
Abstract
C-H activation in the synthesis of medicinally relevant molecules is reviewed. C-H activation is one of the most powerful components in the C-C or C-heteroatom bond formation tool box. This represents such an alternative which has provided the greener solutions to the organometallic/synthetic organic chemistry aiming to the synthesis of complex molecules. In recent years, there has been explosive growth in this area. In this review article we have presented effective methods of C-H activation employed in the synthesis of medicinally relevant molecules since 2010.
Graphical Abstract:
C-H activation in the synthesis of medicinally relevant molecules is reviewed. C-H activation is one of the most powerful components in the C-C or C-heteroatom bond formation tool box. This represents such an alternative which has provided the greener solutions to the organometallic/synthetic organic chemistry aiming to the synthesis of complex molecules. In recent years, there is explosive growth in this area. In this review article we have presented effective methods of C-H activation employed in the synthesis of medicinally relevant molecules.

Similar content being viewed by others
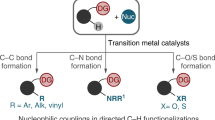

Avoid common mistakes on your manuscript.
1 Introduction
Carbon–carbon bond formation event is one of the most important reactions considering their significance in the key steps to architect more complex molecules from relatively less functionalized starting materials. Among the recent methodological advancement, transition metal mediated C–H functionalization represents a paradigm shift from the regular methods of organic synthesis.[1] This relies on the controlled functionalization of specific and remote C–H bonds, even in the presence of supposedly more reactive functional groups. The classical way of achieving remote C-H functionalization had been either by free-radical pathway or by intramolecular hydrogen abstraction. The recent advancements in organometallic chemistry have made it possible to achieve site-selective C–H functionalization. The initial approaches tend to focus on developing methods for the functionalization of relatively simple hydrocarbons, but in recent years, C–H functionalization has matured to the point where it can be contemplated as a viable strategy for the synthesis of complex targets.[2] In particular, functionalized or un-functionalized aromatic carbons can be used for the construction of biaryls, which are considered to be the important structural motifs in many naturally occurring and medicinally relevant organic molecules. The quest that gained significance over the period of time is to develop carbon-carbon bond formation without functionalization. These strategies are poised to fulfill this need for green and sustainable chemistry.[3]

