
Abstract
A family of signalling proteins called sirtuins is involved in the control of metabolism. The sirtuin family of NAD+-dependent protein lysine deacylases controls a range of physiological processes, including stress reactions and energy metabolism. For ageing-related illnesses such type 2 diabetes, inflammatory diseases, gene repression, metabolic regulation, apoptosis and cell survival, DNA repair, and neurodegenerative disorders, the human sirtuin isoforms (1–7) are thought to be promising therapeutic targets. The search for small compounds that alter the activity of sirtuins is becoming more and more popular since it may have positive implications on treating human ailments. Here, we discussed the sirtuin synthesis, biological importance, potent and specific pharmacological sirtuin activators and inhibitors, isoforms, and the current status of sirtuin-targeted therapeutic research. The rationale behind continued medication development is based on the progressive understanding of the sirtuin modulation processes by such compounds.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Sirtuins are existing in both prokaryotes and eukaryotes, which are NAD+-dependent mono-ADP ribosylases and protein lysine deacylases. The sirtuin family in living things consists of seven isoforms, each with a unique subcellular location and biological functions (Alvarez et al. 2011; Fiorentino et al. 2022b). Sirtuins have established cumulative consideration in the past couple of decades specified their vital roles in a range of biological processes, including cytodifferentiation, transcriptional control, cell cycle progression, apoptosis, swelling, breakdown, brain-related and heart physiology, and cancer. Thus, it has been noted that sirtuin activity regulation represents a prospective therapeutic approach for several illness (Fiorentino et al. 2022a).
Since 2000, sirtuins (SIRT1–7) have drawn increasing interest for their associations with an extensive variety of biological functions, including the control of cellular metabolism, neuroprotection, apoptosis, inflammation, and the growth of cancer (Mellini et al. 2015). In particular, SIRT1 has received the greatest research attention, both for its involvement in calorie restriction and as an eventual path for the therapy of illnesses associated with aging (Yeong et al. 2020). SIRT suppression can have advantageous effects on aging-related conditions including metabolic, cardiovascular, and neurodegenerative illnesses, whereas SIRT inhibitors may be beneficial for the treatment of muscular disorders, HIV infection, or cancer (Mellini et al. 2015; Valente et al. 2016).
The sirtuin route has drawn superior interest since it is linked to the advantages of calorie restriction for anti-aging (Mohamad Nasir et al. 2018). In aging-related laboratory models, pharmacological or genetically overexpression of the sirtuin system has also revealed encouraging outcomes. Mammalian sirtuins, which are related of the yeast Sir2 family, consist of seven members (SIRT1–SIRT7). ADP-ribosyltransferase and a deacetylase activity are two distinct actions that sirtuins have been discovered to exhibit (Liszt et al. 2005; Landry et al. 2000). Histones, transcription factors, and apoptosis modulators are just a few of the targets that sirtuins use to convey out their deacetylase activity (Youcef et al. 2007; Dai et al. 2018).
Similar to this, the revelation which are catalytic action requires NAD+, as opposed to Zn2+ in the situations of other groups of deacetylases, suggests their potential significance in balancing management in the cell’s breakdown and its functional state. Seven sirtuin iso-forms regulate numerous metabolic, anxiety, and aging processes in living things. For illnesses associated with metabolism and aging, such as metabolic syndrome and neurodegenerative disorders, sirtuins are supposed to be promising therapeutic targets. Sirtuins are thought to be prospective therapeutic targets for disorders of metabolism and aging, such as neurodegenerative illnesses and metabolic disorders.
Few powerful and selective compounds have been produced despite intensive attempts to generate minor-molecule sirtuin inhibitors and activators, in part because of the limitations of currently available assays and a lack of mechanistic characterisation of discovered compounds (Schutkowski et al. 2014).
3 Pharmacological Sirtuin Modulation
Sirtuins have fascinated attention as possible therapeutic targets, therefore much work has been put into developing specialised sirtuin agonist and inhibitors, both are utilised in research for understanding sirtuin function and as potential anti-ageing medications. Recent growths in sirtuin biochemistry, analytical test, and crystal structures of sirtuin/modulator composite, which disclose a challenging relationship between numerous chemicals and the structure and function of the enzyme, are currently assisting in the discovery of pharmacological sirtuin modulators (Dai et al. 2018).
3.1 Sirtuin Activators
Since more than 75 years ago, it has been recognised that calorie restriction (CR) increases mammalian longevity and promotes better health. When sirtuins were eliminated, it had been assumed that food restriction did not increase lifespan (Rogina and Helfand 2004; Lin et al. 2000) and that calorie restriction might lengthen mammalian lifetime by bringing SIRT1 expression (Cohen et al. 2004) led researchers to study sirtuins and to discover and develop molecules that could stimulate them.
Despite the fact that the function of sirtuins in enhancing lifespan is currently being called into doubt, based on the beneficial effects of calorie restriction on mammalian health and the consequent rises in SIRT1 (Cohen et al. 2004), sirtuins are gaining a lot of attention and compounds that can activate them due to the identification and formation of medicines that activate SIRT1.
3.1.1 Resveratrol
Two phenyl rings of the polyphenol resveratrol (RSV), which are connected by a methylene bridge (Fig. 15.1a), was the primary substance to be found that can imitate calorie restriction via activating sirtuins (Howitz et al. 2003; Wood et al. 2004). The primary substance identified that may imitate calorie restriction through activating sirtuins is resveratrol, a polyphenol comprising two phenyl rings split apart by a methylene bridge (Fig. 15.1a) (Howitz et al. 2003; Wood et al. 2004).

Sirtuin activators. (a) Resveratrol (3,5,4′-Trihydroxy-trans-stilbene) (b) SRT1720; (c) Oxazolo[4,5-b]pyridines derivative; (d) Imidazo[1,2-b]thiazole derivatives; (e) 1,4-Dihydropyridine (DHP) derivatives are a few examples
RSV significantly reduced ageing symptoms without altering the pattern of expression of any sirtuin genetic factor and mimicked CR-induced gene expression patterns in several organs (Barger et al. 2008; Pearson et al. 2008). RSV’s bioavailability and pharmacokinetics have been updated and reported in the past (Alcain and Villalba 2009; Vang et al. 2011). It has been shown that RSV, in in vivo testing to significantly slow down the ageing process in mice fed a diet loaded with calories (Baur et al. 2006). The occurrences of cataracts and albuminuria are reduced, vascular endothelium inflammation and apoptosis are decreased, aortic flexibility is increased, motor coordination is enhanced, and bone mineral density is maintained (Pearson et al. 2008). RSV’s limited bioavailability has led to modifications to boost bioavailability. A nutraceutical product called resVida, which contains 150 mg of resveratrol daily, has demonstrated success in lowering intrahepatic lipid levels, moving glucose, fatty acids, alanine-aminotransferase, or signs of inflammation in healthy obese males. These outcomes are identical to CRs (Timmers et al. 2011).
