africa, Nepal, and South America. The genus Eleusine is divided into two subspecies namely subsp. coracana (that includes cultivated finger millet) and subsp. africana. This crop has its origin from the diversity center of East Africa (Bisht and Mukai 2002). E. africana is proposed as the wild progenitor of this species. Domestication of finger millet is believed to have happened approximately 5000 years ago in Africa and was introduced to India around 2000 years from then. India is regarded as a secondary center of diversity of this crop. In India it is commonly known as ragi, mandua, nachni etc. The crop is an allotetraploid (AABB) and while the diploid species E. indica is identified as the A genome progenitor (maternal genome donor), B genome contributor is believed to be extinct and is unknown (Hilu 1988; Bisht and Mukai 2001; Liu et al. 2014). The genome Eleusine has around eight species that exists, namely E. africana, E. coracana, E. kigeziensis, E. indica, E. floccifolia, E. intermedia, E. multiflora, and E. jaegeri out of which E. coracana is the only domesticated species.
Finger millet has spectacular adaptive abilities and thrives well from the arid plains in India to up to 2300 m elevation in the Himalayas. Finger millet grains are one of the richest vegetative sources of calcium and it has moderately high contents of iron, manganese, and phosphorus in comparison to other cereals. Its grains are rich in protein (7–8%) and its methionine rich storage protein possesses high biological value and superior quality. With its rich nutrient profile, it is considered as a promising solution for malnutrition and hidden hunger. The grains are rich in dietary fiber and has antioxidant, anti-diabetic, and anti-microbial properties. Its straw serves as an important cattle fodder and grain malt, a nutritious baby food and the grain has superior keeping quality. Finger millet is regarded as a stress hardy crop and can withstand drought and salinity to a greater extent than many other crops. The crop has good survival ability at high temperatures making it an ideal crop for changing climatic conditions. The valuable and unique stress tolerant genes and gene networks harbored by finger millet need to be explored for utilization in other crops and in finger millet itself. Altogether finger millet makes a wise component of sustainable agriculture for the future generations owing to its inherent ability to thrive well in marginal lands with poor nutrient levels and ability to withstand frequent droughts.
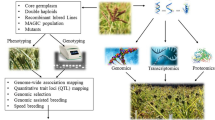
Finger millet so far was considered as an orphan crop having only local significance and lacked well documented scientific information. There was scarcity of germplasm and genetic resources as well as genomic information in comparison to other field crops. The crop is highly self-pollinated with tiny flowers and emasculation of small flowers is often injurious to floral parts. In addition, the crop has only a narrow genetic base of germplasm all of which were impediments in breeding improvement in this crop. Molecular marker technology has revolutionized breeding programs through various tools such as molecular markers, QTL mapping, association mapping, and so on. Molecular tools help in identifying genomic variation and marker-trait association more efficiently and they have become valuable tools in crop improvement. This chapter covers the recent advances in the application of biotechnological tools in this crop and the impact that they had brought in.
14.2 Advances in Molecular Marker Studies
One of the ways to enhance plant productivity is to expand the genetic base by introducing genes from the germplasm pool. Molecular tools will expedite the process and help in developing superior varieties. To make utilization of the germplasm resources manageable and more effective, a “core subset” of finger millet germplasm accessions was developed (Upadhyaya et al. 2006) just like in sorghum, barley maize, wheat etc. Based on the geographical origin and data on 14 quantitative traits, 622 accessions were selected for core subset generation from a total of 5940 global collection of accessions available in ICRISAT, Patancheru. The representation of the five races and the types of genotypes (improved cultivars, breeding materials, landraces, and wild types) were in similar proportion in the core subset as in the entire collection. Much of the diversity of the entire set has been captured in the core, as substantiated by various statistical analyses, and they can be used as a good starting point for any research programs. From this core collection, a “minicore” comprising of 80 accessions was developed later based on multi-location evaluation data of the core collection (Upadhyaya et al. 2010). It was ensured that the minicore captured the entire diversity of the core collection and it serves as an ideal pool of diverse germplasm for exploring new sources of variation and thereby enhancing the genetic potential of the crop.
Development of a genetic map is a steppingstone in any crop improvement program. The first finger millet genetic map was constructed using RFLP, AFLP, EST, and SSR markers (Dida et al. 2007). The map was constructed in an F2 population raised from a cross between E. coracana subsp. africana (accession MD-20) and E. coracana subsp. coracana cv. Okhale-1 and the population consisted of 151 progenies. They developed genomic SSRs by isolating di- and trinucleotide SSRs from genomic libraries of finger millet. The first genetic map covered all 18 finger millet chromosomes, at least partially. In addition, to be used for marker assisted selection, a set of 82 SSR markers was also developed and some of them were mapped. A finger millet-rice comparative map analysis was carried out to create a better finger millet genetic map and interestingly gene orders between rice and finger millet were found to be highly conserved (Srinivasachary et al. 2007). Linkage groups in finger millet had high collinearity to respective rice chromosomes (Srinivasachary et al. 2007). Table 14.1. gives the important marker systems used in finger millet for various purposes.
An essential part of germplasm conservation and utilization in breeding programs is genetic variability studies. Three different marker systems namely RFLP, RAPD, and ISSR were employed for the analysis of 22 accessions of 5 different species of Eleusine (Salimath et al. 1995). The study revealed a very low level of DNA sequence variability in finger millet and among the three, ISSR was identified as the most promising marker system for diversity analysis. Later, RAPD was successfully utilized to understand the genetic variability and relationships between various genotypes in finger millet (Das et al. 2007; Fakrudin et al. 2007; Babu et al. 2007). A variety of 18 RAPD primers, 10 SSR primers, and 10 pairs of cytochrome P450 gene-based markers were employed for a trait based comparative evaluation of genetic diversity in finger millet genotypes collected from different districts of Uttarakhand where the trait considered was calcium content. Although all these marker systems were effective in identifying polymorphism, SSR gave the maximum Polymorphism Information Content (PIC) of 0.505 (Panwar et al. 2010). Using these markers, genotypes could be effectively grouped into high, medium and low calcium categories. Genome-wide association mapping identified 16 significant associations between 13 SSR markers and six agronomic traits in 138 Ethiopian and exotic accessions (Lule et al. 2018). Pandian et al. (2018) developed 56 new genic SSRs from drought responsive ESTs and used them in population structure and genetic diversity analysis.
Appraisal of relationships at the phylogenetic level is important as wild relatives often are repositories of several valuable untapped traits. The first phylogenetic relationship study in finger millet used species-specific chloroplast deoxyribonucleic acid (cpDNA) polymerase chain reaction–restriction fragment length polymorphism (PCR–RFLP) and chloroplast simple sequence repeat (cpSSR) markers/sequences (Agrawal et al. 2013). This study also helped in re-affirming the hypothesis that E. indica is the maternal parent of E. coracana.
