
Abstract
The eco-friendly chemistry approach embraces almost all the main branches of chemistry based on the twelve principles introduced by Anastas as green chemistry rules. During the last decade, C–H bond activation protocols attracted intensive consideration as a powerful plan to create organic building blocks of complex structures in organic synthesis and transformations because of its step- and atom-economic nature. In this chapter, a number of innovative green methods of C–H bond activation and functionalization are highlighted.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- C–H bond activation
- C–H functionalization
- Green chemistry
- Electrochemical C–H bond activation
- Flow condition
- Transition metal-free
- Photocatalytic
- Ball milling
- Green oxidant
- Green solvent
1 Introduction
1.1 Green Chemistry and C–H Bond Activation
The origin of the most majority of organic molecules and compounds is natural gas and petroleum-based unrenewable feedstocks; therefore, one of the most challenging issues in chemistry is how to make these feedstocks useful by breaking and forming the new C–C bonds and modifying the C–H bonds into other functional groups [1]. On the other hand, synthetic chemistry continuously encounters challenges to produce selectively and efficiently organic molecules, whether the synthesis of small or complex structures [2] and also many strategies of organic synthesis are being developed in order to extend the chemical toolbox [3].
The traditional procedures, known as the functional group interconversion strategies [4], have achieved this aim by initial C–H bond functionalizations which are then followed by a modifiable sequence of steps to introduce desired functional groups or C–C bonds building the desired skeletons [5].
Notwithstanding its merited place in organic chemistry that the introductory chapters of organic chemistry textbooks intensely concentrate on radical C–H bond functionalizations (halogenations) which are followed by substitution and elimination reactions, it suffers from a perceptual disadvantage. For example, it leads to profoundly futile processes by requiring several reaction steps from unfunctionalized feedstocks to functionalized products, during which undesired by-products are unfortunately generated that leads to high E factors (kg waste/kg product) [6].
In contrast, a significantly greener and sustainable approach is represented by the direct use of otherwise inert C–H bonds as latent functional groups. C–H bond activations contribute a new path to introducing functional groups by preventing lengthy synthetic operations and reducing the by-products and thereby blocking waste production and facilitating direct access to desired target structures even at the late stages of synthesis (Scheme 1) [7].

C–H bond activation and functionalization vs traditional methods
Synthetic C–H activation catalysts struggle to achieve similar selectivities as remarkable chemoselectivity of enzymatic C–H oxidations. Some critical challenges in catalyst development are considering the significant energies that are needed to directly cleavage of C–H bond [8]. The bond dissociation energies (BDEs) and acidities of common C–H bonds in hydrocarbons are shown in Scheme 2. The quantities of BDEs are very close to each other and are between 361.1 and 552.2 kJ/mol, which are one of the most difficult bonds to cleave among other bonds. Furthermore, an isolated C–H bond in a molecule has a very low reactivity owing to the large kinetic barrier associated with the C–H bond cleavage and a polar nature of this bond which do not possess suitable lone pairs to coordinate with a catalyst (Scheme 2) [9].

Bond dissociation energies and pKa values of selected hydrocarbon C–H bonds
Considering all these critical challenges, the importance of site- and product-selective transformation of unactivated C–H bonds into other functional groups and especially catalytic ones is largely accepted by modern chemists, and it is under active study for several years and still regarded as the Holy Grail in chemistry for its step efficiency, atom economy, and potential as a method for late-stage functionalization of complex organic molecules [10].
2 Proposed Mechanisms for C–H Bond Activation
The promotion of technical facilities and laboratory proficiency has increased the scientific abilities to explore the mechanistic foresight of a chemical reaction. During the last decade, due to the prospering interest on catalytic C–H functionalization and owing to the deep understanding of the elementary steps in homogeneous catalysis systems, many researchers have developed significant improvements in the activity and performances in catalytic systems for C–H activation [11]. There are some well-established mechanisms for C–H bond activation that mainly fall under these categories: (1) oxidative addition (OA); (2) electrophilic aromatic substitution (SE Ar); (3) σ-bond metathesis (σBM); (4) single-electron transfer (SET); concerted metalation deprotonation (CMD); and (5) base-assisted intramolecular electrophilic-type substitution (BIES). Herein, we give a brief explanation for each one of them [12]:
Oxidative addition (OA): This is the most common mechanism by which a R-H bond is cleaved and a M-R bond and a M-H bond are formed. It frequently occurs by having an electron-rich and low-oxidation late transition metal centers (Re, Fe, Ru, Os, Ir, Pt) interacting strongly with the C–H bond. The σ-C–H bond coordinates to the metal and a dπ-back donates to the σ*-C–H orbital, lowering its bond order and resulting in the bond cleavage and oxidizing the reaction center in two units. Oxidative addition reaction leads to the creation of a reactive organometallic species containing a hydride and alkyl/aryl ligands at the oxidized metal center (Scheme 3).

Oxidative addition (OA)
Electrophilic aromatic substitution (SEAr): This classification of electrophilic substitution has emerged from the mechanistic pathway by which the hydrogen atom of the substrate is replaced by a metal and thus it acts as a Lewis acid. This reaction is based on the electronic interaction between the π-electronic cloud of the substrate and the electrophilic metal center which acts as Lewis acid forming a new C(aryl)–M bond. In contrast with the oxidative addition, metal oxidation state stays without any changes (Scheme 4).

Electrophilic aromatic substitution (SEAr)
The vicinal C(aryl)–H bond could be easily lost as a proton by re-aromatization or by the action of a base, as a result of the acidity enhancement on this bond. Under circumstances in which the base is in the coordination area of the metal center, the mechanism is acknowledged as a base-assisted intramolecular electrophilic-type substitution (BIES) which has more recently been put forward to account for the often-preferred reactivity of electron-rich arenes (Scheme 5).