Severe dose-sensitive improvements in vasodilation mediated by the endothelium were also seen by oral RSV supplementation, and these improvements were associated with higher plasma RSV concentrations (Wong et al. 2011). After 3 months of treatment, RSV administered when nutritional preparation Longevinex® increased dilatation mediated by flow, but 3 months later Longevinex® was stopped. This option went back to its default value. The medication had no effect on inflammatory markers, lipid profiles, blood pressure, insulin resistance, or any of these parameters (Fujitaka et al. 2011).
3.1.2 Sirtuin Activators Structurally Unrelated to Resveratrol
The hunt for new compounds that may activate sirtuins more efficiently than RSV has gained attention because sirtuins are important for maintaining metabolic activities and disorders linked to ageing. Milne et al., in 2007 found small molecule SIRT1 activators that are structurally different from RSV but 100 times more powerful.
The most efficient substance was SRT1720, which at 10 M increased SIRT1 activity by 750%. Due to additive SIRT1 activation caused by medication combination, it was shown that SRT1720 (Fig. 15.1b) binds to and activates the enzyme at the same molecular site as RSV. The key domain’s nitrogen-terminal protein sequences 183–225 were significant in identifying the chemical attaching region.
In vivo and in vitro, SRT1720 boosted the deacetylation of SIRT1 substrates such p53, the transcriptional co-activator PGC-1α, and Foxo1a. Innately overweight mice (Lep ob/ob), DIO mice, and the Zucker fa/fa rat were used as in vivo disease models to examine the healing potential of SIRT1720 to cure insulin resistance and type-2 diabetes (Smith et al. 2009).
SRT1720 treatment significantly decreased fasting blood glucose in mice on a high-fat diet to levels close to normal, moderately controlling raised insulin amount, and protected mice from DIO and insulin resistance by improving oxidative metabolism in skeletal muscle, the liver, and brown adipose tissue through a general metabolic change that mimicked low energy levels (Feige et al. 2008).
Mice fed a diet rich in fat received SRT1720, which significantly dropped their fasting blood glucose levels to levels that were almost normal, partially normalised their increased insulin levels, and reduced their feeding glucose levels (Messa et al. 2020). By increasing oxidative metabolism in skeletal muscle, the liver, and brown adipose tissue through a general metabolic adaptation imitating low energy levels, these results effectively protected mice from DIO and insulin resistance (Feige et al. 2008).
Novel small molecule SIRT1 stimulants that are distinct from RSV have been identified, including a series of Oxazolo[4,5-b]pyridines (Fig. 15.1c). Additionally, a series of imidazo(1,2-b)thiazole derivatives bearing an oxazolopyridine core have been synthesised, and they may represent novel healing aims for the treatment of several illnesses.
Compound 29 (Fig. 15.1d), the least effective homologue in this generation, demonstrated oral antidiabetic activity for three distinct forms of type-2 diabetes and had a significant oral bioavailability in mouse and rat models of type-2 diabetes. In the DIO model after 2 weeks of dosage, in the genetic Zucker fa/fa rat model after 3 weeks, and in the ob/ob deficient mice after just 1 week of therapy, an intake of 100 mg/kg of compound 29 administered per day resulted in a considerable drop in fasting blood glucose content. Compound 29 was capable to control a noticeably minor amount of administered insulin and glucose in the DIO mice even after 10 weeks of dosage without having any effects on body weight, overall clinical chemistry, or haematology (Vu et al. 2009) (Fig. 15.2).

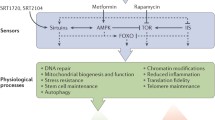
Sirt1 activation pathway by resveratrol
3.2 Sirtuin Blockers
Entirely sirtuins comprise the co-factor nicotinamide adenine dinucleotide (NAD+) (Landry et al. 2000). The acetyl-peptide remains bound by ADP-ribose, which causes the creation of a 0-alkylamidate intermediate, in the first stage of the suggested reaction process, which involves the cleavage of nicotinamide (NIC) from NAD+.
NIC is a powerful process product inhibitor due to its potential to re-bind the enzyme’s intermediate form the 0-alkylamidate and target the intermediate (Sauve and Schramm 2003). The NIC promotes megakaryocyte maturation and ploidy, in part, by inhibiting SIRT1 and by enhancing p53 binding to the NIC consensus DNA binding sequence (Giammona et al. 2009).
Due to up-regulated SIRT1 being reported in cancer cell lines, sirtuin inhibitors may also be effective as therapeutic drugs. That opens up the possibility of sirtuin inhibition can restrict the tumorigenesis. In various animal models, increased SIRT1 activity was discovered to be advantageous, which stimulated the creation of pharmacological sirtuin activators, perhaps as CR mimics. Sirtuin blockers are also suggested for the therapy for other condition like Parkinson’s disease, leishmaniosis, or HIV in along with treatment of cancer (Pagans et al. 2005). It is also crucial to take into account that SIRT3, SIRT4, and SIRT5 are found in the mitochondria, wherever many mitochondrial proteins undergo run of acetylation/deacetylation (Verdin et al. 2010) processes crucial for energy utilisation and apoptotic the beginning, as well as in the situation of certain illnesses such as cancer and the metabolic disorder (Pereira et al. 2012).
High-throughput and computational analysis screens identified sirtuin inhibitors for SIRT1, SIRT2, SIRT3, and SIRT5, in contrast to the activators sector, where only SIRT1 activators have been discovered (Villalba and Alcaín 2012). The majority of sirtuin inhibitors that have been studied till now, only SIRT1 and/or SIRT2 have been inhibited; however, a few of them have lesser affinity inhibitory effects on SIRT3 and SIRT5.