Mechanisms of C-H activation.
The metal catalyst is involved throughout each stage of a C–H transformation and duly plays a fundamental role in determining its mildness.[4] The identity and oxidation state of the metal largely dictates which C–H activation mechanism operates in any given reaction.[5] Mechanisms for common C-H activations mainly fall under four general categories: (i) Oxidative addition, this is the most common mechanism by which a R-H bond cleaves and a M-C bond and a M-H bond are formed. The mechanistic pathway starts by coordination of the R-H bond to the metal vacant site (Figure 1). This mechanism is common for electron-rich, low-valent complexes of the late transition metals (Re, Fe, Ru, Os, Ir, Pt) for which the higher oxidation state of the metal in the product and the required change in geometry upon formation of the two new bonds are energetically favourable. (ii) Sigma-bond metathesis, oxidative addition is not possible with transition metals having with \(\hbox {d}^{0}\) electronic configuration (groups 3 and 4, lanthanides and actinides) and thus preferred mechanism is \(\upsigma \)-bond metathesis involving usually an alkyl or a hydride complex (Figure 1). The concerted mechanistic pathway of \(\upsigma \)-bond metathesis involves concerted formation of \(\hbox {M}\)-\(\hbox {R}^{1}\) and R-H bonds and breaking of M-R and \(\hbox {R}^{1}\)-\(\hbox {H}\) bonds at the transition state. This is usually common for late or post-transition metals (\(\hbox {Pd}^{2+}\), \(\hbox {Pt}^{2+}\) or \(\hbox {Pt}^{4+}\), \(\hbox {Hg}^{2+})\). (iii) Electrophilic addition, the intermediate formed in this mode of C-H activation is by the electrophilic attack of the metal. This classification of electrophilic substitution has emerged from the mechanistic pathway by which the hydrogen atom of the substrate is replaced by a metal and thus it acts as a lewis acid. (iv) 1, 2 addition, in this mechanistic pathway- the C-H bond directly adds across an unsaturated M-X bond in a 1,2 addition fashion (Figure 1). This mechanism is closely similar to \(\upsigma \)-bond metathesis; however the newly formed X-H bond does not lead to release of XH because the M-X \(\upsigma \)-bond is still present in the product. The complexes of early to middle transition metals have been shown to activate C-H bonds by 1, 2-addition.
To position better in terms of carbon-carbon or carbon-heteroatom bond formation it becomes imperative in most of the cases to employ transition metal as a catalyst for cross-dehydrogenative coupling (CDC) reaction with two C-H bonds as a greener choice. CDC is also aimed to avoid the installation of functional groups, thus making synthetic routes shorter, greener and efficient.[6] Here in, we have reviewed the C-H activation followed by carbon-carbon bond formation by using Pd, V, Ru and Rh metals in the synthesis of several medicinally relevant molecules. This review does not necessarily categorize C-H functionalization reactions on the basis of the mechanism by which they occur, but provides a selective overview of C-H functionalization strategies that has been employed for the rapid synthesis of medicinally relevant molecules as pharmaceutical targets since 2010.
2 C-H activation in medicinally relevant molecules
2.1 Palladium catalyzed C-H activation
2.1.1 Synthesis of dragmacidins
Synthetic studies towards the marine alkaloids dragmacidins have been successfully attempted by several research groups and several valuable synthetic methods were developed for the preparation of the homo- or heterodimerization of precursors.[7] Among them, most notably is the asymmetric aminohydroxylation of vinyl indoles in Jiang’s total synthesis of dragmacidin A.[8] However, the lengthy synthetic sequence involved in the preparation of functionalized precursors proved less attractive. Stoltz laboratory has conducted an extensive and systematic study for the synthesis of several analogues of dragmacidin.[9] Among them, it is noteworthy to mention C-H activation-based oxidative coupling strategy applied in Stoltz’s dragmacidin campaign that employed halogen-selective (Scheme 1), sequential Suzuki–Miyaura coupling for the synthesis of the structurally more challenging dragmacidin F 6.[10]

Stolz’s Asymmetric total synthesis of (+)-dragmacidin F.
The concise total synthesis of dragmacidin D 13 was accomplished by Itami and co-workers through C-H coupling reactions in 15 steps synthesis,[11] which is 10 steps shorter than the preceding synthesis that employed Suzuki–Miyaura coupling reactions (Scheme 2). The state of art methodological developments that was employed in this elegant synthesis include (i) Pd-catalyzed thiophene–indole C–H/C–I coupling, (ii) Pd-catalyzed indole–pyrazine N-oxide C–H/C–H coupling, and (iii) acid-catalyzed indole–pyrazinone C–H/C–H coupling. These regio selective catalytic C–H couplings had been used to rapidly assemble simple building blocks to construct the core structure of dragmacidin D 13 in a step-economical fashion.

Itami’s total synthesis of dragmacidin D.

Synthesis of Piperarborenines.

Synthesis of Calothrixin A & B.