Base-assisted intramolecular electrophilic substitution (BIES)
σ-Bond metathesis (σBM): This mechanism is favored for electron-poor metal centers with a high oxidation state. Oxidative addition is not possible with transition metals having d0 electronic configuration (groups 3 and 4, lanthanides and actinides) and thus preferred mechanism is σ-bond metathesis. Cleavage and formation of bonds go through a four-membered square transition state without changing the oxidation state at the metal center (Scheme 6). This is usually common for late or post-transition metals (Pd2+, Pt2+ or Pt4+, Hg2+) and the new C-M and C–H bonds are made without containing any metal hydride species.

σ-bond metathesis (σ-BM)
Single-electron transfer (SET): It contains two steps, that each step involves one electron. Homolytic cleavage of the C–H bond conduce formation of the metal hydride species and a carbon-centered radical (Scheme 7) and construction of the alkyl/aryl-hydride metal oxidized species occurs after recombination reaction between the radical and the metal center.

Single-electron transfer (SET)
Concerted metalation deprotonation (CMD): Close contiguity of this bond to the metal center that is usually promoted by a directing donor group is the key point in this mechanism. At the same time, the metal center possesses a coordinated base that promotes the deprotonation of the C–H bond in a concerted fashion while the formation of the C–M bond is occurring (Scheme 8).

Concerted metalation deprotonation (CMD)
3 Classification of Green C–H Activations
There are some circumstances that affect green principles of C–H activation such as solvent, oxidant, metal catalysts, and so on. For many years, chemists are working on these circumstances to make it greener and safer methodology. From this point of view, we can classify green C–H activation from the traditional green methods to the developed new methods:
-
Green protocols for C–H bond activation:
-
a.
Transition metal-catalyzed C–H activation
-
b.
Transition metal-free C–H activation
-
c.
Green solvent/solvent-free C–H activation
-
d.
Green oxidant/oxidant-free C–H activation
-
e.
Direct C–H functionalization
-
a.
-
Electrochemical C–H bond activation
-
C–H bond activation under flow condition
-
Electrochemical C–H bond activation under flow condition
-
Photocatalytic C–H bond activation
-
C–H bond activation using ball milling and transition metal catalysts
4 Green Protocols for C–H Bond Activation
Past decades have witnessed the emergence of C–H functionalizations as a particularly powerful tool for molecular syntheses, [13] with enabling applications to material sciences, late-stage diversification, natural product synthesis, and pharmaceutical industries, among others [14]. However, most C–H functionalization protocols suffer from stoichiometric amounts of costly and/or toxic transition metal oxidants that make undesired metal-containing by-products [15]. Therefore, applying green methods in C–H activation is highly desirable. In the next parts, we will introduce some green protocols and methods for the aforesaid purpose.
4.1 Applicable Metals in C–H Activation
Metal-catalyzed C–H functionalization chemistry provides the step economical and original construction of C–C, and C-X (X=N, O, etc.) bonds, commencing from hydrocarbon fragments without the necessity of prior non-catalytic oxidation steps and pre-functionalization of substrates [16]. Thus far, the vast majority of C–H functionalization advances continue to heavily rely on precious 4d or 5d transition metal catalysts such as Pd, Ir, Rh, and Ru [17]. Unfortunately, these 4d and 5d transition metals are not only cost-intensive but are generally comparatively toxic. Given the cost-effective and sustainable nature of earth-abundant first row transition metal and also less toxicity of these metals, the evolution of 3d metal catalysts such as Sc, V, Mn, Fe, Ti, Cr, Co, Ni, Cu, Zn, for C–H activation has attained remarkable recent momentum as green alternatives [18].
The C–H activation by 3d-based metals continues to largely undergo single-electron transfer manifolds, setting the platform for more reactivities and selectivities [19]. More widespread applications of these 3d metal catalysts are moderately disturbed by their substantial oxophilicity that leads to reducing chemo-selectivities and functional group tolerance. Manganese-, cobalt-, and iron-catalyzed C– H activations emerged as potent systems for various C–H alkylations, alkenylations, and arylations [20]. Besides, according to the d6-electron configuration of manganese (I) and cobalt (III) complexes, these metals accomplish the C–H transformations, in which the oxidation state of metal remains without changing during the entire catalytic cycle [16]. In terms of versatility, Ni [21] and Cu [22] catalysis were utilized in alkylation and arylation reactions under mild conditions. Furthermore, the Ni and Cu catalysis regimes were not restricted to redox-neutral C–H transformations. Indeed, these manifolds proved particularly powerful for oxidative C–H functionalizations.
The combinations of C–H activation and cross-coupling reactions give countless opportunities to synthesize complex molecules via Mizoroki–Heck and Suzuki-type cross-coupling C–H functionalization reactions [23].
There are many practical methods of metal-catalyzed C–H activation operated in the synthesis of medicinally valuable molecules like lithospermic acid, piperaborenine B, losartan, valsartan, anacetrapib, and oxazolidinone antibacterial using transition metals [24].
4.2 Transition Metal-Free C–H Activation
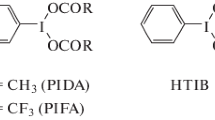
Most of the transition metal catalysts are normally very expensive, and the supporting ligands are generally even more costly and sometimes there are obstacles in their preparation. Also, as aforesaid, several transition metals are toxic. On the other hand, various transition metal catalysts are normally sensitive to oxygen (O2) and moisture. Moreover, in cases which high efficiency and selectivity of transformations are challenging, it is essential to use special additives and co-catalysts. Consequently, transition metal-free conditions became more attractive than classic transition metal-catalyzed reactions [25]. The usage of hypervalent iodine reagents [26], diazonium salts [27], or employing electrochemistry methods [28] are some of the interesting examples of transition metal-free processes.
The mixture of electron-rich arenes and hypervalent iodine(III) reagents results in radical cation as a selective and efficient SET oxidizing agent that provides a series of direct C–H functionalization products under mild conditions [29]. In these reactions, the application of polar and low-nucleophilic protic solvent such as multifluorosubstituted alcohol is crucial because it affects stabilizing the reactive cationic intermediates and preventing other probable side-reactions (Scheme 9) [30].