3.2.1 Splitomicin and Its Derivatives
Bedalov et al., discovered a substance called splitomicin (Fig. 15.3a) that produces a restricted phenocopy of a Sir2 deletions variant in Saccharomyces cerevisiae, provide a fresh method to study the vital function of Sir2 in vivo. IC50 for splitomicin’s inhibition of Sir2 is 60 μM (Bedalov et al. 2001). Splitomicin’s effects on human SIRT1 were, however, only moderately inhibited.

Sirtuin inhibitors. (a) Splitomicin, (b) HR73, (c) sirtinol, (d) AGK2, (e) cambinol, (f) salermide, (g) tenovin, (h) Suramin
One of the two β-phenylsplitomicin stereoisomers was clearly preferred, as shown by docking and free energy computing, and a connection between enhanced enzyme inhibition and antiproliferative action in MCF-7 breast cancer cells was identified (Neugebauer et al. 2008).
By heterochromatinizing the regulator and silencing the gene, SIRT1 contributes significantly to the amplification of the CGG·CCG-repeat tract that causes Fragile X syndrome, one of maximum prevalent genetic form of psychological illness. Its interesting to note that the Fragile X intellectual disabilities syndrome gene silence is reduced by splitomicin-mediated regulation of SIRT1 activity (Biacsi et al. 2008).
The HIV Tat protein is controlled by process of acetylation and deacetylation. Through its acetyl group and the bromodomain of PCAF, acetylated Tat that is linked to the expanding polymerase attracts the transcriptional coactivator PCAF. Tat’s separation from the expanding polymerase and PCAF might result from SIRT1’s deacetylation of the protein. A splitomicin analogue known as HR73 (Fig. 15.3b) was shown by Pagans et al. to decrease SIRT1 functioning in vitro with an IC50 value less than 5 μM, confirming SIRT1 as a new therapeutic aim for HIV infection (Pagans et al. 2005).
The Tat protein of the HIV virus is controlled by processes of acetylation and deacetylation (Chen et al. 2020). Through the polymerases elongating domain and the bromodomain of PCAF, acetylated Tat attracts the transcriptional coactivator PCAF. Tat’s separation from the elongating polymerase and PCAF could occur if SIRT1 deacetylates Tat. A splitomicin analogue (Villalba and Alcaín 2012) identified as HR73 (Fig. 15.3b) was discovered by Pagans et al. (2005) and it suppressed SIRT1 action in vitro with an IC50 value of less than 5 μM, establishing SIRT1 as a new HIV infection treatment target (Pagans et al. 2005).
3.2.2 Sirtinol
An additional sirtuin antagonist, sirtinol (2-[(2-hydroxy-naphthalen-1-ylmethylene)-amino]-N-(1-phenyl-ethyl)-benzamide) (Fig. 15.3c), was also found by Grozinger et al. (2001), in a cell-based screen. This substance decreased both yeast Sir2 and human SIRT2 action in vitro. It was proven that the 2-hydroxyl-1-napthol portion was enough to block (Grozinger et al. 2001). Sirtinol’s two derivatives, m- and p-sirtinol, were two- to tenfold highly effective against human SIRT1 and SIRT-2 than sirtinol (Mai et al. 2005). The activation of SIRT1 may be crucial in fostering cell development. As a result of decrease in apoptosis in consequence of diverse genotoxic stimuli caused by deacetylation of p53, SIRT1 inhibitors like sirtinol have potential to treat cancer (Mirzayans et al. 2017). Sirtinol also led to senescence-like growth arrest in human breast cancer MCF-7 and lung cancer H1299 cells, in addition to raising chemosensitivity to camptothecin and cisplatin in PC3, DU145, and HeLa cells (Carafa et al. 2016). The result of an increase in programmed cell death, this led to a large decrease in viable cells (Kojima et al. 2008; Peck et al. 2010).
It has also been discovered that sirtinol, nicotinamide, and SIRT3 down-regulation reduced cell proliferation and expansion in oral squamous cell carcinoma (OSCC) cell lines in vitro and in vivo, whereas SIRT3 is abundantly expressed comparison to other sirtuins, and triggered apoptosis. Additionally, SIRT3 downregulation increased OSCC cells susceptible to radiotherapy and cis-platin’s cytotoxic effects (Alhazzazi et al. 2011).
Oculopharyngeal muscular dystrophy is modeled using nematodes, a condition brought on via polyalanine increase in the nuclear protein PABPN1, sirtinol therapy proved protective, by boosting the dose of Sir2/SIRT1, increased muscle prognosis (Pasco et al. 2010). Furthermore, sirtinol treatment decreased pain and swelling in human superficial microvascular endothelial cells, modulated the appearance of binding molecules and monocyte sticking in main human cutaneous microvascular endothelial cells and induced cell death in Leishmania infantum, greatly reducing this axenic amastigote’s in vitro proliferation (Orecchia et al. 2011).
3.2.3 AGK2
According to reports, neuroprotection is provided by inhibiting SIRT2 activity. The protein α-synuclein (α-syn) is found in Lewy bodies which are the most prevalent histological characteristic of Parkinson disease (PD). The inhibition of SIRT2 with short interfering RNA or including AGK2 (Fig. 15.3d) prevented the impairment of dopaminergic nerve cells caused on by α-syn toxicity in a Drosophila PD model and lessened the neurotoxicity generated by mutant α-syn in rat primary dopamine-positive neurons (Outeiro et al. 2007).
Treatment with AGK2 increased the amounts of acetylated tubulin, but it also made PC12 cells more susceptible to necrosis without changing autophagy and caused C-6 glioma cells to undergo caspase-3-dependent apoptosis (He et al. 2012; Nie et al. 2011). Additionally, through down-regulated the RNAs necessary for sterol production. In both animal and cell-based models of Huntington’s disease (HD), sirt2 inhibition produced neuroprotection (Luthi-Carter et al. 2010). In contrast to the neuroprotective effects of SIRT2 suppression in PD and HD models, pharmacological inhibition of SIRT1/2 by nicotinamide, AGK2, or cambinol increased multiplication in cultivated megakaryocytic cells, boosting acetylation of nucleosomes and p53 (Giammona et al. 2009).