Blast disease caused by the fungus, Magnaporthe grisea, is the major limiting factor for finger millet production worldwide. It causes huge financial losses when the fungus affects the economically important grains by infecting neck and fingers. Before finger millet genome was sequenced, efforts were underway in gathering information available in rice and other grass species where blast was a serious problem and utilizing the information in identifying disease resistant genes or resistance related markers in finger millet. Finger millet germplasm for blast disease showed a continuous variation in resistance trait indicating a quantitative gene control of resistance. A comparative genomics analysis for blast resistance was carried out by Babu et al. (2014a) using 190 genotypes and 58 functional SSRs developed from blast resistance genes of rice/finger millet. Half of the markers were polymorphic, and they yielded 65 scorable alleles with a mean of 2.4 alleles per marker. Association mapping analysis was also carried out using the phenotypic data (leaf blast, neck blast, finger blast) and the genotypic data of 104 microsatellite markers spread across the chromosomes. The association of SSR marker data with the leaf blast, neck blast, and finger blast data identified four significant QTLs for finger blast and one for neck blast. This is the first report in the development of functional SSR markers for finger millet blast resistance genes. A few of the Nucleotide Binding Site (NBS) Leucine-rich Repeat (LRR) resistance (R) genes were cloned from finger millet and were utilized in developing molecular markers linked to resistance (Panwar et al. 2011). Genetic diversity analysis of 190 global finger millet accessions for blast resistance using 58 genic functional SSR markers identified a few markers that could effectively differentiate the genotypes based on their response to blast pathogen (Babu et al. 2014a). Association mapping has identified some QTLs for blast resistance using a few genomic and genic SSR markers (Babu et al. 2014b; Ramakrishnan et al. 2016). Evaluation of 128 genotypes using 87 genomic SSRs along with phenotyping for agronomic traits identified 7 QTLs associated with agronomic traits including leaf blast (Ramakrishnan et al. 2016). Putative candidate genes associated with these QTLs were identified through cross-species validation. The association of SSR marker data with leaf blast, neck blast, and finger blast data identified five significant QTLs for finger blast and neck blast which are efficient tools for cloning resistant genes and candidates for transgenic development. Various marker systems like SRAP and SSRs were also used to know the extent of genetic polymorphism between resistant and susceptible genotypes to select diverse parents for use in breeding programs (Saha et al. 2016).
Finger millet grains are rich sources of protein and are exceptionally rich in Calcium which is almost five times of that in wheat and ten times of that in rice. Nirgude et al. 2014 developed 36 EST-SSR primers for the opaque2 modifiers and 20 anchored-SSR primers for calcium transporters and calmodulin for analysis of the genetic diversity of 103 finger millet genotypes for grain protein and calcium contents. The opaque2 modifiers specific EST-SSRs could differentiate finger millet genotypes into high, medium, and low protein containing genotypes. However, calcium-dependent candidate gene-based EST-SSRs could only broadly differentiate the genotypes based on the calcium content with a few exceptions. Finger millet being a heavy accumulator of calcium and since its genome was not sequenced, Yadav et al. (2014) explored the possibility of using microsatellite markers designed from calcium signalling and transport genes of rice and sorghum for assessing cross-transferability among grass species. Primers were designed for around 146 genic SSR markers which showed on an average 68% cross transferability to finger millet but failed to generate polymorphism between genotypes, may be indicating the highly conserved behavior of calcium related genes in plants. In a similar line, 23 anchored SSR markers were designed specifically from calcium transporters and sensors to be used in association mapping of finger millet accessions with high or low seed calcium content (Kumar et al. 2015a). They identified nine marker-trait associations related with calcium content that explained up to 41% of the total variation in calcium content. Association mapping approach identified putative QTLs for seedling stage phosphorus starvation response in finger millet (Ramakrishnan et al. 2017).
Many EST-SSRs and SNPs loci belonging to genes involved in calcium transport and sensing were identified from the spike transcriptome data which could be valuable tools for mapping, marker-assisted breeding, and comparative genomics studies in finger millet (Kumar et al. 2015b). As these are gene specific markers, they can be ideal for genotyping finger millet lines for seed calcium and protein traits. SNPs are much more effective than SSRs in revealing the genetic diversity among genotypes. With the advances in Next Generation Sequencing (NGS) technologies, SNP discovery has been more efficient. Around 10,327 SSRs and 23,285 non-homologous SNPs were identified from two finger millet genotypes (Gimode et al. 2016) of which 101 SSRs and 92 SNPs were tested across germplasm for polymorphism. The polymorphic markers gave a mean polymorphism information content (PIC) of 0.42 for the SSRs and 0.29 for SNPs. One hundred and nine SNPs generated through genotyping by sequencing (GBS) were used for association mapping to identify reliable marker(s) linked to grain yield and its component traits (Sharma et al. 2018).
Being a highly self-pollinated plant with tiny flowers and floral parts, makes crossing as well as identification of true hybrids tedious in finger millet. Krishna et al. (2020) used molecular markers for the assessment of the genetic purity of F1 hybrids. RAPD, ISSR, and SSR markers were used in assessing parental polymorphism in a reciprocal cross between PR202 and IE2606. This study found that molecular markers are effective to compare the efficiency of emasculation methods and in identification of true hybrids in the early stages of seedling growth.
14.3 Progress in Identification and Functional Analysis of Candidate Genes
Ragi being a stress hardy crop is a prospective candidate for isolation of genes governing stress tolerance. These genes, so identified, could be recruited as potential candidates for crop improvement through transgenesis, not only in ragi accessions, but even in distant species, also. Identification and characterization of a stress responsive transcription factor, EcNAC1, from finger millet was such a step. Its enhanced expression under low moisture and salinity stress imparted resistance to these stresses in transgenic tobacco plants proving its applicability (Ramegowda et al. 2012). A salinity responsive NAC transcription factor gene from finger millet, EcNAC67, was introduced to rice through agrobacterium mediated transformation and transgenic lines had enhanced tolerance against drought and salinity and maintained a higher relative water content during drought and displayed lesser drought-induced spikelet sterility (Rahman et al. 2016). Transgenic expression of EcGBF3 (G-box Binding Factor) in Arabidopsis imparted enhanced tolerance to abiotic stresses like drought, high salinity, and osmotic stress (Ramegowda et al. 2017). EcbZIP17 transcription factor overexpressed transgenic tobacco plants had better vegetative growth and seed yield and were tolerant to many abiotic stresses by modulating ER (Endoplasmic Reticulum) pathways (Ramakrishna et al. 2018).