Synthesis of Oxazolidinone anti-bacterial agent.
2.1.2 Synthesis of piperarborenine B
Baran laboratory has elegantly applied C-H activation that has culminated in the total synthesis of piperarborenine B 19 and piperarborenine D.[12] A strategy for the construction of unsymmetrical cyclobutanes using C–H functionalization logic by sequential, palladium catalyzed C-H activation followed by arylations with iodobenzene derivatives to afford mono- and di-aryl substituted cyclobutanes with remarkable efficiency and stereochemical control is demonstrated (Scheme 3). These syntheses feature a new preparation of cis-cyclobutanedicarboxylates from commercially available coumalate starting material and the divergent approach to the controlled cis or trans installation of the two distinct aryl rings found in the natural products is the first example of cyclobutane C–H arylation.
2.1.3 Synthesis of calothrixin A and B
The total synthesis of calothrixin A and B reported by Rajagopal Nagarajan features an intramolecular Pd-catalyzed cross coupling reaction via C-H activation (Scheme 4).[13] Total synthesis of calothrixins have been reported by several groups by utilizing simple methods with good yields, however most of them comprise a greater number of steps.[14] The C-H activation route has enabled the synthesis in reduced number of steps so that the overall yield has increased considerably and route has become more concise. The precursor 21 was synthesized in one pot by Friedel - Crafts acylation of 20 with oxalyl chloride in the presence of \(\hbox {AlCl}_{3}\) in dry DCM followed by amidation with aniline. Pd-catalyzed intramolecular oxidative coupling of 21 in the presence of 5 mol% Pd(TFA)\(_{2}\) heated at \(120\,{^{\circ }}\hbox {C}\) for 24 h under an oxygen atmosphere in melted benzoic acid afforded the indolophenanthridinone 22 in 83% yield. Thereafter, 22 was subjected to chemoselective reduction using \(\hbox {Tf}_{2}\hbox {O}/\hbox {Et}_{3}\hbox {SiH}\) to provide indolophenanthridine 23, which was easily transformed into Calothrixin A 26 & B 25 in a couple of steps in a total of 5 steps over 35% overall yield.
2.1.4 Synthesis of oxazolidinone anti-bacterial agent
Michael Pamment and team at Pfizer Global R&D have developed a kilogram scale synthesis of oxazolidinone anti-bacterial agent 32 following the palladium-catalyzed C-H activation strategy to build the key oxindole intermediate 30 (Scheme 5).[15] The overall synthesis is a 6-step linear sequence and the C-H activation chemistry has been exploited in fourth stage of the synthesis. Authors found that the use of trifluorotoluene as solvent minimized the product decomposition and furnished the desired oxindole 32 in 75% yield with facile product isolation. This method was exploited to generate 4.9 kg of the oxindole 32 in 86% yield.
Synthesis of oxindole from chloroacetanilides was originally developed by Buchwald group.[16] They have propose three mechanistic pathways (Scheme 6). The first step is proposed to be oxidative addition of the chloroacetamide to the catalyst. This step is slow and rate determining. Pathway A involves electrophilic aromatic substitution to form a six-membered palladacycle which upon reductive elimination release the desired product with regeneration of Pd(0) catalyst. Alternatively, the aromatic ring can undergo carbo-palladation and subsequent HPdCl elimination generates the oxindole (pathway B). The third possibility is C-H activation which can either proceed by metathesis of C-H sigma bond or through the intermediacy of \(\uppi \upeta ^{1}\) interaction (pathway C).

Mechanistic consideration in the synthesis of oxazolidinone anti-bacterial.

Synthesis of SPT inhibitor.
2.1.5 Synthesis of SPT inhibitor
E. Jason Kiser and his team at Pfizer Inc. have developed a three-step, chromatography-free synthesis of oxyindole 45, which is the key intermediate for the synthesis of a potential drug candidate 46 that has been proposed to elevate the level of HDL cholesterol by inhibition of serine palmitoyltransferase (SPT) enzyme, thus potentially useful in the treatment of heart diseases (Scheme 7).[17] The original medicinal chemistry route for synthesizing the key intermediate 45 consists of a linear six-step synthetic process which was satisfactory for the preparation of small batches of 46. However, for the kilo scale synthesis the team has developed a concise and scalable route by taking advantage of Buchwald’s palladium-catalyzed C-H functionalization to cyclize a \(\upalpha \)-chloroacetanilide to form the five-membered ring. Intermediate 45 was synthesized twice on 5 kg scale and once on 10 kg scale in 76–84% yield following this chemistry.
2.2 Rhodium catalyzed C-H activation
2.2.1 Synthesis of Lithospermic acid
First total synthesis of racemic heptamethyl lithospermate was reported by Jacobson in 1979.[18] In this work, the synthesis was started with isovanillin, which was converted to racemic heptamethyl lithospermate by a sequence that largely involved pedestrian functional group transformations and condensation reactions for coupling of fragments. One of the pioneering work of C-H activation promoted carbon bond formation in the landmark synthesis of (+)-lithospermic acid 52 was reported by Bergman, Ellman and co-workers.[19] In 2001, Ellman and co-workers have reported a rhodium-catalyzed intramolecular alkylation of aromatic imines tethered, through a mechanistic pathway that involve a directed C-H activation.[20] This methodology was later applied in an asymmetric setting with high level of enantio-control to access the benzofuran core of Lithospermic acid by C-H activation (Scheme 8). In a racemic system, the benzaldehyde derived benzylimine 48 was smoothly converted to benzofuran 49 in 89% yield as a single cis isomer. The optically active alkylation product 49 obtained (89% yield, 73% ee) was further enriched to 99% ee after recrystallization (56% overall yield). The side chain of (-)-lithospermic acid was attached onto aldehyde 49 through an analogous Knoevenagel condensation and the methyl ester bearing stereo center was also fortuitously epimerized under the reaction condition to obtain 50. To complete the first total synthesis of (+)-lithospermic acid 52, the final demethylation was ultimately achieved by TMSI-quinoline combination to fulfill this objective in 35% yield.