3.2.4 Cambinol
The chemically stable molecule cambinol (Fig. 15.3e), which suppresses both SIRT1 and SIRT2 in vitro with IC50 values of 56 and 59 μM, consequently, is known as β-naphtol. It shares a pharmacophore with sirtinol and splitomicin. Cambinol has individual marginal suppression effect (42% suppression at 300 μM) against SIRT5 (Heltweg et al. 2006). Mice showed good tolerance to cambinol and prevented the spread of Burkitt lymphoma xenografts by triggering apoptosis through hyperacetylation of the BCL6 oncoprotein and p53 (Heltweg et al. 2006).
Although the efficacy against SIRT2 improved in vitro when the substituent at the N1-position was utilised or this was not the case for SIRT1, changes to the phenyl ring of cambinol improved activity and increased selectivity for SIRT1, leading to the identification of a variety of SIRT2 selective analogues (Medda et al. 2009).
3.2.5 Suramin
Suramin, which has an IC50 value of 22 M, is a more powerful blocker of SIRT5 NAD+-dependent deacetylase activity than cambinol (Fig. 15.3h). Suramin has a strong inhibitory effect on SIRT1 and SIRT2 (IC50 = 0.297 μM and 1.15 μM, respectively) (Trapp et al. 2007). In addition to its antiproliferative and antiviral properties, the polyanionic naphthylurea known as suramin was first used to treat trypanosomiasis (Perabo and Müller 2005). A stronger inhibition of SIRT5 NAD+-dependent deacetylase activity than cambinol is suramin (Fig. 15.3h), with an IC50 value of 22 μM. SIRT1 and SIRT2 are both significantly inhibited by suramin (IC50 values of 0.297 μM and 1.15 μM, respectively) (Trapp et al. 2007). Originally used to treat trypanosomiasis, suramin is a polyanionic naphthylurea that also has antiproliferative and antiviral properties (Perabo and Müller 2005).
3.2.6 Tenovin
Two SIRT1 inhibitors were discovered by Lain et al. (2008) looking for tiny molecules that might stimulate p53 and stop cancer development utilising a cell-based screening method: tenovin-1 and its more water-soluble counterpart, tenovin-6 (Fig. 15.3g). Both substances inhibited tumour development in vivo at one-digit micromolar doses in vitro, all without producing appreciable over-all toxicity. In chronic myelogenous leukaemia (CML), SIRT1 activation enhances cell survival, and proliferation was linked to the deacetylation of many SIRT1 substrates, including FOXO1, p53, and KU70. Tenovin-6, an inhibitor of SIRT1, was administered to mice to stop the course of the illness (Yuan et al. 2012).
Tenovin-1 and tenovin-6 have recently undergone a series of more water-soluble counterparts that, generally, kept the required biological activity. In the presence of a solution, tenovin-1 analogues take on a preferred shape that includes an intramolecular hydrogen bond necessary for SIRT1 binding. Additionally, When the 4-tert-butyl substituent in tenovin-6 was replaced with shorter alkyl chains (4-propyl or 4-iso-propyl substituent), analogues with longer n-alkyl chains (4-n-butyl or 4-n-pentyl substituent) showed hazardous or ineffective in cells (Mccarthy et al. 2012).
3.2.7 Salermide
A reverse amide with a strong in vitro inhibitory effect on SIRT1 and SIRT2 is salermide is (N-{3-[(2-hydroxy-1-naphthalenylmethylene)-amino]-phenyl2}-phenyl-propionamide) (Fig. 15.3f). At dosages up to 100 μM, salermide was tolerated effectively by mice and induced p53-independent apoptosis in cancer cells but not in healthy cells. This was accomplished by regenerating proapoptotic genes that SIRT1 had been epigenetically suppressed in cancer cells (Lara et al. 2009). A further SIRT1 and SIRT2 inhibitor called sirtinol requires p53, as well as salermide-induced apoptosis, according to a different research utilising breast cancer cell lines and p53-insufficient mice fibroblasts (Peck et al. 2010). The salermide impact in human non-small cell lung cancer cells could be mediated by an increase in death receptor 5 expression (Liu et al. 2012).
3.2.8 Other Inhibitors of Human Sirtuins
There are many of other SIRT1 and SIRT2 inhibitors that have been found and thoroughly explored. Tripeptide analogues based on lysine, N-thioacetyl lysine found in non-peptides, thiobarbiturates, indole derivatives (EX-527), and other inhibitors with various structural cores are among them.
4 Natural Sirtuin Inhibitors and Modulators Beneficial Effects on Health
See Table 15.2.
5 Conclusions
Sirtuins are a significant group of histone deacetylases that take part in NAD+-dependent deacetylation processes. Activation, inhibition, and regulation of sirtuins are engaged in a variety of key metabolic processes that are connected to conditions including type 2 diabetes, ageing, and inflammation. As a result, they have anti-cancer, anti-oxidant, anti-microbial, and anti-viral effects and are a significant drug target class. Natural products are renowned for being significant sources of lead compounds. In this study, we’ve made an effort to give a quick rundown of the most current findings about natural products that have been found to interact with sirtuins.
Natural sirtuin modulators and inhibitors may have positive effect on health in addition to well-recognised mechanisms of action and individuals being researched in clinical studies. Alkaloids, bichalcones, resveratrol, xanthone, tanikolide, and flavonoids are a few examples of natural substances and chemical types that have been demonstrated to have actions against sirtuins. The next generation sirtuins modulators are anticipated to be natural product derivates beginning from the discovered lead compounds.
