
Abstract
Interstitial lung diseases (ILD) are a heterogeneous group of parenchymal pulmonary disorders that result from varying degrees of inflammation or fibrosis in the lung interstitium, that is, the septum between alveoli and the blood capillaries. The clinical presentation of ILD is complex and the diagnosis is often challenging. Therefore, the need to establish disease-specific molecular fingerprints to better understand the underlying pathogenesis is well realized. “Omics” is a powerful tool that collectively depicts and quantifies biomolecules, including key genomic, transcriptomic, proteomic, and metabolomic signatures, and discloses their dynamic interactions within an organism. Metabolomics is a branch of omics that identifies numerous small molecules from body fluids or tissues and holds immense potential for early diagnosis, therapeutic monitoring, and understanding of disease pathophysiology. Another evolving popular omic field is transcriptomics, which identifies key genetic regulations and posttranscriptional modifications triggering diseases. The findings of 17 original articles on metabolomics and 63 on transcriptomics of ILD reported are discussed. Though each omic dataset provides valuable information, integrating these platforms offers an overall snapshot of the interplay between the candidate molecules and genes, thereby paving the path for highlighting the genotype-to-phenotype relationship and assisting in making more effective treatment decisions for complex diseases.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Interstitial lung disease (ILD) is an umbrella term that encompasses about 300 parenchymal pulmonary disorders, resulting from varying degrees of inflammation or fibrosis in the lung interstitium, that is, the septum between alveoli and the blood capillaries. A schematic diagram of a healthy vs. ILD lung is shown in Fig. 1.

Healthy lung vs. interstitial lung disease (created using BioRender.com)
Numerous studies across the globe have reported the incidence, prevalence, and relative frequency of ILD. The annual incidence of ILD varies between 1 and 31.5 per 100,000 [1]. The incidence and prevalence vary among populations, likely due to differences in study design, data collection, and incorrect recognition of the disease subtypes [2]. ILD is classified based on clinical, radiological, and histopathological features. The latest classification focuses on recognizing the underlying etiology since this often impacts both prognostication and management decisions. ILD mainly consists of disorders of known causes [collagen vascular disease, hypersensitivity pneumonitis (HP)] as well as disorders of unknown/idiopathic causes [idiopathic interstitial pneumonia (IIP), sarcoidosis] [3]. ILD registries comprising patients from Western countries suggest that idiopathic pulmonary fibrosis (IPF) and sarcoidosis are the most common phenotypes. However, the ILD registry of India indicates HP to be the most common, which accounts for nearly 50% of all ILD cases [4].
The emerging field of metabolomics, in which many small molecules from body fluids or tissues can be identified, holds immense potential for early diagnosis, therapeutic monitoring, and understanding of disease pathophysiology. Over the past two decades, nuclear magnetic resonance (NMR) spectroscopy and gas chromatography (GC)/liquid chromatography (LC) coupled with mass spectrometry (MS) combined with chemometric analysis have emerged as principal analytical techniques for use in metabolomics. Several biofluids including cerebrospinal fluid (CSF), bronchoalveolar lavage fluid (BALF), bile, seminal fluid, amniotic fluid, synovial fluid, gut aspirate, serum/plasma, saliva, exhaled breath condensate (EBC), and urine contain hundreds to thousands of detectable metabolites which have been extensively studied so far [5]. More recently, metabolic profiling of intact tissue and extracts of lipid and aqueous metabolites are gaining increasing importance for detection of biomarkers.
Another branch of popular omic science is transcriptomics, which provides detailed information about gene regulation in normal and diseased conditions. Two key contemporary techniques commonly used for transcriptomic analysis are hybridization-based microarray techniques, which quantify a set of predetermined sequences, and next-generation sequencing (NGS), which uses high-throughput sequencing to capture all sequences [6]. In the last decade, these two transcriptomic approaches have been utilized most widely to understand the underlying disease pathogenesis at both molecular and genetic levels and also for molecular diagnosis and clinical therapy. Human biofluids including amniotic fluid, aqueous humor, ascites, bile, BALF, breast milk, CSF, colostrum, gastric fluid, pancreatic cyst fluid, plasma, saliva, seminal fluid, serum, sputum, stool, synovial fluid, sweat, tears, urine, and tissues are widely used for transcriptomic studies to identify biomarkers of several diseases [7,8,9].
2 Types of ILD
ILD, as mentioned earlier, refers to a group of lung diseases ranging from occasional self-limited inflammatory processes to severe debilitating fibrosis of the lung parenchyma. There are varied causes of ILD, which generally result from a range of environmental, occupational, recreational, or drug-related exposures or could arise from the various systemic autoimmune or connective tissue diseases (CTD) [10]. Classification of different types of ILD is shown in Fig. 2. A few of the common ILD subtypes are described in the present section.

Classification of different types of interstitial lung disease (Cottin et al. 2018) [3]. ILD interstitial lung disease, IIP idiopathic interstitial pneumonia, IPF idiopathic pulmonary fibrosis, iNSIP idiopathic nonspecific interstitial pneumonia, RB-ILD respiratory bronchiolitis-associated ILD, COP cryptogenic organizing pneumonia, RA-ILD rheumatoid arthritis-associated ILD, SSC-ILD systemic sclerosis-associated ILD, HP hypersensitivity pneumonitis
2.1 Idiopathic Interstitial Pneumonia (IIP)
The cause of IIP, comprising of diffuse parenchymal lung diseases, remains unknown. IIP is characterized by varying degrees of inflammation and fibrosis in the lung interstitium. These characteristics split IIP into eight clinicopathologic entities, that is, IPF, nonspecific interstitial pneumonia (NSIP), cryptogenic organizing pneumonia (COP), acute interstitial pneumonia, respiratory bronchiolitis-associated interstitial lung disease, desquamative interstitial pneumonia, lymphoid interstitial pneumonia, and idiopathic pleuroparenchymal fibroelastosis [11]. Among all IIPs, IPF is the most common phenotype characterized by fibroblastic foci and the presence of inflammation and honeycombing in the lung parenchyma.
2.2 Autoimmune ILD
Autoimmune ILD is caused specifically by autoimmune disorders, which involve the body’s immune system attacking the lungs. This ILD group gradually develops and emerges over a long period of time. The symptoms of this ILD include difficulty in breathing, dry cough, and shortness of breath. Connective tissue disease-related ILD (CTD-ILD), rheumatoid arthritis-associated ILD (RA-ILD), and systemic sclerosis-associated ILD (SSC-ILD) are the common types of autoimmune ILD [12, 13].
2.3 Hypersensitivity Pneumonitis (HP)
HP, also referred to as extrinsic alveolar alveolitis, is a complex subtype of ILD arising from repeated exposure to certain antigens, most commonly avian, microbial (especially molds), or chemical. HP is the third most prevalent ILD after IPF and CTD-ILD. The inhaled antigen triggers type III and type IV hypersensitivity reactions, which causes the damage of alveolar epithelial cells. An impaired repair mechanism may result in fibroblast activation, deposition of collagen by the destruction of extracellular matrix, and parenchymal architecture [14]. The major forms of HP are acute, subacute, and chronic. Acute and subacute HP is mainly characterized by influenza-like symptoms, such as cough, dyspnea, and fever, developing after 2–9 h of antigen exposure. The chronic form of HP arises from repetitive, low-level exposure to the causative agent. Still, the identity of the causative antigen may remain unknown in more than half the cases. Chronic HP patients slowly develop fibrosis in the lung interstitium and are associated with a significantly high mortality rate [15].