Asymmetric total synthesis of (+)-lithospermic acid by Bergman and Ellman.
Through a highly ingenious approach, the Yu group have demonstrated a highly convergent total synthesis of (+)-lithospermic acid 52 by two successive C-H activation reactions as key steps.[21] Rh-catalyzed carbene C-H insertion reaction utilizing Davies’s catalyst was used to forge dihydrobenzofuran core 54, and a late-stage intermolecular C-H olefination coupled the olefin unit with the dihydrobenzofuran core to construct the molecule in a highly convergent manner (Scheme 9). The directing group is a chiral non-racemic imine capable of intramolecular alkylation.

Asymmetric total synthesis of (+)-lithospermic acid by Yu group.

Synthesis of Vildagliptin Analogue.

Synthesis of angiotensin II receptor blockers (ARBs).

Synthesis of advanced intermediate of Anacetrapib.
2.2.2 Synthesis of Vildagliptin analogues
Radim Hrdina et al., have developed a straightforward way to prepare 1,2-disubstituted diamondoids by C–H bond amination reactions on a rigid tricyclic systems and applied this strategy to the synthesis of an enantiopure N-protected \(\upbeta \)-amino acid and new analogues of Vildagliptin.[22] Traditionally, the selective C–H functionalization of diamondoids have been achieved by undirected functionalization reactions, such as oxidation reactions, which mostly leads to tertiary products as the selectivity is based on the different C-H dissociation energies or thermodynamic stabilities of corresponding ions or radicals.[23] However, the C-H activation based approach adopted by Radim Hridina laboratory ensured high reactivity of metal-bound nitrene species that would allow functionalization of un-activated rigid diamondoids and regioselectivity of nitrene insertions towards the formation of six-membered rings (Scheme 10). The synthesis of Vildagliptin analogue 62, as new antidiabetic drug candidates (DPP-4 inhibitors) has been achieved by intramolecular C–H functionalization using dirhodium-acetatecatalyzed (1 mol%) nitrene insertion reaction. This work is focused on C–H amination reactions of conformationally rigid tricyclic frameworks and presents the synthesis of two bioisosters of the antihyperglycemic agent DPP-4 inhibitor.

Synthesis of LY2784544.

Mechanistic consideration in the synthesis of LY2784544.
2.3 Ruthenium catalyzed C-H activation
2.3.1 Synthesis of angiotensin II receptor blockers (ARBs)
Masahiko Seki and Masaki Nagahama have developed an efficient catalytic system for C-H activation to functionalize aryltetrazoles that involves inexpensive \(\hbox {RuCl}_{3}\cdot \hbox {xH}_{2}\hbox {O}\) and have successfully applied to the practical synthesis of angiotensin II receptor blockers (ARBs) Losartan 67 and Valsartan 68 (Scheme 11).[24]\(^{\mathrm{a}}\) The previous synthetic approaches have critical drawbacks due to the need of stoichiometric amounts of expensive and/or hazardous organometallics such as Grignard reagents and boronic acids to install the fundamental biphenyltetrazole framework.[25] The robust catalytic system used for C-H activation route has been worked out that involves inexpensive \(\hbox {RuCl}_{3}\cdot \hbox {xH}_{2}\hbox {O}\) and a specific amount of \(\hbox {PPh}_{3}\). The residual ruthenium that existed in the reaction mixture was thoroughly removed by treatment with properly selected metal scavengers. The new process afforded ready access to the important class of drugs in a highly atom-economical and sustainable manner. Lutz Auckermann have also similarly demonstrated C-H functionalizations of the aryltetrazoles to set the stage for the preparation of various angiotensin II receptor blockers (ARBs) in a concise and atom-economical fashion.[24]\(^{\mathrm{b}}\)

