
Abstract
An important aspect of sustainable drug development is drug-target interaction. In cancer cell lines, the drug response target ratio is critical. It is important to estimate the drug reaction in a cancer cell line. In prior research, we employed ensemble algorithms with voting methods to predict medication response and achieved 97.5% accuracy. A hybrid ensemble algorithm for the revised drug response (HBEA) method is developed to improve drug-target strategy in cell lines. Rather than generating several homogeneous weak learners to generate a single model in the ensemble, this enhanced algorithm uses a diverse collection of weak learners such as random forest, Naive Bayes, and decision tree to create a strong meta-classifier. Cross-validation of hard and soft data would be used to accomplish this. The concentrations of various drugs are used as inputs, and the cell line predicts the relevant drug response. The goal of this enhanced ensemble algorithm is to suggest a new medicine based on a single licensed drug or a combination of drugs. This approach increased the drug responsiveness from 97.5 to 100%, according to our findings. The proposed method is applied in an open-source and freely available at https://decrease.fimm.fi.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
1 Introduction
People across the globe are dying from cancer at a record rate. Anti-cancer treatments are an essential part of cancer treatment, and their proper regulation can help prolong the patient’s life. Many clinical studies have shown that cancers with different genetic characteristics respond differently to the same treatment or drug [1,2,3]. Precision medicine tries to precisely pick cancer treatments based on each patient’s genetic information [4]. It is difficult to predict the response to anti-cancer treatments for individual patients in precision medicine [5,6,7]. Several cell line-based datasets are publicly available, including the National Cancer Institute 60, the Cancer Therapeutics Response Portal (CTRP), the Genomics of Drug Sensitivity in Cancer, the Cancer Cell line Encyclopedia, and Genentech Cell Line Screening Initiative (gCSI). This dataset can help to get better outcomes. Medicine’s physiochemical properties strongly influence their therapeutic index. In aqueous physiological conditions at pH 7.4, the majority of anti-cancer drugs are poorly soluble. Paclitaxel and docetaxel are axene-based medicines, and 9-nitrocamptothecin is a camptothecin derivative. To develop delivery techniques as well as lung clearance, fundamental features of anticancer drugs, such as log P and pKa values, are critical [8]. Medicinal drug’s solubility determined both the permeability and potency of their cancer therapeutic effect, according to Lipinski’s rule [9, 10]. Early cancer detection and prediction of survivorship depth can assist patients and healthcare providers in better controlling expenditures, treatment intensity, and time spent in the medical care setting. When such a condition is detected early on, the chances of a positive outcome increase. Although considerable progress has been made in the early detection of cancer [11, 12], much more research is needed to discover strategies for estimating survivorship that is both common and feasible in medical practice [13]. The introduction of targeted anti-cancer therapies based on gene-specific effects may prove a useful tool in the fight against cancer. Many clinical studies are required to develop particular targeted therapy for cancer patients in clinical treatment. However, there are other challenges, including sample limitations, difficult procedures, severe environmental standards, and expensive costs, which prevent the supply from matching demand [14]. To limit the risk of drug-target interactions, computational methods were used for these predictions. Therefore, the focus of research on the drug target in cell lines might be more effective [15, 16]. In general, drugs interact with their target molecules in three ways: (1) Machine learning-based prediction, (2) deep learning prediction, and (3) network-based prediction [16,17,18,19,20,21,22,23]. Recently, multiple classifiers have become popular in research. It is well established that by integrating multiple classifiers, classification performance can be improved over single classifiers [24,25,26]. Over the last few years, hybrid and ensemble machine learning algorithms have attracted the scientific community’s interest [27]. There is strong evidence that multiple, ensemble models perform better than single weak learners in both theory and practice, especially when dealing with multidimensional, difficult regression, and classification problems [28]. The paper is structured as follows: The literature study on similar recent techniques is described in Sect. 2. The proposed algorithm is analyzed in Sect. 3. Section 4 consists of architecture diagram which includes various components involved in drug response prediction. Materials and methodology are discussed in Sect. 5. The result has been analyzed in Sect. 6. Finally, the conclusion is presented in Sect. 7.
2 Literature Survey
Ensemble Learning
Creating and combining multiple inducers to solve a specific machine learning problem is called ensemble learning. The natural reason for the ensemble process comes from human nature and our proclivity to collect and weigh multiple viewpoints to make a complex decision. The primary premise is that evaluating and aggregating multiple individual viewpoints is preferable to choosing one individual’s opinion. Matching a medical treatment to sickness is an example of such a decision [29]. The limitation is homogeneous ensemble learning. The absence of a strong classifier to predict drug response [29].