2.4 Sarcoidosis
Sarcoidosis is a systemic, inflammatory disease resulting from an unknown origin. Chronic immune response to an idiopathic antigen may lead to sarcoidosis in genetically susceptible subjects. Almost 90% of sarcoidosis patients have pulmonary involvement. Dry cough, chest tightness, chronic dyspnea on exertion, shortness of breath, wheezing, hypoxemia, and decline in pulmonary function are the common signs and symptoms of sarcoidosis. Near about 20% of sarcoidosis patients develop pulmonary fibrosis, that is, stage IV sarcoidosis which is associated with high mortality [16].
2.5 Occupational and Environmental Exposure-Related Other ILDs
Long-term exposure to occupational or environmental antigens could cause certain types of ILD via pulmonary and systemic inflammation and oxidative stress. Many different types of mineral dust, such as silica, asbestos, beryllium, coal mine dust, metal, and organic dust, including mold spores, can also affect the lung airways, either by a direct wound or through reactive oxygen molecules. Common conditions include asbestosis, which is associated with asbestos fibers, and silicosis, which is caused by free crystalline silicon dioxide or silica particles [17, 18].
3 Metabolomics: An Emerging Tool in Clinical Research
Metabolomics, one of the newest omics science, is an evolving field in clinical research. Metabolomics is the scientific study of metabolic fingerprints that all cellular processes leave behind in a biological sample [19]. It provides a snapshot of the metabolic state of an individual at a given point in time. On the other hand, “metabonomics,” a term first coined by Jeremy Nicholson, refers to “the quantitative measurement of the dynamic multiparametric metabolic response of living systems to pathophysiological stimuli or genetic modification” [20, 21]. The terms “metabolomics” and “metabonomics” are often used interchangeably. Among the different omic approaches, metabolomics is considered to modulate best and depict the molecular phenotype of health and disease [22]. Thus, it is increasingly becoming a useful and powerful tool for the investigation of complex diseases with unclear etiology, enabling the discovery of novel biomarkers, which, in turn, aid in the prevention and early diagnosis of diseases. Metabolomics can also monitor the effect of pharmacotherapy, allowing clinicians to choose the best treatment option for patients suffering from potentially devastating disorders. The two analytical techniques popularly used have their own advantages and disadvantages. While mass spectrometry can analyze a wider range of metabolites and is more sensitive, it results in the destruction of the analyzed sample. NMR spectroscopy, on the other hand, is highly reproducible and does not destroy the sample; however, sensitivity is limited [23, 24]. Over the years, application of metabolomics in diseases is rapidly growing, and recent studies exploring the metabolic profiles of various human samples, including but not limited to plasma, serum, urine, BALF, exhaled breath, saliva, and tissues, bring this technology closer to the patients’ bedside, thereby enhancing its clinical utility. A schematic representation of the metabolomic workflow is shown in Fig. 3.

Schematic representation of the metabolomic workflow (created using BioRender.com)
3.1 Metabolomics in ILD
Several attempts have been made to understand the metabolic status of ILD patients and identify prospective biomarkers in lung tissues and various body fluids using a nontargeted and targeted metabolomic approach. Studies utilizing the metabolomic approaches to investigate ILD are summarized in Table 1.
4 Transcriptomics: A Promising Omic Approach
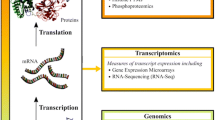
Transcriptome analysis utilizes high-throughput methods to study the complete set of RNA transcripts produced by the genome under specific circumstances. It covers all types of transcripts, including mRNAs, miRNAs, and different types of long noncoding RNAs (lncRNAs). Transcriptome analysis gives us an overview of all genes’ expression levels and enables us to understand the physiology of the cell. More precisely, it also discloses key regulations of biological processes triggering diseases. While microarrays are generally less complex and easier to use than NGS, the latter is associated with greater flexibility, high throughput, and high discovery potential. A schematic representation of the transcriptomic workflow is shown in Fig. 4.

Schematic representation of the transcriptomic workflow (created using BioRender.com)
4.1 Transcriptomics in ILD
Various studies have been performed to understand the transcriptomic signatures of ILD patients and identify prospective biomarkers in lung tissues and various biofluids using NGS and microarray techniques. Despite increasing interest and effort invested by clinicians and scientists during the last decade, the etiology of ILD remains elusive and controversial.
As mentioned earlier, IPF is characterized by remodeling or scarring of the airway epithelium. The activated extracellular matrix (ECM)-produced myofibroblasts play a key role in the process of fibrotic tissue remodeling. Advances in transcriptomic techniques have allowed high-throughput analysis and discovery of gene deregulation in IPF. Several studies using lung tissues have reported that IPF is associated with variances in the expression levels of genes such as CCL8 [42], CXCL14 [43], CXCL4 and CXCL12 [44], NOTCH2 [45], TGF-β1 and RhoA kinase [46], REVERBα [47], IL-1β [48], FLIL33 and POU2AF1 [49], FOXL1 [50], COL6A3, and POSTN [51]. Microarray analysis of peripheral blood by Abe et al. (2020) has shown dysregulated PDGF B, VEGF B, and FGF 2. The authors confirmed their findings using ELISA, western blot, immunofluorescence, and 3H thymine uptake assays. Xia and co-workers (2021) recently utilized weighted gene co-expression network analysis (WGCNA) of BALF samples and could associate four genes, TLR2, CCR2, HTRA1, and SFN, with disease prognosis.
Pathway enrichment analysis based on dysregulated genes highlights the associated biological pathways, molecular functions, and cellular components. This method identifies all biological pathways enriched in a gene list more than would be expected by chance. The KEGG pathway tool maps the pathways associated with dysregulated genes in a specific disease. Pathway enrichment analysis of IPF patients revealed that the differentially expressed genes were majorly associated with myofibroblast differentiation and massive ECM deposition. The transcriptomic signatures of fibroblasts suggest that characterization of lung proteins, specifically lung fibrotic ECM, helps determine its composition and define targetable molecules for advanced stages of fibrosis. Boesch and his team (2020) isolated fibrosis-specific mesenchymal stem cell-like cells from lung tissue of IPF subjects and observed that the differentially expressed genes were enriched with hypoxia, fibrosis, and bacterial colonization factors which are the typical hallmarks of pulmonary fibrosis. They found that the cells isolated from IPF patients express genes associated with activating canonical TGF-β, HIPPO/YAP, PI3K/AKT, p53, and WNT signaling cascades, which are activated in an integrated network. Another interesting study by Hsu and co-authors (2011) suggested that IPF lungs enriched in fibrosis-related genes, insulin-like growth factor signaling, and caveolin-mediated endocytosis. This microarray analysis also highlighted the common molecular signatures between lung tissue and fibroblasts of these patients.