Synthesis of Dictyodendrin B.
2.3.2 Synthesis of Anacetrapib
C-H biaryl coupling assisted by directing groups was applied to access the core structure 71 of anacetrapib 72 by Ouelett et al., at Merck by using ruthenium catalyzed direct C-H arylation as the key step.[26] Lutz Auckermann inspired C-H arylation of oxazoline 69 with bromoanisole 70 under their robust ruthenium-based catalyst system was applied to obtain biaryl intermediate 71, followed by reductive cleavage to afford the anacetrapib biaryl core.[27] Notably, this practical, scalable, and chromatography-free synthesis was successfully demonstrated on a kilogram scale (Scheme 12). The effect of low level impurity found in solvent is exploited to the high yielding arylation protocol demonstrated in multigram scale using carboxylate as the cocatalyst. Importantly, this C-H activation strategy was not only advantageous because of its economical and ecological benefits but also allowed streamlining the organic syntheses.
2.4 Vanadium catalyzed C-H activation
2.4.1 Synthesis of LY2784544
Researchers of Lilly Research Laboratories have reported an alternative synthetic route to a functionalized imidazopyridazine intermediate via sequential C-H bond functionalization for the synthesis of JAK2 inhibitor LY2784544 79 that was undergoing clinical trials for the treatment of several myeloproliferative disorders.[28] The previously reported synthesis have several liabilities, including the use of amino acetal to form the pyridazine framework which was not readily available and usage trifluoroacetic acid (TFA) and triethylsilane liability from a waste disposal perspective, among others.[29] Key to the success of this alternative route is the use of two C-H functionalization reactions: a Pd-catalyzed direct benzylation reaction to functionalize a C-H bond with a substituted benzyl group and a V-catalyzed NMO addition reaction to install a benzylic morpholine moiety (Scheme 13). Thus team tasked to optimize the synthetic process developed a C-H activation driven synthetic route by successfully circumventing the major challenges associated with the previous route, namely fluoride and silane waste incineration. Furthermore, using the route described in this report, the synthesis of the imidazopyridazine core is accomplished in a single step and avoided the use of the amino acetal which had required long lead times to secure (Scheme 14).[29]
2.5 Various metal catalyzed C-H activation
2.5.1 Synthesis of dictyodendrin B
The complex poly(hetero)aromatic architecture of marine natural alkaloid dictyohydrin has inspired a number of elegant total syntheses from the groups of Fürstner, Iwoa and Ishibashi, Tokuyama and Jia having often lengthy chemical sequences.[30] However, Matthew J. Gaunt and his team have elegantly employed a sequential C-H functionalization strategy for the synthesis of structurally intriguing dictyodendrin B in concise synthetic approach.[31] The total synthesis of dictyodendrin B 94 starting from a commercially available 4-bromoindole and involves six direct functionalization around the heteroarene core as part of a gram-scale strategy towards the natural product (Scheme 15). The inherent nucleophilicity of indole makes reactions through the C3 position an ideal starting point for copper-catalyzed C-H arylation using diaryliodonium salt. Inexpensive \(\hbox {Cu}^{\mathrm{I}}\hbox {Cl}\) (5 mol%) functioned effectively as the catalyst to combine 4-bromoindole 85 with bis(4-methoxyphenyl)iodonium