Agarwal et al. used ensemble voting to predict lung cancer survival after 6-months, 9-months, 1-year, 2-years, and 5-years of diagnosis. Five decision tree algorithms were created using data from the SEER program. By taking the average of the probabilities generated by each classifier, they integrated. The researchers used tenfold cross-validation for training and testing and compared the performance of ensemble voting with that of individual classifiers to demonstrate how ensemble data mining can help weak classifiers perform better. The limitation of this research is only based on the clinical outcome [30, 31]. In a study published in science [32], Matlock et al. examined a variety of ensembles that were trained to predict drug sensitivity, including a deep learning method for gene expression data. There were only a limited number of cell lines and compounds used in this study.
Ensemble learning is a machine learning method in which multiple models, referred to as “weak learners,” are taught to tackle the same issue and then integrated to improve results, according to the survey on ensemble learning. The core concept is that we can generate more accurate and/or resilient models by correctly integrating weak models.
Machine Learning
Zhang [33] stated that the machine learning approach saves time and effort by eliminating the need for specialists to establish rules and threshold values because the algorithm performs these activities internally. Choosing features simplifies the model and makes it easier to understand and implement. It enhances accuracy while also cutting down on training time. It helps to support generalization by reducing overfitting. It lowers the chances of data mistakes during installation. Algorithms for machine learning are self-improving, which means they learn from examples and experiences. Even if a machine-based method is beneficial, it is necessary to focus on key features for medication analysis. Only cell line data is concentrated in feature selection in this case. It is necessary to keep an eye on the omics analysis. As a result, the reaction is still not clinically meaningful. Complementary ML models, according to Costello et al. [32], improve the predictability of drug response prediction models. It improves the model’s robustness. The work’s limitation is the tiny number of cell lines. In this research [34], the author Guosheng Lianga et al. examined that artificial intelligence plays a significant part in the discovery of new materials and intensely accelerates anti-cancer drug development. The author stated that machine learning analysis was used to predict the sensitivity of the drug. The drawback of this method is that it is tough to formulate the best treatment.
There are three types of machine learning-based methods:
Feature vector-based approach
Feature vectors are n-dimensional vectors that represent the characteristics of an object. In the field of machine learning, numerical representations of objects are commonly used because they make statistical analysis and processing easier. As an example, feature values can represent pixels of an image, and term occurrences in texts. Feature vectors are the same as explanatory variable vectors, which are utilized in statical techniques like linear regression. By pairing feature vectors with weights, a linear predictor function is constructed, which is used to calculate a prediction score. This is known as the feature space or vector space. A variety of dimensionality reduction approaches can be used to lower the dimensionality of feature space. Feature creation has long been thought to be a useful approach for improving structure accuracy and comprehension, especially in high-dimensional issues [35]. Analyzing which input features contributed most to a given prediction could help identify potential biomarkers of drug response or characteristics of drugs that would induce better drug response. Identifying new biomarkers for precision medicine is particularly important [36].
Similarity-based approach
Treating given similarities as inner products in some Hilbert space or treating dissimilarities as distances in some Euclidean space is a prominent technique for similarity-based categorization. There are a number of common techniques for similarity-based categorization, such as treating given similarity as an inner product in some Hilbert space or treating dissimilarity as distance in some Euclidean space. To begin, the samples are explicitly embedded in a Euclidean space based on the differences (dis)similarities using multidimensional scaling [37]. A second option is to embed the samples implicitly in a Euclidean space based on their similarity or dissimilarity. Santini and Jain [38] found similar functions might fail to satisfy the other mathematical requirements for metrics or inner products—especially when they are asymmetric. Similarity-based classification can be useful in computer vision, bioinformatics, information retrieval, natural language processing, and more. Some simple examples of similarity functions (edit distance) are travel time from one location to another, the compressibility of the random process given a code corresponding to another, and the minimum number of steps needed to transform one sequence into another. As part of bioinformatics, the Smith–Waterman algorithm [39], the FASTA algorithm [40], and the BLAST algorithm [41] are commonly used to determine amino acid sequence similarity in protein classification. Term frequency and inverse document frequency vector (tf-IDF) are commonly utilized in information retrieval and text mining for document categorization. There is a limitation to similarity networks in that they are only based on genome-wide gene expression profiles, but do not take into consideration somatic mutants and copy number variations within cell lines.
