
Abstract
The significant rise in the global incidence of neurological diseases, such as Alzheimer’s disease (AD) and Parkinson’s disease (PD), has been attributed to the aging population. There are no efficient therapies for these disorders, regardless of medical improvements. Therefore, there is an urgent need for innovative treatments for these diseases. Over the years, stem cell-based therapy has transformed regenerative medicine by providing crucial and compelling possibilities to treat a variety of diseases (e.g., cancer), including neurological diseases. Stem cell therapy, referred to as regenerative therapy, uses stem cells or their derivatives to enhance the curative response of dysfunctional and injured tissue. Hence, mesenchymal stem cells (MSCs) and embryonic stem cells (ESCs) are two stem cell types that are used. Recent evidence supports stem cell transplantation as promising therapeutic potential, which could be regarded as stem cells’ mechanistic actions. In addition to stem cells, exosomes are a type of nanovesicles with a wide range of functionalities and possibilities for diagnosing and treating. It is shown that exosomes are implicated in cell-cell communication and have been explored as candidates for possible biomarkers, which are particularly relevant in AD and PD. Exosomes are also employed as a drug delivery vehicle at a target site; thus, their inherent ability to cross the blood-brain barrier and selectively adorn it with the ligand depends on the treatments of the targeted brain regions. This chapter aims to shed light on the different roles of stem cells and derived exosomes as therapeutic agents in the treatment of neurodegenerative disorders.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
Keywords
10.1 Introduction
Two of the most prevalent neurodegenerative diseases are Alzheimer’s disease (AD) and Parkinson’s disease (PD), with PD affecting 1% of adults over 60 years of age, while AD has affected approximately 50 million people in the United States (Han et al. 2018). AD or PD currently has no known treatments for complete cure; however, there are treatments to reduce the disease’s progression and relieve some symptoms (e.g., rivastigmine, a cholinesterase inhibitor for PD) (Sun and Armstrong 2021). The common clinical symptoms of AD and PD include memory loss, motor dysfunction, and cognitive decline. Aggregates of proteins in cells cause the death of neurons in both PD and AD, although distinct brain regions are affected in each condition (Kim et al. 2003). For the creation of functional human neural cells for cell-based therapy and in vitro modeling, stem cells represent an unlimited cell source. Exosomes alone have a neuroprotective significance, as seen when oligodendrocyte-derived exosomes are added to cultured neurons, increasing cell viability even under stressful circumstances (Soares Martins et al. 2021).
This chapter highlights the pathophysiology of AD and PD, stem cell-derived cells, exosome technologies, and, the most recent, cutting-edge approaches to treating the conditions. Particularly, it focuses on the promising role or functions of stem cells in the differentiation of the neurons and implantation in vivo for potential treatment and drug testing of neurodegenerative diseases (Fig. 10.1). Additionally, derived exosomes serve as targeted cargoes and biomarkers in both AD and PD (Lakshmi et al. 2020).

The general method for reprogramming somatic/blood samples to differentiate ESCs from blastocysts is by destroying the trophoblast layer with antibodies or lasers and iPSCs by different transcription factors. The most often used differentiation factors to form neural rosettes are sodium selenite GSK3 inhibitor, RA, insulin, transferrin, TGF-β, and BMP inhibitors. Additionally, N2, bFGF, Noggin, B27, RA, trophic factors, and dorsomorphin promote neuronal development. FGF8, SHH, RA, ascorbic acid, BDNF, and HAG influence the distinct differentiation by the lineage of iPSC-NSCs into the motor neurons, glial cells, and astrocytes (SHH, FGF2, and PDGF, respectively). These models and reprogramming methods can advance the research of neurodegenerative diseases, drug discovery, and therapeutic applications (Amoroso et al. 2013; Hayashi et al. 2011; Kim et al. 2012; Nutt et al. 2013)
10.1.1 What Are Stem Cells?
Stem cells are progenitor cells with the capability to regenerate or self-renew unlimitedly and the ability to develop further into differentiated cell types under appropriate conditions (Chagastelles and Nardi 2011). Stem cells can be characterized into totipotent stem cells which have the potential to form into every cell type of an organism (extraembryonic and embryonic structures) and have the highest capacity for differentiation. Pluripotent stem cells usually give rise to the cells of the three germ layers (endoderm, mesoderm, and ectoderm), and multipotent stem cells develop into multiple specialized cell types which exist in a particular tissue or organ (e.g., hematopoietic stem cells) (Zakrzewski et al. 2019). Unipotent stem cells, on the other hand, are distinct by having the most limited capability for differentiation. Having a special tendency to divide repeatedly, these cells can form only one cell type, e.g., satellite cells of skeleton muscles, making them a prime requisite for therapeutic agents (Dulak et al. 2015). After fertilization, the embryo divides and reaches the stage of the blastocyst, at which point it loses totipotency and acquires pluripotency, from which embryonic stem cells (ESCs) can be isolated (Zakrzewski et al. 2019). By acquiring the inner cell mass (ICM), which results in the potential destruction of the embryos, ESCs are obtained. The cells keep on dividing until it reaches the multipotent stage, thus becoming adult stem cells with the constrained potential to differentiate into cell types within a specific lineage (Vatsa et al. 2022). Adult cells such as the fibroblast or peripheral blood mononuclear cells which are isolated from the blood, can be reprogrammed into pluripotent stem cells termed as induced pluripotent stem cells (iPSCs). Reprogramming is established by the expression of certain reprogramming transcription factors, also termed as “Yamanaka factors” or “OKSM factors,” Oct 3/4 (octamer-binding transcription factor 3/4), Sox2 (Sex-determining region Y) box 2, and Klf4 (Kruppel-like factor 4) (Takahashi et al. 2007; Takahashi and Yamanaka 2006). These transcription factors modulate the stemness and differentiation potential of stem cells (Yamanaka et al. 2007).
Adult or somatic stem cells are undifferentiated and are present in the differentiated cells of different tissues or organs. These cells serve to enhance the growth, repair, and replacement of the cells that are shed daily. These cells are multipotent or unipotent stem cells because they have limited differentiation potential into different cell kinds of their genesis tissue (Prochazkova et al. 2015; Romito and Cobellis 2016). Somatic stem cells exist in a range of distinct types, such as MSCs, neural stem cells (NSCs), hematopoietic stem cells, and skin stem cells. The bone marrow is where these cells largely mature into fat, bone, or even cartilage cells. The NSCs develop into oligodendrocytes and astrocytes, which are cells that support nerve cells. Compared to ESCs, somatic stem cells multiply longer although adult stem cells can also be reprogrammed to regain their pluripotency. The first cellular reprogramming was achieved by Sir John Gurdon in 1962 by transplanting the nucleus from the intestinal epithelial somatic cells of tadpoles into enucleated unfertilized frog egg cells and reported generation of tadpoles (Gurdon 1962). This unique technique is termed somatic nuclear transfer (SCNT) for reprogramming of the somatic stem cells to the pluripotent embryonic state with a similar or the same genetic makeup; as a result, it led to the invention of cloning. The first mammal to be generated through somatic cloning was Dolly the sheep, which was cloned in the year 1997 by Sir Ian Wilmut and his team using the same method (Wilmut et al. 1997).
10.1.2 Exosomes
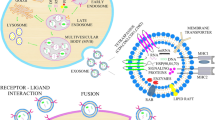
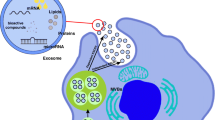
About 50 years ago, Peter Wolf first referred to extracellular vesicles (EVs) in plasma as “platelet dust” (Wolf 1967). Exosomes, one of the three forms of EVs, are distinguished by their varied sizes as well as by their biogenesis and release mechanisms. All cell types, including blood cells, neurons, epithelial cells, immunological or cancer cells, etc., release exosomes. Several biological fluids, such as cell culture supernatants, cerebrospinal fluid (CSF), serum, plasma, saliva, semen, urine, breast milk, and amniotic fluid, can be used to extract exosomes (Théry et al. 2006). Exosomal vesicles are formed by the inward growth of early endosomes that restrict membranes, which grow into multivesicular bodies (MVBs) (Raposo and Stoorvogel 2013). Their density ranges from around 1.13 g/mL to 1.19 g/mL (Bobrie et al. 2011; Zakharova et al. 2007). The sorting of protein as well as recycling, storage, transportation, and release is facilitated by exosomes in their intercellular signaling. MVBs are either destroyed with all their elements in the lysosome or may get fused with the plasma membrane of the cell in order to liberate their materials, including the exosomes, into the extracellular space (Simons and Raposo 2009). Exosomes were discovered to be released in a similar manner by the B-lymphocytes as well as dendritic cells. Eventually, it was revealed that exosomes were also released in an analogous manner by platelets, cytotoxic T cells, neurons, Schwann cells, mast cells, oligodendrocytes, intestinal epithelial cells, and endothelial cells, as well as stem cells; however, some cells, like the immune cells and MSCs, release more than others (Budnik et al. 2016; Jahan et al. 2022; Muller 2020).
Exosomes are particularly promising in regenerative medicine because of the range of components they contain, and their lipid bilayer membranes ensure stability and durability as well as include distinct marker proteins that link them to specific cells. The endosomal sorting complex required for transport (ESCRT) process initiated through the ubiquitin-binding subunits of ESCRT-0 to identify and sequester complex will merge with ESCRT-III, which is a protein complex involved in encouraging the process of budding, after contact with ESCRT-I and II complexes. After cleaving the buds to create intraluminal vesicles (ILVs), the ESCRT-III complex subsequently separates from the MVB membrane with the aid of energy given by the sorting protein Vps4 (Henne et al. 2011). The microdomains based on the raft seem to be required for the ESCRT-independent lateral segregation of the cargo present inside the endosomal membrane. These microdomains have a large number of sphingomyelinases, which can hydrolyze the phosphocholine moiety to produce ceramides (Airola and Hannun 2013). Furthermore, both ESCRT-dependent and ESCRT-independent pathways engage in exosome formation as well as the packing of biological cargo into exosomes, therefore including the utilization of cellular signals like phosphatidic acid, diglycerides, and ceramides as well as exosomal membrane lipid messengers (Stahl and Barbieri 2002). As lipid rafts, which are highly concentrated regions of sphingolipids and cholesterol in the membrane that are necessary for cell communication and endocytosis, are a component of exosome packaging, their presence on exosomal membranes makes them easily distinguishable as endosomes and can be used to detect exosomes rather than other vesicular products (Théry 2011). Exosomes can transport many molecules, such as particular proteins, RNA, and miRNA. The exosomal system also supports the horizontal transfer of the mRNA as well as proteins, with the genetic information successfully translated into suitable proteins, according to various studies (Bruno et al. 2009; Ratajczak et al. 2006).