C–H functionalization with azides, acetate, and 1,3-dicarbonyl compounds
Kita and co-workers made significant progress in hypervalent iodine(III)-promoted metal-free C–H functionalization reactions on a variety of heteroaromatic compounds such as thiophenes and pyrroles (Scheme 10) [31].

C–H functionalization with thiophenes and pyrroles
Direct fluorination to get the mono- and difluoromethylated arenes in the presence of selectfluor and potassium persulfate was disclosed by Yi and co-workers [32] (Scheme 11).

Transition metal-free direct benzylic C–H fluorination
The ligand- and transition metal-free direct C–H functionalization of quinones and naphthoquinones with diaryliodonium salts through the radical pathway which led to the synthesis of aryl naphthoquinones as β-secretase inhibitors in moderate to good yields was introduced by Wang et al. [33] (Scheme 12).

Transition metal-free direct C–H functionalization of Quinones
In another example, a transition metal-free regio-selective coupling reaction of indoles and aryl halides using KOtBu and degassed solvent was reported (Scheme 13) [34].

Transition metal-free C3 arylation of indoles with aryl halides
Recently, many chemists are working on expanding modern metal-free aerobic C–H bond functionalization reactions which have utilized aldehydes, ethers, benzylamines, and glycine derivatives [35].
4.3 Green Solvent/solvent-Free C–H Activation
It has been determined that most of the waste produced in the chemical synthesis originates from the usage of solvents which are frequently used in large quantities in comparison with the other reactants. Accordingly, it is not unexpected that significant attention has been dedicated to the definition of “green solvents” [36]. In general, bio-based solvents coming from the biomass feedstock can be recognized as “greener” alternatives [37]. But none of the alternative solvents are as green as water. However, there is still a gap between the use of water as a reaction medium and practical green chemistry. Another alternative procedure is conducting reactions under solvent-free conditions [38].
During the past five years, employing greener solvents such as water, dialkyl carbonates, and PEGs in the ruthenium and palladium-catalyzed direct arylation of aromatic heterocycles have increased significantly [39]. Using dialkyl carbonates resulted in the facilitated work-up procedure and decreased the production of wastes and PEGs as a solvent provided the recycling of the catalyst and using water led to higher rates and cleaner reactions in several cases particularly with ruthenium. So far, there are no examples of these reactions that have been described in other alternative media such as ionic liquids or supercritical CO2. Also, carbonates have been determined as a suitable solvent for palladium-catalyzed direct arylation reactions and the reaction was found to be more selective than in DMF, dioxane, and other previously used toxic solvents since using this solvent resulted in fewer traces of side products [40].
Wang and Wu developed a green step-economic and sustainable approach to construct a C–C bond without any organic solvents or additives. They reported the example of water-mediated C–H activation of arenes using sulfoxonium ylides (Scheme 14) [41].

Water-mediated C–H activation
In the same year, Yao and co-workers developed a method for C–C bond formation via a terminal alkyne C–H bond activation and synthesizing quinoline derivatives using stable and inexpensive Fe(OTf)3 as a catalyst under solvent-free condition (Scheme 15) [42].

Green synthesis of quinoline derivatives
Recently, major advances have been represented in using biomass-derived solvents such as; glycerol, 2-methyl tetrahydrofuran, ethyl lactate, and γ-valerolactone (GVL), in transition metal-catalyzed couplings, including Suzuki–Miyaura, Mizoroki–Heck, Sonogashira–Hagihara reactions, and C–H functionalizations [37]. Ackermann, Vaccaro, and co-workers reported the first example of a palladium-catalyzed Catellani reaction using GVL as a solvent instead of the frequently employed DMF, DMA, or acetonitrile (Scheme 16) [43].

Catellani reaction catalyzed by palladium/Al2O3 in GVL
4.4 Green Oxidant/Oxidant-Free C–H Activation
One of the recognized and extremely attractive conversions from environmental and economical viewpoints is the construction of C–C bonds from two C–H bonds under oxidative conditions that reduced waste and reaction steps. Despite excellent improvements being made, key difficulties remain [44]. Therefore, the development of green and user-friendly C–H bond activation procedures in the lack of chemical oxidants would be instantly desirable. Oxygen gas is well known for an ideal and readily available green oxidant and its solo by-product is desired for cross-coupling C–H activation but suffers from weak reactivity.
The coupling reaction of 2,6-lutidine and internal alkynes which begun with C(sp3)-H bond activation via σ-bond metathesis and then mediated by a non-metallocene cationic alkylhafnium complex to give five-membered carbocyclic compounds was firstly developed by Mashima and co-workers (Scheme 17) [45].

Coupling reaction of 2,6-lutidine and internal alkynes
Four years later Hu and co-workers reported a palladium-catalyzed oxidative Suzuki coupling reaction of 3-unsubstituted indolizines at the 3-position with aryl boronic acids to produce 3-aryl-indolizine. The distinguished advantage of this method was using O2 as a green oxidant (Scheme 18) [46].