Network-based Prediction method
Stanfield et al. [42] propose a network-based prediction method that combines cell line genetic mutation and drug responses with protein–protein interaction (PPI) network and uses random walk with restart (RWR) to calculate feature vectors for drugs and cell lines in order to predict missing response values. Other researchers, on the other hand, are focusing on using similarities across cell lines or medications to make predictions [42, 43]. Zhang et al. [44], e.g., project multiple side information from a cell line and a drug into similarity networks to create an integrated dual-layer network that fills in the gaps [44].
While the methods described above have been proved to outperform previous methods, they do have certain drawbacks. Stanfield's method, e.g., solely takes into account gene mutation information and ignores similarity information, whereas Zhang et al. method ignores genetic variants and does not take gene correlations into account.
Cross-Validation
According to Pedregosa et al. [45], datasets were separated into training and testing sets after dimensionality reduction. To avoid overfitting and account for variance in each classifier, tenfold cross-validation was utilized, in which the dataset was randomly partitioned into training and test sets ten times. Hastie et al. and Duda et al. [46, 47] stated that cross-validation is a data resampling technique. It is used for evaluating prediction model generalization and avoiding overfitting. In [48], Efron et al. stated that cross-validation is like a bootstrap that belongs to the Monte Carlo method family. This research introduces cross-validation and resampling procedures that go with it. Because every observation is used for both training and testing, this technique uses data more “efficiently” than other machine learning algorithms.
Heterogeneous-Based Ensemble Algorithm (HBEA)
In [45], Zhang et al. stated that for solving real-world problems, ensemble machine learning algorithms are extremely powerful and adaptable. Ensemble learning improved the predictability of decision-making systems by increasing accuracy by minimizing variation. The author explained the feature selection, missing features, and data imbalance. Classifiers are integrated into a variety of ways to improve performance measurements [27], including bagging, boosting, and stacking. According to [49,50,51,52], Bhardwaj et al. developed a double ensemble machine learning algorithm to predict the pathological response after neoadjuvant chemotherapy. This double ensemble was used to predict multi-criteria decision-making. Based on the above reference to develop a strong learner from a weak learner by generating a more accurate model to predict the drug response, the HBEA technique is introduced. When compared to the homogeneous ensemble approach, the accuracy improvement was significantly higher. In addition, to improve the precision of the analysis, the drug responses in cell lines were absorbed. A specific pair index graph is plotted for a certain drug concentration ratio.
In the following section, pseudocode of the proposed methodology is discussed.
3 Pseudocode
-
1.
Extract drug data set # contains similarity matrix of each pair of drugs and targets
-
2.
Input x = conc1 && conc2 # drug concentration
-
3.
For i = 1 to n # n-number of concentration count in drug dataset
-
4.
Applied random \(Ei\) forest
$$f_{rf}^{n} \left( x \right) = 1/n \mathop \sum \limits_{i = 1}^{n} T_{i} \left( x \right)$$(1)# frfn(x)—Drug Response
-
5.
Applied Gaussian NB
$$P\left( {x_{i} /y} \right) = 1{\text{/sqrt }}\left( {2*3.14*\sigma_{y}^{2} } \right)$$(2)# P(xi/y)—probability of drug response
-
6.
Applied Logistic Regression
$$(x) = 1/(1 + \exp ( - f(x)))$$(3)# f(x) = b0 + b1x
-
7.
Applied SVM y = f(x) # x ∊ RD, RD here is a vector space with D dimension.
-
8.
Applied Decision Irie Classifier
$$y = - \mathop \sum \limits_{i = 1}^{n} x_{i} \log_{2} \left( {x_{i} } \right)$$(4)# y = Drug Response for x-drug concentration
# Enhanced HBEA algorithm
-
9.
Chosen the best result and algorithm among RF, GNB, LR, SVM & DT #Random Forest, Gaussian NB, Logistic Regression, Support Vector Machine, Decision Tree
-
10.
Extract higher accuracy
$${\text{ Extract higher accuracy}} $$(5)# Hard Voting to obtain drug response
-
11.
Calculate the average
$$(RF, GNB, LR, SVM\, \&\, DT)/5$$(6)# Soft voting to obtain drug response
# Cross validation
-
12.
Split data set # k number of subsets
-
13.
K-fold cross-validation set # k-1 training parts
-
14.
Cross-validation of all models E = 1/K \(\sum\nolimits_{k = 1}^{k} {Ei}\) # Ei—HBEA of random forest && Gaussian NB && Decision Tree
-
15.
Made more evaluation # training more evaluation on all the subsets
-
16.
Used model stacking
-
17.
Work with dependent/Grouped Data
-
18.
Parameters fine-tuning
-
19.
Cross validation of (5) && (6)
-
20.