Like IPF, HP is associated with matrix remodeling and formation of fibrosis. There exist only two studies where transcriptomics has been used to explore genetic alterations in HP. Sarcoidosis, as mentioned earlier, is an immune-mediated multisystem disease characterized by the formation of non-caseating granuloma. Multiple pro-inflammatory signaling pathways, including IFN-γ/STAT-1, IL-6/STAT-3, and NF-κB, have been implicated in mediating macrophage activation and granuloma formation in sarcoidosis. Utilizing RT-PCR, Christophi et al. (2014) have demonstrated that IL-6, COX-2, MCP-1, IFN-γ, T-bet, IRF-1, Nox2, IL-33, and eotaxin-1 hold potential for differential diagnosis between sarcoidosis, suture, and fungal granulomas. In another recent study, Lepzien and co-workers (2021) have shown that allogeneic T cell proliferation increased after coculture with monocytes and dendritic cells of sarcoidosis patients. The authors also found that mainly T-bet and RORγt-expressing T cells produce IFN-γ. Monocytes from sarcoidosis patients can activate and polarize T cells towards Th1 and Th17.1 cells. In a comparative study between sarcoidosis and IPF, cluster analysis of BALF cells showed elevated mRNA expression of genes associated with ribosome biogenesis in sarcoidosis patients. Clusters formed by genes with altered mRNA expression in patients with IPF could be implicated in cell migration and adhesion processes, metalloproteinase expression, and negative regulation of cell proliferation. Various studies highlighting the transcriptome fingerprints and associated pathways in different ILD subtypes are summarized in Table 2.
5 Integration of Metabolomic and Transcriptomic Fingerprints
As mentioned earlier, clinical metabolomics is primarily used to identify low molecular weight compounds differentially expressed in a particular disease. In contrast, transcriptomics identifies the complete set of dysregulated RNAs associated with a disease. Integration of metabolomic and transcriptomic signatures has emerged as a popular application-driven method for investigating underlying disease mechanisms, monitoring disease progression, and identifying potential biomarkers [101,102,102]. The omic tools highlight alterations in genotype and phenotype and provide complementary information about genetic alterations, protein synthesis, metabolism, and cellular function. Pathways and network connections further reflect the association between key metabolites and candidate transcripts.
Biological pathway networks reveal hidden patterns in unstructured data by converting them into logically structured and visually evident representations, with nodes representing genes and metabolites and edges suggesting relationships between nodes and clusters with similar chemical activities. VANTED [103], VisAnt [104], Impala [105], and Metscape2 [106] are some of the network-based visualization tools that interface with public databases. In addition, Arena3D allows users to envision three-dimensional biological networks [107]. Interactive editing is frequently performed for small biological networks. However, for major networks, automated layout web tools, that is, Cytoscape [108], NAViGaTOR [109], and Cerebral [110], are more convenient. Alternatively, pathway visualization tools highlight the biochemical activities and different interactive pathways in experimental datasets. Pathguide offers an overview of nearly 190 web-usable network databases and biological pathways [110]. Arakawa and his team have developed a pathway visualization tool for KEGG-based pathways. Users can capture systematic features of biological activity by visualizing pathways at the level of different omic data representations [111]. Paintomics, another software program, analyzes the expression of genes and concentration of metabolite data and displays it on KEGG pathway maps [112]. ProMeTra can display dynamic data and accept annotated images in SVG format [113]. In plants, KaPPa-View and MapMan show the number of metabolites and transcripts for preset route blocks [114, 115]. Other tools like MAYDAY enable viewing expression data in a genomic context with any metadata [116], and PaVESy creates personalized pathways using proteins and metabolites provided by the user [117]. A schematic representation of integrated metabolomic and transcriptomic workflow is shown in Fig. 5.

Schematic representation of integrated metabolomic and transcriptomi data (created using BioRender.com, STITCH database, and Graph pad prism version 7)
In a recent study, our group used NMR coupled with chemometric analysis to identify the unique metabolic fingerprints in BALF of HP subjects. A total of six metabolites were found to be significantly altered in HP compared to non-HP controls [35]. Next, we considered NGS data of lung tissues from HP patients and controls, reported in the NCBI-GEO public database by Furusawa et al., and performed bioinformatic analysis. A total of 555 genes were dysregulated (373 upregulated and 182 downregulated) in HP cases. An interaction network between the six candidate metabolites and most significantly altered genes (five upregulated and five downregulated) was established utilizing the Search Tool for Interactions of Chemicals (STITCH) database. The metabolite-gene interaction by STITCH demonstrated 19 nodes connected via 16 edges. The clustering coefficient of the network was found to be 0.768 (protein-protein interaction enrichment p-value: 0.0838). Overall pathway overrepresentation analysis was performed by integrating the candidate metabolites and transcripts utilizing IMPaLA version 12. Glycolysis and phosphatidylinositol 3-kinase-protein kinase B (PI3K-AKT) signaling pathways emerged to be most significantly associated with the pathogenesis of HP. These findings are encouraging, since association of these pathways in chronic HP is well established. Since glycolysis is the key energy driving force for myofibroblast differentiation and formation of fibrosis, perturbation of glycolysis seems likely [118]. The involvement of PI3K-AKT pathway is also evidenced in bleomycin-induced pulmonary fibrosis. It is hypothesized that PI3K-AKT plays a central role in fibrosis development [119, 120]. A novel insight into the pathogenesis of HP is envisioned by integrating the findings of the two omic platforms.
6 Challenges and Future Scope
Most of the omic-driven studies conducted on ILD so far have included a small number of patients, which is quite understandable considering that ILD is a severe condition with a short average life expectancy. Power and sample size estimation, however difficult, would be useful because the low sample size is connected with statistical errors and risks of overfitting and misleading calculations. Since omic output is highly dynamic, clinical variables such as physiological status, age, gender, and treatment may influence the findings. Hence, baseline characteristics of recruited ILD subjects need to be closely matched. Lack of a rigorous subject selection approach could also result in discovering markers that are not exclusive to ILD subtypes. It is observed that only a few groups have included healthy controls in their omic-driven research on ILD. Also, nonuniformity in including smokers and nonsmokers is frequently observed while comparing disease populations with healthy controls. This makes unbiased comparisons and conclusions impossible. A few groups were also unable to validate ILD candidate markers, which is crucial for biomarker identification. In fact, one of the main reasons why most of the omic-based disease markers identified so far have not made it to clinical practice is due to a lack of adequate validation trials. Another observation that warrants attention while using omics is that different research groups identify different biomarkers in the same biofluid for a particular disease. This is not surprising given the fact that factors such as sampling methods, sample collection, handling and preparation, instrumentation, and data mining protocols tend to vary from one setup to another. To generate robust and reproducible data, the practices and procedures should be standardized and rigorously followed across all clinics and research laboratories. Metabolic flux analysis is crucial to obtain insight into dysregulated cellular metabolism caused by disease perturbations. It is expected that stratifying ILD patients based on disease severity and subtypes will significantly improve metabolome and transcriptome coverage. Assessment of sensitivity, specificity, and clinical relevance of the differentially expressed molecules is also recommended. For a reliable and unbiased diagnosis of this severe pulmonary disease, large-scale, well-designed, multicentric clinical studies and recruitment of suitable controls are recommended.