Palladium-catalyzed oxidative Suzuki coupling reaction
Gong and Meggers have also introduced the first example of an asymmetric photoredox dehydrogenative cross-coupling of two Csp3-H groups catalyzed by a chiral rhodium complex and with molecular oxygen as the oxidant in 2015 (Scheme 19) [47].

Asymmetric green cross-coupling of two Csp3-H groups
The rhodium-catalyzed oxidative dehydrogenative cross-coupling of arylamines with electron-rich arenes under mild aerobic conditions resulting in the synthesis of non-symmetrical biaryl amines, in excellent yields and high selectivities, is another example of using green oxidant (Scheme 20) [48].

Rhodium-catalyzed synthesis of non-symmetrical biaryl amines
In 2018, Bhanage reported an effective protocol for the synthesis of valuable p-hydroxybenzoates directly from phenols by palladium-catalyzed aerobic oxidative carbonylation of phenolic C–H bond, proceeding through oxidative iodination. Using O2, high selectivity, no co-catalyst, co-solvent, and external ligand are some advantages of this method (Scheme 21) [49].

Synthesis of p-hydroxybenzoates
4.5 Direct C–H Functionalization
Owing to the complexity of organic substrates, several types of C–H bonds can be found in their chemical skeletons. However, in terms of selectivity, controlling reactivity on one single bond is very challenging. For this reason, the use of a donor group (DG) as a directing group is a very broadly applied strategy to selectively activate C–H bonds [50]. Direct C–H functionalization procedures are based on the use of directing Lewis bases covalently linked to the substrate. Heteroatom-based groups are the most used directing groups, although alkenes can also be effective. This field has been much studied, especially for arene C–H activation [51]. For the past fifty years, ortho-selectivity via cyclometalation is the most utilized way of C–H activation; more recently devised special directing groups allow also meta-selectivity [52]. Prominently, directed C–H activation allows to activate unactivated C(sp3)-H bonds, too [53].
5 Electrochemical C–H Bond Activation
Combining metal-catalyzed C–H activation with electrocatalysis, have resurfaced as a viable platform for sustainable transformations due to its inherent advantages and unique characters such as replacement of dangerous and toxic chemicals by electric current, less waste production, applying few amounts of chemicals, and affording fewer reaction steps than traditional methods [54]. Moreover, a majority of the preparative electrochemical reactions even with high activation energies, can be carried out at ambient temperature since the energy of the electrochemical system is controlled by the applied electrode potential [55]. There are two kinds of cell setups in electrochemistry systems: (1) divided cell, the anode and cathode are separated by a porous and ion-exchange membrane; (2) undivided cell setup the anode and cathode are being placed in the same cell in a significantly more user-friendly manner [56]. Also, organic electrochemistry can be classified into two categories; direct electrolysis without redox mediator and indirect electrolysis [57] with redox mediators.
Advantages of redox-mediated electrolysis are avoiding obstacles related to heterogeneous electron transfer such as overpotentials and conducting electrolysis at lower potentials which lead to accelerating the reaction rate and can feature beneficial effects in terms of chemoselectivity and robustness with bypassing probable side reactions [58]. The low atom economy for this strategy was the most important motivation for applying direct metallaelectrocatalytic C–H activations. Recently the scope of useful direct electrochemical ruthenium (II) catalysis was extended by Ackermann, Qiu, Mei and Xu [59]. Also, there is plenty of reported rhodium-catalyzed oxidative C–H activation reaction with stoichiometric metal oxidants during the last decades [60]. More recently Ackermann reported the novel rhodaelectro-catalyzed C–H activation in which there was no need for stoichiometric chemical oxidants [61]. Mei and co-workers recently reported a notable palladaelectro-catalyzed C(sp3)-H oxygenation of substituted oxime ethers [62]. Inspired from this approach, further palladium-catalyzed electrochemical C(sp2)-H activations, including direct acetoxylations [63], acylations [64], methylations, and alkylations [65] have been reported. In 2017, the Ackermann group [66] made a significant success in reaching full resource economy through direct metallaelectrocatalytic C–H oxygenation using inexpensive alcohols as a coupling partner with 3d transition metals and in undivided cell setups. Significant progress was achieved by Ackermann [67], Lei [68], and Ye [69] in this field (Scheme 22). Lately, there are a few reports of copper-catalyzed electrocatalytic C–H amination [70] and also a new electrooxidative nickel catalysis without redox mediators with a full resource economy [71].

Co-catalyzed electrocatalytic C–H activations
6 C–H Bond Activation Under Flow Condition
Flow technologies are one of the most assuring tools for providing more extended applicability of C–H bond functionalization reactions [72]. Some features of flow condition are listed as follows: (a) more reliable and safer [73]; (b) easy temperature controlling [74]; (c) efficient mixing of two phases and contacting between reagents in different phases (mainly gases and liquids or solids and liquids) [75]; (d) the high surface area-to-volume ratio of flow reactors [76]; (e) easier scale-up of the reaction; and (f) simple separation of the catalyst from the reaction products [77].
As an example, the ortho-selective C–H alkenylation of acetanilides in heterogeneous continuous flow conditions was discovered by Ackermann, Vaccaro, and co-workers. The reaction was catalyzed by Pd/C, required benzoquinone (BQ) as the terminal oxidant and a strong Brønsted acid as an additive in biomass-derived \(\gamma\)-valerolactone (GVL). The advantages of performing this reaction in flow were the shorter reaction times and improvement in the stability of the catalyst compared to batch conditions (Scheme 23) [78].