Retrieve Drug Response # target mapping
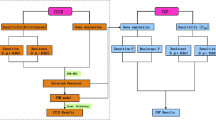
4 Architecture Diagram
The process flow diagram of the proposed approach is presented in Fig. 1. This model comprises two modules as genomic module (Phase #1) and the computational module (Phase #2).

Architecture diagram of proposed HBEA model
Genomic Module
Here, bio genomic and biomechanical features are the input dataset of the model. This dataset is freely available on the web for research purposes [1] and carries drug and cell line features. A model can predict a particular drug response using multiple molecular features and their combinations due to the considerable molecular heterogeneity observed across tumors [53]. Hence, the genetic expression has been considered an important feature in cell lines. As well as target protein, mechanical, and electrical properties are also considered drug line features.
Detailed process is explained in the materials and methods section.
Computational Models
A computational model incorporates several variables that describe the system under investigation. Adjusting each of the variables individually or in combination will monitoring the effect of changes in the outcomes. Five different types of weak learners in machine learning models are combined to fabricate an HBEA model which are logistic regression, decision tree, support vector machine, K-nearest neighbor, and Naïve Bayes. Cross-validation helps to estimate the performance of the model where N-fold cross-validation is used in this analysis. N-fold cross-validation is performed by partitioning the original training dataset into N equal subsets [54]. Each subset is called a fold which is denoted as fold 1, fold 2, …, fold n. Generally, the value of k is taken to be 10, whereas k can be any value. The voting classifier estimator is assembled by combining different classification models which turn out to be a strong meta-classifier that stabilizes the individual classifier’s weakness on a particular dataset. The hard voting classifier takes the majority voting based on weights, and the soft voting classifier takes the probabilities of all the predictions made by different classifiers. The target attribute is a binary variable indicating whether the drug is responding in a cell line or not [55, 56].
5 Materials and Methodology
A comprehensive set of genetic, molecular, and electro-mechanical features for cancer cell lines are collected from CCLE, GDSC, and DECREASE [1] dataset and it has been tested in the proposed machine learning framework.
Heterogeneous-Based Ensemble Algorithm (HBEA) Model:
Data acquisition and selection: Data acquisition is made up of two words: data and acquisition. Data refers to raw facts and numbers that can be structured or unstructured, and acquisition refers to gathering data for a specific goal. This web link carries 23,595 drug combination metrics [57].
Preprocessing
The models discussed in this review utilize past biological knowledge like route data to filter out less relevant variables and optimize the models. This drug response data is multidimensional and highly noisy, so some preprocessing and filtering is required, particularly for omics datasets that characterize the cell lines [58].
Applying algorithms
HBEA enhanced ensemble learning model is created using five different types of machine learning algorithms. The most common of these are logistic regression, decision tree, support vector machine, K-nearest neighbor, and Naive Bayes. In other ensemble models, a homogeneous collection is used. However, in HBEA, a heterogeneous collection is used. In other ensemble models, a homogeneous collection is used. However, in HBEA, a heterogeneous collection is used.
Analysis
Drug response prediction from a cancer patient is used to predict the response of a patient’s cell line to a drug. This dataset comprises the cancer patient’s details including the drug names and their concentration level and also the response to the concentration level. It comprises the record of 1152 cancer patient details with 3 different attributes in three different cell lines. The target attribute is a way of predicting drug response in a specific cell line. It indicates whether the patient has cancer or not.
In Hex, Hela, and Hep cell lines, two different drug concentrations are compared with a specific pair index. In comparison to Hex cell lines, the medication appears to respond quickly in Hela and Hep cell lines.
Confusion Matrix
The confusion matrix displays the number of patients with various actual and predicted drug responses in the cell line. The classification findings can be interpreted in a variety of ways. For example, as demonstrated in the confusion matrices (Figs. 2 and 3), “misclassified” samples for a given medicine could be an indicator of its potential for novel usage, or repurposing, in these “incorrectly” assigned conditions. As a result, misclassification may lead to surprising new findings. This method covers the way for the use of machine learning in the field of drug response.

a Homogeneous confusion matrix output. b Accuracy measure of homogeneous

Heterogeneous confusion matrix
6 Experimentation
The heterogeneous and homogeneous outputs are compared in this study. When compared to the homogeneous model, the heterogeneous ensemble model produces more accurate findings. The next sections detail the experimentation and analysis of homogeneous and heterogeneous output.
Homogeneous Output
The accuracy prediction analysis utilizing the homogeneous ensemble method is shown in Fig. 2. In this study, weak learners, logistic regression, decision trees, support vector machines, K-NN, and the Naive Bayes model were employed.