The ultimate focus of metabolomic and transcriptomic data integration is identifying key metabolic and genetic factors that contribute significantly to disease etiology. Integrated omics is more than a collection of tools; it is a comprehensive paradigm for interpreting multi-omic datasets in a way that can provide new insights into basic biology, as well as health and disease. Machine learning approaches for multi-omic data analyses is an emerging trend for exploring molecular pathways in detail and drawing a holistic representation of a given phenotype using all biological and clinical information of an individual. One of the major advantages is incorporating biological domain knowledge into the machine learning models as inductive biases to reduce data overfitting. Additionally, as omic tools evolve, they need to be user-friendly, interoperable, and effective for computationally intensive analyses. Machine learning methods offer novel techniques to integrate such omic datasets. With the emerging precision medicine initiative, where disease prevention and management take into account the variability in genes, environment, and lifestyle of each individual in contrast to the conventional one-size-fits-all approach, integration of clinical data with the patients’ metabolome and genetic makeup will provide an in-depth understanding of disease pathophysiology and facilitate designing of targeted therapies for individuals, thereby revolutionizing precision medicine-based decision-making in the clinic.
References
Rivera-Ortega P, Molina-Molina M. Interstitial lung diseases in developing countries. Ann Glob Health. 2019;85(1):4.
ERS. Interstitial lung diseases. In: ERS European lung White-book. Chapter 22. ERS; 2015. http://www.erswhitebook.org/chapters/interstitial-lung-diseases/. Accessed 15 Sep 2016.
Cottin V, Hirani NA, Hotchkin DL, Nambiar AM, Ogura T, Otaola M, Skowasch D, Park JS, Poonyagariyagorn HK, Wuyts W, Wells AU. Presentation, diagnosis and clinical course of the spectrum of progressive-fibrosing interstitial lung diseases. Eur Respir Rev. 2018;27(150):180076.
Singh S, Collins BF, Sharma BB, Joshi JM, Talwar D, Katiyar S, Singh N, Ho L, Samaria JK, Bhattacharya P, Gupta R. Interstitial lung disease in India. Results of a prospective registry. Am J Respir Crit Care Med. 2017;195(6):801–13.
Gowda GN, Zhang S, Gu H, Asiago V, Shanaiah N, Raftery D. Metabolomics-based methods for early disease diagnostics. Expert Rev Mol Diagn. 2008;8(5):617–33.
Hulstaert E, Morlion A, Cobos FA, Verniers K, Nuytens J, Eynde EV, Yigit N, Anckaert J, Geerts A, Hindryckx P, Jacques P. Charting extracellular transcriptomes in the human biofluid RNA atlas. Cell Rep. 2020;33(13):108552.
El-Mogy M, Lam B, Haj-Ahmad TA, McGowan S, Yu D, Nosal L, Rghei N, Roberts P, Haj-Ahmad Y. Diversity and signature of small RNA in different bodily fluids using next generation sequencing. BMC Genomics. 2018;19(1):1–24.
Ferrero G, Cordero F, Tarallo S, Arigoni M, Riccardo F, Gallo G, Ronco G, Allasia M, Kulkarni N, Matullo G, Vineis P. Small non-coding RNA profiling in human biofluids and surrogate tissues from healthy individuals: description of the diverse and most represented species. Oncotarget. 2018;9(3):3097.
Godoy PM, Bhakta NR, Barczak AJ, Cakmak H, Fisher S, MacKenzie TC, Patel T, Price RW, Smith JF, Woodruff PG, Erle DJ. Large differences in small RNA composition between human biofluids. Cell Rep. 2018;25(5):1346–58.
Guler SA, Corte TJ. Interstitial lung disease in 2020: a history of progress. Clin Chest Med. 2021;42(2):229–39.
Oliveira DS, Araújo JDA, Paiva AFL, Ikari ES, Chate RC, Nomura CH. Idiopathic interstitial pneumonias: review of the latest American Thoracic Society/European Respiratory Society classification. Radiol Bras. 2018;51(5):321–7.
Antoniou KM, Margaritopoulos G, Economidou F, Siafakas NM. Pivotal clinical dilemmas in collagen vascular diseases associated with interstitial lung involvement. Eur Respir J. 2009;33(4):882–96.
Mathai SC, Danoff SK. Management of interstitial lung disease associated with connective tissue disease. BMJ. 2016;352:h6819.
Takemura T, Akashi T, Ohtani Y, Inase N, Yoshizawa Y. Pathology of hypersensitivity pneumonitis. Curr Opin Pulm Med. 2008;14(5):440–54.
Sforza GGR, Marinou A. Hypersensitivity pneumonitis: a complex lung disease. Clin Mol Allergy. 2017;15(1):1–8.
Nardi A, Brillet PY, Letoumelin P, Girard F, Brauner M, Uzunhan Y, Naccache JM, Valeyre D, Nunes H. Stage IV sarcoidosis: comparison of survival with the general population and causes of death. Eur Respir J. 2011;38(6):1368–73.
Gulati M, Redlich CA. Asbestosis and environmental causes of usual interstitial pneumonia. Curr Opin Pulm Med. 2015;21(2):193.
Leung CC, Yu ITS, Chen W. Silicosis. Lancet. 2012;379(9830):2008–18.
Oliver SG, Winson MK, Kell DB, Baganz F. Systematic functional analysis of the yeast genome. Trends Biotechnol. 1998;16(9):373–8.
Nicholson JK, Lindon JC, Holmes E. ‘Metabonomics’: understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica. 1999;29(11):1181–9.
Faber JH, Malmodin D, Toft H, Maher AD, Crockford D, Holmes E, Nicholson JK, Dumas ME, Baunsgaard D. Metabonomics in diabetes research. J Diabetes Sci Technol. 2007;1(4):549–57.
Guijas C, Montenegro-Burke JR, Warth B, Spilker ME, Siuzdak G. Metabolomics activity screening for identifying metabolites that modulate phenotype. Nat Biotechnol. 2018;36(4):316–20.
Li S, Todor A, Luo R. Blood transcriptomics and metabolomics for personalized medicine. Comput Struct Biotechnol J. 2016;14:1–7.
Schreier C, Kremer W, Huber F, Neumann S, Pagel P, Lienemann K, Pestel S. Reproducibility of NMR analysis of urine samples: impact of sample preparation, storage conditions, and animal health status. Biomed Res Int. 2013;2013:878374.
Kottmann RM, Kulkarni AA, Smolnycki KA, Lyda E, Dahanayake T, Salibi R, Honnons S, Jones C, Isern NG, Hu JZ, Nathan SD. Lactic acid is elevated in idiopathic pulmonary fibrosis and induces myofibroblast differentiation via pH-dependent activation of transforming growth factor-β. Am J Respir Crit Care Med. 2012;186(8):740–51.
Kang YP, Lee SB, Lee JM, Kim HM, Hong JY, Lee WJ, Choi CW, Shin HK, Kim DJ, Koh ES, Park CS. Metabolic profiling regarding pathogenesis of idiopathic pulmonary fibrosis. J Proteome Res. 2016;15(5):1717–24.