Palladium-catalyzed trifluoromethylthiolation of amides in flow
7 Electrochemical C–H Bond Activation Under Flow Condition
Very recently, there are some examples of merging organic electrochemistry with flow technologies. For instance, Xu and co-workers reported an electrochemical protocol to synthesize 4H-1,3-benzoxazines from the cyclization of readily available N-benzylamides. Most of the researches were conducted in batch, with only a single reaction being conducted in an electrochemical microreactor. The reactor was containing a platinum foil as the cathode and a graphite layer as the anode, separated by a fluorinated ethylene propylene (FEP) membrane. Notably, the use of flow conditions made it possible to reduce the amount of the supporting electrolyte, to perform the reaction at ambient temperature and to scale-up the process in high yields (Scheme 24) [79].

Electrochemical synthesis of a 4H-1,3-benzoxazine in flow
8 Photocatalytic C–H Bond Activation
Visible light photoredox catalysis has rebirthed due to the improvement of radical chemistry in organic synthesis. The simplicity of reaction setups, as well as mild reaction conditions, low price, green and clean energy sources, and the broad applicability of them led to resolve some of the contemporary challenges and scarcities in several synthetic methodologies using transition metals or strong oxidants. One of the most influential accomplishments in photocatalysis was the activation of molecular oxygen [80]. Despite its poor reactivity as a diradical, it can be transformed into the superoxide radical anion upon single-electron transfer (SET), which can undergo H-radical abstraction or lead to the in situ formation of hydrogen peroxide (Scheme 25, top) [81].

Photocatalytic C–H bond activation pathways
Additionally, transition metal chromophores as photocatalysts [82] have sufficient potential in their excited states and can induce C–H functionalization reactions by the direct oxidation of the substrates to the corresponding radical or radical cation without the necessity of a stoichiometric amount of an oxidant or a pre-functionalization of the substrate (Scheme 25, bottom). Generally used photoorganocatalysts are mostly heterocycles and organic dyes such as Eosin Y, methylene blue, and riboflavin [83].
Although most of the reported visible light-mediated catalytic C–H functionalization methods have been performed by expensive metal-based Ru or Ir photoredox complexes but their mild reaction conditions and the use of stable and cheap organic dyes could justify its green manner [84]. In 2019, an efficient photoredox-induced decarboxylative C2-alkylation of benzothiazoles was produced by Wang and co-workers [85]. The reaction was carried out by using a catalytic amount of 9-mesityl-10-methyl acridinium perchlorate as photocatalyst and O2 as an oxidant with a blue LED under transition metal free conditions at 25 °C. Broad scope, high yields, mild reaction conditions, and easy work-up were its highlighted advantages (Scheme 26).

Mes-Acr+ catalyzed C–H alkylation reaction
In 2017, the group of Nicewicz reported the direct C–H cyanation of electron-rich arenes via visible light photoredox catalysis [86]. The authors applied an acridinium salt derivative (NZ) as the photocatalyst for the single electron oxidation of aromatic compounds into the radical cationic species, which were subsequently trapped by TMSCN to produce nitrile. The employed oxygen atmosphere made the addition of external oxidants unnecessary. Also, one year later, the same group, applied a similar photocatalytic methodology for the visible light-mediated C–H bond azidation of aliphatic compounds with acridinium (Scheme 27) [87].

Mechanochemical C–H bond alkylation of indoles
9 C–H Bond Activation Using Ball Milling and Transition Metal Catalysts
Mechanochemistry has attracted increasing attention from chemists. Mechanochemistry methods are applicable to distribute energy for chemical processes as efficient as possible via grinding, ball milling, shearing, and kneading [88].
Ball-milling-induced reactions are cost-effective, efficient, and green methods which are typically operated in a stainless-steel jar with plenty of balls rotating at high speed (60–800 rpm). These reactions are performed under mild conditions, without any organic solvents (or minimum amounts of solvent), at approximately ambient temperature and in relatively short reaction times. The first mechanochemical C–H functionalization was reported by Bolm and co-workers [89]. Although the discovery was remarkable, the solventless process was limited in terms of catalyst efficiency. Since then chemists have been trying to expand the scope of mechanochemical C–H functionalizations in the presence of transition metals such as Pd, Rh, Ru, Co, and Ir and apply it for a variety of important functionalizations, including halogenation, amidation, alkynylation, and dehydrogenative coupling and also utilize it for other important organic reactions [90]. As a ball-milling reaction example, Bolm and co-workers reported a procedure for the direct mechanochemical rhodium-(III)-catalyzed C(sp2)-H bond amidation of the arenes using a 1,4,2-dioxazol-5-one as the nitrogen source and amidating agent and using ball milling in a 25 mL ZrO2 milling jar with one ZrO2 ball of 15 mm diameter at 30 Hz. The reaction proceeds in the presence of [Cp*RhCl2]2, AgSbF6, and AgOAc under solvent-free conditions without additional heating. The ortho amidated products were formed in great yields and in shorter reactions times (99 min.) in comparison with the solution and exhibited benefits of mechanistic techniques to the standard solvent-mediated protocols (Scheme 28) [91].