tetrafluoroborate to afford 86 in 68% yield on 42 gram scale. Next, the intrinsic reactivity of indole for the C2 acylation was exploited and a bismuth(III) triflate catalyzed Friedel–Crafts type acylation afforded the 2,3-disubstituted indole product 87 as a single isomer in 57% yield. C7 substitution of Indole moiety 87 was achieved by utilizing the indole N-H motif to direct an iridium-catalyzed C-H borylation. With the indole N-H moiety having served its purpose for directing the C-H arylation, next N-alkylation was achieved by utilizing commercial 4-methoxyphenethyl bromide and \(\hbox {K}_{2}\hbox {CO}_{3}\) to afford the desired N-alkylated product 89 in 83% yield. Thereafter, C4 arylation was achieved by Suzuki–Miyaura coupling of the C4 bromide and aryl boronic ester using a mixture of [\(\hbox {PdCl}_{2}\)(dppf)] (5 mol%) and an aqueous solution of \(\hbox {K}_{2}\hbox {CO}_{3}\) (2M) in dioxane at \(90 \,{^{\circ }}\hbox {C}\) to furnish the C4-arylated product 90 in 93% yield on a 1.5 gram scale. Electrophilic bromination at the C6 position of the indole was achieved with exclusive selectivity at room temperature using a slight excess of N-bromosuccinimide. Direct addition of a sodium methoxide solution in methanol (4M) and copper(I) iodide to the bromination reaction mixture led to the formation of the methyl ether, completing a two-step one-pot etherification process to obtain 91 in 81% yield on a 1.5 gram scale. The functional group transformation of nitro group into the azide was done by a two-step reduction/diazotization/azidation process in 95% yield on 1.5 gram scale to obtain 92. This followed by C-H amination, presumably via the formation of nitrene intermediate, in super-heated dioxane at \(180 \,{^{\circ }}\hbox {C}\) in a continuous-flow process led to desired the carbazole product 93 in 62% yield. Finally, four-step deprotection and sulfonylation sequence afforded dictohydrin B.
3 Conclusions
In summary, we have presented effective methods of C-H activation employed in the synthesis of medicinally relevant molecules like lithospermic acid, dragmacidin D, piperaborenine B, vildagliptin analogue, losartan, valsartan, anacetrapib, dictyodendrin B, calothrixin A&B, oxazolidinone antibacterial, serinpalmitoyltranferase (SPT) enzyme inhibitor and LY2784544 (JAK2-V617F inhibitor) using transition metals which appeared in the literature since 2010.
References
(a) Selected reviews on C-H activation: (a) Wencel-Delord J, Dröge T, Liu F and Glorius F 2011 Towards mild metal-catalyzed C–H bond activation Chem. Soc. Rev. 40 4740; (b) Yeung C S and Dong V M 2011 Catalytic Dehydrogenative Cross-Coupling: Forming Carbon-Carbon Bonds by Oxidizing Two Carbon-Hydrogen Bonds Chem. Rev. 111 1215; (c) Jia C, Kitamura T and Fujiwara Y 2001 Catalytic Functionalization of Arenes and Alkanes via C-H Bond Activation Acc. Chem. Res. 34 633; (d) Chen D Y-K and Youn S W 2012 C-H Activation: A Complementary Tool in the Total Synthesis of Complex Natural Products Chem. Eur. J. 18 9452; (e) Yamaguchi J, Yamaguchi A D and Itami K 2012 C-H bond functionalization: emerging synthetic tools for natural products and pharmaceuticals Angew. Chem. Int. Edit. 51 8960
Wencel-Delord J and Glorius F 2013 C–H bond activation enables the rapid construction and late-stage diversification of functional molecules Nature Chem. 5 369
(a) Li C-J and Trost B M 2008 Green chemistry for chemical synthesis Proc. Natl. Acad. Sci. USA 105 13197; (b) Anastas P T and Warner J C 1998 Green Chemistry: Theory and Practice (New York: Oxford University Press)