Another transcription factor, EcDof (DNA binding with one finger) from Eleusine coracana, was identified as the key regulators of protein accumulation in the grains of ragi even at zero nitrogen input (Gupta et al. 2011, 2012). The ratio of the two transcripts Dof1/Dof2 was also identified as a pointer to the NUE (Nitrogen Use Efficiency) of crops (Gupta et al. 2014). Dof1 also consistently expressed itself in high grain protein ragi genotypes along with light inducible genes involved in carbon metabolism and can even be used as a biomarker of traits like grain protein content and yield (Kanwal et al. 2014). Amplification of five genes namely, EcHNRT2, EcLNRT1, EcNADH-NR, EcGS, and EcFd-GOGAT involved in nitrate uptake and assimilation helped in understanding the molecular basis of high nitrogen use efficiency of finger millet (Gupta et al. 2013). Candidate gene approach also elucidated how light regulated the expression of genes involved in photosynthesis and carbon metabolism in two finger millet genotypes differing in grain protein content (Kanwal et al. 2014). Co-expression pattern of seven carbon metabolism related genes and a transcription factor, Dof1, showed that Dof1 differentially regulated the carbon metabolism genes and thereby controlled grain protein content in finger millet genotypes. OPAQUE2 transcription factor gene was characterized in finger millet (EcO2) (Gaur et al. 2018) and its expression levels were found different in genotypes that differed in their seed protein content. Depending on the nitrogen status in soil and thereby in plant, nitrogen responsive regulatory elements modulated EcO2 expression and in turn regulated seed protein status. Four Pht genes (inorganic phosphate transporter) were also identified from finger millet which helps in phosphate acquisition especially in symbiotic relationship with arbuscular mycorrhizae (Pudake et al. 2017). One of the genes, EcPT1-4, was consistent in its expression levels even under Pi (inorganic phospohorus) stress in seedlings showing promise in utilizing them in enhancing the phosphorus use efficiency of the crop.
From among the many genes involved in calcium absorption and transport in the plant, a calmodulin gene that is strongly expressed during grain development was cloned and characterized at the molecular and protein level and the study could infer that a high expression of this gene might cause enhanced calcium accumulation in the grains (Nath et al. 2010; Kumar et al. 2014). Yet another gene involved in calcium metabolism, EcCIPK24 was cloned and characterized (Chinchole et al. 2017). It is a calcium sensor gene having a role in calcium transport regulation by binding with a calcium exchanger, CAX. Based on the expression studies in two contrasting finger millet genotypes for calcium content and in silico study, EcCIPK24 was found activating EcCAX1b protein and thereby playing an important role in high seed Ca2+ accumulation.
Finger millet is known to be tolerant to abiotic stress such as heat and drought and the molecular machinery behind it needs to be worked out completely. A stress responsive bHLH (basic helix-loop-helix) transcription factor gene was isolated and characterized in finger millet whose transcript was induced by ABA, NaCl, PEG, methyl viologen, and drought stress (Babitha et al. 2015). An abiotic-stress responsive protein kinase, EcCIPK31-like, was also cloned and characterized which was upregulated in multiple stress conditions such as salinity, desiccation, oxidative, and high temperature stress as well as drought (Nagarjuna et al. 2016). Overexpression of EcbHLH57 in tobacco imparted tolerance to salinity and drought stress and the transgenic plants showed improved root growth, higher photosynthetic rate, and altered stomatal conductance under stress as well as enhanced the expression of several stress responsive genes including antioxidant genes. Antioxidant genes of finger millet were also shown to be involved in controlling blast fungus growth in finger millet plants (Jacob et al. 2019). Differential expression of these genes was found to determine the extent of ROS (reactive oxygen species) accumulation in the tissues and compatibility of a plant-pathogen interaction deciding the fate of the plant, whether to be resistant or susceptible. Candidate gene identification and characterization has remarkable impact on trait characterization and improvement through breeding. As in many studies described above, many genes from finger millet when expressed in other species, imparted tolerance to various kinds of stresses implying cross-species utility of candidate genes.
14.4 Progress in Genome Sequencing
Advances in NGS (Next generation Sequencing) techniques have facilitated whole genome sequencing (WGS) of many plant species starting from Arabidopsis, the first plant whose genome was completely sequenced (Kaul et al. 2000). Finger millet was considered as an orphan crop genomically till recently and genetic as well as genomic studies were missing in the crop due to non-availability of sufficient genome sequence information. Even when genome of other millets like sorghum and foxtail millet have been sequenced, there wasn’t much progress in finger millet genomics which was an impediment in breeding and crop improvement efforts in the crop. The polyploidy of finger millet was another hindrance in its genome sequencing and annotation efforts. Genome size in finger millet is around 1500 Mb. First de novo genome assembly of finger millet was reported in 2017 in a short duration, high yielding, and drought tolerant ML-365 variety (Hittalmani et al. 2017). It was accomplished using a combination of Illumina and SOLiD sequencing technologies. There were 525,759 scaffolds of ~200 bp length with an N50 value of around 24 kb. A total of 85,000 genes were predicted from the sequenced data, majority of which could be functionally annotated. It had shared gene families with other major grass species and had highly conserved genomic regions with these species. Collinearity of finger millet genome was highest with that of foxtail millet and rice, followed by sorghum, and maize. Synteny relationships of finger millet will help in comparative genomics studies and in utilizing the QTLs and genes of interest from related plant species. Like most other plant genomes, finger millet genome also contained repetitive DNA elements, which accounted for ~50% of the total genome dominated by LTRs.
In an attempt to overcome the issues of polyploid genome sequencing and to develop a high-quality assembled genome, Hatakeyama et al. (2018) sequenced finger millet genome using diverse technologies with sufficient coverage and assembled it using a multiple hybrid assembly workflow. The high yielding national check from India, that is resilient to high heat and drought, PR202 was sequenced, and the sequenced genome was 1.2 Gb in size with around 63,000 predicted genes. They observed a significant number of single-copy genes which was not expected considering the tetraploid status of the crop. This may indicate that polyploidization happened prior to domestication and many of the duplicated genes might be lost. Sequenced genome had 2387 scaffolds having an N50 value >2.5 Mb. Genome sequence information generated in finger millet is expected to expedite identification of many SSRs, SNPs, candidate genes, alleles, and molecular breeding programs in general. Having the genomic information enables us to understand the molecular and genetic basis of traits better. The sequence information of finger millet can be accessed at https://phytozome-next.jgi.doe.gov/info/Ecoracana_v1_1.
14.5 Advances in Transcriptomics, Proteomics, and Metabolomics
High throughput techniques have enabled the development and utilization of multiple omics modules in plant biology research. Functional genomics involving multiple omics techniques have enabled the dissection of traits and their understanding more effective. There have been a few omics based trait characterization in finger millet lately and Table. 14.2 lists the major omics studies undertaken in the crop.