Kim HS, Yoo HJ, Lee KM, Song HE, Kim SJ, Lee JO, Hwang JJ, Song JWW. Stearic acid attenuates profibrotic signalling in idiopathic pulmonary fibrosis. Respirology. 2021;26(3):255–63.
Zhao YD, Yin L, Archer S, Lu C, Zhao G, Yao Y, Wu L, Hsin M, Waddell TK, Keshavjee S, Granton J. Metabolic heterogeneity of idiopathic pulmonary fibrosis: a metabolomic study. BMJ Open Respir Res. 2017;4(1):e000183.
Rindlisbacher B, Strebel C, Guler S, Kollár A, Geiser T, Fiedler GM, Leichtle AB, Bovet C, Funke-Chambour M. Exhaled breath condensate as a potential biomarker tool for idiopathic pulmonary fibrosis—a pilot study. J Breath Res. 2017;12(1):016003.
Gaugg MT, Engler A, Bregy L, Nussbaumer-Ochsner Y, Eiffert L, Bruderer T, Zenobi R, Sinues P, Kohler M. Molecular breath analysis supports altered amino acid metabolism in idiopathic pulmonary fibrosis. Respirology. 2019;24(5):437–44.
Yan F, Wen Z, Wang R, Luo W, Du Y, Wang W, Chen X. Identification of the lipid biomarkers from plasma in idiopathic pulmonary fibrosis by lipidomics. BMC Pulm Med. 2017;17(1):1–12.
Nambiar S, Tan DBA, Clynick B, Bong SH, Rawlinson C, Gummer J, Corte TJ, Glaspole I, Moodley YPP, Trengove R. Untargeted metabolomics of human plasma reveal lipid markers unique to chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Proteom Clin Appl. 2021;15(2–3):e2000039.
Nambiar S, Clynick B, How BS, King A, Walters EH, Goh NS, Corte TJ, Trengove R, Tan D, Moodley Y. There is detectable variation in the lipidomic profile between stable and progressive patients with idiopathic pulmonary fibrosis (IPF). Respir Res. 2021;22(1):1–8.
Rindlisbacher B, Schmid C, Geiser T, Bovet C, Funke-Chambour M. Serum metabolic profiling identified a distinct metabolic signature in patients with idiopathic pulmonary fibrosis–a potential biomarker role for LysoPC. Respir Res. 2018;19(1):1–12.
Dasgupta S, Ghosh N, Choudhury P, Joshi M, Chowdhury SR, Bhattacharyya P, Chaudhury K. NMR metabolomic and microarray-based transcriptomic data integration identifies unique molecular signatures of hypersensitivity pneumonitis. Mol Omics. 2022;18(2):101–11.
Geamanu A, Gupta SV, Bauerfeld C, Samavati L. Metabolomics connects aberrant bioenergetic, transmethylation, and gut microbiota in sarcoidosis. Metabolomics. 2016;12(2):35.
Banoei MM, Iupe I, Bazaz RD, Campos M, Vogel HJ, Winston BW, Mirsaeidi M. Metabolomic and metallomic profile differences between veterans and civilians with pulmonary sarcoidosis. Sci Rep. 2019;9(1):1–12.
Furukawa H, Oka S, Shimada K, Hashimoto A, Komiya A, Matsui T, Fukui N, Tohma S. Serum metabolomic profiles of rheumatoid arthritis patients with acute-onset diffuse interstitial lung disease. Biomark Insights. 2019;14:1177271919870472.
Furukawa H, Oka S, Shimada K, Okamoto A, Hashimoto A, Komiya A, Saisho K, Yoshikawa N, Katayama M, Matsui T, Fukui N. Serum metabolomic profiling in rheumatoid arthritis patients with interstitial lung disease: a case–control study. Front Med. 2020;7:599794.
Xue C, Wu N, Fan Y, Ma J, Ye Q. Distinct metabolic features in the plasma of patients with silicosis and dust-exposed workers in China: a case–control study. BMC Pulm Med. 2021;21(1):1–10.
Sun Y, Gu X, Zhang E, Park MA, Pereira AM, Wang S, Morrison T, Li C, Blenis J, Gerbaudo VH, Henske EP. Estradiol promotes pentose phosphate pathway addiction and cell survival via reactivation of Akt in mTORC1 hyperactive cells. Cell Death Dis. 2014;5(5):e1231.
Lee JU, Cheong HS, Shim EY, Bae DJ, Chang HS, Uh ST, Kim YH, Park JS, Lee B, Shin HD, Park CS. Gene profile of fibroblasts identify relation of CCL8 with idiopathic pulmonary fibrosis. Respir Res. 2017;18(1):1–12.
Boesch M, Baty F, Brutsche MH, Tamm M, Roux J, Knudsen L, Gazdhar A, Geiser T, Khan P, Hostettler KE. Transcriptomic profiling reveals disease-specific characteristics of epithelial cells in idiopathic pulmonary fibrosis. Respir Res. 2020;21(1):1–9.
Guillotin D, Taylor AR, Platé M, Mercer PF, Edwards LM, Haggart R, Miele G, McAnulty RJ, Maher TM, Hynds RE, Jamal-Hanjani M. Transcriptome analysis of IPF fibroblastic foci identifies key pathways involved in fibrogenesis. Thorax. 2021;76(1):73–82.
Luzina IG, Salcedo MV, Rojas-Peña ML, Wyman AE, Galvin JR, Sachdeva A, Clerman A, Kim J, Franks TJ, Britt EJ, Hasday JD. Transcriptomic evidence of immune activation in macroscopically normal-appearing and scarred lung tissues in idiopathic pulmonary fibrosis. Cell Immunol. 2018;325:1–13.
Checa M, Hagood JS, Velazquez-Cruz R, Ruiz V, Garcia-De-Alba C, Rangel-Escareño C, Urrea F, Becerril C, Montaño M, García-Trejo S, Lira JC. Cigarette smoke enhances the expression of profibrotic molecules in alveolar epithelial cells. PLoS One. 2016;11(3):e0150383.
Carraro G, Mulay A, Yao C, Mizuno T, Konda B, Petrov M, Lafkas D, Arron JR, Hogaboam CM, Chen P, Jiang D. Single-cell reconstruction of human basal cell diversity in normal and idiopathic pulmonary fibrosis lungs. Am J Respir Crit Care Med. 2020;202(11):1540–50.
Hsu E, Shi H, Jordan RM, Lyons-Weiler J, Pilewski JM, Feghali-Bostwick CA. Lung tissues in patients with systemic sclerosis have gene expression patterns unique to pulmonary fibrosis and pulmonary hypertension. Arthritis Rheum. 2011;63(3):783–94.
Kwapiszewska G, Gungl A, Wilhelm J, Marsh LM, Puthenparampil HT, Sinn K, Didiasova M, Klepetko W, Kosanovic D, Schermuly RT, Wujak L. Transcriptome profiling reveals the complexity of pirfenidone effects in idiopathic pulmonary fibrosis. Eur Respir J. 2018;52(5):1800564.