10.2 Role of Stem Cells and Derived Exosomes in Neurodegenerative Disease
The progressive degradation of neurons in the central and peripheral nervous systems is the characteristic feature of the heterogeneous group of disorders known as neurodegenerative diseases (Przedborski et al. 2003). They manifest as a consequence of loss of function, structure, and/or several neurons, including the death of neurons in the spinal cord or brain, which results in loss of cognition and different degrees of motor disability (Poddar et al. 2021). Neurodegeneration is linked with the disruption of a neural network, the synapse, and the accumulation of physiochemically altered proteins (Lamptey et al. 2022).
The most common neurodegenerative conditions are Huntington’s disease, spinal muscular atrophy, Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), motor neuron disease, prion disease, and spinocerebellar ataxia (SCA) (Lamptey et al. 2022). According to reports, AD is the sixth most prevalent cause of death in the United States (Kumar et al. 2021). Up to 5.8 million Americans were expected to have AD in 2020 ((Matthews et al. 2019). Up to 24 million people worldwide are estimated to have dementia; by 2050, that number suffered from PD, most of which are idiopathic (Zafar and Yaddanapudi 2021). Around 10% of them have a genetic cause and affect the young age group. ALS, also known by the name “Lou Gehrig,” exists in two forms, familial, comprising 90–95% of cases, and sporadic. It has been estimated that ALS has a prevalence of 5.2 per 100,000 within the United States and 1.6 cases per 100,000 persons annually worldwide (Brotman et al. 2022). SCA is a subset of hereditary cerebellar ataxia, is a comparatively rare disease, and has a global prevalence of 1 to 5 in 10,000. It has many subtypes, with SCA3 the most common worldwide and SCA2 the most common subtype in South Korea and India (Bhandari et al. 2022).
According to a report by Feigin et al., neurological diseases were the second-most common incidence of deaths globally in 2016 and the leading cause of DALYs (disability-adjusted life years; the sum of the years of life lost and the years spent with disability) (Feigin and Vos 2019). Migraine, stroke, Alzheimer’s, and other dementias were the four largest contributors to neurological DALY. In general, over the past 27 years, it was shown that the prevalence of neurological disorders has grown and it is anticipated to continue to increase due to the exponential growth of the population as well as aging, placing even higher pressure on heavily overburdened resources and services for people with neurological disorders (Feigin and Vos 2019).
The absence of effective treatments for neurodegenerative diseases places a heavy burden on society, as well as the cost of care, and has a significant influence on the quality of life. Current clinical approaches focus on addressing symptom alleviation and disease management but fail to halt the progress of neurodegeneration (Poddar et al. 2021). Neither pharmaceutical nor neurosurgical treatments are effective at arresting the course of the neurodegenerative processes (Sakthiswary and Raymond 2012). Furthermore, due to variations in mammalian genomes and embryonic development, numerous therapies developed in animal models have not yet been successfully translated into clinical trials (Dawson et al. 2018). On this basis, human-based studies involving stem cells present a clear model for studying the pathophysiological process, signaling pathways, growth control, and disease mechanism in previously inaccessible human brain tissue.
10.2.1 Stem Cells Therapy in Neurodegenerative Diseases
Regenerative cell therapy, otherwise called stem cell therapy, over the past two decades has provided a significant opportunity to explore potentially powerful innovative strategies to treat diseases associated with neurodegeneration. This is because stem cells have the ability to restore damaged neural tissue by mostly replacing lost or damaged cells with differentiated ones, creating an environment that promotes regeneration, stabilizes neuronal networks, or protects already healthy neurons and glial cells from more harm. Different stem cell types have a differential capacity to descend into a specific cell lineage/type, as discussed above. This seemingly unlimited potential of stem cells has opened unprecedented opportunities for developing novel medical therapies for degenerative diseases and injuries, and diseases like AD, PD, HD, spinal cord injury, diabetes, and a few heart diseases have few or no treatment options; hence, stem cell-based therapy is a beneficial option, as stem cell uses their derivatives to enhance the repair reaction of damaged and dysfunctional tissue (Institute of Medicine 2002). Since every stem cell type comes with its own unique traits and benefits, the rationale for using a specific type depends on the applications as well as its results that are desired. Subsequently, human embryonic stem cell (hESC) research involves destruction of human embryos, thus raising ethical as well as political concern (Lo and Parham 2009a). Because of these limitations and ethical concerns surrounding ESCs, scientists have developed techniques for inducing pluripotency in non-pluripotent cells or somatic cells. The generated cells, thus named iPSCs, open the possibility of offering customized models to study and treat neurodegenerative diseases using patients’ own somatic cells through reprogramming (Dantuma Elise et al. 2010).
The primary types of stem cells utilized for neurodegenerative therapies are embryonic, progenitors, mesenchymal, and iPSCs (Singh et al. 2016). ESCs being pluripotent hold remarkable potential to restore the damage that occurred due to injury or neurodegeneration (Singh et al. 2016). An additional restriction is an immunological incompatibility between donor and recipient cells, which can lead to the of transplanted cells. The spectrum of their clinical application is however constrained by their capacity for unrestricted self-renewal, which carries a significant danger for the development of tumors after engraftment (Sivandzade and Cucullo 2021).
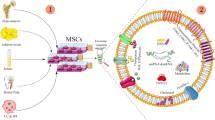
MSCs are immunomodulating, that is, derived from the same source (and hence do not elicit host immune response) and multipotent and find high applicability in neurodegenerative diseases, as it promotes neural growth, reduce free radicals, decrease apoptosis, and repress inflammation (Sakthiswary and Raymond 2012). However, their use gets restricted in genetic diseases because the autologous source holds the same genetic predisposition to the disease. In the next part, the prevalence of stem cells is discussed in different neurodegenerative diseases.
10.2.1.1 Huntington’s Disease
Transplantation of median spiny neurons (MSNs) derived from both iPSCs and ESCs has shown successful integration and neural circuit development (Aubry et al. 2008), but most studies are restricted to animal models. Reliable data indicates that three out of five participants had the advantage of transplantation in the Créteil pilot experiment, nonrandomized, open-label, monocentric cell transplant trial employing fetal donor cells of humans obtained from the fetal ganglionic eminence (GE). Therefore, the two participants exhibited the typical HD deterioration; however, at the end it was hypothesized that among one of them, the disease was too advanced in order to allow for effective graft vascularization (Bachoud-Lévi et al. 2021).
10.2.1.2 Amyotrophic Lateral Sclerosis
Stem cell transplantation has demonstrated great potential in recent clinical trials in ALS patients. Six studies have shown proof that stem cell therapy largely has a positive effect in slowing the progression of disease (L. Xu et al. 2006). Numerous studies have revealed the efficacy of NSC therapy on ALS rats, as a result of the transplanted NSCs’ ability to differentiate into neurons and form synaptic connections in addition to their potential to prevent the disease’s onset and progress, thereby increasing the survival of animal models (Xu et al. 2006). Considering the preclinical evidence for NSC-based treatments, the Food and Drug Administration (FDA) approved a clinical trial in 2009 regarding the safety as well as tolerability of the surgical introduction of stem cells and any therapy following cell toxicity. In this first-in-human phase I clinical trial, researchers injected fetal-derived NSCs into the lumber spinal cord of 12 ALS patients (Glass et al. 2012). Clinical evaluations performed around 6–18 months after the transplantation showed no indications of further progression of the disease. By exploring the intraspinal injections in the cervical spinal cord, the researchers aim to advance this clinical trial and potentially prolong the lives of ALS patients by protecting the motor neuron groups that affect respiratory function. The therapeutic potential of MSCs has been evaluated in several research employing ALS animal models by either injecting the cells peripherally or directly into the spinal cord (Mao et al. 2015). In 2003, Mazzini et al. examined the safety as well as tolerability of MSC transplantation directly intraparenchymal for the treatment of ALS (Mazzini et al. 2003). Although there was no functional improvement, follow-up studies also did not show detrimental effects.
10.2.1.3 Spinocerebellar Ataxia
MSCs serve as good candidates for SCA treatment because of their ability to differentiate between lines and immunomodulatory properties. In a study done by Chang et al., the human MSCs were administered intravenously and intracranially to transgenic mice having a poly-glutamine mutation in the ataxin-2 gene before and after the onset of loss of motor function. Intravenous transplantation successfully enhanced the rotarod function of the SCA2 mice as well as postponed the onset of neuronal deterioration, whereas intracranial transplantation was unable to procure any kind of neuroprotective effect (Chang et al. 2011). In phase I/IIa of the clinical trials conducted in Taiwan, six patients having type 3 SCA and one with multiple-system atrophy-cerebellar were intravenously administered MSC derived from allogenic adipose tissue from healthy donors (Tsai et al. 2017). Upon a year of follow-up, the intravenously injected MSCs seemed to be well tolerated and no adverse effects were observed. The study concludes the safety and tolerability of allogenic MSCs through intravenous injection.
10.2.2 Exosomes in Neurodegenerative Diseases
Recent preclinical research has indicated that EVs produced from stem cells can be utilized as a possible alternative to stem cell therapy to treat brain disorders. EV-based therapy outperforms cell therapy by means of biodistribution, scalability, and safety profiles; it can be used to treat neurological disorders as a potential substitute to stem cell-based therapy. Furthermore, EVs produced from stem cells have superior biocompatibility, immunogenicity, and safety characteristics compared to minute chemicals and macromolecules (Bang and Kim 2022).
Stem cell-derived exosomes are considered an inherent drug delivery system and a natural therapeutic agent for the potential treatments of brain diseases. EVs consist of molecules with heterogeneous functions such as cellular proteins, DNA and RNA. EVs have been revealed to be able to cross the BBB as well as can target specific cell types effectively (Fig. 10.2). Contrary to cell-based therapeutics, EVs usually are not affected by the first-pass effect or by cell-mediated side effects like coagulopathy, tumor growth, or arterial blockage. Additionally, the several components of the EVs’ copious nucleic acid are protected from RNase in the blood because lipids are required for the development of EVs’ membranes (Mirzaaghasi et al. 2021; Wen et al. 2019).

Biogenesis of exosomes from different cell sources and acting as drug delivery vehicle and biomarkers in AD and PD
In a study incorporating MSC transplant, it was shown that the number or amount of circulating EVS increased rapidly in number after the transplantation of MSCs (Bang et al. 2022). Also, in patients receiving the same dosage of MSCs, the quantity of the circulating EVS differed among patients, which was further associated with the improvement of motor function. Given the quantity of EVs that influence the results of MSC-based therapy, these studies suggested the possibility of using MSC-EVs rather than MSCs in and of themselves (Allan et al. 2020).
Henceforth, EVs have a role in maintaining CNS cellular function, also contributing to the pathophysiology underlying neurodegenerative disease. EVS that are derived from native stem cells can cure neurological disorders, but their shot half-lives, restricted targeting, quick clearance after the application, and insufficient payload make them difficult to use successfully in clinical settings (Khan et al. 2021). The heterogeneity of the donors is a significant problem that hinders the clinical applicability of EVs. Due to the usage of different donors, the cell variability is huge which might affect the therapeutic potential of EVs due to differences in donor age, comorbidity, artificial niches of MSCs, and culture conditions. In addition to creating a production/developmental process that reduces the donor-to-donor as well as batch-to-batch variances, each EV production lot needs to have a strong quality control system (Bang and Kim 2022).