The confusion matrix output of homogeneous ensemble weak learners and their accuracy level are depicted in Fig. 2a and b. The drug concentration in cell response is predicted using the confusion matrix in Fig. 2a. Figure 2b shows the accuracy of logistic regression, decision trees, support vector machines, naive Bayes, and K-NN. In a homogeneous mode, each learner's accuracy is evaluated individually.
HBEA Model Result Outcome
Figure 3 shows the HBEA model prediction between drug concentration and the response of the cancer cell line.
Figure 3 illustrates the confusion matrix of heterogeneous output and its accuracy level. According to Fig. 3, the confusion matrix predicts the concentration of drugs in the cell. In this Fig. 3, accuracy of logistic regression, decision trees, support vector machines, naive Bayes, and K-NN is combined. In this graph, it can be seen that all algorithm accuracies are combined and evaluated as one plot in strong learner mode.
The confusion matrix of Figs. 2a and 3 gave information on data visualization techniques through heat maps. A heatmap is a two-dimensional graphic representation of data. Each data value is represented in a matrix by a separate color. Confusion matrix describes the performance of a classification model (or “classifier”) on a set of test data which already contains the true values. The output of the heatmap is explained in Table 1. It has been shown that 1152 patient’s medication concentrations and cell line responses were taken as an input. 15% of the testing data is used in the classification after training. As a result, 173 data points were provided for testing. The following findings were discovered throughout the testing. In this cell line, 164 patients had a good response to the drugs. Nine patients showed a poor response to the drugs. We calculated the categorization model’s performance based on that observation. The findings below prove that the HBEA algorithm is 100% accurate. The explanation is given in Table 1.
Table 1 has given that out of 173 cancer patients, 164 are diagnosed with cancer. The remaining nine patients are cancer free. True and false positives and negatives are used to assess this, where
True Positives (TP) indicate that these are the cell lines in which we predicted yes (), and they are not leaving the network. The value is 164.
True Negatives (TN) indicate that no cancer prediction in cell lines, and they are not leaving the network. The value is 9.
False Positives (FP): We predicted yes, but they are not leaving the network (a drug not responding to the cell line). It is also known as a “Type 1 error.” Here no one meets this criterion.
False Negatives FN: Although we predicted no, they left the network (drug responding to the cell line). Type 2 errors occur when this happens. None of the records in this dataset meet this criterion.
Therefore, accuracy obtained in this validation is given below in Eq. (7).
From the above validation, it is clear that the heterogeneous ensemble is obtaining good accuracy in the cancer prediction.
Inputs and Outputs
The input data for this proposed approach is given in Table 2. Two medications are used as inputs, and their concentrations are taken into account using a specific pair index. Finally, the drug response in a specific cell line is anticipated. The cell lines HEK 293, HeLa, and HepG2 are showing rapid pharmacological responsiveness. Table 3 gives the drug response in different cell lines for particular concentration level for a particular pair index.
The graph between drugs (BMS-754807, BGB324, Cisplatin, and NVP-LCL161) and drug concentration is shown in Fig. 4a. A graph is plotted between drugs (LY3009120, Trametinib, Everolimus, Ipatasertib) and drug concentration in Fig. 4b. Pair index and their numbers are displayed in Fig. 4c. Figure 4d depicts the drug response in HEK 293, HeLa cell, and HepG2 cell lines. The maximal drug response in the HEK cell line is 91.78%, as shown in Fig. 4d. In Hela and Hep G2, the maximum medication response was 100%. In the HEK293 cell line, the pair index below 7 indicates that there is no pharmacological response. If the pair index was 7 or above, the drug response in HeLa cells was zero. If the pair index was greater than 13, the medication response was nil in Hep G2.

a and b drug versus drug concentration count in nm(nanomole). c Pair index versus and pair index count in cell line. d Cell line versus drug response
Considering the context of the present research, ensemble models are considered to be useful tools to enhance anti-cancer drug response. In contrast to previous studies of the use of homogeneous ensembles for such types of problems, the present research concentrates on the use of heterogeneous ensembles.
The results obtained suggest that the application of HBEA modeling for anti-cancer drug prediction is effective. For cancer diagnostics, heterogeneous ensembles provide reliable predictions.
It is obvious from the output that inhomogeneous mode, not all of the classifiers perform well in the drug response prediction analysis. As a result, it has been demonstrated that HBEA (heterogeneous) classifiers are effective in predicting anti-cancer medication response in cell lines.