Lee JU, Son JH, Shim EY, Cheong HS, Shin SW, Shin HD, Baek AR, Ryu S, Park CS, Chang HS, Park JS. Global DNA methylation pattern of fibroblasts in idiopathic pulmonary fibrosis. DNA Cell Biol. 2019;38(9):905–14.
Nance T, Smith KS, Anaya V, Richardson R, Ho L, Pala M, Mostafavi S, Battle A, Feghali-Bostwick C, Rosen G, Montgomery SB. Transcriptome analysis reveals differential splicing events in IPF lung tissue. PLoS One. 2014;9(3):e92111.
Tan J, Tedrow JR, Nouraie M, Dutta JA, Miller DT, Li X, Yu S, Chu Y, Juan-Guardela B, Kaminski N, Ramani K. Loss of Twist1 in the mesenchymal compartment promotes increased fibrosis in experimental lung injury by enhanced expression of CXCL12. J Immunol. 2017;198(6):2269–85.
Rodriguez LR, Emblom-Callahan M, Chhina M, Bui S, Aljeburry B, Tran LH, Novak R, Lemma M, Nathan SD, Grant GM. Global gene expression analysis in an in vitro fibroblast model of idiopathic pulmonary fibrosis reveals potential role for CXCL14/CXCR4. Sci Rep. 2018;8(1):1–12.
Tzouvelekis A, Ntolios P, Karameris A, Vilaras G, Boglou P, Koulelidis A, Archontogeorgis K, Kaltsas K, Zacharis G, Sarikloglou E, Steiropoulos P. Increased expression of epidermal growth factor receptor (EGF-R) in patients with different forms of lung fibrosis. Biomed Res Int. 2013;2013:654354.
Vukmirovic M, Herazo-Maya JD, Blackmon J, Skodric-Trifunovic V, Jovanovic D, Pavlovic S, Stojsic J, Zeljkovic V, Yan X, Homer R, Stefanovic B. Identification and validation of differentially expressed transcripts by RNA-sequencing of formalin-fixed, paraffin-embedded (FFPE) lung tissue from patients with idiopathic pulmonary fibrosis. BMC Pulm Med. 2017;17(1):1–12.
Senavirathna LK, Huang C, Pushparaj S, Xu D, Liu L. Hypoxia and transforming growth factor β1 regulation of long non-coding RNA transcriptomes in human pulmonary fibroblasts. Physiol Rep. 2020;8(1):e14343.
Morse C, Tabib T, Sembrat J, Buschur KL, Bittar HT, Valenzi E, Jiang Y, Kass DJ, Gibson K, Chen W, Mora A. Proliferating SPP1/MERTK-expressing macrophages in idiopathic pulmonary fibrosis. Eur Respir J. 2019;54(2):1802441.
Miyashita N, Horie M, Suzuki HI, Saito M, Mikami Y, Okuda K, Boucher RC, Suzukawa M, Hebisawa A, Saito A, Nagase T. FOXL1 regulates lung fibroblast function via multiple mechanisms. Am J Respir Cell Mol Biol. 2020;63(6):831–42.
McDonough JE, Ahangari F, Li Q, Jain S, Verleden SE, Herazo-Maya J, Vukmirovic M, DeIuliis G, Tzouvelekis A, Tanabe N, Chu F. Transcriptional regulatory model of fibrosis progression in the human lung. JCI Insight. 2019;4(22):e131597.
Luzina IG, Fishelevich R, Hampton BS, Courneya JP, Parisella FR, Lugkey KN, Baleno FX, Choi D, Kopach P, Lockatell V, Todd NW. Full-length IL-33 regulates Smad3 phosphorylation and gene transcription in a distinctive AP2-dependent manner. Cell Immunol. 2020;357:104203.
Hadjicharalambous MR, Roux BT, Csomor E, Feghali-Bostwick CA, Murray LA, Clarke DL, Lindsay MA. Long intergenic non-coding RNAs regulate human lung fibroblast function: implications for idiopathic pulmonary fibrosis. Sci Rep. 2019;9(1):1–15.
Beisang DJ, Smith K, Yang L, Benyumov A, Gilbertsen A, Herrera J, Lock E, Racila E, Forster C, Sandri BJ, Henke CA. Single-cell RNA sequencing reveals that lung mesenchymal progenitor cells in IPF exhibit pathological features early in their differentiation trajectory. Sci Rep. 2020;10(1):1–12.
Roach KM, Sutcliffe A, Matthews L, Elliott G, Newby C, Amrani Y, Bradding P. A model of human lung fibrogenesis for the assessment of antifibrotic strategies in idiopathic pulmonary fibrosis. Sci Rep. 2018;8(1):1–15.
Zhang Y, Jiang M, Nouraie M, Roth MG, Tabib T, Winters S, Chen X, Sembrat J, Chu Y, Cardenes N, Tuder RMM. GDF15 is an epithelial-derived biomarker of idiopathic pulmonary fibrosis. Am J Phys Lung Cell Mol Phys. 2019;317(4):L510–21.
Adams TS, Schupp JC, Poli S, Ayaub EA, Neumark N, Ahangari F, Chu SG, Raby BA, DeIuliis G, Januszyk M, Duan Q. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci Adv. 2020;6(28):eaba1983.
Jonigk D, Stark H, Braubach P, Neubert L, Shin HO, Izykowski N, Welte T, Janciauskiene S, Warnecke G, Haverich A, Kuehnel M. Morphological and molecular motifs of fibrosing pulmonary injury patterns. J Pathol Clin Res. 2019;5(4):256–71.
Mullenbrock S, Liu F, Szak S, Hronowski X, Gao B, Juhasz P, Sun C, Liu M, McLaughlin H, Xiao Q, Feghali-Bostwick C. Systems analysis of transcriptomic and proteomic profiles identifies novel regulation of fibrotic programs by miRNAs in pulmonary fibrosis fibroblasts. Genes. 2018;9(12):588.
Patel NM, Kawut SM, Jelic S, Arcasoy SM, Lederer DJ, Borczuk AC. Pulmonary arteriole gene expression signature in idiopathic pulmonary fibrosis. Eur Respir J. 2013;41(6):1324–30.
Xu Y, Mizuno T, Sridharan A, Du Y, Guo M, Tang J, Wikenheiser-Brokamp KA, Perl AKT, Funari VA, Gokey JJ, Stripp BR. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight. 2016;1(20):e90558.
Yang IV, Coldren CD, Leach SM, Seibold MA, Murphy E, Lin J, Rosen R, Neidermyer AJ, McKean DF, Groshong SD, Cool C. Expression of cilium-associated genes defines novel molecular subtypes of idiopathic pulmonary fibrosis. Thorax. 2013;68(12):1114–21.
Paplińska-Goryca M, Goryca K, Misiukiewicz-Stępień P, Nejman-Gryz P, Proboszcz M, Górska K, Maskey-Warzęchowska M, Krenke R. mRNA expression profile of bronchoalveolar lavage fluid cells from patients with idiopathic pulmonary fibrosis and sarcoidosis. Eur J Clin Investig. 2019;49(9):e13153.