10.3 Therapeutic Role of Stem Cells and Derived Exosomes in AD
Alzheimer’s disease (AD) is a fatal neurological disorder characterized by behavioral problems, progressive cognitive decline, and loss of daily functioning. Globally, between 50% and 70% of cases with dementia are caused by AD. Moreover, dementia affects 50 million individuals worldwide and costs around $818 billion. By 2050, this number is expected to increase to 132 million because of age being the primary risk factor and the fact that national populations are gradually aging (Association 2016; Ferri et al. 2005). Clinical symptoms of AD develop rapidly. Early neuroinflammation, learning, and memory issues are prominent features, followed by visuospatial function, executive function, complex attention, praxis (learned motor activity), gnosis (recognized previously learned information), language, behavior, and/or social abnormalities (Si and Wang 2021).
Additionally, the two primary neuropathological markers for the diagnosis of AD are beta-amyloid extracellular deposits (senile plaques) and hyperphosphorylated tau intercellular deposits (neurofibrillary tangles) (Kent et al. 2020). Many cases of AD have a late onset and are sporadic. Moreover, there are established risk factors for the illness besides age, such as the apolipoprotein-E4 (ApoE4) gene, cardiovascular disease, depression, and low education levels (Piers et al. 2021). In addition, under 5% of AD cases are familial and are caused by highly penetrant autosomal mutations in the PSEN1, PSEN2, and, less frequently, the APP genes (Sabayan and Sorond 2017). Moreover, genome-wide association studies (GWAS) revealed that mutations in various genes can promote the development of AD (Kang et al. 2016). Positron emission tomography (PET), CSF biomarkers, and some comparatively recent clinical standards can all be used to diagnose living patients, even though only a postmortem autopsy can provide a diagnosis with certainty. These criteria underline that etiological diagnosis still relies heavily on neuropathological evaluation (Kang et al. 2016). Numerous pharmaceutical strategies, including vaccination and secretase inhibition, have been investigated to improve amyloid clearance and reduce production (Duncan and Valenzuela 2017).
New therapies and medications are suggested every year to decrease the cognitive decline and neuronal death linked to AD. Nonetheless, only five drugs, including the glutamate receptor antagonist memantine and cholinesterase inhibitors galantamine, rivastigmine, tacrine, and donepezil, have received FDA approval for the clinical treatment of AD (Si and Wang 2021). Sadly, these drugs can only treat symptoms and do nothing to alter the primary pathologic aspects of AD (Si and Wang 2021). To ameliorate the pathogenic state of the disease, enhance neural precursors, prevent nerve death, and promote structural plasticity, effective, novel treatments must be developed. These therapies may include eliminating toxic deposits and replacing damaged neurons (Liu et al. 2020). Hope for the treatment of refractory neurodegenerative disorders like Alzheimer’s has been ignited by recent developments in stem cell preclinical research and clinical trials. For providing the uniform and cell replacement therapy that call required, stem cells are the greatest alternative (Liu et al. 2020).
10.3.1 Stem Cells as Therapeutics for AD
In AD research today, iPSCs, MSCs, brain-derived NSCs, and ESCs are most frequently exploited. Since the main reason for neurodegenerative diseases, for example, AD, is aging, it appears counterintuitive to research AD using stem cells. iPSC-derived neurons can develop electrophysiologically active synaptic networks and are structurally and functionally mature. It is also feasible to control the differentiation of iPSCs into various neuronal subgroups, including dopaminergic neurons, by using additional transcription factors during the induction phase (Duncan and Valenzuela 2017). Among multipotent stem cells, NSCs are a subset which can differentiate into oligodendrocytes, astrocytes, neurons, and microglia (Si and Wang 2021). In a study, neurons generated using iPSCs with FAD mutations or from AD patients displayed similar AD characteristics at the earliest possible developmental stages. The amyloid precursor protein (APP) V717I gene mutation increased Aβ and tau phosphorylation in neurons, whereas the APP A673T mutation suppressed β-secretase cleavage of APP and Aβ synthesis (Maloney et al. 2014; Muratore et al. 2014). Astrocytes with the PSEN1 ΔE9 mutation also produced more reactive oxygen species (ROS), had defective fatty acid oxidation, and elevated the production of Aβ42. When compared to astrocytes with APOE3 mutations, those with APOE4 mutations show substantial changes in gene expression and a decreased capacity to absorb amyloid beta 42. Lipopolysaccharide treatment increased the release of certain cytokines and altered phagocytosis in iPSC-derived microglia from SAD patients (Xu et al. 2019; Si and Wang 2021). When compared to isogenic APOE3 controls, microglia with mutated APOE4 had a decreased impaired capacity to internalize Aβ and morphologic complexity. The phenotypes of SAD-derived iPSCs and FAD mutation carriers are frequently similar. Another recent research discovered that human iPSC-derived cholinergic neuronal progenitors recovered from intra-hippocampal transplantation into transgenic AD mice model matured into phenotypically adult cholinergic neurons and repaired the spatial memory loss (Liu et al. 2020).
Additionally, human iPSC-derived NSCs improved neurological function and decreased pro-inflammatory markers in a mouse model of ischemic stroke through a neurotrophin-associated bystander effect (Duncan and Valenzuela 2017). The secreted neurotrophic factors by transplanted NSCs improved the memory function and overexpression of a β-degrading enzyme by NSCs reduced Aβ aggregation. Alleviated Aβ synthesis and acetylcholinesterase activity were seen following NSC transplantation into these Tg2576 mice who carry human Swedish APP mutation (isoform 695; KM670/671NL). Additionally, early-stage NSC transplantation enhanced anti-inflammatory cytokine levels in microglial cells and may reduce the generation of Aβ, increasing the Aβ clearance rate. It also increased vascular endothelial growth factor (VEGF), synaptic density, and neurogenesis. However, prompt action is required because later NSC transplantation into Tg2576 mice did not provide comparable outcomes (Liu et al. 2020). Also, in NSC administration in APP/PS1 mice, tropomyosin receptor kinase B and BDN levels increased. NSC-derived cholinergic neurons were also introduced into APP/PS1 mice showing an increase in the concentration of cholinergic acetyltransferase and its activity, and there were enhanced operational dendrites (Gu et al. 2015). The genetic modification of NSCs to optimally release the Aβ-degrading enzyme neprilysin may promote synaptic plasticity and potential Aβ-pathogenic characteristics (Liu et al. 2020).
MSCs are not predicted to replace injured neurons and integrate into neural network, in contrast to iPSCs and NSCs, because it is not known if they can differentiate into endodermal or ectodermal cells. The neuroprotective properties of MSCs are mediated by a variety of pathways. To increase brain cell survival, MSCs can secrete neurotrophic growth factors, including brain-derived neurotrophic factor (BDNF) and glial cell-derived neurotrophic factors. Ucb-MSCs have been demonstrated in prior research utilizing AD murine models to enhance spatial learning and prevent memory loss. Numerous additional pathways have also been proposed, such as the decrease in Aβ plaques, the hyperphosphorylation of BACE and tau, the restoration of microglial inflammation, and the activation of anti-inflammatory cytokines (Lee et al. 2012; Liu et al. 2020). Two Aβ-degrading factors including neprilysin and insulin-degrading enzyme rose following treatment with neuron-like cells generated by MSCs of the umbilical cord (human). Bone marrow-derived MSCs were implanted in the APP/PS1 mouse model of AD by tail vein injection. These mice had fewer microglia but the amounts of amyloid plaques remained unchanged (Naaldijk et al. 2017). Contrarily, Cartel et al. discovered that intracerebral injection of bone marrow-derived MSCs resulted in a substantial decrease over the course of 2 months compared to PBS-treated controls (Bae et al. 2013). In a study that evaluated Aβ therapy alone, brain progenitor cells co-cultured with MSCs showed considerably greater expression of GFAP, nestin, Ki-67, HuD, and SOX2. Furthermore, treatment with Aβ showed increased expression of β-catenin and Ngn1 in neural progenitor cells co-cultured with MSCs (Oh et al. 2015).
An in vivo study showed that ESCs can be differentiated into astrocytes and cells that mimic neurons, which can be used to treat neurodegenerative diseases. Researchers have demonstrated that ESCs, when transformed into gamma-aminobutyric acid and basal forebrain cholinergic neurons, can enhance spatial learning and memory in AD-affected mice (Liu et al. 2020). Nonetheless, direct ESC transplantation resulted in the development of teratomas in vivo rather than neurons, limiting their clinical utility (Liu et al. 2020). With and without pretreatment, NPCs derived from mESCs were transplanted to the unilateral Meynert basal nucleus, enhancing learning and memory in the AD rat model. The transplanted NPC cells show a cholinergic phenotype to a degree of about 40%, whereas the remaining cells continue to maintain a neuronal phenotype (Moghadam et al. 2009). Contrarily, the control group’s ESCs produced teratomas, which are tumors incapable of generating neurons, resulting in a drastic decline in working memory (Wang et al. 2006). Despite the limitation of evidence regarding hESCs’ ability to treat AD, hESCs can be considered a new therapeutic alternative for various neurological conditions and degenerative diseases. However, ethical issues should be addressed prior to the administration in clinical trials that have acquired approval from the FDA (Liu et al. 2020).
10.3.2 Exosomes as AD Biomarkers
Exosomes derived from the brain in peripheral blood have shown remarkable promise as the perfect “liquid biopsy” for Alzheimer’s. It is interesting to note that brain-derived exosomes can cross the BBB and enter the peripheral blood circulation; however, their quantity is lower than in CSF. Researchers use immunoprecipitation techniques to enrich plasma brain-derived exosomes to overcome these restrictions. According to earlier studies, the lysosomal and synaptic protein concentrations of neuron-derived exosomes (NDEs) can be used to predict dementia before it manifests as mild cognitive impairment (MCI) (Winston et al. 2016). Reduced levels of synaptic proteins with specific functions in NDE may signify the progression degree of AD. Additionally, complement protein levels in astrocyte-derived exosomes (ADEs) seem to be correlated with the disease’s stage (Goetzl et al. 2018). Because plasma ADEs contain far more cargo proteins than NDEs, BACE-1 inhibitors may be able to specifically target ADEs. A systematic method for the preparation and qualification of biomarkers remains extremely difficult, and to evaluate the diagnostic relevance of exosomes, large cohort studies are needed (Guo et al. 2020).