The next-generation sequencing platform has been used in finger millet in a genome-wide transcriptional analysis of two finger millet genotypes differing in their level of salinity tolerance (Rahman et al. 2014). The leaf transcripts were sequenced, and reads were mapped and annotated against rice gene models leading to the identification of several useful candidate salinity responsive genes. Two genotypes differing in their sensitivity to salinity were used (tolerant “Trichy” and sensitive “CO12”) and the study found groups of genes like transcription factors, transporters, aquaporins, genes involved in osmotic homeostasis and biosynthesis of compatible solutes to be upregulated in the tolerant genotype. This study helped in the identification of various salinity responsive transcription factors and signalling elements in finger millet.
Drought responsive genes were identified from ML-365 genotype of finger millet using transcriptomics approach (Hittalmani et al. 2017). The transcripts for ATP-binding and zinc ion types were plenty and transcripts coding for membrane integral components were found to be enriched in the cellular and biological process component. The upregulated genes in low moisture conditions included protein kinases and phosphatases, Myb-like protein binding and zinc binding genes, pectinacetylesterase, protein tyrosine kinase, and late embryogenesis abundant proteins. Cytochrome P450, NB-ARC, UDP-glucoronosyl, and UDP-glucosyl transferase proteins were among downregulated genes. In a later study using GPU-28, several pathways were found to be regulated under water stress in finger millet (Parvathi et al. 2019). Several drought stress signalling genes were found to act in the process including serine threonine protein phosphatase 2A (PP2A), calcineurin B-like interacting protein kinase31 (CIPK31), farnesyl pyrophosphate synthase (FPS), signal recognition particle receptor α (SRPR α) etc. Interestingly, basal regulatory genes like TATA-binding protein associated factors were also responsive to drought indicating the crucial involvement of housekeeping genes too in stress regulation. In a combined transcriptome and proteome study (Li et al. 2021), several pathways associated with photosynthesis, response to water stress, translation, ribosome process, and carbon metabolism. Significantly differentially expressed proteins belonged to glycosyl hydrolase family 17 (GHL17), the thaumatin family, aquaporins, glutathione sulpha-transferase and peroxidase gene families, all of which are involved in various abiotic stress responses.
Finger millet is a treasure trove of calcium. It is one of the richest plant-based sources of calcium in nature. The exceptionally high calcium accumulation in ragi grains is intriguing as it is almost 10 to 20 times more than that in other cereals and millets. Hence it is interesting to find out the exact function of this mineral in this crop. Calcium uptake and transport are genetically and epigenetically regulated traits and there are various transporters and calcium sensor proteins that are involved in calcium content regulation in plants. One of the earliest studies in this line used transcriptome sequencing of spike tissue in two genotypes of finger millet (GP-1 and GP-45) differing in their calcium content (Singh et al. 2014). They identified and annotated calcium sensor gene families using the transcript data and were classified in to eight calcium sensor gene families such as CaM, CaMLs, CBLs, CIPKs, CRKs, PEPRKs, CDPKs, CaMKs, and CCaMK. There was differential expression of the genes belonging to these classes and most of them belonged to stress adaptation, hormonal changes, and biotic stress, mostly pathogen resistance. Differential expression of a few chosen genes (Ca2 +/H+ antiporter (CAX1), two pore channel (TPC1), CaM-stimulated type IIB Ca2 + ATPase and two CaM dependent protein kinase (CaMK1 and 2) homologs) involved in calcium metabolism was studied in the same genotypes (Mirza et al. 2014) and for most of the genes the high accumulating genotype had a higher expression. Kumar et al. (2015b) reported complete grain-filling stage transcriptome of GP-1 and GP-45, two contrasting genotypes for grain calcium and protein content. This study helped in identifying the genes responsible for high grain calcium and protein content. Several gene families involved in calcium transport and signalling such as calcium channel, calcium ATPase, calcium exchanger (CaX), calcium-dependent protein kinase (CDPKs), and calcium-binding proteins (CBPs) during grain development were identified. Transcripts and expression levels for main storage proteins like prolamin, globulin, gliadin, kafirin, albumin, glutemin, and legumin were also identified. The contrasting genotypes were found to express many of the gene family members differentially and the information will be of great help in breeding programs aiming at biofortification in the crop.
Through transcriptomics, an extensive analysis of Ca2+ transporter gene families in relation to grain calcium was done from the developing spikes of genotypes contrasting for their grain calcium content (Singh et al. 2015). A total of 19 transporter genes were identified and a high correlation was established between the expression of EcCAX3 gene, a Ca2+/H+ exchanger, and the amount of calcium accumulated in spike. Anti-nutritional compounds such as oxalates are reported in finger millet that will adversely affect calcium availability and absorption by humans. Developing spike transcriptomics was employed to study the molecular mechanism of high calcium accumulation in line with oxalate metabolism (Akbar et al. 2018). More than one pathway for oxalate synthesis was established in finger millet and the crop could be a prospective candidate for studying the nutrient-anti-nutrient interactions.
A combined transcriptome and metabolome approach to know the effect of silicon (Si) amendment in overcoming osmotic stress in finger millet proved that Si improved seed germination as well as growth parameters under stress (Mundada et al. 2021). Enhanced silicon mediated the diversion of an enhanced pool of acetyl coA to lipid biosynthesis and membrane lipid damage was reduced significantly. The metabolite abundance was in line with relative expression of transcripts, the study found. A genome-wide identification of nutrient transporters in finger millet was undertaken computationally to have a better understanding of the nutrient transport pathways and to improve the pathways using the genomic information (Maharajan et al. 2022). Many of the health benefits of finger millet grains and the stress tolerance of the crop is related to its antioxidant properties. To have a systematic analysis of the ROS producing and scavenging genes in this crop, Avashthi et al. (2018) undertook a transcriptome analysis to identify the genes of the ascorbate-glutathione cycle (Halliwell-Asada pathway) and related pathways. Several key genes of this pathway were identified (such as APX, DHAR, MDHAR, GR, and SOD) from a low and high Ca2+ genotype (GP1 and GP45 respectively). The key genes identified using rice as a reference genome are hoped to open new avenues for the systematic functional analysis of antioxidant genes in finger millet which has direct/indirect effects in human health and nutrition.
14.6 Status of Genetic Transformation and Transgenics Development
In the literature reports available on genetic transformation in finger millet, Agrobacterium mediated method is found to be more widely used as compared to ballistic method. It could be due to the development of superior strains of agrobacterium and tissue culture protocols developed in the recent years. Effective transformation protocols have been developed and standardized by different research groups.
Co-cultivation of shoot apex explants obtained on the 16th day after callus induction with hygromycin phosphotransferase (hptII) as selectable marker was used to obtain 3.8% stable transformation in two finger millet genotypes, GPU45 and CO14 (Ceasar and Ignacimuthu 2011). A rapid protocol using shoot apical meristems (SAMs) as explants enabled transgenic plantlet production in the greenhouse within 45 days through recovery of transgenic plants via direct plant regeneration without a callus phase (Satish et al. 2017). Forty-five-day old calli obtained from the scutellum of the mature seeds was used by Hema et al. (2014). Calli from mature seeds was used for infection with Agrobacterium (Anjaneyulu et al. 2014).