References
Alcain FJ, Villalba JM (2009) Sirtuin activators. Expert Opin Ther Pat 19(4):403–414
Alhazzazi TY, Kamarajan P, Joo N, Huang JY, Verdin E, D’Silva NJ, Kapila YL (2011) Sirtuin-3 (SIRT3), a novel potential therapeutic target for oral cancer. Cancer 117(8):1670–1678
Alvarez R, Altucci L, Gronemeyer H, de Lera AR (2011) Epigenetic multiple modulators. Curr Top Med Chem 11(22):2749–2787
Amirchaghmaghi M, Delavarian Z, Iranshahi M, Shakeri MT, Mosannen Mozafari P, Mohammadpour AH, Farazi F, Iranshahy M (2015) A randomized placebo-controlled double blind clinical trial of quercetin for treatment of oral lichen planus. J Dent Res Dent Clin Dent Prospects 9:23–28
An F, Wang S, Tian Q, Zhu D (2015) Effects of orientin and vitexin from Trollius chinensis on the growth and apoptosis of esophageal cancer EC-109 cells. Oncol Lett 10:2627–2633
Anekonda TS, Reddy PH (2006) Neuronal protection by sirtuins in Alzheimer’s disease. J Neurochem 96(2):305–313
Bae JS (2015) Inhibitory effect of orientin on secretory group IIA phospholipase A2. Inflammation 38:1631–1638
Barger JL, Kayo T, Pugh TD, Prolla TA, Weindruch R (2008) Short-term consumption of a resveratrol-containing nutraceutical mixture mimics gene expression of long-term caloric restriction in mouse heart. Exp Gerontol 43(9):859–866
Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, Pistell PJ (2006) Resveratrol improves health and survival of mice on a high-calorie diet. Nature 444(7117):337–342
Baur JA, Ungvari Z, Minor RK, Le Couteur DG, De Cabo R (2012) Are sirtuins viable targets for improving healthspan and lifespan? Nat Rev Drug Discov 11(6):443–461
Bedalov A, Gatbonton T, Irvine WP, Gottschling DE, Simon JA (2001) Identification of a small molecule inhibitor of Sir2p. Proc Natl Acad Sci U S A 98(26):15113–15118
Biacsi R, Kumari D, Usdin K (2008) SIRT1 inhibition alleviates gene silencing in Fragile X mental retardation syndrome. PLoS Genet 4(3):e1000017
Bonda DJ, Lee HG, Camins A, Pallàs M, Casadesus G, Smith MA, Zhu X (2011) The sirtuin pathway in ageing and Alzheimer disease: mechanistic and therapeutic considerations. Lancet Neurol 10(3):275–279
Boominathan SP, Sarangan G, Srikakulapu S, Rajesh S, Duraipandian C, Srikanth P, Jo L (2014) Antiviral activity of bioassay guided fractionation of Plumbago zeylanica roots against herpes simplex virus type 2. World J Pharm Sci 3:1003–1017
Carafa V, Rotili D, Forgione M, Cuomo F, Serretiello E, Hailu GS, Jarho E, Lahtela-Kakkonen M, Mai A, Altucci L (2016) Sirtuin functions and modulation: from chemistry to the clinic. Clin Epigenetics 8:1–21
Chen J, Liu Q, Zeng L, Huang X (2020) Protein acetylation/deacetylation: a potential strategy for fungal infection control. Front Microbiol 11:574736
Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, Howitz KT, Gorospe M, de Cabo R, Sinclair DA (2004) Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science 305(5682):390–392
Currais A, Prior M, Dargusch R, Armando A, Ehren J, Schubert D, Quehenberger O, Maher P (2014) Modulation of p25 and inflammatory pathways by fisetin maintains cognitive function in Alzheimer’s disease transgenic mice. Aging Cell 13:379–390
Dai H, Sinclair DA, Ellis JL, Steegborn C (2018) Sirtuin activators and inhibitors: promises, achievements, and challenges. Pharmacol Ther 188:140–154
Dali-Youcef N, Lagouge M, Froelich S, Koehl C, Schoonjans K, Auwerx J (2007) Sirtuins: the ‘magnificent seven’, function, metabolism and longevity. Ann Med 39(5):335–345. https://doi.org/10.1080/07853890701408194
Feige JN, Lagouge M, Canto C, Strehle A, Houten SM, Milne JC, Lambert PD, Mataki C, Elliott PJ, Auwerx J (2008) Specific SIRT1 activation mimics low energy levels and protects against diet-induced metabolic disorders by enhancing fat oxidation. Cell Metab 8(5):347–358
Ferry DR, Smith A, Malkhandi J, Fyfe DW, deTakats PG, Anderson D, Baker J, Kerr DJ (1996) Phase I clinical trial of the flavonoid quercetin: pharmacokinetics and evidence for in vivo tyrosine kinase inhibition. Clin Cancer Res 2:659–668
Fiorentino F, Castiello C, Mai A, Rotili D (2022a) Therapeutic potential and activity modulation of the protein lysine deacylase sirtuin 5. J Med Chem 65(14):9580–9606
Fiorentino F, Mautone N, Menna M, D’Acunzo F, Mai A, Rotili D (2022b) Sirtuin modulators: past, present, and future perspectives. Future Med Chem 14(12):915–939
Fujitaka K, Otani H, Jo F, Jo H, Nomura E, Iwasaki M, Nishikawa M, Iwasaka T, Das DK (2011) Modified resveratrol Longevinex improves endothelial function in adults with metabolic syndrome receiving standard treatment. Nutr Res 31(11):842–847
Giammona LM, Panuganti S, Kemper JM, Apostolidis PA, Lindsey S, Papoutsakis ET, Miller WM (2009) Mechanistic studies on the effects of nicotinamide on megakaryocytic polyploidization and the roles of NAD+ levels and SIRT inhibition. Exp Hematol 37(11):1340–1352
Grozinger CM, Chao ED, Blackwell HE, Moazed D, Schreiber SL (2001) Identification of a class of small molecule inhibitors of the sirtuin family of NAD-dependent deacetylases by phenotypic screening. J Biol Chem 276(42):38837–38843
Hamdy AA, Ibrahem MA (2010) Management of aphthous ulceration with topical quercetin: a randomized clinical trial. J Contemp Dent Pract 11:E009–E016
He X, Nie H, Hong Y, Sheng C, Xia W, Ying W (2012) SIRT2 activity is required for the survival of C6 glioma cells. Biochem Biophys Res Commun 417(1):468–472