Xia Y, Lei C, Yang D, Luo H. Construction and validation of a bronchoalveolar lavage cell-associated gene signature for prognosis prediction in idiopathic pulmonary fibrosis. Int Immunopharmacol. 2021;92:107369.
Abe S, Sato S, Aono Y, Azuma M, Kishi M, Koyama K, Takahashi N, Kagawa K, Kawano H, Nishioka Y. Functional analysis of human fibrocytes derived from monocytes reveals their profibrotic phenotype through paracrine effects. J Med Investig. 2020;67(1.2):102–12.
Di Mauro S, Scamporrino A, Fruciano M, Filippello A, Fagone E, Gili E, Scionti F, Purrazzo G, Di Pino A, Scicali R, Di Martino MT. Circulating coding and long non-coding RNAs as potential biomarkers of idiopathic pulmonary fibrosis. Int J Mol Sci. 2020;21(22):8812.
Nakanishi T, Cerani A, Forgetta V, Zhou S, Allen RJ, Leavy OC, Koido M, Assayag D, Jenkins RG, Wain LV, Yang IV. Genetically increased circulating FUT3 level leads to reduced risk of idiopathic pulmonary fibrosis: a Mendelian randomisation study. Eur Respir J. 2021;59:2003979.
Fraser E, Denney L, Antanaviciute A, Blirando K, Vuppusetty C, Zheng Y, Repapi E, Iotchkova V, Taylor S, Ashley N, St Noble V. Multi-modal characterization of monocytes in idiopathic pulmonary fibrosis reveals a primed type I interferon immune phenotype. Front Immunol. 2021;12:226.
Sala MA, Balderas-Martínez YI, Buendía-Roldan I, Abdala-Valencia H, Nam K, Jain M, Bhorade S, Bharat A, Reyfman PA, Ridge KM, Pardo A. Inflammatory pathways are upregulated in the nasal epithelium in patients with idiopathic pulmonary fibrosis. Respir Res. 2018;19(1):1–10.
Boon K, Bailey NW, Yang J, Steel MP, Groshong S, Kervitsky D, Brown KK, Schwarz MI, Schwartz DA. Molecular phenotypes distinguish patients with relatively stable from progressive idiopathic pulmonary fibrosis (IPF). PLoS One. 2009;4(4):e5134.
Furusawa H, Cardwell JH, Okamoto T, Walts AD, Konigsberg IR, Kurche JS, Bang TJ, Schwarz MI, Brown KK, Kropski JA, Rojas M. Chronic hypersensitivity pneumonitis, an interstitial lung disease with distinct molecular signatures. Am J Respir Crit Care Med. 2020;202(10):1430–44.
De Sadeleer LJ, McDonough JE, Schupp JC, Yan X, Vanstapel A, Van Herck A, Everaerts S, Geudens V, Sacreas A, Goos T, Aelbrecht C. Lung microenvironments and disease progression in fibrotic hypersensitivity pneumonitis. Am J Respir Crit Care Med. 2021;205(1):60–74.
Christophi GP, Caza T, Curtiss C, Gumber D, Massa PT, Landas SK. Gene expression profiles in granuloma tissue reveal novel diagnostic markers in sarcoidosis. Exp Mol Pathol. 2014;96(3):393–9.
Casanova NG, Gonzalez-Garay ML, Sun B, Bime C, Sun X, Knox KS, Crouser ED, Sammani N, Gonzales T, Natt B, Chaudhary S. Differential transcriptomics in sarcoidosis lung and lymph node granulomas with comparisons to pathogen-specific granulomas. Respir Res. 2020;21(1):1–12.
Tanaka H, Yamaguchi E, Asai N, Yokoi T, Nishimura M, Nakao H, Yoneda M, Ohtsuka Y, Konno S, Yamada N. Cathepsin S, a new serum biomarker of sarcoidosis discovered by transcriptome analysis of alveolar macrophages. Sarcoidosis Vasc Diffuse Lung Dis. 2019;36(2):141.
Vukmirovic M, Yan X, Gibson KF, Gulati M, Schupp JC, DeIuliis G, Adams TS, Hu B, Mihaljinec A, Woolard TN, Lynn H. Transcriptomics of bronchoalveolar lavage cells identifies new molecular endotypes of sarcoidosis. Eur Respir J. 2021;58(6):2002950.
Lepzien R, Nie M, Czarnewski P, Liu S, Yu M, Ravindran A, Kullberg S, Eklund A, Grunewald J, Smed-Sörensen A. Pulmonary and blood dendritic cells from sarcoidosis patients more potently induce IFNγ-producing Th1 cells compared with monocytes. J Leukoc Biol. 2021;2021:1–10.
Lepzien R, Liu S, Czarnewski P, Nie M, Österberg B, Baharom F, Pourazar J, Rankin G, Eklund A, Bottai M, Kullberg S. Monocytes in sarcoidosis are potent TNF producers and predict disease outcome. Eur Respir J. 2021;58(1):2003468.
Bloom CI, Graham CM, Berry MP, Rozakeas F, Redford PS, Wang Y, Xu Z, Wilkinson KA, Wilkinson RJ, Kendrick Y, Devouassoux G. Transcriptional blood signatures distinguish pulmonary tuberculosis, pulmonary sarcoidosis, pneumonias and lung cancers. PLoS One. 2013;8(8):e70630.
Kachamakova-Trojanowska N, Jazwa-Kusior A, Szade K, Kasper L, Soja J, Andrychiewicz A, Jakiela B, Plutecka H, Sanak M, Jozkowicz A, Sladek K. Molecular profiling of regulatory T cells in pulmonary sarcoidosis. J Autoimmun. 2018;94:56–69.
Garman L, Pelikan RC, Rasmussen A, Lareau CA, Savoy KA, Deshmukh US, Bagavant H, Levin AM, Daouk S, Drake WP, Montgomery CG. Single cell transcriptomics implicate novel monocyte and T cell immune dysregulation in sarcoidosis. Front Immunol. 2020;11:567342.
Locke LW, Crouser ED, White P, Julian MW, Caceres EG, Papp AC, Le VT, Sadee W, Schlesinger LS. IL-13–regulated macrophage polarization during granuloma formation in an in vitro human sarcoidosis model. Am J Respir Cell Mol Biol. 2019;60(1):84–95.
Jung SM, Park KS, Kim KJ. Integrative analysis of lung molecular signatures reveals key drivers of systemic sclerosis-associated interstitial lung disease. Ann Rheum Dis. 2021;81(1):108–16.
Valenzi E, Bulik M, Tabib T, Morse C, Sembrat J, Bittar HT, Rojas M, Lafyatis R. Single-cell analysis reveals fibroblast heterogeneity and myofibroblasts in systemic sclerosis-associated interstitial lung disease. Ann Rheum Dis. 2019;78(10):1379–87.
Valenzi E, Tabib T, Papazoglou A, Sembrat J, Trejo Bittar HE, Rojas M, Lafyatis R. Disparate interferon signaling and shared aberrant basaloid cells in single-cell profiling of idiopathic pulmonary fibrosis and systemic sclerosis-associated interstitial lung disease. Front Immunol. 2021;12:960.