Mechanochemical Rh(III)-catalyzed C–H bond amidation of benzamides
10 Conclusion
During the last decade, considerable advances in C–H functionalization reactions have witnessed the importance of these transformations. These protocols give us powerful tools to create organic building blocks of complex structures in molecular science. On the other hand, their step- and atom-economic nature, are completely in agreement with green chemistry protocols. The applicability of C–H bond activation reactions is currently under remarkable investigation in related chemistries which will be reported in due course.
References
Hess J, Bednarz D et al (2011) Petroleum and health care: evaluating and managing health care’s vulnerability to petroleum supply shifts. Am J Publ Health 101:1568–1579
(1) Chen K, Baran PS (2009) Total synthesis of eudesmane terpenes by site-selective C–H oxidations. Nature 459:824–828; (2) Jørgensen L, McKerrall SJ et al (2013) 14-step synthesis of (+)-ingenol from (+)-3-carene. Science 341:878–882
Hesp KD, Bergman RG, Ellman JA (2011) Expedient synthesis of N-acyl anthranilamides and β-enamine amides by the Rh (III)-catalyzed amidation of aryl and vinyl C–H bonds with isocyanates. J Am Chem Soc 133:11430–11433
Das P, Dutta A et al (2014) Heterogeneous ditopic ZnFe 2 O 4 catalyzed synthesis of 4 H-pyrans: further conversion to 1, 4-DHPs and report of functional group interconversion from amide to ester. Green Chem 16:1426–1435
Walling C, Jacknow BB (1960) Positive halogen compounds. I. The radical chain halogenation of hydrocarbons by t-butyl hypochlorite1. J Am Chem Soc 82:6108–6112
Davies HML, Du Bois J et al (2011) C–H Functionalization in organic synthesis. Chem Soc Rev 40:1855–1856
Potavathri S, Pereira KC et al (2010) Regioselective oxidative arylation of indoles bearing N-alkyl protecting groups: dual C−H functionalization via a concerted metalation−deprotonation mechanism. Am Chem Soc 132:14676–14681
Blanksby SJ, Ellison GB (2003) Bond dissociation energies of organic molecules. Acc Chem Res 36:255–263
Roudesly F, Oble J, Poli G (2017) Metal-catalyzed CH activation/functionalization: the fundamentals. J Mol Catal a: Chem 426:275–296
Shang R, Ilies L, Nakamura E (2017) Iron-catalyzed C–H bond activation. Chem Rev 117:9086–9139
Li JJ (2015) CH bond activation in organic synthesis. CRC press
(1) Goldman AS, Goldberg KI (2004) Organometallic C–H bond activation: an introduction, ACS; (2) Lapointe D, Fagnou K (2010) Overview of the mechanistic work on the concerted metallation–deprotonation pathway. Chem Lett 39:1118–1126; (3) Ackermann L (2011) Carboxylate-assisted transition-metal-catalyzed C−H bond functionalizations: mechanism and scope. Chem Rev 111:1315–1345; (4) Balcells D, Clot E, Eisenstein O, (2010). C–H bond activation in transition metal species from a computational perspective. Chem Rev 110:749–823; (5) Gallego D, Baquero EA, (2018) Recent advances on mechanistic studies on C–H activation catalyzed by base metals. Open Chem 16: 1001–1058
He J, Wasa M, Chan KS et al (2017) Palladium-catalyzed alkyl C–H bond activation. Chem Rev 117:8754–8786
Park Y, Kim Y et al (2017) Transition metal-catalyzed C–H lamination: scope, mechanism, and applications. Chem Rev 117:9247–9301
Hickman AJ, Sanford MS (2012) High-valent organometallic copper and palladium in catalysis. Nature 484:177–185
Gandeepan P, Müller T et al (2019) 3d transition metals for C–H activation. Chem Rev 119(4):2192–2452
Choy PY, Wong SM et al (2018) Recent developments in palladium-catalysed non-directed coupling of (hetero) arene C–H bonds with C–Z (Z= B, Si, Sn, S, N, C, H) bonds in bi (hetero) aryl synthesis. Org Chem Front 5:288–321
Egorova KS, Ananikov VP (2017) Toxicity of metal compounds: knowledge and myths. Organometallics 36:4071–4090
Gallego D, Baquero EA (2018) Recent advances on mechanistic studies on C–H activation catalyzed by base metals. Open Chem 16:1001–1058
(a) Liu W, Ackermann L (2016) Manganese-catalyzed C–H activation. ACS Catal 6:3743–3752; (b) Shang R, Ilies L, Nakamura E, (2017) Iron-catalyzed C–H bond activation. Chem Rev 117:9086−9139; (c) Wang H, Moselage M et al (2016) Selective synthesis of indoles by cobalt (III)-catalyzed C–H/N–O functionalization with nitrones. ACS Catal 6:2705–2709
Castro LCM, Chatani N (2015) Nickel catalysts/N, N′-bidentate directing groups: an excellent partnership in directed C–H activation reactions. Chem Lett 44:410–421
Daugulis O, Do HQ et al (2009) Palladium-and copper-catalyzed arylation of carbon−hydrogen bonds. Acc Chem Res 42:1074–1086
Shi S, Nawaz KS et al (2018) Advances in enantioselective C–H activation/mizoroki-heck reaction and Suzuki reaction. Catalysts 8:90
Basu D, Kumar S et al (2018) Transition metal catalyzed CH activation for the synthesis of medicinally relevant molecules: a review. J Chem Sci 130:71
Sun CL, Shi ZJ (2014) Transition-metal-free coupling reactions. Chem Rev 114:9219–9280
Gu Y, Wang D (2010) Direct C-3 arylation of N-acetylindoles with anisoles using phenyliodine bis (trifluoroacetate)(PIFA). Tetrahedron Lett 51:2004–2006