Optimum conditions for particle bombardment of finger millet were defined as 1100 psi rupture disk pressure with 3 cm distance from rupture disk to macrocarrier and 12 cm microprojectile travel distance, double bombardment with gold particles of 1.0 μm size and osmotic treatment of callus with 0.4 M sorbitol (Jagga-Chugh et al. 2012). Calli were bombarded and placed on regeneration medium containing hygromycin as the selection marker that led to 45.3% transformation efficiency.
Blast disease affects the leaf, neck, and spikes of finger millet and sometimes it causes average yield losses of 20–50% and even complete crop loss. Rice chitinase (chi11) gene was introduced in the genotype GPU45 for resistance to leaf blast disease (Ignacimuthu and Ceasar 2012). Chitinase was transformed through Agrobacterium-mediated transformation under the control of maize ubiquitin promoter. Somatic embryogenesis and regeneration of shoot apex explant was employed. Bioassay showed fewer lesions in transgenic plants compared to control plants. A gene coding for an antifungal protein (PIN) of prawn driven by CaMV 35S promoter was chemically synthesized and introduced into the callus of shoot-tip explant (Latha et al. 2005). Transgenic plants expressing pin gene exhibited marked resistance to leaf blast disease (0–4 scale) while control plants showed heavy damage (5–9 scale) in bioassay.
Mutant alpha-tubulin gene (TUAm 1) isolated from R-biotype goosegrass (Eleusine indica L.) conferred resistance to dinitroaniline herbicides. It is a modified tubulin gene that can be used as a selectable marker. Trifluralin, the key constituent of dinitroaniline herbicides, is used as selection agent for differentiating genetically transformed cells. Trifluralin at a concentration of 10 microM was found optimum as selection agent in the production of transgenic finger millet for dinitroaniline-resistance (Bayer et al. 2014). Transgenic finger millet plants expressing the mannitol biosynthetic pathway gene from bacteria, mannitol-1-phosphate dehydrogenase (mtlD) were developed through Agrobacterium tumefaciens-mediated genetic transformation (Hema et al. 2014). These progenies had better growth under drought and salinity stress compared to wild type. A vacuolar proton pyrophosphatase from Sorghum bicolor (SbVPPase) was introduced into embryogenetic calli through Agrobacterium and it enhanced the plant’s performance under salt stress (Anjaneyulu et al. 2014). Relative water content (RWC), plant height, leaf expansion, finger length and width, and grain weight were more compared to control plants and relative changes in enzyme activities were identified in transgenic plants. Salinity tolerant finger millet transgenic plants were developed using a double gene construct consisting of PgNHX1 from pearl millet and AVP1 from Arabidopsis using Agrobacterium mediated transformation (Jayasudha et al. 2014). This was the first time a double gene construct was used for producing finger millet transgenic.
XvAld1gene that encodes aldose reductase was introduced into finger millet via Agrobacterium-mediated transformation and the transgenic events regenerated through direct organogenesis using shoot apical meristems to impart resistance to drought and salinity stress, where the transgenic plants were found tolerant as compared to wild plant types (Mukami 2019). OsSOS1 gene from Oryza sativa was overexpressed through Agrobacterium tumerfaciens – mediated transformation using direct plant regeneration by culturing shoot apical meristems that lead to high salt tolerance, promoted seed germination, and increased root length, shoot length, chlorophyll, membrane stability index, and reduction in reactive oxygen species (ROS) relative to wild type plants (Pushpa et al. 2020).
14.7 Research Gaps and Future Strategies
In the light of increasing world population, climate change and reduction in ground water table as well as soil nutrient levels, there is a dire need to improve minor/orphan crops like finger millet as they have proven their drought hardiness and nutritional superiority. As there is a gradual decline in the cultivated area under finger millet in recent years, the only alternative is to increase the productivity which is possible only through improved breeding tools. Since the crop has been genetically and genomically under-exploited, concerted efforts are needed in the development of advanced molecular breeding tools for crop improvement. There is huge potential in identification of QTLs associated with characteristic features of millets like drought, biotic stress resistance, and high calcium and mineral contents. This would further facilitate the marker assisted back cross transfer of mapped QTLs to the genetic background of economically important parental lines. Finger millet is an ideal candidate for basic studies on the biochemical and molecular mechanisms underlying the ability of this crop and millets in general, to survive under low soil moisture and low soil fertility to be used further in other crops that are susceptible to these conditions. The cellular pathways leading to their high physiological efficiency to make them the hardy crops that they are is a potential area yet to be explored. There is huge scope for genomic technologies and high throughput phenotyping in breeding strategies to better utilize natural and induced genetic variations existing in germplasm collections and wild relatives. The genome sequencing has given a fresh boost to the genetic research in finger millet. The accessibility of the genome sequence in the public domain is a potential tool for more research into the structural and functional genetics of the crop and this area needs to be exploited more. Considering the severity and economic significance of blast disease in finger millet, there is immediate need for elaborate studies on resistance gene identification against the fungus and host-pathogen interactions which might yield unique and useful insights.
14.8 Conclusion
Advances in molecular techniques and their effective application in breeding programs have made crop improvement rapid and much more efficient in comparison to conventional methods. Even though it took several years to utilize the modern techniques in the orphan crop like finger millet, the recent developments have been promising. Whole genome sequencing has given a new direction to genomics-aided breeding programs in finger millet. Still, there are several research gaps that need to be addressed starting from the characterization of the germplasm resources, mapping population development, identification of candidate genes for important traits, SNP identification, association mapping, and NGS based allele discovery. Improvements in molecular breeding can make the exploitation of valuable traits owned by this nutritionally superior and stress tolerant crop for its own development as well as for the betterment of other crop species.