7 Result and Discussion
HBEA was developed with the goal of predicting drug response in cancer cell lines, assuming that cancer pathways would properly represent drug therapeutic effects (Fig. 5). We estimated drug responses for 1152 drugs across 3 cell lines collected from the DREAMZ dataset using available drug concentration and several pair indices. In our prediction model, we need two drug-based data types and three cell line-based data types to predict drug response values.

a Predicted versus actual drug therapeutic effects. b Number of observation. c True positive rates (TPR) and false negative rates (FNR). d Positive predicted values (PPV) and false discovery rates (FDR)
The actual drug response is compared to the predicted drug response in the cell line in Fig. 5. The number of observations in the cell line, TPR with FNR, and PPV with FDR can all be seen in the same fig. The model was dubbed HBEA after a study of five machine learning algorithms: logistic regression, decision tree, support vector machine (SVM), Naïve Bayes, and K-NN. We compared the results of subsets of the data categories as well. Drug concentration1 and 2 are drug-based qualities for a specific pair index. The cell line-based features include pathway enrichment scores in Hep G2, Hela, and HEK 293 cells. Figure 6 shows that it is working.

a Dataset. b Pair index versus response. c Cell line response of Drug 1. d Cell line response of Drug 2. e Drug 1 and Drug 2 responses in cell line
Figure 6 shows the drug reactions in cell lines, including the pair index, drug1 concentration level, and drug2 concentration level. As a result, in Fig. 6 for the particular pair index, the use of a specific drug and a specific concentration to improve drug response is clearly detailed.
The main metrics in the performance test results relating to the application’s stability are standard deviation, range, L2 norm, zero mean, and unit variance are explained in Fig. 7.

a Standard deviation analysis. b L2 norm evaluation. c Zero mean calculation. d Unit variance. e Different colour indication of cell line
As shown in Fig. 7, standard deviation is a key performance test result analysis metric that is related to application stability. There is a possibility of making a mistake when calculating standard deviation when a large number of data points are available.
The standard deviation is a metric for determining the range of values in a sample. The standard deviation of a sample is calculated using the following formula:
where: xI is the I-th value in the sample; xb is the sample mean; n is the sample size xI The I-th value in the sample; xb. From the calculation, it is observed that the higher the standard deviation, the more uniformly dispersed the data in the sample. Figure 7b shows the L2 norm. It is known as least squares. It is basically minimizing the sum of the square of the differences (D) between the target value (Xi) and the estimated values (f(Yi)
Figure 7c shows the zero mean calculation. Figure 7d explains the unit variance. Variance is expressed in significantly bigger units (e.g., meters squared). Because the variance units are much greater than the units of a normal dataset value, it is more difficult to grasp the variance number intuitively. As a result, the standard deviation is frequently used as a primary measure of variability. Figure 7e explained the varied color indications in the graph for each cell line.
8 Conclusion
Customized medicine seeks to determine the most effective way to treat each patient while minimizing side effects. Several machine learning-based methods have been proposed to solve the problem for currently available data, but the problem remains challenging in terms of predictability and interpretability. There is a need for a better classification of anti-cancer medication response prediction using complex networks. HBEA is proposed for this purpose and is based on not only the neighboring drugs and cell lines, but also all other drugs and cell lines. Therefore, it can be used to examine the similarities between drugs, cell lines, and known drug responses around the world. Using the HBEA technique, the suggested method was tested on the DREAMZ dataset, which contains 1152 cancer patient details with 5 different features in three different cell lines. The proposed method's findings were compared to ensemble approaches. When applied to the DREAMZ dataset with the heterogeneous-based ensemble algorithm, the accuracy results of the comparison with other methods revealed that the proposed technique was more aggressive than any other methods.
In this challenge, various types of machine learning algorithms were put together to solve a categorization problem. From the output, it is observed that the HBEA ensemble algorithm improves the drug response prediction in HeLa and HepG2 Cell lines. In this research, heterogeneous ensemble models were more appropriate than single homogenous ensembles. Source code and output graph are available at https://github.com/KartheeswariRamasamy/drug_response.git.
Future Scope
In forecasting anti-cancer treatment response, the heterogeneous ensemble is critical. Using the methodology, HeLa and Hep G2 cell lines respond favorably to the medication when compared to HEK 293 cell lines in this research. Plan to compare medication responses in several cancer cell lines using the same heterogeneous ensemble approach in the future.