Small compounds and macromolecules are only partially effective against brain diseases because of the single method of action and intricate pathophysiology of these conditions. For instance, despite promising results in preclinical investigations, around 1000 neuroprotective drugs for acute stroke failed in human trials. Consequently, pleiotropic multi-target therapies may be more effective if they employ a variety of strategies to obstruct various stages. Additionally, almost no macromolecules and nearly all small compounds cross the BBB. Adeno-associated viral capsids and polymer- or lipid-based nanoparticles were employed to get around this restriction. Contrarily, using a drug delivery method raises the risk of infection, immunogenicity, and toxicity. For the treatment of brain diseases, stem cell-derived EVs are regarded as inherent drug delivery mechanisms and naturally therapeutic molecules (Guo et al. 2020; Kang et al. 2016; Liu et al. 2020).
The first theory linking exosomes to AD postulates that exosomes can produce Aβ peptides from cultured cells, and exosomal proteins including Alix and flotillin-1 have been observed to aggregate within amyloid plaques in the brains of AD patients. The full-length amyloid precursor protein (flAPP), Aβ peptide, amyloid precursor protein C-terminal fragments (APP-CTFs), and amyloid intracellular domain (AICD), as well as enzymes that cleave flAPP and APP CTFs, have been discovered to be present in exosomes from humans with the autosomal dominant Swedish mutation (BACE1, PS1, PS2, and ADAM10) (Laulagnier et al. 2017). According to these results, APP and its catabolites may be transported across cells via neuronal exosomes. More importantly, several in vivo experiments showed linkage of exosomes to AD. Exosomes from AD patients’ bodily fluids, such as blood and CSF, show an increase in soluble Aβ1-42, p-T181-tau, and p-S396, among other things. Exosomes can one day be employed as a diagnostic tool for AD as this rise could be observed many years before a diagnosis (Guo et al. 2020; Lee et al. 2019).
Exosomes derived from plasma neuronal membranes of AD patients cause AD-like neuropathology in normal mouse brains by increasing tau aggregation. Exosomes from AD patients with BIN1-related genetic variations in their CSF encourage tau spreading in mouse, but microglia depletion or exosome synthesis suppression greatly reduces tau propagation in vivo and in vitro. Furthermore, these toxic species can be transferred to recipient neurons in culture by brain-derived exosomes of AD patients with high amounts of Aβ oligomer, leading to neurotoxicity. It was revealed that ESCRT proteins TSG101 and VPS4A knockdown decreased toxicity and oligomer spread by blocking exosome synthesis, secretion, or uptake (Sardar Sinha et al. 2018).
Exosomes have a role in the etiology of AD by spreading amyloid beta and tau, causing neuroinflammation, affecting neuronal functioning, and ultimately resulting in cell death. Neuron-derived exosomes from AD patients substantially contained less heat shock factor-1 (HFS1), repressor element 1-silencing transcription factor (REST), and low-density lipoprotein receptor-related protein 6 (LRP6) compared to controls. As compared to controls, AD patients had considerably greater amounts of the pro-inflammatory substances IL-1, TNF-α, and IL-1 in ADEs as well as the complement components C1q, C4b, and C3d. The subsequent research showed that patients at the CE 2 stage had higher levels of complement proteins and lower regulatory proteins than those in the CE 1 preclinical stage.
The exosomes released by astrocytes that have been exposed to Aβ contain the protein known as the proapoptotic prostate apoptosis 4 (PAR-4), which causes astrocytes in the culture to undergo apoptosis (Liu et al. 2020). Additionally, serum from AD patients as well as brain tissue and serum from the 5XFAD mice model includes ceramide-enriched and astrocyte-derived exosomes, both of which are linked to the Aβ pathology. These exosomes migrated to the mitochondria both in vivo and in vitro, caused mitochondrial clustering, and elevated the amount of the fission protein DRP1 in neurons. Further, Aβ-associated exosomes encouraged Aβ-binding to voltage-dependent anion channel (VDAC1), which led to neurite rupturing, caspase activation, and neuronal death (Elsherbini et al. 2020a, b; Zhang et al. 2021).
Even though exosomes first seemed to be damaging to AD, a growing corpus of current research indicates that exosomes may be beneficial against the condition. Exosomes may include a range of molecules that primarily work by restoring neuronal function or Aβ clearance to mediate protective effects against AD. For instance, a protein with significant neuroprotective effects like cystatin-C is present in exosomes (Pérez-González et al. 2019; Zhang et al. 2021). Statins were acknowledged in lowering the possibility of AD and might change the content as well as secretion of exosomes. Exosomes produced by BV-2 microglial cells and neuroblastoma cells treated with statins have been shown to encourage extracellular Aβ degradation through exosome IDE (Zhang et al. 2021). Exosomes can release metalloproteases to the extracellular environment to help in the breakdown of Aβ, such as endothelin-converting enzymes (ECE) 1 and 2. The inhibition of metalloprotease disrupts Aβ catabolism, which raises Aβ levels, and promotes the production of Aβ oligomers intracellularly (Pacheco-Quinto et al. 2019). Aβ oligomerization has been demonstrated to be inhibited in vitro by exosome secretion from neuronal cells via enhancing microglia-mediated Aβ clearance.
10.4 Therapeutic Role of Stem Cells and Derived Exosomes in PD
In 1817, James Parkinson described “shaking palsy,” which is known as Parkinson’s disease (PD) and is associated with dyskinesias and dystonias, and several other motor and nonmotor symptoms (Jankovic and Tan 2020). In addition to multiple-system atrophy, progressive supranuclear palsy, chorea, and ataxia, PD is the most prevalent mobility disorder. PD has affected more than ten million people globally, and it was reported that approximately 6.2 million individuals have PD in 2015 (Dorsey et al. 2018). Although its prevalence increases with age, sex being a contributing factor as men are predominantly afflicted by PD, research also indicated that the load of PD would improve significantly in the coming decades (Van Den Eeden et al. 2003; Wanneveich et al. 2018). Consequently, this chronic neurodegenerative disease PD has severely challenged healthcare systems. It is linked with several risk factors, such as oxidative stress, the production of free radicals, and several environmental pollutants, and genetic abnormalities (Chen and Ritz 2018; Zhou et al. 2008). The clinical hallmarks of PD are a premature selective loss of midbrain dopamine neurons (DA) and a buildup of Lewy bodies, which are composed of misfolded α-synuclein and accumulate in several systems in patients with PD (Rizek et al. 2016). Before the 1990s, PD was doubted significantly to be heritable due to sporadic genesis; nonetheless, it was discovered that 5–10% of PD patients have a conventional Mendelian inheritance pattern and that 15% of patients have a family history of the disease (Duvoisin 1984; Lesage and Brice 2009). Mutations in SNCA, LRRK2, and VPS35 are the root causes of inherited monogenic and idiopathic cases of PD. Premature PD cases, before 40 years of age, are linked to autosomal recessive variations such as PARKIN, PINKI, and DJ1, although autosomal dominant mutation variations such as LRRK2 and GBA are associated with delayed PD, after 50 years of age (Cook et al. 2021). The commencement age, diagnosis age, ethnicity, and family history are significant considerations in the genetic diagnosis of PD. Therefore, single-gene screening is only useful in certain situations, such as those with a family history of Gaucher disease and African-Berber ancestry (as they have an increased risk of carrying LRRK2 mutations); in other cases, it is not reliable.
The phenotype of PD cannot often be articulated, even though a multigene panel can determine the cause (Cook et al. 2021). The list keeps growing as more genes are found to be correlated to the onset of PD. As a result, screening of only these genes is available in commercial laboratories. Additionally, whether the patient has the known mutation or not, there is currently no difference in the therapy or administration of PD. It could be achieved by the advent of clinical trials targeting specific mutations. Deaths attributed to PD rise with age. Deaths frequently occur before advanced disease stages, and causes (such as aspiration pneumonia) of death of PD patients are like non-PD patients’ cohorts, as written on the death certificates (Armstrong and Okun 2020). The L-DOPA treatment for PD was developed by Hornykiewicz et al. based on Carlsson’s discoveries (Lees et al. 2015). This strategy remunerates for lower dopaminergic levels via encouraging dopaminergic synthesis in dopaminergic neurons in the midbrain; however, effects of L-DOPA were frequently erratic, even among the same patients, and frequently brought on deep and unbearable side effects like emotional disturbances, motor fluctuations, dyskinesia, and psychiatric conditions (Iarkov et al. 2020). Although several drugs can reduce the disease’s motor symptoms, therapies for PD do not cure the condition. To look over this, many therapeutics are emerging for the prognosis of PD; however, they always result in side effects or are less effective (Clarke 2008; Jagadeesan et al. 2017). So the need for a novel targeted therapy is required for efficient outcomes; in this part, we will talk about the novelty of stem cells and derived exosomes in treating PD.
10.4.1 Stem Cells as Therapeutic for PD
There is a potential pool of cells that can be employed for neural grafting since it is possible to influence the outcome of these cells to become dopaminergic neurons. ESCs and iPSCs are the most promising stem cell types when it comes to treating PD (Stoker 2018). By using in vitro fertilization techniques, many human ESC cell lines have been generated that are derived using the early blastocyst’s ICM. Techniques to direct the development of cells into dopaminergic neurons emerged over the following decade (Kriks et al. 2011). Since ESCs are believed to be the most pluripotent, they can, under specific circumstances, develop cells from all three primary germ layers (Murry and Keller 2008). Although the yield of TH-positive cells was quite variable, studies have indicated that expression of tyrosine hydroxylase (TH), the rate-limiting enzyme for the synthesis of dopamine, could be increased. However, it was shown that these cells can be transplanted into rodents and generate some degree of motor recovery (Brederlau et al. 2006; Grealish et al. 2014; Roy et al. 2006). PD treatment was recently reported to use the first dopamine neuron cell product produced from ESCs. Scientists have long been a driving force in the creation of methods to create dopamine neurons from hPSCs (human pluripotent stem cells) (Piao et al. 2021). These characteristics result from telomerase, which extends telomeres and slows the aging in hESCs (Hiyama and Hiyama 2007). When it comes to developmental potential, murine ESCs and human ESCs differ from each other. hESCs have a limited capacity for development compared to murine ESCs, which can differentiate into tissues from the three germ layers (Pera et al. 2000). Additionally, ESCs cannot be implanted directly into the substantia nigra due to their strong tumorigenic potential in undifferentiated conditions. It is generally believed to use a combination of fibroblast growth-8, sonic hedgehog, and brain-derived neurotrophic factor to develop ECS into DA neurons. Furthermore, MS5 cells and ESCs must be co-cultured (Kriks et al. 2011; Guo et al. 2021). However, due to involvement in the destruction of human embryos, ethical and political controversies are in hESC research (Lo and Parham 2009b).
iPSCs provide a platform to investigate how genetic defects affect the likelihood of developing the disease as they have the complete genomic sequence of a patient. By adopting methods like those used with ESCs, the iPSCs produced in a manner that can be differentiated into DA, which could serve as the foundation for efficient cell-based therapy for PD (Soldner et al. 2009). iPSC-derived grafts have an advantage over ESC-derived grafts in that a patient’s fibroblasts can be used to generate a neural graft product, negating the need for immunosuppression in ESC-derived grafts (Stoker 2018). Several investigations have demonstrated the potential of autologous iPSC-derived dopaminergic neurons or precursors to survive in vivo through transplantation to primates or murine PD models (Song et al. 2020; Hallett et al. 2015). Improvements in motor function were seen after iPSC-derived dopaminergic neurons that had undergone cultured differentiation were implanted into the putamen of Parkinsonian Cygnus monkeys (Hallett et al. 2015). From an iPSC cell line, Song et al. produced clinical-grade dopaminergic neural progenitors of the midbrain. Following the implantation of 100,000–300,000 of these cells into the striatum, the motor function of mice with immunodeficiency-induced PD significantly improved after 14 weeks; further improvement persisted for at least 52 weeks. These cells were created under good manufacturing practices (GMPs) with special procedure following the crucial components: an episomal vector and specific miRNA improved the efficiency of converting fibroblasts to iPSCs (Song et al. 2020).