Gensch T, Hopkinson M N, Glorius F and Wencel-Delord J 2016 Mild metal-catalyzed C–H activation: examples and concepts Chem. Soc. Rev. 45 2900
Balcells D, Clot E and Eisenstein O 2010 C-H Bond Activation in Transition Metal Species from a Computational Perspective Chem. Rev. 110 749
(a) Li C J 2009 Cross-Dehydrogenative Coupling (CDC): Exploring C-C Bond Formations beyond Functional Group Transformations Acc. Chem. Res. 42 335; (b) Ashenhurst J A 2010 Intermolecular oxidative cross-coupling of arenes Chem. Soc. Rev. 39 540; (c) Bugaut X and Glorius F 2011 Palladium-catalyzed selective dehydrogenative cross-couplings of heteroarenes Angew. Chem. Int. Edit. 50 7479
(a) Jiang B, Smallheer J M, Amaral-Ly C and Wuonola M A 1994 Total Synthesis of (\(\pm )\)-Dragmacidin: A Cytotoxic Bis(indole)alkaloid of Marine Origin J. Org. Chem. 59 6823; (b) Kawasaki T, Ohno K, Enoki H, Umemoto Y and Sakamoto M 2002 Syntheses of bis (indolyl)-piperazine alkaloids, dragmacidin B and C, and dihydrohamacanthin A Tetrahedron Lett. 43 4245; (c) Kawasaki T, Enoki H, Matsumura K, Ohyama M, Inagawa M and Sakamoto M 2000 First Total Synthesis of Dragmacidin A via Indolylglycines Org. Lett. 2 3027; (d) Tonsiengsom F, Miyake F Y, Yakushijin K and Horne D A 2006 Reduction of 2,5-Bis (\(3^{\prime }\)-indolyl) pyrazines to 2,5-Bis (\(3^{\prime }\)-indolyl) piperazines: Synthesis of Bisindolylpiperazine Marine Alkaloids Dragmacidin A, B, and C Synthesis 49; (e) Miyake F Y, Yakushijin Kand Horne D A 2000 A Facile Synthesis of Dragmacidin B and 2,5-Bis (6\(^{\prime }\)-bromo-3\(^{\prime }\)-indolyl) piperazine Org. Lett. 2 3185
(a) Yang C G, Wang J, Tang X X and Jiang B 2002 Asymmetric aminohydroxylation of vinyl indoles: a short enantioselective synthesis of the bisindole alkaloids dihydrohamacanthin A and dragmacidin A Tetrahedron: Asymmetr. 13 383; (b) Jiang B and Gu X H 2000 Syntheses of Bis (3\(^{\prime }\)-indolyl)-2(1H)-pyrazinones Heterocycles 53 1559
(a) Garg N K and Stoltz B M 2006 A unified synthetic approach to the pyrazinone dragmacidins Chem. Commun. 3769; (b) Garg N K, Sarpong R and Stoltz B M 2002 The First Total Synthesis of Dragmacidin D J. Am. Chem. Soc. 124 13179; (c) Garg N K, Caspi D D and Stoltz B M 2006 The Utility of the Classical and Oxidative Heck Reactions in Natural Product Synthesis: Studies Directed toward the Total Synthesis of Dragmacidin F Synlett 3081
Garg N K, Caspi D D and Stoltz B M 2005 Development of an Enantiodivergent Strategy for the Total Synthesis of (+)- and (-)-Dragmacidin F from a Single Enantiomer of Quinic Acid J. Am. Chem. Soc. 127 5970
Mandal D, Yamaguchi A D, Yamaguchi J and Itami K 2011 Synthesis of Dragmacidin D via Direct C-H Couplings J. Am. Chem. Soc. 133 19660
Gutekunst W R and Baran P S 2011 Total Synthesis and Structural Revision of the Piperarborenines via Sequential Cyclobutane C–H Arylation J. Am. Chem. Soc. 133 19076
Ramkumar N and Nagarajan R 2013 Total Synthesis of Calothrixin A and B via C–H Activation J. Org. Chem. 78 2802
(a) Choshi T and Hibino S 2009 Progress towards the Total Synthesis of the Bioactive Calothrixins A and B Heterocycles 77 85; (b) Tohyama S, Choshi T, Matsumoto K, Yamabuki A, Hieda Y, Nobuhiro J and Hibino S 2010 Total Synthesis of Bioactive Indolo[3,2-j]phenanthridine Alkaloid, Calothrixin B Heterocycles 82 397
Choy A, Colbry N, Huber C, Pamment M and Duine J V 2008 Development of a Synthesis For a Long-Term Oxazolidinone Antibacterial Org. Process Res. Dev. 12 884