References
Agrawal R, Agrawal N, Tandon R, Raina SN (2013) Chloroplast genes as genetic markers for inferring patterns of change, maternal ancestry and phylogenetic relationships among Eleusine species. AoB Plants. https://doi.org/10.1093/aobpla/plt056. Print 2014
Akbar N, Gupta S, Tiwari A, Singh KP, Kumar A (2018) Characterization of metabolic network of oxalic acid biosynthesis through RNA seq data analysis of developing spikes of finger millet (Eleusine coracana): deciphering the role of key genes involved in oxalate formation in relation to grain calcium accumulation. Gene 649:40–49
Anjaneyulu E, Reddy PS, Sunita MS, Kavi Kishor PB, Meriga B (2014) Salt tolerance and activity of antioxidative enzymes of transgenic finger millet overexpressing a vacuolar H+-pyrophosphatase gene (SbVPPase) from Sorghum bicolor. J Plant Physiol 171:789–798. https://doi.org/10.1016/j.jplph.2014.02.001
Avashthi H, Pathak RK, Pandey N et al (2018) Transcriptome-wide identification of genes involved in ascorbate–glutathione cycle (Halliwell–asada pathway) and related pathway for elucidating its role in antioxidative potential in finger millet (Eleusine coracana (L.)). 3 Biotech 8:499. https://doi.org/10.1007/s13205-018-1511-9
Babitha KC, Vemanna RS, Nataraja KN, Udayakumar M (2015) Overexpression of EcbHLH57 transcription factor from Eleusine coracana L. in tobacco confers tolerance to salt, oxidative and drought stress. PLoS One 10(9):e0137098
Babu BK, Senthil N, Gomez SM, Biji KR, Rajendraprasad NS, Kumar SS et al (2007) Assessment of genetic diversity among finger millet (Eleusine coracana (L.) Gaertn.) accessions using molecular markers. Genet Resour Crop Evol 54:399–404. https://doi.org/10.1007/s10722-006-0002-8
Babu BK, Dinesh P, Agrawal PK, Sood S, Chandrashekara C, Bhatt JC, Kumar A (2014a) Comparative genomics and association mapping approaches for blast resistant genes in finger millet using SSRs. PLoS One 9(6):e99182
Babu BK, Agrawal PK, Pandey D, Jaiswal JP, Kumar A (2014b) Association mapping of agro-morphological characters among the global collection of finger millet genotypes using genomic SSR markers. Mol Biol Rep 41:5287–5297. pmid:24861452
Bayer GY, Yemets AI, Blume YB (2014) Obtaining the transgenic lines of finger millet Eleusine coracana (L.). With dinitroaniline resistance. Cytol Genet 48:139–144. https://doi.org/10.3103/S0095452714030025
Bisht MS, Mukai Y (2001) Genomic in situ hybridization identifies genome donor of finger millet (Eleusine coracana). Theor Appl Genet 102:825–832
Bisht MS, Mukai Y (2002) Genome organization and polyploid evolution in the genus Eleusine (Poaceae). Plant Syst Evol 233:243–258
Ceasar SA, Ignacimuthu S (2011 Sep) Agrobacterium-mediated transformation of finger millet (Eleusine coracana (L.) Gaertn.) using shoot apex explants. Plant Cell Rep 30(9):1759–1770. https://doi.org/10.1007/s00299-011-1084-0
Chinchole M, Pathak RK, Singh UM, Kumar A (2017) Molecular characterization of EcCIPK24 gene of finger millet (Eleusine coracana) for investigating its regulatory role in calcium transport. 3 Biotech 7(4):1–10
Das S, Mishra RC, Rout GR, Aparajita S (2007) Genetic variability and relationships among thirty genotypes of finger millet (Eleusine coracana L. Gaertn.) using RAPD markers. Z Naturforsch C 62(1-2):116–122
Dida MM, Ramakrishnan SS, Bennetzen JL, Gale MD, Devos KM (2007) The genetic map of finger millet, Eleusine coracana. Theor Appl Genet 114:321–332
Dida MM, Wanyera N, Harrison Dunn MLN, Bennetzen JL, Devos KM (2008) Population structure and diversity in finger millet (Eleusine coracana) germplasm. Trop Plant Biol 1:31–141
Fakrudin B, Kulkarni RS, Shashidhar HE, Hittalmani S (2007) Genetic diversity assessment of finger millet, Eleusine coracana, germplasm through RAPD analysis. PGR Newslett 138:52–54
Gaur VS, Kumar L, Gupta S, Jaiswal JP, Pandey D, Kumar A (2018) Identification and characterization of finger millet OPAQUE2 transcription factor gene under different nitrogen inputs for understanding their role during accumulation of prolamin seed storage protein. 3 Biotech 8(3):1–11
Gimode D, Odeny DA, de Villiers EP, Wanyonyi S, Dida MM, Mneney EE et al (2016) Identification of SNP and SSR markers in finger millet using next generation sequencing technologies. PLoS One 11(7):e0159437. https://doi.org/10.1371/journal.pone.0159437
Gupta N, Gupta AK, Singh NK, Kumar A (2011) Differential expression of PBF Dof transcription factor in different tissues of three finger millet genotypes differing in seed protein content and color. Plant Mol Biol Report 29:69–76
Gupta N, Gupta AK, Kumar A (2012) Spatial distribution pattern analysis of Dof1 transcription factor in different tissues of three Eleusine coracana genotypes differing in their grain protein, yield and photosynthetic efficiency. Mol Biol Rep 39:2089–2095
Gupta AK, Gaur VS, Gupta S, Kumar A (2013) Nitrate signals determine the sensing of nitrogen through differential expression of genes involved in nitrogen uptake and assimilation in finger millet. Funct Integr Genom 13(2):179–190
Gupta S, Gupta SM, Gupta AK, Gaur VS, Kumar A (2014) Fluctuation of Dof1/Dof2 expression ratio under the influence of varying nitrogen and light conditions: involvement in differential regulation of nitrogen metabolism in two genotypes of finger millet (Eleusine coracana L.). Gene 546:327–335
Hatakeyama M, Aluri S, Balachadran MT, Sivarajan SR, Patrignani A, Grüter S et al (2018) Multiple hybrid de novo genome assembly of finger millet, an orphan allotetraploid crop. DNA Res 25:39–47. https://doi.org/10.1093/dnares/dsx036
Hema R, Vemanna RS, Sreeramulu S, Reddy CP, Senthil-Kumar M, Udayakumar M (2014) Stable expression of mtlD gene imparts multiple stress tolerance in finger millet. PLoS One 9(6):e99110
Hilu KW (1988) Identification of the “a” genome of finger millet using chloroplast DNA. Genetics 11:163–167
Hittalmani S, Mahesh H, Shirke MD, Biradar H, Uday G, Aruna Y et al (2017) Genome and transcriptome sequence of finger millet (Eleusine coracana (L.) Gaertn.) provides insights into drought tolerance and nutraceutical properties. BMC Genomics 18:465. https://doi.org/10.1186/s12864-017-3850-z
Ignacimuthu S, Ceasar SA (2012 Mar) (2012) development of transgenic finger millet (Eleusine coracana (L.) Gaertn.) resistant to leaf blast disease. J Biosci 37(1):135–147. https://doi.org/10.1007/s12038-011-9178-y
Jacob J, Madhu P, Balakrishna D, Das IK (2019) Magnaporthe grisea infection modifies expression of antioxidant genes in finger millet. J Plant Pathol 101(1):129–134
Jagga-Chugh S, Kachhwaha S, Sharma M et al (2012) Optimization of factors influencing microprojectile bombardment-mediated genetic transformation of seed-derived callus and regeneration of transgenic plants in Eleusine coracana (L.) Gaertn. Plant Cell Tissue Organ Cult 109:401–410. https://doi.org/10.1007/s11240-011-0104-7
Jayasudha BG, Sushma AM, Prashantkumar S. Hanjagi, Sashidhar VR (2014) An efficient in-vitro agrobacterium-mediated transformation protocol for raising salinity tolerant transgenic plants in finger millet [Eleusine coracana (L.) Gaertn.].