Zhang YP, Feng XL (2016) Metal-free, C–H arylation of indole and its derivatives with aryl diazonium salts by visible-light photoredox catalysis. Tetrahedron Lett 57:2298–2302
Morofuji T, Shimizu A et al (2012) Metal-and chemical-oxidant-free C–H/C–H cross-coupling of aromatic compounds: the use of radical-cation pools. Angew Chem Int Ed 51:7259–7262
Kita Y, Tohma H et al (1994) Hypervalent iodine-induced nucleophilic substitution of para-substituted phenol ethers. Generation of cation radicals as reactive intermediates. J Am Chem Soc 116:3684–3691
Eberson L, Hartshorn MP et al (1996) Making radical cations live longer. Chem Commun 18:2105–2112
Kita Y, Takada, et al (1996) Hypervalent iodine reagents in organic synthesis: nucleophilic substitution of p-substituted phenol ethers. Pure Appl Chem 68:627
Ma JJ, Yi WB et al (2015) Transition-metal-free C–H oxidative activation: persulfate-promoted selective benzylic mono-and difluorination. Org Biomol Chem 13:2890–2894
Wang D, Ge B et al (2014) Transition metal-free direct C–H functionalization of quinones and naphthoquinones with diaryliodonium salts: synthesis of aryl naphthoquinones as β-secretase inhibitors. J Org Chem 79:8607–8613
Chen J, Wu J (2017) Transition-metal-free C3 arylation of indoles with aryl halides. Angew Chem Int Ed 56:3951
Shamsabadi A, Chudasama V (2019) Recent advances in metal-free aerobic C–H activation. Org Biomol Chem 17:2865–2872
Jimenez-Gonzalez C, Ponder CS et al (2011) Using the right green yardstick: why process mass intensity is used in the pharmaceutical industry to drive more sustainable processes. Org Process Res Dev 15:912–917
Santoro S, Ferlin F et al (2017) Biomass-derived solvents as effective media for cross-coupling reactions and C–H functionalization processes. Green Chem 19:1601–1612
Fu XP, Liu L et al (2011) “On water”-promoted direct alkynylation of isatins catalyzed by NHC–silver complexes for the efficient synthesis of 3-hydroxy-3-ethynylindolin-2-ones. Green Chem 13:549–553
Fischmeister C, Doucet H (2011) Greener solvents for ruthenium and palladium-catalysed aromatic C–H bond functionalisation. Green Chem 13:741–753
Schäffner B, Schäffner F et al (2010) Organic carbonates as solvents in synthesis and catalysis. Chem Rev 110:4554–4581
Nie R, Lai R et al (2019) Water-mediated C–H activation of arenes with secure carbene precursors: the reaction and its application. Chem Commun 55:11418–11421
Yao C, Qin B et al (2012) One-pot solvent-free synthesis of quinolines by C–H activation/C–C bond formation catalyzed by recyclable iron (III) triflate. RSC Adv 2:3759–3764
Rasina D, Kahler-Quesada A et al (2016) Heterogeneous palladium-catalysed Catellani reaction in biomass-derived γ-valerolactone. Green Chem 18:5025–5030
Sambiagio C, Schönbauer D et al (2018) A comprehensive overview of directing groups applied in metal-catalysed C–H functionalisation chemistry. Chem Soc Rev 47:6603–6743
Tsurugi H, Yamamoto K et al (2010) Oxidant-free direct coupling of internal alkynes and 2-alkylpyridine via double C−H activations by alkylhafnium complexes. J Am Chem Soc 133:732–735
Hu H, Liu Y et al (2014) Palladium catalyzed oxidative Suzuki coupling reaction of indolizine at the 3-position using oxygen gas as the only oxidant. RSC Adv 4:24389–24393
Tan Y, Yuan W et al (2015) Aerobic Asymmetric dehydrogenative cross-coupling between two C−H groups catalyzed by a chiral-at-metal rhodium complex. Angew Chem Int Ed 54:13045–13048
Matsumoto K, Yoshida M et al (2016) Heterogeneous rhodium-catalyzed aerobic oxidative dehydrogenative cross-coupling: nonsymmetrical biaryl amines. Angew Chem 128:5358
Gaikwad VV, Bhanage BM (2018) Palladium-catalyzed aerobic oxidative carbonylation of C–H bonds in phenols for the synthesis of p-hydroxybenzoates. Eur J Org Chem 22:2877–2881
Rostami A, Khakyzadeh V et al (2018) Co (II)-catalyzed regioselective clean and smooth synthesis of 2-(aryl/alkyl-thio) phenols via sp2 CH bond activation. Molecular Catalysis 452:260–263
Albrecht M (2010) Cyclometalation using d-block transition metals: fundamental aspects and recent trends. Chem Rev 110:576–623
Patra T, Watile R et al (2016) Sequential meta-C–H olefination of synthetically versatile benzyl silanes: effective synthesis of meta-olefinated toluene, benzaldehyde and benzyl alcohols. Chem Commun 52:2027–2203
Herrmann P, Bach T (2011) Diastereotopos-differentiating C–H activation reactions at methylene groups. Chem Soc Rev 40:2022–2038
Yoshida JI, Kataoka K et al (2008) Modern strategies in electroorganic synthesis. Chem Rev 108:2265–2299
Frontana-Uribe BA, Little RD et al (2010) Organic electrosynthesis: a promising green methodology in organic chemistry. Green Chem 12:2099–2119
Cardoso DS, Šljukić B et al (2017) Organic electrosynthesis: from laboratorial practice to industrial applications. Org Process Res Dev 21:1213–1226
Kärkäs MD (2018) Electrochemical strategies for C–H functionalization and C–N bond formation. Chem Soc Rev 47:5786–5865
Meyer TH, Finger LH et al (2019) Trends in Chemistry 1:63–76
Qiu Y, Tian C et al (2018) Electrooxidative ruthenium-catalyzed C−H/O−H annulation by weak O-coordination. Angew Chem Int Ed 57:5818–5822