References
Ashley EA (2015) The precision medicine initiative: a new national effort. JAMA 313:2119–2120
Collins FS, Varmus H (2015) A new initiative on precision medicine. N Engl J Med 372:793–795
Adam G, Rampášek L, Safikhani Z, Smirnov P, HaibeKains B, Goldenberg A (2020) Machine learning approaches to drug response prediction: challenges and recent progress, NPJ Precis Oncol
Wang L, Li X, Zhang L, Gao Q (2017) Improved anticancer drug response prediction in cell lines using matrix factorization with similarity regularization. BMC Cancer 17:513
Lu X, Gu H, Wang Y, Wang J, Qin P (2019) Autoencoder based feature selection method for classification of anticancer drug response. Front Genet 10:233
Azuaje F (2017) Computational models for predicting drug responses in cancer research. Brief Bioinf 18:820–829
Costello JC et al (2014) A community effort to assess and improve drug sensitivity prediction algorithms. Nat Biotechnol 32:1202
Lee W-H, Loo C-Y, Daniela Traini P, Young M (2015) Inhalation of nanoparticlebased drug for lung cancer treatment: advantages and challenges. Asian J Pharma Sci
Patton JS, Fishburn CS, Weers JG (2004) The lungs as a portal of entry for systemic drug delivery. Proc Am Thorac Soc 1:338–344
Schanker LS, Less MJ (1977) Lung pH and pulmonary absorption of nonvolatile drugs in the rat. Drug Metabol Dispos 5:174–178
Lai Y-H, Chen W-N, Hsu T-C, Lin C, Tsao Y, Semon W (2020) Overall survival prediction of non-small cell lung cancer by integrating microarray and clinical data with deep learning. Sci Rep 10(1):1–11
Doppalapudi S, Qiu RG, Badr Y, Lung cancer survival period prediction and understanding: deep learning
SEER Program (2019) National Cancer Institute (NCI), SEER Incidence Data, 1975–2017. Available https://seer.cancer.gov/data/
Fangfang Xia “A cross-study analysis of drug response prediction in cancer cell lines”, 23(1), 2022, 1–12.
Yıldırım MA et al (2007) Drug—target network. Nat Biotechnol 25:1119
Heba E-B, Attia A-F, Nawal E-F, Torkey H (2021) Efficient machine learning model for predicting drug-target interactions with case study for Covid-19. Comput Biol Chem
Ding H et al (2014) Similarity-based machine learning methods for predicting drug-target interactions: a brief review. Brief Bioinform 15(5):734–747
Nath A, Kumari P, Chaube R (2018) Prediction of human drug targets and their interactions using machine learning methods: current and future perspectives. Methods Mol Biol 1762:21–30
Ezzat A et al (2018) Computational prediction of drug-target interactions using chemogenomic approaches: an empirical survey. Brief Bioinform 20(4):1337–1357
Sachdev K, Gupta MK (2019) A comprehensive review of feature-based methods for drug-target interaction prediction. J Biomed Inform 93:103159
Zhou L et al (2019) Revealing drug-target interactions with computational models and algorithms. Molecules 24(9):1714
Zhang W et al (2019) Recent advances in machine learning-based drug-target interaction prediction. Curr Drug Metab 20(3):194–202
Thafar M, Raies AB, Albaradei S, Essack M, Bajic VB (2019) Comparison study of computational prediction tools for drug–target binding affinities. Front Chem 7:782
Kuncheva LI (2004) Combining pattern classifiers: methods and algorithms. Wiley, Hoboken, NJ
Windeatt T, Roli F (eds) (2003) Multiple classifier systems, in Lecture Notes in Computer Science, vol 2709, Springer
Roli F, Kittler J, Windeatt T (eds) (2004) Multiple classifier systems, in Lecture Notes in Computer Science, vol 3077, Springer
Zhou Z-H (2012) Ensemble methods: foundations and algorithms (Chapman & Hall/CRC data mining and knowledge discovery series). Chapman & Hall/CRC, Boca Raton, FL
Brazil P, Giraud-Carrier C, Soares C (2009) Meta-learning: applications to data mining. Springer, Berlin Heidelberg
Polikar R (2006) Ensemble-based systems in decision making. IEEE Circ Syst Mag 6(3):21–45
Agrawal SM, Narayanan R, Polepeddi L, Choudhary A (2012) Lung cancer survival prediction using ensemble data mining on SEER data. Sci Progr 20(1):29–42
Safiyari A, Javidan R (2017) Predicting lung cancer survivability using ensemble learning methods. Intell Syst Conf (IntelliSys)
Costello JC, Heiser LM, Georgii E et al (2014) A community effort to assess and improve drug sensitivity prediction algorithms. Nat Biotechnol 32(12):1202–1212