Heinz SA, Henson DA, Austin MD, Jin F, Nieman DC (2010) Quercetin supplementation and upper respiratory tract infection: a randomized community clinical trial. Pharm Res 62:237–242
Heltweg B, Gatbonton T, Schuler AD, Posakony J, Li H, Goehle S, Kollipara R, DePinho RA, Gu Y, Simon JA, Bedalov A (2006) Antitumor activity of a small-molecule inhibitor of human silent information regulator 2 enzymes. Cancer Res 66(8):4368–4377
Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, Scherer B (2003) Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 425(6954):191–196
Kershaw J, Kim K-H (2017) The therapeutic potential of piceatannol, a natural stilbene, in metabolic diseases: a review. J Med Food 20:427–438
Khan N, Syed DN, Ahmad N, Mukhtar H (2013) Fisetin: a dietary antioxidant for health promotion. Antioxid Redox Signal 19:151–162
Kojima K, Ohhashi R, Fujita Y, Hamada N, Akao Y, Nozawa Y, Deguchi T, Ito M (2008) A role for SIRT1 in cell growth and chemoresistance in prostate cancer PC3 and DU145 cells. Biochem Biophys Res Commun 373(3):423–428
Kwon GT, Jung JI, Song HR, Woo EY, Jun JG, Kim JK, Her S, Park JH (2012) Piceatannol inhibits migration and invasion of prostate cancer cells: possible mediation by decreased interleukin-6 signaling. J Nutr Biochem 23:228–238
Lain S, Hollick JJ, Campbell J, Staples OD, Higgins M, Aoubala M, McCarthy A, Appleyard V, Murray KE, Baker L, Thompson A (2008) Discovery, in vivo activity, and mechanism of action of a small-molecule p53 activator. Cancer Cell 13(5):454–463
Lall RK, Adhami VM, Mukhtar H (2016) Dietary flavonoid fisetin for cancer prevention and treatment. Mol Nutr Food Res 60:1396–1405
Lam KY, Ling AP, Koh RY, Wong YP, Say YH (2016) A review on medicinal properties of orientin. Adv Pharm Sci 2016:4104595
Landry J, Sutton A, Tafrov ST, Heller RC, Stebbins J, Pillus L, Sternglanz R (2000) The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc Natl Acad Sci U S A 97(11):5807–5811. https://doi.org/10.1073/pnas.110148297
Lara E, Mai A, Calvanese V, Altucci L, Lopez-Nieva P, Martinez-Chantar ML, Varela-Rey M, Rotili D, Nebbioso A, Ropero S, Montoya G (2009) Salermide, a Sirtuin inhibitor with a strong cancer-specific proapoptotic effect. Oncogene 28(6):781–791
Li YL, Ma SC, Yang YT, Ye SM, But PP (2002) Antiviral activities of flavonoids and organic acid from Trollius chinensis Bunge. J Ethnopharmacol 79:365–368
Lin SJ, Defossez PA, Guarente L (2000) Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science 289(5487):2126–2128
Lin Q, Feng S, Cen Y, Yang Y, Wang L (2004) Study on the antibacterial and antiviral activity compositions of Trollium chinensis Bunge. J Zhejiang Univ (Sci Ed) 31:412–415
Liszt G, Ford E, Kurtev M, Guarente L (2005) Mouse Sir2 homolog SIRT6 is a nuclear ADP-ribosyltransferase. J Biol Chem 280(22):21313–21320. https://doi.org/10.1074/jbc.m413296200
Liu G, Su L, Hao X, Zhong N, Zhong D, Singhal S, Liu X (2012) Salermide up-regulates death receptor 5 expression through the ATF4-ATF3-CHOP axis and leads to apoptosis in human cancer cells. J Cell Mol Med 16(7):1618–1628
Luthi-Carter R, Taylor DM, Pallos J, Lambert E, Amore A, Parker A, Moffitt H, Smith DL, Runne H, Gokce O, Kuhn A (2010) SIRT2 inhibition achieves neuroprotection by decreasing sterol biosynthesis. Proc Natl Acad Sci U S A 107(17):7927–7932
Magyar K, Halmosi R, Palfi A, Feher G, Czopf L, Fulop A, Battyany I, Sumegi B, Toth K, Szabados E (2012) Cardioprotection by resveratrol: a human clinical trial in patients with stable coronary artery disease. Clin Hemorheol Microcirc 50:179–187
Maher P, Dargusch R, Ehren JL, Okada S, Sharma K, Schubert D (2011) Fisetin lowers methylglyoxal dependent protein glycation and limits the complications of diabetes. PLoS One 6:e21226
Mai A, Massa S, Lavu S, Pezzi R, Simeoni S, Ragno R, Mariotti FR, Chiani F, Camilloni G, Sinclair DA (2005) Design, synthesis, and biological evaluation of sirtinol analogues as class III histone/protein deacetylase (Sirtuin) inhibitors. J Med Chem 48(24):7789–7795
Mazloom Z, Abdollahzadeh SM, Dabbaghmanesh MH, Rezaianzadeh A (2014) The effect of quercetin supplementation on oxidative stress, glycemic control, lipid profile and insulin resistance in type 2 diabetes: a randomized clinical trial. J Health Sci Surveill Syst 2:8–14
Mccarthy AR, Pirrie L, Hollick JJ, Ronseaux S et al (2012) Synthesis and biological characterisation of sirtuin inhibitors based on the tenovins. Bioorg Med Chem 20:1779–1793
Medda F, Russell RJ, Higgins M, McCarthy AR, Campbell J, Slawin AM, Lane DP, Lain S, Westwood NJ (2009) Novel cambinol analogs as sirtuin inhibitors: synthesis, biological evaluation, and rationalization of activity. J Med Chem 52(9):2673–2682
Mellini P, Valente S, Mai A (2015) Sirtuin modulators: an updated patent review (2012–2014). Expert Opin Ther Pat 25(1):5–15
Messa GA, Piasecki M, Hurst J, Hill C, Tallis J, Degens H (2020) The impact of a high-fat diet in mice is dependent on duration and age, and differs between muscles. J Exp Biol 223(6):jeb217117
Miles SL, McFarland M, Niles RM (2014) Molecular and physiological actions of quercetin: need for clinical trials to assess its benefits in human disease. Nutr Rev 72:720–734
Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, Jin L, Boss O, Perni RB, Vu CB, Bemis JE (2007) Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 450(7170):712–716
Mirzayans R, Andrais B, Kumar P, Murray D (2017) Significance of wild-type p53 signaling in suppressing apoptosis in response to chemical genotoxic agents: impact on chemotherapy outcome. Int J Mol Sci 18(5):928