Song G, Wang F et al (2012) C-C, C–O and C–N bond formation via rhodium (iii)-catalyzed oxidative C–H activation. Chem Soc Rev 41:3651–3678
Qiu Y, Kong WJ et al (2018) Electrooxidative rhodium-catalyzed C−H/C−H activation: electricity as oxidant for cross-dehydrogenative alkenylation. Angew Chem Int Ed 57:5828
Yang QL, Li YQ et al (2017) Palladium-catalyzed C (sp3)−H oxygenation via electrochemical oxidation. J Am Chem Soc 139:3293–3298
Li YQ, Yang QL et al (2017) Palladium-catalyzed C (sp2)–H acetoxylation via electrochemical oxidation. Org Lett 19:2905–2908
Ma C, Zhao CQ et al (2017) Palladium-catalyzed C–H activation/C–C cross-coupling reactions via electrochemistry. Chem Commun 53:12189–12192
Yang QL, Li CZ et al (2018) Palladium-catalyzed electrochemical C–H alkylation of arenes. Organometallics 38:1208–1212
Sauermann N, Meyer TH et al (2017) Electrochemical cobalt-catalyzed C–H oxygenation at room temperature. J Am Chem Soc 139:18452–18455
Tian C, Massignan L et al (2018) Electrochemical C−H/N−H activation by water-tolerant cobalt catalysis at room temperature. Angew Chem Int Ed 57:2383
Tang S, Wang D et al (2018) Cobalt-catalyzed electrooxidative CH/NH [4+2] annulation with ethylene or ethyne. Nat Commun 9:798
Yu Y, Zheng P et al (2018) Electrochemical cobalt-catalyzed C–H or N–H oxidation: a facile route to synthesis of substituted oxindoles. Org Biomol Chem 16:8917–8921
Yang QL, Wang XY et al (2018) Copper-catalyzed electrochemical C–H amination of arenes with secondary amines. J Am Chem Soc 140:11487–11494
Zhang SK, Samanta RC et al (2018) Nickel-catalyzed electrooxidative C−H amination: support for nickel (IV). Chem Eur J 24:19166
Santoro S, Ferlin F et al (2019) C–H functionalization reactions under flow conditions. Chem Soc Rev 48:2767–2782
Gutmann B, Cantillo D et al (2015) Continuous-flow technology—a tool for the safe manufacturing of active pharmaceutical ingredients. Angew Chem Int Ed 54:6688–6728
Mandrelli F, Buco A et al (2017) The scale-up of continuous biphasic liquid/liquid reactions under super-heating conditions: methodology and reactor design. Green Chem 19:1425–1430
Lévesque F, Seeberger PH (2012) Continuous-flow synthesis of the anti-malaria drug artemisinin. Angew Chem Int Ed 51:1706–1709
Su Y, Straathof NJ et al (2014) Photochemical transformations accelerated in continuous-flow reactors: basic concepts and applications. Chem Eur J 20:10562–10589
Vaccaro L, Curini M et al (2018) Definition of green synthetic tools based on safer reaction media, heterogeneous catalysis, and flow technology. Pure Appl Chem 90:21–33
Ferlin F, Santoro S et al (2017) Heterogeneous C–H alkenylations in continuous-flow: oxidative palladium-catalysis in a biomass-derived reaction medium. Green Chem 19:2510–2514
Xu F, Qian XY et al (2017) Synthesis of 4 H-1, 3-benzoxazines via metal-and oxidizing reagent-free aromatic C–H oxygenation. Org Lett 19:6332–6335
Fabry DC, Rueping M (2016) Merging visible light photoredox catalysis with metal catalyzed C–H activations: on the role of oxygen and superoxide ions as oxidants. Acc Chem Res 49:1969–1979
Zeitler K (2009) Photoredoxkatalyse mit sichtbarem Licht. Angew Chem 121:9969–9974
Karkas MD, Porco JA Jr et al (2016) Photochemical approaches to complex chemotypes: applications in natural product synthesis. Chem Rev 116:9683–9747
Ravelli D, Fagnoni M et al (2013) Photoorganocatalysis. What for? Chem Soc Rev 42:97–113
Romero NA, Nicewicz DA (2016) Organic photoredox catalysis. Chem Rev 116:10075–10166
Wang B, Li P et al (2019) Visible-light induced decarboxylative C2-alkylation of benzothiazoles with carboxylic acids under metal-free conditions. Org Biomol Chem 17:115–121
McManus JB, Nicewicz DA (2019) Direct C–H cyanation of arenes via organic photoredox catalysis. J Am Chem Soc 139:2880–2883
Margrey KA, Czaplyski WL et al (2018) A general strategy for aliphatic C–H functionalization enabled by organic photoredox catalysis. J Am Chem Soc 140:4213–4217
Wang GW (2013) Mechanochemical organic synthesis. Chem Soc Rev 42:7668–7700
Cheng H, Hernández JG et al (2017) Mechanochemical ruthenium-catalyzed hydroarylations of alkynes under ball-milling conditions. Org Lett 19:6284–6287
Howard JL, Cao Q et al (2018) Mechanochemistry as an emerging tool for molecular synthesis: what can it offer? Chem Sci 9:3080–3094
Hermann GN, Bolm C (2017) Mechanochemical rhodium (III)-catalyzed C–H bond amidation of arenes with dioxazolones under solventless conditions in a ball mill. ACS Catal 7:4592–4596
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Khakyzadeh, V., Sheikhaleslami, S. (2021). Green Chemistry on C–H Activation. In: Anilkumar, G., Saranya, S. (eds) Green Organic Reactions. Materials Horizons: From Nature to Nanomaterials. Springer, Singapore. https://doi.org/10.1007/978-981-33-6897-2_11
Download citation
DOI: https://doi.org/10.1007/978-981-33-6897-2_11
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-33-6896-5
Online ISBN: 978-981-33-6897-2
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)