Kadri SU, Mulla SI, Suchithra B, Bilal M, Ameen F, Bharagava RN, Saratale GD, Ferreira LF, Américo-Pinheiro JH (2022) Transcriptome-wide identification and computational insights into protein modeling and docking of CAMTA transcription factors in Eleusine coracana L (finger millet). Int J Biol Macromol 206(2022):768–776., ISSN 0141-8130. https://doi.org/10.1016/j.ijbiomac.2022.03.073
Kanwal P, Gupta S, Arora S, Kumar A (2014) Identification of genes involved in carbon metabolism from Eleusine coracana (L.) for understanding their light-mediated entrainment and regulation. Plant Cell Rep 33:1403–1411
Kaul S, Koo HL, Jenkins J, Rizzo M, Rooney T, Tallon LJ, Feldblyum T, Nierman W, Benito MI, Lin XY et al (2000) Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 408:796–815
Krishna TA, Maharajan T, Roch GV, Ramakrishnan M, Ceasar SA, Ignacimuthu S (2020) Hybridization and hybrid detection through molecular markers in finger millet [Eleusine coracana (L.) Gaertn.]. J Crop Improv 34(3):335–355
Kumar A, Mirza N, Charan T, Sharma N, Gaur VS (2014) Isolation, characterization and immunolocalization of a seed dominant CaM from finger millet (Eleusine coracana L. Gartn.) for studying its functional role in differential accumulation of calcium in developing grains. Appl Biochem Biotechnol 172:2955–2973
Kumar A, Yadav S, Panwar P et al (2015a) Identification of anchored simple sequence repeat markers associated with calcium content in finger millet (Eleusine coracana). Proc Natl Acad Sci India Sect B Biol Sci 85:311–317. https://doi.org/10.1007/s40011-013-0296-1
Kumar A, Gaur VS, Goel A et al (2015b) De novo assembly and characterization of developing spikes transcriptome of finger millet (Eleusine coracana): a minor crop having nutraceutical properties. Plant Mol Biol Report 33:905–922. https://doi.org/10.1007/s11105-014-0802-5
Latha MK, Venkateswara Rao V, Reddy D (2005) Production of transgenic plants resistant to leaf blast disease in finger millet (Eleusine coracana (L.) Gaertn.). Plant Sci 169(4):657–667
Li J, Wang Y, Wang L, Zhu J, Deng J, Tang R et al (2021) Integration of transcriptomic and proteomic analyses for finger millet [Eleusine coracana (L.) Gaertn.] in response to drought stress. PLoS One 16(2):e0247181
Liu Q, Jiang B, Wen J, Peterson PM (2014) Low-copy nuclear gene and McGISH resolves polyploid history of Eleusine coracana and morphological character evolution in Eleusine. Turkish J Bot 38(1–12):3
Lule D, de Villiers S, Fetene M, Odeny DA, Rathore A, Das RR, Tesfaye K (2018) Genetic diversity and association mapping of Ethiopian and exotic finger millet accessions. Crop Pasture Sci 69(9):879–891
Maharajan T, Stanislaus Antony Ceasar, Thumadath Palayullaparambil Ajeesh Krishna (2022) Finger millet (Eleusine coracana (L.) Gaertn): nutritional importance and nutrient transporters, Crit Rev Plant Sci, 41:1, 1-31. DOI: https://doi.org/10.1080/07352689.2022.2037834
Manyasa E, Tongoona P, Shanahan P, Mgonja M, De Villiers S (2015) Genetic diversity in east African finger millet (Eleusine coracana (L.) Gaertn) landraces based on SSR markers and some qualitative traits. Plant Genetic Resources 13(1):45–55. https://doi.org/10.1017/S1479262114000628
Mirza N, Taj G, Arora S, Kumar A (2014) Transcriptional expression analysis of genes involved in regulation of calcium translocation and storage in finger millet (Eleusine coracana L. Gartn.). Gene 550:171–179. https://doi.org/10.1016/j.gene.2014.08.005
Mukami NA (2019) Genetic engineering of finger millet (Eleusine coracana) with aldose reductase gene isolated from Xerophyta viscosa to enhance drought and salinity tolerance. Post graduate thesis submitted to South Eastern. Kenya University, Kenya
Mundada PS, Barvkar VT, Umdale SD, Kumar SA, Nikam TD, Ahire ML (2021) An insight into the role of silicon on retaliation to osmotic stress in finger millet (Eleusine coracana (L.) Gaertn). J Hazard Mater 403
Nagarjuna KN, Parvathi MS, Sajeevan RS, Pruthvi V, Mamrutha HM, Nataraja KN (2016) Full-length cloning and characterization of abiotic stress responsive CIPK31-like gene from finger millet, a drought-tolerant crop. Curr Sci:890–894
Nath M, Goel A, Taj G, Kumar A (2010) Molecular cloning and comparative in silico analysis of calmodulin genes from cereals and millets for understanding the mechanism of differential calcium accumulation. Journal of Proteomics Bioinformatics 3:294–301
Neves SS, Swire-Clark G, Hilub KW, Baird WV (2005) Phylogeny of Eleusine (Poaceae: Chloridoideae) based on nuclear ITS and plastid trnT–trnF sequences. Mol Phylogenet Evol 35:395–419
Nirgude M, Babu BK, Shambhavi Y, Singh UM, Upadhyaya HD, Kumar A (2014) Development and molecular characterization of genic molecular markers for grain protein and calcium content in finger millet (Eleusine coracana (L.) Gaertn.). Mol Biol Rep 41(3):1189–1200
Pandian S, Satish L, Rameshkumar R, Muthuramalingam P, Rency AS, Rathinapriya P, Ramesh M (2018) Analysis of population structure and genetic diversity in an exotic germplasm collection of Eleusine coracana (L.) Gaertn. Using genic-SSR markers. Gene 653:80–90
Panwar P, Nath M, Yadav VK, Kumar A (2010) Comparative evaluation of genetic diversity using RAPD, SSR and cytochrome P450 gene-based markers with respect to calcium content in finger millet (Eleusine coracana L. Gaertn.). J Genet 89(2):121–133
Panwar P, Jha AK, Pandey PK, Gupta AK, Kumar A (2011) Functional markers based molecular characterization and cloning of resistance gene analogs encoding NBS-LRR disease resistance proteins in finger millet (Eleusine coracana). Mol Biol Rep 38:3427–3436. https://doi.org/10.1007/s11033-010-0452-0
Parvathi MS, Nataraja KN, Reddy YAN et al (2019) Transcriptome analysis of finger millet (Eleusine coracana (L.) Gaertn.) reveals unique drought responsive genes. J Genet 98:46. https://doi.org/10.1007/s12041-019-1087-0