The first iPSCs harboring SNCA triple replication and iPSCs that have been differentiated into dopaminergic neurons were obtained by Devine et al. These iPSC-derived dopaminergic neurons successfully reproduced the α-syn accumulation characteristic of PD that was not seen in PD patient skin fibroblasts (Devine et al. 2011). PD is characterized by the accumulation of α-syn, the intrinsic upregulation of oxidative stress markers, and peroxide-induced oxidation in SNCA triplication is linked to increased expression and accumulation of α-syn (Byers et al. 2011). SNCA triplication iPSC-derived neural stem cells are more vulnerable and sensitive to oxidative stress under environment toxins or oxidative stress conditions. Importantly, knocking down endogenous α-syn allows for the reversal of this phenotype (Flierl et al. 2014).
10.4.2 Derived Exosomes in PD
Scientists are using new targeted strategies, such as derived exosomes, to overcome the problem of neurodegenerative diseases, as cell-cell communication is the main feature of it. Also, exosomes can be used as biomarkers for metabolic diseases to diagnose disease risk factors for disease and perhaps treat or prevent disease, and they are also important in the treatment of other diseases such as cancer (Dai et al. 2020).
The key diagnostic criteria for PD are the apparent clinical motor symptoms. However, several non-motor signs are present prior to the onset of motor symptoms (Chaudhuri et al. 2006). Many drugs are used to treat PD, but provide only temporary relief and have adverse side effects as the disease progresses (Müller 2012). Most drugs tested for CNS conditions during clinical trials failed because they would not be able to cross the BBB (Banks et al. 2020; Pardridge 2012). In both in vivo and in vitro experiments, the BBB was crossed by human blood exosomes and delivered dopamine to the brain due to a connection between transferrin and its receptor. When administered intravenously or systemically, dopamine-containing exosomes were less toxic than free dopamine and more therapeutically effective in the PD mouse model (Qu et al. 2018).
MSC-derived exosomes have demonstrated efficacy in treating many clinical conditions such as PD, multiple sclerosis, and osteoarthritis (Li et al. 2019; Vilaça-Faria et al. 2019). In 6-OHDA mouse models of PD, MSC-derived exosomes were found to repair dopaminergic neurons, offering a potential therapy for PD. MSC-derived exosomes associate with neuron cells in the PD animal model to reduce neuroinflammation and enhance neurogenesis along with the expression of advantageous miRNAs. In MSC-derived exosomes, miR-21 and miR-143 significantly influenced immune regulation and neural death (Mianehsaz et al. 2019). As one of the miRNAs downregulated in PD, miR-133b can be supplied to neural cells via MSC-derived exosomes to promote neurite outgrowth. Furthermore, mimic-miR-7 can enhance the neuroinflammation response in PD by modifying MSC-derived exosomes to reduce α-syn aggregation and repress NLRP3 inflammasome activation in SNpc and striatum. The modification in antago-miR-155 can also lessen neuroinflammation and microglia cell activation, therefore perchance beneficial for PD (Mianehsaz et al. 2019).
10.5 Conclusion
Recent research revealing therapeutic advantages for several neurodegenerative diseases in stem cells have promising translational significance. Numerous studies have described the fundamental processes, which range from cell replacement and paracrine actions at the region of neurodegeneration to proliferation, differentiation, and immunomodulation. Depending on the nature and origins of stem cells, preclinical investigations have revealed a variety of impacts. But there are some restrictions that prevent employing of regenerative cell therapy derived from stem cells. Studies indicated that grafts failed to show an advantage of transplantation (J. Y. Li and Li 2021). However, the advantages are noted in countless animal research and human pilot trials; therefore, an in-depth analysis of the sources, types, stages, dosages, and methods is needed to establish the optimum treatment outcome for stem cell transplantation in AD and PD models (Reuter et al. 2008). The different stages of AD and PD progression, along with other related diseases, may significantly affect how cell transplantation functions. Additionally, exosomes are subcellular components that contribute to the start, progression, and spread of AD and PD by releasing harmful substances including misfolded α-syn and inflammatory mediators into the environment. Through the isolation and identification of EV cargo, new biomarkers for AD and PD diagnosis have been discovered. Two main factors embody exosomes as the ideal drug delivery platforms in treating AD and PD and their ability to cross the BBB and low immunogenic activities. Nonetheless, further investigations are needed into the molecular mechanism in the pathology of AD and PD and directly categorize α-syn in PD.
References
Airola MV, Hannun YA (2013) Sphingolipid metabolism and neutral sphingomyelinases. Handb Exp Pharmacol 215(215):57–76. https://doi.org/10.1007/978-3-7091-1368-4_3
Allan D, Tieu A, Lalu M, Burger D (2020) Mesenchymal stromal cell-derived extracellular vesicles for regenerative therapy and immune modulation: progress and challenges toward clinical application. Stem Cells Transl Med 9(1):39. https://doi.org/10.1002/SCTM.19-0114
Amoroso MW, Croft GF, Williams DJ, O’Keeffe S, Carrasco MA, Davis AR, Roybon L, Oakley DH, Maniatis T, Henderson CE, Wichterle H (2013) Accelerated high-yield generation of limb-innervating motor neurons from human stem cells. J Neurosci 33(2):574–586. https://doi.org/10.1523/JNEUROSCI.0906-12.2013
Armstrong MJ, Okun MS (2020) Diagnosis and treatment of Parkinson disease: a review. JAMA–J Am Med Assoc 323(6):548–560. https://doi.org/10.1001/JAMA.2019.22360
Association A (2016) 2016 Alzheimer’s disease facts and figures. Alzheimers Dement 12(4):459–509
Aubry L, Bugi A, Lefort N, Rousseau F, Peschanski M, Perrier AL (2008) Striatal progenitors derived from human ES cells mature into DARPP32 neurons in vitro and in quinolinic acid-lesioned rats. Proc Natl Acad Sci U S A 105(43):16707–16712. https://doi.org/10.1073/PNAS.0808488105/SUPPL_FILE/0808488105SI.PDF
Bachoud-Lévi A-C, Massart R, Rosser A, Anne-Catherine Bachoud-Lévi C, de Neurologie S, Henri Mondor H (2021) Cell therapy in Huntington’s disease: taking stock of past studies to move the field forward. Stem Cells 39(2):144–155. https://doi.org/10.1002/STEM.3300
Bae J, Jin HK, Lee JK, Richardson JC, Carter JE (2013) Bone marrow-derived mesenchymal stem cells contribute to the reduction of amyloid-β deposits and the improvement of synaptic transmission in a mouse model of pre-dementia Alzheimer’s disease. Curr Alzheimer Res 10(5):524–531
Bang OY, Kim EH, Cho YH, Oh MJ, Chung J-W, Chang WH, Kim Y-H, Yang SW, Chopp M (2022) Circulating extracellular vesicles in stroke patients treated with mesenchymal stem cells: a biomarker analysis of a randomized trial. Stroke 53(7). https://doi.org/10.1161/STROKEAHA.121.036545
Bang OY, Kim JE (2022) Stem cell-derived extracellular vesicle therapy for acute brain insults and neurodegenerative diseases. BMB Rep 55(1):20–29. https://doi.org/10.5483/BMBREP.2022.55.1.162
Banks WA, Sharma P, Bullock KM, Hansen KM, Ludwig N, Whiteside TL (2020) Transport of extracellular vesicles across the blood-brain barrier: brain pharmacokinetics and effects of inflammation. Int J Mol Sci 21(12):1–21. https://doi.org/10.3390/IJMS21124407
Bhandari J, Thada PK, Samanta D (2022) Spinocerebellar ataxia. StatPearls. https://www.ncbi.nlm.nih.gov/books/NBK557816/
Bobrie A, Colombo M, Raposo G, Théry C (2011) Exosome secretion: molecular mechanisms and roles in immune responses. Traffic 12(12):1659–1668. https://doi.org/10.1111/J.1600-0854.2011.01225.X
Brederlau A, Correia AS, Anisimov SV, Elmi M, Paul G, Roybon L, Morizane A, Bergquist F, Riebe I, Nannmark U, Carta M, Hanse E, Takahashi J, Sasai Y, Funa K, Brundin P, Eriksson PS, Li J-Y (2006) Transplantation of human embryonic stem cell-derived cells to a rat model of Parkinson’s disease: effect of in vitro differentiation on graft survival and Teratoma formation. Stem Cells 24(6):1433–1440. https://doi.org/10.1634/STEMCELLS.2005-0393
Brotman RG, Moreno-Escobar MC, Joseph J, Pawar G (2022) Amyotrophic lateral sclerosis. StatPearls. https://www.ncbi.nlm.nih.gov/books/NBK556151/
Bruno S, Grange C, Deregibus MC, Calogero RA, Saviozzi S, Collino F, Morando L, Busca A, Falda M, Bussolati B, Tetta C, Camussi G (2009) Mesenchymal stem cell-derived microvesicles protect against acute tubular injury. J Am Soc Nephrol: JASN 20(5):1053–1067. https://doi.org/10.1681/ASN.2008070798
Budnik V, Ruiz-Cañada C, Wendler F (2016) Extracellular vesicles round off communication in the nervous system. Nat Rev Neurosci 17(3):160–172. https://doi.org/10.1038/NRN.2015.29