Mohamad Nasir NF, Zainuddin A, Shamsuddin S (2018) Emerging roles of Sirtuin 6 in Alzheimer’s disease. J Mol Neurosci 64:157–161
Nagai S, Matsumoto C, Shibano M, Fujimori K (2018) Suppression of fatty acid and triglyceride synthesis by the flavonoid orientin through decrease of C/EBPδ expression and inhibition of PI3K/Akt-FOXO1 signaling in adipocytes. Nutrients 10:130
Nayak V, Devi PU (2005) Protection of mouse bone marrow against radiation-induced chromosome damage and stem cell death by the ocimum flavonoids orientin and vicenin. Radiat Res 163:165–171
Nayak V, Uma P (2006) Antioxidant and radioprotective effects of ocimum flavonoids orientin and vicenin in Escherichia coli. Def Sci J 56:179
Neugebauer RC, Uchiechowska U, Meier R, Hruby H, Valkov V, Verdin E, Sippl W, Jung M (2008) Structure-activity studies on splitomicin derivatives as sirtuin inhibitors and computational prediction of binding mode. J Med Chem 51(5):1203–1213
Nguyen AV, Martinez M, Stamos MJ, Moyer MP, Planutis K, Hope C, Holcombe RF (2009) Results of a phase I pilot clinical trial examining the effect of plant-derived resveratrol and grape powder on Wnt pathway target gene expression in colonic mucosa and colon cancer. Cancer Manag Res 1:25–37
Nie H, Chen H, Han J, Hong Y, Ma Y, Xia W, Ying W (2011) Silencing of SIRT2 induces cell death and a decrease in the intracellular ATP level of PC12 cells. Int J Physiol Pathophysiol Pharmacol 3(1):65
Orecchia A, Scarponi C, Di Felice F, Cesarini E, Avitabile S, Mai A, Mauro ML, Sirri V, Zambruno G, Albanesi C, Camilloni G (2011) Sirtinol treatment reduces inflammation in human dermal microvascular endothelial cells. PLoS One 6(9):e24307
Outeiro TF, Kontopoulos E, Altmann SM, Kufareva I et al (2007) Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science 317:516–519
Outeiro TF, Marques O, Kazantsev A (2008) Therapeutic role of sirtuins in neurodegenerative disease. Biochim Biophys Acta Mol Basis Dis 1782(6):363–369
Pagans S, Pedal A, North BJ, Kaehlcke K, Marshall BL, Dorr A, Hetzer-Egger C, Henklein P, Frye R, McBurney MW, Hruby H (2005) SIRT1 regulates HIV transcription via Tat deacetylation. PLoS Biol 3(2):e41
Pasco MY, Rotili D, Altucci L, Farina F, Rouleau GA, Mai A, Neri C (2010) Characterization of sirtuin inhibitors in nematodes expressing a muscular dystrophy protein reveals muscle cell and behavioral protection by specific sirtinol analogues. J Med Chem 53(3):1407–1411
Pearson KJ, Baur JA, Lewis KN, Peshkin L, Price NL, Labinskyy N, Swindell WR, Kamara D, Minor RK, Perez E, Jamieson HA (2008) Resveratrol delays age-related deterioration and mimics transcriptional aspects of dietary restriction without extending life span. Cell Metab 8(2):157–168
Peck B, Chen CY, Ho KK, Di Fruscia P, Myatt SS, Coombes RC, Fuchter MJ, Hsiao CD, Lam EW (2010) SIRT inhibitors induce cell death and p53 acetylation through targeting both SIRT1 and SIRT2SIRT inhibitors target SIRT1/2 to activate p53. Mol Cancer Ther 9(4):844–855
Perabo FG, Müller SC (2005) New agents in intravesical chemotherapy of superficial bladder cancer. Scand J Urol Nephrol 39(2):108–116
Pereira CV, Lebiedzinska M, Wieckowski MR, Oliveira PJ (2012) Regulation and protection of mitochondrial physiology by sirtuins. Mitochondrion 12(1):66–76
Poulsen MM, Vestergaard PF, Clasen BF, Radko Y, Christensen LP, Stødkilde-Jørgensen H, Møller N, Jessen N, Pedersen SB, Jørgensen JO (2013) High-dose resveratrol supplementation in obese men: an investigator-initiated, randomized, placebo-controlled clinical trial of substrate metabolism, insulin sensitivity, and body composition. Diabetes 62:1186–1195
Rezvan N, Moini A, Janani L, Mohammad K, Saedisomeolia A, Nourbakhsh M, Gorgani-Firuzjaee S, Mazaherioun M, Hosseinzadeh-Attar MJ (2017) Effects of quercetin on adiponectin-mediated insulin sensitivity in polycystic ovary syndrome: a randomized placebo-controlled double-blind clinical trial. Horm Metab Res 49:115–121
Rimando AM, Nagmani R, Feller DR, Yokoyama W (2005) Pterostilbene, a new agonist for the peroxisome proliferator-activated receptor alpha-isoform, lowers plasma lipoproteins and cholesterol in hypercholesterolemic hamsters. J Agric Food Chem 53:3403–3407
Rogina B, Helfand SL (2004) Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc Natl Acad Sci U S A 101(45):15998–16003
Sauve AA, Schramm VL (2003) Sir2 regulation by nicotinamide results from switching between base exchange and deacetylation chemistry. Biochemistry 42(31):9249–9256
Schutkowski M, Fischer F, Roessler C, Steegborn C (2014) New assays and approaches for discovery and design of Sirtuin modulators. In: Expert opinion on drug discovery, vol 9(2). Informa UK Limited, pp 183–199. https://doi.org/10.1517/17460441.2014.875526
Serban M-C, Sahebkar A, Zanchetti A, Mikhailidis DP, Howard G, Antal D, Andrica F, Ahmed A, Aronow WS, Muntner P et al (2016) Effects of quercetin on blood pressure: a systematic review and meta-analysis of randomized controlled trials. J Am Heart Assoc 5:e002713
Smith JJ, Kenney RD, Gagne DJ, Frushour BP, Ladd W, Galonek HL, Israelian K, Song J, Razvadauskaite G, Lynch AV, Carney DP (2009) Small molecule activators of SIRT1 replicate signaling pathways triggered by calorie restriction in vivo. BMC Syst Biol 3(1):1–4
Sun A, Ren G, Deng C, Zhang J, Luo X, Wu X, Mani S, Dou W, Wang Z (2016) C-glycosyl flavonoidorientin improves chemically induced inflammatory bowel disease in mice. J Funct Foods 21:418–430