Pudake RN, Mehta CM, Mohanta TK et al (2017) Expression of four phosphate transporter genes from Finger millet (Eleusine coracana L.) in response to mycorrhizal colonization and Pi stress. 3 Biotech 7:17. https://doi.org/10.1007/s13205-017-0609-9
Pushpa BN, Kiranmai K, Shankar AG (2020) Development of finger millet (Eleusine coracana (L.) Gaertn.) transgenic for salt tolerance by overexpressing antiporter gene OsSOS1 involved in sodium extrusion. Ind J Pure Appl Biosci 8(6):598–610
Rahman H, Jagadeeshselvam N, Valarmathi R, Sachin B, Sasikala R, Senthil N, Sudhakar D, Robin S, Muthurajan R (2014) Transcriptome analysis of salinity responsiveness in contrasting genotypes of finger millet (Eleusine coracana L.) through RNA sequencing. Plant Mol Biol 85:485–503
Rahman H, Ramanathan V, Nallathambi J et al (2016) Over-expression of a NAC 67 transcription factor from finger millet (Eleusine coracana L.) confers tolerance against salinity and drought stress in rice. BMC Biotechnol 16(Suppl. 1):35. https://doi.org/10.1186/s12896-016-0261-1
Ramakrishna C, Singh S, Raghavendrarao S et al (2018) The membrane tethered transcription factor EcbZIP17 from finger millet promotes plant growth and enhances tolerance to abiotic stresses. Sci Rep 8:2148. https://doi.org/10.1038/s41598-018-19766-4
Ramakrishnan M, Antony Ceasar S, Duraipandiyan V, Vinod KK, Kalpana K, Al-Dhabi NA et al (2016) Tracing QTLs for leaf blast resistance and agronomic performance of finger millet (Eleusine coracana (L.) Gaertn.) genotypes through association mapping and in silico comparative genomics analyses. PLoS One 11(7):e0159264. https://doi.org/10.1371/journal.pone.0159264
Ramakrishnan M, Ceasar SA, Vinod KK, Duraipandiyan V, Ajeesh Krishna TP, Upadhyaya HD et al (2017) Identification of putative QTLs for seedling stage phosphorus starvation response in finger millet (Eleusine coracana L. Gaertn.) by association mapping and cross species synteny analysis. PLoS One 12(8):e0183261. https://doi.org/10.1371/journal.pone.0183261
Ramegowda V, Senthil-Kumar M, Nataraja KN, Reddy MK, Mysore KS, Udayakumar M (2012) Expression of a finger millet transcription factor, EcNAC1, in tobacco confers abiotic stress tolerance. PLoS One 7(7):e40397
Ramegowda V, Gill US, Sivalingam PN et al (2017) GBF3 transcription factor imparts drought tolerance in Arabidopsis thaliana. Sci Rep 7:9148. https://doi.org/10.1038/s41598-017-09542-1
Saha D, Rana RS, Arya L, Verma M, Gowda MC, Upadhyaya HD (2016) Genetic polymorphisms among and between blast disease resistant and susceptible finger millet, Eleusine coracana (L.) Gaertn. Plant genetic. Resources 15(4):355–365
Salimath SS, Olivera ACD, Godwin ID, Bennetzen JL (1995) Assessment of genome origins and diversity in the genus Eleusine with DNA markers. Genome 38:757–763
Satish L, Ceasar SA, Ramesh M (2017) Improved agrobacterium-mediated transformation and direct plant regeneration in four cultivars of finger millet (Eleusine coracana (L.) Gaertn.). Plant Cell Tissue Organ Cult 131:547–565. https://doi.org/10.1007/s11240-017-1305-5
Sharma D, Tiwari A, Sood S, Jamra G, Singh NK, Meher PK, Kumar A (2018) Genome wide association mapping of agro-morphological traits among a diverse collection of finger millet (Eleusine coracana L.) genotypes using SNP markers. PLoS One 13(8):e0199444
Singh UM, Chandra M, Shankhdhar SC, Kumar A (2014) Transcriptome wide identification and validation of calcium sensor gene family in the developing spikes of finger millet genotypes for elucidating its role in grain calcium accumulation. PLoS One 9(8):e103963. https://doi.org/10.1371/journal.pone.0103963
Singh UM, Metwal M, Singh M, Taj G, Kumar A (2015) Identification and characterization of calcium transporter gene family in finger millet in relation to grain calcium content. Gene 566(1):37–46
Singh M, Metwal M, Kumar VA, Kumar A (2016) Identification and molecular characterization of 48 kDa calcium binding protein as calreticulin from finger millet (Eleusine coracana) using peptide mass finger printing and transcript profiling. J Sci Food Agric 96:672–679. https://doi.org/10.1002/jsfa.7139
Srinivasachary DMM, Gale MD, Devos KM (2007) Comparative analyses reveal high levels of conserved colinearity between the finger millet and rice genomes. Theor Appl Genet 115:489–499
Upadhyaya HD, Gowda CL, Pundir RP, Reddy VG, Singh S (2006) Development of core subset of finger millet germplasm using geographical origin and data on 14 quantitative traits. Genet Resour Crop Evol 53(4):679–685
Upadhyaya HD, Sarma NDRK, Ravishankar CR, Albrecht T, Narasimhudu Y, Singh SK, Varshney SK, Reddy VG, Singh S, Dwivedi SL, Wanyera N (2010) Developing a mini-core collection in finger millet using multilocation data. Crop Sci 50(5):1924–1931
Yadav S, Gaur VS, Jaiswal JP et al (2014) Simple sequence repeat (SSR) analysis in relation to calcium transport and signaling genes reveals transferability among grasses and a conserved behavior within finger millet genotypes. Plant Syst Evol 300:1561–1568. https://doi.org/10.1007/s00606-014-0982-3
Zhang H, Hall N, Goertzen LR, Chen CY, Peatman E, Jinesh Patel J, McElroy S (2019) Transcriptome analysis reveals unique relationships among Eleusine species and heritage of Eleusine coracana. G3 Genes Genomes Genet 9(6):2029–2036. https://doi.org/10.1534/g3.119.400214
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2024 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Jacob, J., Visarada, K.B.R.S., Malathi, V.M., Venkateswarlu, R., Karunakaran, B., Kannababu, N. (2024). Advanced Biotechnological Tools for Genetic Improvement of Finger Millet. In: Mishra, S., Kumar, S., Srivastava, R.C. (eds) Genetic improvement of Small Millets. Springer, Singapore. https://doi.org/10.1007/978-981-99-7232-6_14
Download citation
DOI: https://doi.org/10.1007/978-981-99-7232-6_14
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-99-7231-9
Online ISBN: 978-981-99-7232-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)