Byers B, Cord B, Nguyen HN, Schüle B, Fenno L, Lee PC, Deisseroth K, Langston JW, Pera RR, Palmer TD (2011) SNCA triplication Parkinson’s Patient’s iPSC-derived DA neurons accumulate α-Synuclein and are susceptible to oxidative stress. PLoS One 6(11):e26159. https://doi.org/10.1371/JOURNAL.PONE.0026159
Chagastelles PC, Nardi NB (2011) Biology of stem cells: an overview. Kidney Int Suppl 1(3):63–67. https://doi.org/10.1038/KISUP.2011.15
Chang YK, Chen MH, Chiang YH, Chen YF, Ma WH, Tseng CY, Soong BW, Ho JH, Lee OK (2011) Mesenchymal stem cell transplantation ameliorates motor function deterioration of spinocerebellar ataxia by rescuing cerebellar Purkinje cells. J Biomed Sci 18(1). https://doi.org/10.1186/1423-0127-18-54
Chaudhuri KR, Healy DG, Schapira AHV (2006) Non-motor symptoms of Parkinson’s disease: diagnosis and management. Lancet Neurol 5(3):235–245. https://doi.org/10.1016/S1474-4422(06)70373-8
Chen H, Ritz B (2018) The search for environmental causes of Parkinson’s disease: moving forward. J Parkinsons Dis 8(Suppl 1):S9. https://doi.org/10.3233/JPD-181493
Clarke CE (2008) Medical Management of Parkinson’s disease. Pharm Ther 33(10):590. https://doi.org/10.1136/jnnp.72.suppl_1.i22
Cook L, Schulze J, Kopil C, Hastings T, Naito A, Wojcieszek J, Payne K, Alcalay RN, Klein C, Saunders-Pullman R, Simuni T, Foroud T (2021) Genetic testing for Parkinson disease. Neurol: Clin Pract 11(1):69–77. https://doi.org/10.1212/CPJ.0000000000000831
Dai J, Su Y, Zhong S, Cong L, Liu B, Yang J, Tao Y, He Z, Chen C, Jiang Y (2020) Exosomes: key players in cancer and potential therapeutic strategy. Signal Transduct Target Ther 5(1):1–10. https://doi.org/10.1038/s41392-020-00261-0
Elise D, Stephanie M, Kiminobu S (2010) Stem cells for the treatment of neurodegenerative diseases. Stem Cell Res Ther. https://stemcellres.biomedcentral.com/track/pdf/10.1186/scrt37.pdf
Dawson TM, Golde TE, Lagier-Tourenne C (2018) Animal models of neurodegenerative diseases. Nat Neurosci 21(10):1370. https://doi.org/10.1038/S41593-018-0236-8
Devine MJ, Ryten M, Vodicka P, Thomson AJ, Burdon T, Houlden H, Cavaleri F, Nagano M, Drummond NJ, Taanman JW, Schapira AH, Gwinn K, Hardy J, Lewis PA, Kunath T (2011) Parkinson’s disease induced pluripotent stem cells with triplication of the α-synuclein locus. Nat Commun 2(1):1–10. https://doi.org/10.1038/ncomms1453
Dorsey ER, Sherer T, Okun MS, Bloemd BR (2018) The emerging evidence of the Parkinson pandemic. J Parkinsons Dis 8(s1):S3–S8. https://doi.org/10.3233/JPD-181474
Dulak J, Szade K, Szade A, Nowak W, Józkowicz A (2015) Adult stem cells: hopes and hypes of regenerative medicine. Acta Biochim Pol 62(3):329–337. https://doi.org/10.18388/abp.2015_1023
Duncan T, Valenzuela M (2017) Alzheimer’s disease, dementia, and stem cell therapy. Stem Cell Res Ther 8(1):111. https://doi.org/10.1186/s13287-017-0567-5
Duvoisin RC (1984) Is Parkinson’s disease acquired or inherited? Can J Neurol Sci 11:151–155. https://doi.org/10.1017/S031716710004631X
Elsherbini A, Kirov AS, Dinkins MB, Wang G, Qin H, Zhu Z, Tripathi P, Crivelli SM, Bieberich E (2020a) Association of Aβ with ceramide-enriched astrosomes mediates Aβ neurotoxicity. Acta Neuropathol Commun 8(1):1–19
Elsherbini A, Qin H, Zhu Z, Tripathi P, Crivelli SM, Bieberich E (2020b) In vivo evidence of exosome-mediated Aβ neurotoxicity. Acta Neuropathol Commun 8(1):1–3
Feigin VL, Vos T (2019) Global burden of neurological disorders: from global burden of disease estimates to actions. Neuroepidemiology 52:1–2. https://doi.org/10.1159/000495197
Ferri CP, Prince M, Brayne C, Brodaty H, Fratiglioni L, Ganguli M, Hall K, Hasegawa K, Hendrie H, Huang Y (2005) Global prevalence of dementia: a Delphi consensus study. Lancet 366(9503):2112–2117
Flierl A, Oliveira LMA, Falomir-Lockhart LJ, Mak SK, Hesley J, Soldner F, Arndt-Jovin DJ, Jaenisch R, Langston JW, Jovin TM, Le BS (2014) Higher vulnerability and stress sensitivity of neuronal precursor cells carrying an alpha-Synuclein gene triplication. PLoS One 9(11):e112413. https://doi.org/10.1371/JOURNAL.PONE.0112413
Glass JD, Boulis NM, Johe K, Rutkove SB, Federici T, Polak M, Kelly C, Feldman EL (2012) Lumbar intraspinal injection of neural stem cells in patients with amyotrophic lateral sclerosis: results of a phase I trial in 12 patients. Stem Cells (Dayton, Ohio) 30(6):1144–1151. https://doi.org/10.1002/STEM.1079
Goetzl EJ, Schwartz JB, Abner EL, Jicha GA, Kapogiannis D (2018) High complement levels in astrocyte-derived exosomes of Alzheimer’s disease. Ann Neurol 83(3):544. https://doi.org/10.1002/ANA.25172
Grealish S, Diguet E, Kirkeby A, Mattsson B, Heuer A, Bramoulle Y, Van Camp N, Perrier AL, Hantraye P, Björklund A, Parmar M (2014) Human ESC-derived dopamine neurons show similar preclinical efficacy and potency to fetal neurons when grafted in a rat model of Parkinson’s disease. Cell Stem Cell 15(5):653–665. https://doi.org/10.1016/J.STEM.2014.09.017/ATTACHMENT/47BFF55A-6C68-46BB-B83D-B2C73155AAFD/MMC1.PDF
Gu G, Zhang W, Li M, Ni J, Wang P (2015) Transplantation of NSC-derived cholinergic neuron-like cells improves cognitive function in APP/PS1 transgenic mice. Neuroscience 291:81–92
Guo M, Yin Z, Chen F, Lei P (2020) Mesenchymal stem cell-derived exosome: a promising alternative in the therapy of Alzheimer’s disease. Alzheimers Res Ther 12(1):109. https://doi.org/10.1186/s13195-020-00670-x
Guo X, Tang L, Tang X (2021) Current developments in cell replacement therapy for Parkinson’s disease. Neuroscience 463:370–382. https://doi.org/10.1016/J.NEUROSCIENCE.2021.03.022
Gurdon JB (1962) The developmental capacity of nuclei taken from intestinal epithelium cells of feeding tadpoles. Development 10(4):622–640. https://doi.org/10.1242/DEV.10.4.622
Hallett PJ, Deleidi M, Astradsson A, Smith GA, Cooper O, Osborn TM, Sundberg M, Moore MA, Perez-Torres E, Brownell AL, Schumacher JM, Spealman RD, Isacson O (2015) Successful function of autologous iPSC-derived dopamine neurons following transplantation in a non-human primate model of Parkinson’s disease. Cell Stem Cell 16(3):269–274. https://doi.org/10.1016/J.STEM.2015.01.018/ATTACHMENT/A5BD7CDE-4426-4170-89E7-7F91F73CE3F6/MMC1.PDF
Han Z, Tian R, Ren P, Zhou W, Wang P, Luo M, Jin S, Jiang Q (2018) Parkinson’s disease and Alzheimer’s disease: a Mendelian randomization study. BMC Med Genet 19(Suppl 1). https://doi.org/10.1186/S12881-018-0721-7
Hayashi K, Hashimoto M, Koda M, Naito AT, Murata A, Okawa A, Takahashi K, Yamazaki M (2011) Increase of sensitivity to mechanical stimulus after transplantation of murine induced pluripotent stem cell–derived astrocytes in a rat spinal cord injury model: laboratory investigation. J Neurosurg Spine 15(6):582–593. https://doi.org/10.3171/2011.7.SPINE10775
Henne WM, Buchkovich NJ, Emr SD (2011) The ESCRT pathway. Dev Cell 21(1):77–91. https://doi.org/10.1016/J.DEVCEL.2011.05.015
Hiyama E, Hiyama K (2007) Telomere and telomerase in stem cells. Br J Cancer 96(7):1020–1024. https://doi.org/10.1038/sj.bjc.6603671
Iarkov A, Barreto GE, Grizzell JA, Echeverria V (2020) Strategies for the treatment of Parkinson’s disease: beyond dopamine. Front Aging Neurosci 12:4. https://doi.org/10.3389/FNAGI.2020.00004/BIBTEX
Institute of Medicine (2002) 2002. The National Academies Press, Stem Cells and the Future of Regenerative Medicine. https://doi.org/10.17226/10195
Jagadeesan AJ, Murugesan R, Vimala Devi S, Meera M, Madhumala G, Vishwanathan Padmaja M, Ramesh A, Banerjee A, Sushmitha S, Khokhlov AN, Marotta F, Pathak S (2017) Current trends in etiology, prognosis and therapeutic aspects of Parkinson’s disease: a review. Acta Bio Medica : Atenei Parmensis 88(3):249. https://doi.org/10.23750/abm.v%vi%i.6063
Jahan S, Mukherjee S, Ali S, Bhardwaj U, Choudhary RK, Balakrishnan S, Naseem A, Mir SA, Banawas S, Alaidarous M, Alyenbaawi H, Iqbal D, Siddiqui AJ (2022) Pioneer role of extracellular vesicles as modulators of cancer initiation in progression, drug therapy, and vaccine prospects. Cell 11(3). https://doi.org/10.3390/CELLS11030490
Jankovic J, Tan EK (2020) Parkinson’s disease: etiopathogenesis and treatment. J Neurol Neurosurg Psychiatry 91(8):795–808. https://doi.org/10.1136/JNNP-2019-322338
Kang JM, Yeon BK, Cho S-J, Suh Y-H (2016) Stem cell therapy for Alzheimer’s disease: a review of recent clinical trials. J Alzheimers Dis 54(3):879–889
Kent SA, Spires-Jones TL, Durrant CS (2020) The physiological roles of tau and Aβ: implications for Alzheimer’s disease pathology and therapeutics. Acta Neuropathol 140(4):417–447
Khan H, Pan JJ, Li Y, Zhang Z, Yang GY (2021) Native and bioengineered exosomes for ischemic stroke therapy. Front Cell Dev Biol 9:619565. https://doi.org/10.3389/FCELL.2021.619565
Kim DS, Lee DR, Kim HS, Yoo JE, Jung SJ, Lim BY, Jang J, Kang HC, You S, Hwang DY, Leem JW, Nam TS, Cho SR, Kim DW (2012) Highly pure and expandable PSA-NCAM-positive neural precursors from human ESC and iPSC-derived neural rosettes. PLoS One 7(7):e39715. https://doi.org/10.1371/JOURNAL.PONE.0039715