Zhang C, Ma Y (2012) Ensemble machine learning: methods and applications, Springer
Lianga G, Fanb W, Luo H, Zhua X (2020) The emerging roles of artificial intelligence in cancer drug development and precision therapy, vol 128, p 110255
Iorio F, Knijnenburg TA, Vis DJ et al (2016) A landscape of pharmacogenomic interactions in cancer. Cell 166(3):740–754
Borg I, Groenen PJF (2005) Modern multidimensional scaling: theory and applications, 2nd edn. Springer, New York
Santini S, Jain R (1999) Similarity measures. IEEE Trans Patt Anal Mach Intel 21(9):871–883
Smith F, Waterman MS (1981) Identification of common molecular subsequences. J Mole Biol 147(1):195–197
Lipman J, Pearson WR (1985) Rapid and sensitive protein similarity searches. Science 227(4693):1435–1441
Gish AW, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mole Biol 215(3):403–410
Stanfield Z et al (2017) Drug response prediction as a link prediction problem. Sci Rep 7:40321
Yang J, Li A, Li Y, Guo X, Wang M (2019) A novel approach for drug response prediction in cancer cell lines via network representation learning. Bioinformatics
Zhang N et al (2015) Predicting anticancer drug responses using a dual-layer integrated cell line-drug network model. PLoS Comput Biol 11:e1004498
Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Durbourg V, Vanderplas J, Passos A, Cournapeau D, Brucher M, Perrot M, Duchesnay E (2011) Scikit-learn: machine learning in python. J Mach Learn Res 12:2825–2830
Hastie T, Tibshirani R, Friedman J (2008) The elements of statistical learning, 2nd edn. Springer, New York/Berlin/Heidelberg
Duda R, Hart P, Stork D (2001) Pattern classification. John Wiley & Sons
Efron B, Tibshirani R (1993) An Introduction to the Bootstrap. Chapman & Hall
Bhardwaj R, Hooda N (2009) Prediction of pathological complete response after Neoadjuvant Chemotherapy for breast cancer using ensemble machine learning, pp 2352–9148
Chen JIZ, Hengjinda P (2021) Early prediction of coronary artery disease (CAD) by machine learning method-a comparative study. J Artif Intell 3(01):17–33
Manoharan S (2019) Study on Hermitian graph wavelets in feature detection. J Soft Comput Paradigm (JSCP) 1(1):24–32
Kening Li, Yuxin Du, Lu Li, Dong-Qing Wei. “Bioinformatics Approaches for Anti-cancer Drug Discovery” , Current Drug Targets, 2019.
Kuenzi BM, Park J, Fong SH, Sanchez KS, Lee J, Kreisberg JF, Ma J, Ideker T (2020) Predicting drug response and synergy using a deep learning model of human cancer cells. Cancer Cell
Anithaashri TP, Rajendran PS, Ravichandran G (2022) Novel intelligent system for medical diagnostic applications using artificial neural network, Lecture Notes on Data Engineering and Communications Technologiesthis link is disabled, vol 101, pp 93–101
Niroomand N, Bach C, Elser M (2021) Segment-based CO emission evaluations from passenger cars based on deep learning techniques. IEEE Access
Prajwal K, Tharun K, Navaneeth P, Anand Kumar M (2022) Cardiovascular disease prediction using machine learning. In: International conference on innovative trends in information technology (ICITIIT)
lanevski A, Giri AK, Gautam P, Kononov A, Potdar S, Saarela J, Wennerberf K, Aittokallio T (2019) Prediction of drug combination effects with a minimal set of experiments, vol 1, pp. 568–577
Baptista D, Ferreira PG, Rocha M (2020) Deep learning for drug response prediction in cancer. Brief Bioinform
Rajendran PS, Kartheeswari KR (2022) Anti-cancer drug response prediction system using stacked ensemble approach, Lecture Notes in Networks, vol 436, pp 205–218
Acknowledgements
This research is funded by the Indian Council of Medical Research (ICMR). (Sanction no: ISRM/12(125)/2020 ID NO.2020-5128 dated 10/01/21).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this paper

Cite this paper
Rajendran, P.S., Kartheeswari, K.R. (2023). Implementation of HBEA for Tumor Cell Prediction Using Gene Expression and Dose Response. In: Rajakumar, G., Du, KL., Rocha, Á. (eds) Intelligent Communication Technologies and Virtual Mobile Networks. ICICV 2023. Lecture Notes on Data Engineering and Communications Technologies, vol 171. Springer, Singapore. https://doi.org/10.1007/978-981-99-1767-9_46
Download citation
DOI: https://doi.org/10.1007/978-981-99-1767-9_46
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-99-1766-2
Online ISBN: 978-981-99-1767-9
eBook Packages: EngineeringEngineering (R0)