Lindahl GE, Stock CJ, Shi-Wen X, Leoni P, Sestini P, Howat SL, Bou-Gharios G, Nicholson AG, Denton CP, Grutters JC, Maher TM. Microarray profiling reveals suppressed interferon stimulated gene program in fibroblasts from scleroderma-associated interstitial lung disease. Respir Res. 2013;14(1):1–14.
Renaud L, da Silveira WA, Takamura N, Hardiman G, Feghali-Bostwick C. Prominence of IL6, IGF, TLR, and bioenergetics pathway perturbation in lung tissues of scleroderma patients with pulmonary fibrosis. Front Immunol. 2020;11:383.
Assassi S, Wu M, Tan FK, Chang J, Graham TA, Furst DE, Khanna D, Charles J, Ferguson EC, Feghali-Bostwick C, Mayes MD. Skin gene expression correlates of severity of interstitial lung disease in systemic sclerosis. Arthritis Rheum. 2013;65(11):2917–27.
Hutter C, Kauer M, Simonitsch-Klupp I, Jug G, Schwentner R, Leitner J, Bock P, Steinberger P, Bauer W, Carlesso N, Minkov M. Notch is active in Langerhans cell histiocytosis and confers pathognomonic features on dendritic cells. Blood. 2012;120(26):5199–208.
Pang J, Luo Y, Wei D, Cao Z, Qi X, Song M, Liu Y, Li Z, Zhang J, Li B, Chen J. Comparative transcriptome analyses reveal a transcriptional landscape of human silicosis lungs and provide potential strategies for silicosis treatment. Front Genet. 2021;12:652901.
Yao W, Yang P, Qi Y, Jin L, Zhao A, Ding M, Wang D, Li Y, Hao C. Transcriptome analysis reveals a protective role of liver X receptor alpha against silica particle-induced experimental silicosis. Sci Total Environ. 2020;747:141531.
Chen J, Zhang R, Xie M, Luan C, Li X. Transcriptome sequencing identifies PLAUR as an important player in patients with dermatomycitis-associated interstitial lung disease. Front Genet. 2021;12:784215.
Johnson CH, Ivanisevic J, Siuzdak G. Metabolomics: beyond biomarkers and towards mechanisms. Nat Rev Mol Cell Biol. 2016;17(7):451–9.
Chambers DC, Carew AM, Lukowski SW, Powell JE. Transcriptomics and single-cell RNA-sequencing. Respirology. 2019;24(1):29–36.
Junker BH, Klukas C, Schreiber F. VANTED: a system for advanced data analysis and visualization in the context of biological networks. BMC Bioinform. 2006;7(1):1–13.
Kamburov A, Cavill R, Ebbels TM, Herwig R, Keun HC. Integrated pathway-level analysis of transcriptomics and metabolomics data with IMPaLA. Bioinformatics. 2011;27(20):2917–8.
Hu Z, Hung JH, Wang Y, Chang YC, Huang CL, Huyck M, DeLisi C. VisANT 3.5: multi-scale network visualization, analysis and inference based on the gene ontology. Nucleic Acids Res. 2009;37(Web Server issue):W115–21.
Karnovsky A, Weymouth T, Hull T, Tarcea VG, Scardoni G, Laudanna C, Sartor MA, Stringer KA, Jagadish HV, Burant C, Athey B. Metscape 2 bioinformatics tool for the analysis and visualization of metabolomics and gene expression data. Bioinformatics. 2012;28(3):373–80.
Pavlopoulos GA, O’Donoghue SI, Satagopam VP, Soldatos TG, Pafilis E, Schneider R. Arena3D: visualization of biological networks in 3D. BMC Syst Biol. 2008;2(1):1–7.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–504.
McGuffin MJ, Jurisica I. Interaction techniques for selecting and manipulating subgraphs in network visualizations. IEEE Trans Vis Comput Graph. 2009;15(6):937–44.
Barsky A, Munzner T, Gardy J, Kincaid R. Cerebral: visualizing multiple experimental conditions on a graph with biological context. IEEE Trans Vis Comput Graph. 2008;14(6):1253–60.
Arakawa K, Kono N, Yamada Y, Mori H, Tomita M. KEGG-based pathway visualization tool for complex omics data. In Silico Biol. 2005;5(4):419–23.
García-Alcalde F, García-López F, Dopazo J, Conesa A. Paintomics: a web based tool for the joint visualization of transcriptomics and metabolomics data. Bioinformatics. 2011;27(1):137–9.
Neuweger H, Persicke M, Albaum SP, Bekel T, Dondrup M, Hüser AT, Winnebald J, Schneider J, Kalinowski J, Goesmann A. Visualizing post genomics datasets on customized pathway maps by ProMeTra–aeration-dependent gene expression and metabolism of Corynebacterium glutamicum as an example. BMC Syst Biol. 2009;3(1):1–14.
Tokimatsu T, Sakurai N, Suzuki H, Ohta H, Nishitani K, Koyama T, Umezawa T, Misawa N, Saito K, Shibata D. KaPPA-view. A web-based analysis tool for integration of transcript and metabolite data on plant metabolic pathway maps. Plant Physiol. 2005;138(3):1289–300.
Thimm O, Bläsing O, Gibon Y, Nagel A, Meyer S, Krüger P, Selbig J, Müller LA, Rhee SY, Stitt M. MAPMAN: a user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 2004;37(6):914–39.
Symonsy S, Zipplies C, Battke F, Nieselt K. Integrative systems biology visualization with MAYDAY. J Integr Bioinform. 2010;7(3):1–14.
Lüdemann A, Weicht D, Selbig J, Kopka J. PaVESy: pathway visualization and editing system. Bioinformatics. 2004;20(16):2841–4.
Xie N, Tan Z, Banerjee S, Cui H, Ge J, Liu RM, Bernard K, Thannickal VJ, Liu G. Glycolytic reprogramming in myofibroblast differentiation and lung fibrosis. Am J Respir Crit Care Med. 2015;192(12):1462–74.
Hsu HS, Liu CC, Lin JH, Hsu TW, Hsu JW, Su K, Hung SC. Involvement of ER stress, PI3K/AKT activation, and lung fibroblast proliferation in bleomycin-induced pulmonary fibrosis. Sci Rep. 2017;7(1):1–11.
Zhao H, Li C, Li L, Liu J, Gao Y, Mu K, Chen D, Lu A, Ren Y, Li Z. Baicalin alleviates bleomycin induced pulmonary fibrosis and fibroblast proliferation in rats via the PI3K/AKT signaling pathway. Mol Med Rep. 2020;21(6):2321–34.
Acknowledgments
Figures were created with BioRender.com.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Dasgupta, S. et al. (2023). Metabolomics and Transcriptomic Approach to Understand the Pathophysiology of Interstitial Lung Disease. In: Abdel Rahman, A.M. (eds) Clinical Metabolomics Applications in Genetic Diseases. Springer, Singapore. https://doi.org/10.1007/978-981-99-5162-8_14
Download citation
DOI: https://doi.org/10.1007/978-981-99-5162-8_14
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-99-5161-1
Online ISBN: 978-981-99-5162-8
eBook Packages: MedicineMedicine (R0)