Kim SW, Jang YJ, Chang JW, Hwang O (2003) Degeneration of the nigrostriatal pathway and induction of motor deficit by tetrahydrobiopterin: an in vivo model relevant to Parkinson’s disease. Neurobiol Dis 13(2):167–176. https://doi.org/10.1016/S0969-9961(03)00037-8
Kriks S, Shim JW, Piao J, Ganat YM, Wakeman DR, Xie Z, Carrillo-Reid L, Auyeung G, Antonacci C, Buch A, Yang L, Beal MF, Surmeier DJ, Kordower JH, Tabar V, Studer L (2011) Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature 480(7378):547–551. https://doi.org/10.1038/nature10648
Kumar A, Sidhu J, Goyal A, Tsao JW (2021) Alzheimer disease. StatPearls:1–27. https://www.ncbi.nlm.nih.gov/books/NBK499922/
Lakshmi S, Essa MM, Hartman RE, Guillemin GJ, Sivan S, Elumalai P (2020) Exosomes in Alzheimer’s disease: potential role as pathological mediators, biomarkers and therapeutic targets. Neurochem Res 45(11):2553–2559. https://doi.org/10.1007/S11064-020-03111-1
Lamptey RNL, Chaulagain B, Trivedi R, Gothwal A, Layek B, Singh J (2022) A review of the common neurodegenerative disorders: current therapeutic approaches and the potential role of nanotherapeutics. Int J Mol Sci 23(3):1851. https://doi.org/10.3390/ijms23031851
Laulagnier K, Javalet C, Hemming FJ, Chivet M, Lachenal G, Blot B, Chatellard C, Sadoul R (2017) Amyloid precursor protein products concentrate in a subset of exosomes specifically endocytosed by neurons. Cell Mol Life Sci 75(4):757–773. https://doi.org/10.1007/S00018-017-2664-0
Lee HJ, Lee JK, Lee H, Carter JE, Chang JW, Oh W, Yang YS, Suh J-G, Lee B-H, Jin HK (2012) Human umbilical cord blood-derived mesenchymal stem cells improve neuropathology and cognitive impairment in an Alzheimer’s disease mouse model through modulation of neuroinflammation. Neurobiol Aging 33(3):588–602
Lee S, Mankhong S, Kang J-H (2019) Extracellular vesicle as a source of Alzheimer’s biomarkers: opportunities and challenges. Int J Mol Sci 20(7):1728
Lees AJ, Tolosa E, Olanow CW (2015) Four pioneers of L-dopa treatment: Arvid Carlsson, Oleh Hornykiewicz, George Cotzias, and Melvin Yahr. Mov Disord 30(1):19–36. https://doi.org/10.1002/MDS.26120
Lesage S, Brice A (2009) Parkinson’s disease: from monogenic forms to genetic susceptibility factors. Hum Mol Genet 18(R1):R48–R59. https://doi.org/10.1093/HMG/DDP012
Li JY, Li W (2021) Postmortem studies of fetal grafts in Parkinson’s disease: what lessons have we learned? Front Cell Dev Biol 9. https://doi.org/10.3389/FCELL.2021.666675/FULL
Li Z, Liu F, He X, Yang X, Shan F, Feng J (2019) Exosomes derived from mesenchymal stem cells attenuate inflammation and demyelination of the central nervous system in EAE rats by regulating the polarization of microglia. Int Immunopharmacol 67:268–280. https://doi.org/10.1016/J.INTIMP.2018.12.001
Liu X-Y, Yang L-P, Zhao L (2020) Stem cell therapy for Alzheimer’s disease. World J Stem Cells 12(8):787
Lo B, Parham L (2009a) Ethical issues in stem. Cell Res. https://doi.org/10.1210/er.2008-0031
Lo B, Parham L (2009b) Ethical issues in stem cell research. Endocr Rev 30(3):204. https://doi.org/10.1210/ER.2008-0031
Maloney JA, Bainbridge T, Gustafson A, Zhang S, Kyauk R, Steiner P, van der Brug M, Liu Y, Ernst JA, Watts RJ (2014) Molecular mechanisms of Alzheimer disease protection by the A673T allele of amyloid precursor protein. J Biol Chem 289(45):30990–31000
Mao Z, Zhang S, Chen H (2015) Stem cell therapy for amyotrophic lateral sclerosis. Cell Regen 4(1):11. https://doi.org/10.1186/S13619-015-0026-7
Matthews KA, Xu W, Gaglioti AH, Holt JB, Croft JB, Mack D, McGuire LC (2019) Racial and ethnic estimates of Alzheimer’s disease and related dementias in the United States (2015–2060) in adults aged ≥65 years. Alzheimers Dement 15(1):17–24. https://doi.org/10.1016/J.JALZ.2018.06.3063
Mazzini L, Fagioli F, Boccaletti R, Mareschi K, Oliveri G, Olivieri C, Pastore I, Marasso R, Madon E (2003) Stem cell therapy in amyotrophic lateral sclerosis: a methodological approach in humans. Amyotroph Lateral Scler Other Motor Neuron Disord 4(3):158–161. https://doi.org/10.1080/14660820310014653
Mianehsaz E, Mirzaei HR, Mahjoubin-Tehran M, Rezaee A, Sahebnasagh R, Pourhanifeh MH, Mirzaei H, Hamblin MR (2019) Mesenchymal stem cell-derived exosomes: a new therapeutic approach to osteoarthritis? Stem Cell Res Therapy 10(1):1–13. https://doi.org/10.1186/S13287-019-1445-0
Mirzaaghasi A, Han Y, Ahn SH, Choi C, Park JH (2021) Biodistribution and Pharmacokinectics of liposomes and exosomes in a mouse model of sepsis. Pharmaceutics 13(3). https://doi.org/10.3390/PHARMACEUTICS13030427
Moghadam FH, Alaie H, Karbalaie K, Tanhaei S, Esfahani MHN, Baharvand H (2009) Transplantation of primed or unprimed mouse embryonic stem cell-derived neural precursor cells improves cognitive function in Alzheimerian rats. Differentiation 78(2–3):59–68
Muller L (2020) Exosomes: nanodust? HNO 68(1):56–59. https://doi.org/10.1007/S00106-019-00786-Z
Müller T (2012) Drug therapy in patients with Parkinson’s disease. Transl Neurodegen 1(1):1–13. https://doi.org/10.1186/2047-9158-1-10/TABLES/5
Muratore CR, Rice HC, Srikanth P, Callahan DG, Shin T, Benjamin LNP, Walsh DM, Selkoe DJ, Young-Pearse TL (2014) The familial Alzheimer’s disease APPV717I mutation alters APP processing and tau expression in iPSC-derived neurons. Hum Mol Genet 23(13):3523–3536
Murry CE, Keller G (2008) Differentiation of embryonic stem cells to clinically relevant populations: lessons from embryonic development. Cell 132(4):661–680. https://doi.org/10.1016/J.CELL.2008.02.008
Naaldijk Y, Jaeger C, Fabian C, Leovsky C, Blüher A, Rudolph L, Hinze A, Stolzing A (2017) Effect of systemic transplantation of bone marrow-derived mesenchymal stem cells on neuropathology markers in APP/PS 1 Alzheimer mice. Neuropathol Appl Neurobiol 43(4):299–314
Nutt SE, Chang EA, Suhr ST, Schlosser LO, Mondello SE, Moritz CT, Cibelli JB, Horner PJ (2013) Caudalized human iPSC-derived neural progenitor cells produce neurons and glia but fail to restore function in an early chronic spinal cord injury model. Exp Neurol 248:491–503. https://doi.org/10.1016/J.EXPNEUROL.2013.07.010
Oh SH, Kim HN, Park H-J, Shin JY, Lee PH (2015) Mesenchymal stem cells increase hippocampal neurogenesis and neuronal differentiation by enhancing the Wnt signaling pathway in an Alzheimer’s disease model. Cell Transplant 24(6):1097–1109
Pacheco-Quinto J, Clausen D, Pérez-González R, Peng H, Meszaros A, Eckman CB, Levy E, Eckman EA (2019) Intracellular metalloprotease activity controls intraneuronal Aβ aggregation and limits secretion of Aβ via exosomes. FASEB J 33(3):3758–3771
Pardridge WM (2012) Drug transport across the blood-brain barrier. J Cereb Blood Flow Metab 32(11):1959–1972. https://doi.org/10.1038/jcbfm.2012.126
Pera MF, Reubinoff B, Trounson A (2000) Human embryonic stem cells. J Cell Sci 113(1):5–10. https://doi.org/10.1242/JCS.113.1.5
Pérez-González R, Sahoo S, Gauthier SA, Kim Y, Li M, Kumar A, Pawlik M, Benussi L, Ghidoni R, Levy E (2019) Neuroprotection mediated by cystatin C-loaded extracellular vesicles. Sci Rep 9(1):1–10
Piao J, Zabierowski S, Dubose BN, Hill EJ, Navare M, Claros N, Rosen S, Ramnarine K, Horn C, Fredrickson C, Wong K, Safford B, Kriks S, El Maarouf A, Rutishauser U, Henchcliffe C, Wang Y, Riviere I, Mann S et al (2021) Preclinical efficacy and safety of a human embryonic stem cell-derived midbrain dopamine progenitor product, MSK-DA01. Cell Stem Cell 28(2):217–229.e7. https://doi.org/10.1016/J.STEM.2021.01.004/ATTACHMENT/6B7AA27F-658E-4E60-99B0-F5AB59AD1824/MMC1.PDF
Piers RJ, Liu Y, Ang TFA, Tao Q, Au R, Qiu WQ (2021) Association between elevated depressive symptoms and cognitive function moderated by APOE4 status: framingham offspring study. J Alzheimers Dis: JAD 80(3):1269. https://doi.org/10.3233/JAD-200998
Poddar MK, Chakraborty A, Banerjee S (2021) Neurodegeneration: diagnosis, prevention, and therapy. Oxidoreductase. https://doi.org/10.5772/INTECHOPEN.94950
Prochazkova M, Chavez MG, Prochazka J, Felfy H, Mushegyan V, Klein OD (2015) Embryonic versus adult stem cells. Stem Cell Biol Tissue Engg Dental Sci 249–262. https://doi.org/10.1016/B978-0-12-397157-9.00020-5
Przedborski S, Vila M, Jackson-Lewis V (2003) Series introduction: neurodegeneration: what is it and where are we? J Clin Investig 111(1):3. https://doi.org/10.1172/JCI17522
Qu M, Lin Q, Huang L, Fu Y, Wang L, He S, Fu Y, Yang S, Zhang Z, Zhang L, Sun X (2018) Dopamine-loaded blood exosomes targeted to brain for better treatment of Parkinson’s disease. J Control Release 287:156–166. https://doi.org/10.1016/J.JCONREL.2018.08.035
Raposo G, Stoorvogel W (2013) Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol 200(4):373–383. https://doi.org/10.1083/JCB.201211138
Ratajczak J, Miekus K, Kucia M, Zhang J, Reca R, Dvorak P, Ratajczak MZ (2006) Embryonic stem cell-derived microvesicles reprogram hematopoietic progenitors: evidence for horizontal transfer of mRNA and protein delivery. Leukemia 20(5):847–856. https://doi.org/10.1038/sj.leu.2404132