Syed DN, Adhami VM, Khan MI, Mukhtar H (2013) Inhibition of Akt/mTOR signaling by the dietary flavonoid fisetin. Anti Cancer Agents Med Chem 13:995–1001
Tang Y-L, Chan S-W (2014) A review of the pharmacological effects of piceatannol on cardiovascular diseases. Phytother Res 28:1581–1588
Timmers S, Konings E, Bilet L, Houtkooper RH, van de Weijer T, Goossens GH, Hoeks J, van der Krieken S, Ryu D, Kersten S, Moonen-Kornips E (2011) Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab 14(5):612–622
Trapp J, Meier R, Hongwiset D, Kassack MU, Sippl W, Jung M (2007) Structure–activity studies on suramin analogues as inhibitors of NAD+-dependent histone deacetylases (sirtuins). ChemMedChem 2(10):1419–1431
Tripathi R, Samadder T, Gupta S, Surolia A, Shaha C (2011) Anticancer activity of a combination of cisplatin and fisetin in embryonal carcinoma cells and xenograft tumors. Mol Cancer 10:255–268
Uma Devi P, Ganasoundari A, Rao BS, Srinivasan KK (1999) In vivo radioprotection by ocimum flavonoids: survival of mice. Radiat Res 151:74–78
Valente S, Mellini P, Spallotta F, Carafa V, Nebbioso A, Polletta L, Carnevale I, Saladini S, Trisciuoglio D, Gabellini C, Tardugno M (2016) 1,4-Dihydropyridines active on the SIRT1/AMPK pathway ameliorate skin repair and mitochondrial function and exhibit inhibition of proliferation in cancer cells. J Med Chem 59(4):1471–1491
Vang O, Ahmad N, Baile CA, Baur JA, Brown K, Csiszar A, Das DK, Delmas D, Gottfried C, Lin HY, Ma QY (2011) What is new for an old molecule? Systematic review and recommendations on the use of resveratrol. PLoS One 6(6):e19881
Verdin E, Hirschey MD, Finley LW, Haigis MC (2010) Sirtuin regulation of mitochondria: energy production, apoptosis, and signaling. Trends Biochem Sci 35(12):669–675
Villalba JM, Alcaín FJ (2012) Sirtuin activators and inhibitors. Biofactors 38(5):349–359
Vion E, Page G, Bourdeaud E, Paccalin M, Guillard J, Rioux Bilan A (2018) Trans ε-viniferin is an amyloid-β disaggregating and anti-inflammatory drug in a mouse primary cellular model of Alzheimer’s disease. Mol Cell Neurosci 88:1–6
Vu CB, Bemis JE, Disch JS, Ng PY, Nunes JJ, Milne JC, Carney DP, Lynch AV, Smith JJ, Lavu S, Lambert PD (2009) Discovery of imidazo [1, 2-b] thiazole derivatives as novel SIRT1 activators. J Med Chem 52(5):1275–1283
Wątroba M, Szukiewicz D (2016) The role of sirtuins in aging and age-related diseases. Adv Med Sci 61(1):52–62
Wong RH, Howe PR, Buckley JD, Coates AM, Kunz I, Berry NM (2011) Acute resveratrol supplementation improves flow-mediated dilatation in overweight/obese individuals with mildly elevated blood pressure. Nutr Metab Cardiovasc Dis 21(11):851–856
Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL, Tatar M, Sinclair D (2004) Sirtuin activators mimic caloric restriction and delay ageing in metazoans. Nature 430(7000):686–689
Yáñez M, Fraiz N, Cano E, Orallo F (2006) (−)-Trans-epsilon-viniferin, a polyphenol present in wines, is an inhibitor of noradrenaline and 5-hydroxytryptamine uptake and of monoamine oxidase activity. Eur J Pharmacol 542:54–60
Yeong KY, Berdigaliyev N, Chang Y (2020) Sirtuins and their implications in neurodegenerative diseases from a drug discovery perspective. ACS Chem Neurosci 11(24):4073–4091
Yoo H, Ku SK, Lee T, Bae JS (2014) Orientin inhibits HMGB1-induced inflammatory responses in HUVECs and in murine polymicrobial sepsis. Inflammation 37:1705–1717
Yousefzadeh MJ, Zhu YI, McGowan SJ, Angelini L, Fuhrmann-Stroissnigg H, Xu M, Ling YY, Melos KI, Pirtskhalava T, Inman CL, McGuckian C (2018) Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 36:18–28
Yu B, Jiang Y, Zhang B, Yang H, Ma T (2018) Resveratrol dimer trans-ε-viniferin prevents rotaviral diarrhea in mice by inhibition of the intestinal calcium-activated chloride channel. Pharm Res 129:453–461
Yuan Z-P, Chen L-J, Fan L-Y, Tang M-H, Yang G-L, Yang H-S, Du X-B, Wang G-Q, Yao W-X, Zhao Q-M et al (2006) Liposomal quercetin efficiently suppresses growth of solid tumors in murine models. Clin Cancer Res 12:3193–3199
Yuan H, Wang Z, Li L, Zhang H, Modi H, Horne D, Bhatia R, Chen W (2012) Activation of stress response gene SIRT1 by BCR-ABL promotes leukemogenesis. Blood 119(8):1904–1914
Zahedi M, Ghiasvand R, Feizi A, Asgari G, Darvish L (2013) Does quercetin improve cardiovascular risk factors and inflammatory biomarkers in women with type 2 diabetes: a double-blind randomized controlled clinical trial. Int J Prev Med 4:777–785
Zhao H, Ma T, Fan B, Yang L, Han C, Luo J, Kong L (2016) Protective effect of trans-δ-viniferin against high glucose-induced oxidative stress in human umbilical vein endothelial cells through the SIRT1 pathway. Free Radic Res 50:68–83
Zhu Y, Doornebal EJ, Pirtskhalava T, Giorgadze N, Wentworth M, Fuhrmann-Stroissnigg H, Niedernhofer LJ, Robbins PD, Tchkonia T, Kirkland JL (2017) New agents that target senescent cells: the flavone, fisetin, and the BCL-X(L) inhibitors, A1331852 and A1155463. Aging (Albany NY) 9:955–963
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Kori, S.K. et al. (2023). Sirtuin Modulator: Design, Synthesis, and Biological Evaluation. In: Sharma, A., Modi, G.P. (eds) Natural Product-based Synthetic Drug Molecules in Alzheimer's Disease. Springer, Singapore. https://doi.org/10.1007/978-981-99-6038-5_15
Download citation
DOI: https://doi.org/10.1007/978-981-99-6038-5_15
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-99-6037-8
Online ISBN: 978-981-99-6038-5
eBook Packages: MedicineMedicine (R0)