Reuter I, Tai YF, Pavese N, Chaudhuri KR, Mason S, Polkey CE, Clough C, Brooks DJ, Barker RA, Piccini P (2008) Long-term clinical and positron emission tomography outcome of fetal striatal transplantation in Huntington’s disease. J Neurol Neurosurg Psychiatry 79(8):948–951. https://doi.org/10.1136/JNNP.2007.142380
Rizek P, Kumar N, Jog MS (2016) An update on the diagnosis and treatment of Parkinson disease. CMAJ 188(16):1157–1165. https://doi.org/10.1503/CMAJ.151179/-/DC1
Romito A, Cobellis G (2016) Pluripotent stem cells: current understanding and future directions. Stem Cells Int 2016. https://doi.org/10.1155/2016/9451492
Roy NS, Cleren C, Singh SK, Yang L, Beal MF, Goldman SA (2006) Functional engraftment of human ES cell–derived dopaminergic neurons enriched by coculture with telomerase-immortalized midbrain astrocytes. Nat Med 12(11):1259–1268. https://doi.org/10.1038/nm1495
Sabayan B, Sorond F (2017) Reducing risk of dementia in older age. JAMA 317(19):2028
Sakthiswary R, Raymond AA (2012) Stem cell therapy in neurodegenerative diseases: from principles to practice. Neural Regen Res 7(23):1822. https://doi.org/10.3969/J.ISSN.1673-5374.2012.23.009
Sardar Sinha M, Ansell-Schultz A, Civitelli L, Hildesjö C, Larsson M, Lannfelt L, Ingelsson M, Hallbeck M (2018) Alzheimer’s disease pathology propagation by exosomes containing toxic amyloid-beta oligomers. Acta Neuropathol 136(1):41–56. https://doi.org/10.1007/s00401-018-1868-1
Si Z, Wang X (2021) Stem cell therapies in Alzheimer’s disease: applications for disease modeling. J Pharmacol Exp Ther 377(2):207–217
Simons M, Raposo G (2009) Exosomes–vesicular carriers for intercellular communication. Curr Opin Cell Biol 21(4):575–581. https://doi.org/10.1016/J.CEB.2009.03.007
Singh S, Srivastava A, Srivastava P, Dhuriya YK, Pandey A, Kumar D, Rajpurohit CS (2016) Advances in stem cell research- A ray of hope in better diagnosis and prognosis in neurodegenerative diseases. Front Mol Biosci 3(NOV):72. https://doi.org/10.3389/FMOLB.2016.00072/BIBTEX
Sivandzade F, Cucullo L (2021) Regenerative stem cell therapy for neurodegenerative diseases: an overview. Int J Mol Sci 22(4):1–21. https://doi.org/10.3390/IJMS22042153
Soares Martins T, Trindade D, Vaz M, Campelo I, Almeida M, Trigo G, da Cruz e Silva OAB, Henriques AG (2021) Diagnostic and therapeutic potential of exosomes in Alzheimer’s disease. J Neurochem 156(2):162–181. https://doi.org/10.1111/JNC.15112
Soldner F, Hockemeyer D, Beard C, Gao Q, Bell GW, Cook EG, Hargus G, Blak A, Cooper O, Mitalipova M, Isacson O, Jaenisch R (2009) Parkinson’s disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell 136(5):964–977. https://doi.org/10.1016/J.CELL.2009.02.013/ATTACHMENT/DCDE63C5-0DCA-4577-8025-E336FDFAE083/MMC2.XLS
Song B, Cha Y, Ko S, Jeon J, Lee N, Seo H, Park KJ, Lee IH, Lopes C, Feitosa M, Luna MJ, Jung JH, Kim J, Hwang D, Cohen BM, Teicher MH, Leblanc P, Carter BS, Kordower JH, Kim KS et al (2020) Human autologous iPSC–derived dopaminergic progenitors restore motor function in Parkinson’s disease models. J Clin Invest 130(2):904–920. https://doi.org/10.1172/JCI130767
Stahl PD, Barbieri MA (2002) Multivesicular bodies and multivesicular endosomes: the “ins and outs” of endosomal traffic. Sci STKE 2002(141). https://doi.org/10.1126/STKE.2002.141.PE32
Stoker TB (2018) Stem cell treatments for Parkinson’s disease. Parkinson’s Disease: Pathogenesis and Clinical Aspects 161–175. https://doi.org/10.15586/CODONPUBLICATIONS.PARKINSONSDISEASE.2018.CH9
Sun C, Armstrong MJ (2021) Treatment of Parkinson’s disease with cognitive impairment: current approaches and future directions. Behav Sci 11(4). https://doi.org/10.3390/BS11040054
Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131(5):861–872. https://doi.org/10.1016/J.CELL.2007.11.019
Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126(4):663–676. https://doi.org/10.1016/J.CELL.2006.07.024
Théry C (2011) Exosomes: secreted vesicles and intercellular communications. F1000 Biol Rep 3(1):15. https://doi.org/10.3410/B3-15
Théry C, Amigorena S, Raposo G, Clayton A (2006) Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol 30(1):3.22.1-3.22.29. https://doi.org/10.1002/0471143030.CB0322S30
Tsai Y-A, Liu R-S, Lirng J-F, Yang B-H, Chang C-H, Wang Y-C, Wu Y-S, Hui-Chun Ho J, Lee OK, Soong B-W (2017) Treatment of spinocerebellar ataxia with mesenchymal stem cells: a phase I/IIa clinical study. Cell Transplant 26:503–512. https://doi.org/10.3727/096368916X694373
Van Den Eeden SK, Tanner CM, Bernstein AL, Fross RD, Leimpeter A, Bloch DA, Nelson LM (2003) Incidence of Parkinson’s disease: variation by age, gender, and race/ethnicity. Am J Epidemiol 157(11):1015–1022. https://doi.org/10.1093/AJE/KWG068
Vatsa P, Jahan S, Ansari UA, Khan A, Mir SA, Alshehri B, Choudhary RK, Siddiqui AJ (2022) Stem cell safety and sterility testing: a promising approach in regenerative medicine. Stem Cell Prod 205–232. https://doi.org/10.1007/978-981-16-7589-8_9
Vilaça-Faria H, Salgado AJ, Teixeira FG (2019) Mesenchymal stem cells-derived exosomes: a new possible therapeutic strategy for Parkinson’s disease? Cells 8:118. https://doi.org/10.3390/CELLS8020118
Wang Q, Matsumoto Y, Shindo T, Miyake K, Shindo A, Kawanishi M, Kawai N, Tamiya T, Nagao S (2006) Neural stem cells transplantation in cortex in a mouse model of Alzheimer’s disease. J Med Investig 53(1, 2):61–69
Wanneveich M, Moisan F, Jacqmin-Gadda H, Elbaz A, Joly P (2018) Projections of prevalence, lifetime risk, and life expectancy of Parkinson’s disease (2010-2030) in France. Movement Dis 33(9):1449–1455. https://doi.org/10.1002/MDS.27447
Wen S, Dooner M, Papa E, del Tatto M, Pereira M, Borgovan T, Cheng Y, Goldberg L, Liang O, Camussi G, Quesenberry P (2019) Biodistribution of mesenchymal stem cell-derived extracellular vesicles in a radiation injury bone marrow murine model. Int J Mol Sci 20(21). https://doi.org/10.3390/IJMS20215468
Wilmut I, Schnieke AE, McWhir J, Kind AJ, Campbell KHS (1997) Viable offspring derived from fetal and adult mammalian cells. Nature 385(6619):810–813. https://doi.org/10.1038/385810a0
Winston CN, Goetzl EJ, Akers JC, Carter BS, Rockenstein EM, Galasko D, Masliah E, Rissman RA (2016) Prediction of conversion from mild cognitive impairment to dementia with neuronally derived blood exosome protein profile. Alzheimers Dement 3:63. https://doi.org/10.1016/J.DADM.2016.04.001
Wolf P (1967) The nature and significance of platelet products in human plasma. Br J Haematol 13(3):269–288. https://doi.org/10.1111/J.1365-2141.1967.TB08741.X
Xu L, Yan J, Chen D, Welsh AM, Hazel T, Johe K, Hatfield G, Koliatsos VE (2006) Human neural stem cell grafts ameliorate motor neuron disease in SOD-1 transgenic rats. Transplantation 82(7):865–875. https://doi.org/10.1097/01.TP.0000235532.00920.7A
Xu M, Zhang L, Liu G, Jiang N, Zhou W, Zhang Y (2019) Pathological changes in Alzheimer’s disease analyzed using induced pluripotent stem cell-derived human microglia-like cells. J Alzheimers Dis 67(1):357–368
Yamanaka S, Takahashi K, Okita K, Nakagawa M (2007) Induction of pluripotent stem cells from fibroblast cultures. Nat Protoc 2(12):3081–3089. https://doi.org/10.1038/NPROT.2007.418
Zafar S, Yaddanapudi SS (2021) Parkinson Disease. StatPearls 8:1–13. https://www.ncbi.nlm.nih.gov/books/NBK470193/
Zakharova L, Svetlova M, Fomina AF (2007) T cell exosomes induce cholesterol accumulation in human monocytes via phosphatidylserine receptor. J Cell Physiol 212(1):174–181. https://doi.org/10.1002/JCP.21013
Zakrzewski W, Dobrzyński M, Szymonowicz M, Rybak Z (2019) Stem cells: past, present, and future. Stem Cell Res Therapy 10(1):1–22. https://doi.org/10.1186/S13287-019-1165-5/FIGURES/8
Zhang T, Ma S, Lv J, Wang X, Afewerky HK, Li H, Lu Y (2021) The emerging role of exosomes in Alzheimer’s disease. Ageing Res Rev 68:101321
Zhou C, Huang Y, Przedborski S (2008) Oxidative stress in Parkinson’s disease: a mechanism of pathogenic and therapeutic significance. Ann N Y Acad Sci 1147:93–104. https://doi.org/10.1196/ANNALS.1427.023
Acknowledgments
The authors thank Mr. Manish Kumar for taking the necessary time and effort to review the chapter. We sincerely acknowledge your insightful comments and recommendations, which enabled us to improve the quality of the chapter.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Ali, S., Mukherjee, S., Goel, D., Ghosh, A., Faruq, M. (2023). Role of Stem Cells and Derived Exosomes as a Novel Therapeutic Agent against Alzheimer’s and Parkinson’s Disease. In: Jahan, S., Siddiqui, A.J. (eds) Applications of Stem Cells and derived Exosomes in Neurodegenerative Disorders. Springer, Singapore. https://doi.org/10.1007/978-981-99-3848-3_10
Download citation
DOI: https://doi.org/10.1007/978-981-99-3848-3_10
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-99-3847-6
Online ISBN: 978-981-99-3848-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)