Hennessy E J and Buchwald S L 2003 Synthesis of Substituted Oxindoles from \(\upalpha \)-Chloroacetanilides via Palladium-Catalyzed C-H Functionalization J. Am. Chem. Soc. 125 12084
Kiser E J, Magano J, Shine R J and Chen M H 2012 Kilogram-Lab-Scale Oxindole Synthesis via Palladium-Catalyzed C–H Functionalization Org. Process Res. Dev. 16 255
Jacobson R M and Raths R A 1979 Total synthesis of heptamethyl lithospermate J. Org. Chem. 44 4013
Malley S J O, Tan K L, Watzke A, Bergman R G and Ellman J A 2005 Total Synthesis of (+)-Lithospermic Acid by Asymmetric Intramolecular Alkylation via Catalytic C-H Bond Activation J. Am. Chem. Soc. 127 13496
(a) Thalji R K, Ahrendt K A, Bergman R G and Ellman J A 2001 Annulation of Aromatic Imines via Directed C-H Activation with Wilkinson’s Catalyst J. Am. Chem. Soc. 123 9692; (b) Ahrendt K A, Bergman R G and Ellman J A 2003 Synthesis of a Tricyclic Mescaline Analogue by Catalytic C-H Bond Activation Org. Lett. 5 1301
Wang D H and Yu J Q 2011 Highly Convergent Total Synthesis of (+)-Lithospermic Acid via a Late-Stage Intermolecular C-H Olefination J. Am. Chem. Soc. 133 5767
Hrdina R, Metz F M, Larrosa M, Berndt J–P, Zhygadlo Y, Becker S and Becker J 2015 Intramolecular C–H Amination Reaction Provides Direct Access to 1,2-Disubstituted Diamondoids Eur. J. Org. Chem. 28 6231
(a) Fokin A A, Schreiner P R, Gunchenko P A, Peleshanko S A, Shubina T E, Isaev S D, Tarasenko P V, Kulik N I, Schiebel H M and Yurchenko A G 2000 Oxidative Single-Electron Transfer Activation of \(\upsigma \)-Bonds in Aliphatic Halogenation Reactions J. Am. Chem. Soc. 122 7317; (b) Roizen J L, Zalatan D N and Du Bois J 2013 Selective intermolecular amination of C-H bonds at tertiary carbon centers Angew. Chem. Int. Edit. 52 11343
(a) Seki M and Nagahama M 2011 Synthesis of angiotensin II receptor blockers by means of a catalytic system for C-H activation J. Org. Chem. 76 10198; (b) Lutz A 2015 Robust Ruthenium(II)-Catalyzed C-H Arylations: Carboxylate Assistance for the Efficient Synthesis of Angiotensin-II-Receptor Blockers Org. Process Res. Dev. 19 260
(a) Larsen R D, King A O, Chen C Y, Corley E G, Foster B S, Roberts F E, Yang C, Lieberman D R, Reamer R A, Tschaen D M, Verhoeven T R, Reider P J, Lo Y S, Rossano L T, Brookes S, Meloni D, Moore J R and Arnett J F 1994 Efficient Synthesis of Losartan, A Nonpeptide Angiotensin II Receptor Antagonist J. Org. Chem. 59 6391; (b) Beutler U, Boehm M, Fuenfschilling P C, Heinz T, Mutz J P, Onken U, Mueller M and Zaugg W 2007 A High-Throughput Process for Valsartan Org. Process Res. Dev. 11 892; (c) Kumar N, Reddy S B, Sinha B K, Mukkanti K and Dandala R 2009 New and Improved Manufacturing Process for Valsartan Org. Process Res. Dev. 13 1185; (d) Wang G X, Sun B P and Peng C H 2011 An Improved Synthesis of Valsartan Org. Process Res. Dev. 15 986
Ouellet S G, Roy A, Molinaro C, Angelaud R, Marcoux J F, O’Shea P D and Davies I W 2011 Preparative scale synthesis of the biaryl core of anacetrapib via a ruthenium-catalyzed direct arylation reaction: unexpected effect of solvent impurity on the arylation reaction J. Org. Chem. 76 1436
Lutz A, Vicente R, Potukuchi H K and Pirovano V 2010 Mechanistic Insight into Direct Arylations with Ruthenium(II) Carboxylate Catalysts Org. Lett. 12 5032
Campbell A N, Cole K P, Martinelli J R, May S A, Mitchell D, Pollock P M and Sullivan K A 2013 Development of an Alternate Synthesis for a Key JAK2 Inhibitor Intermediate via Sequential C-H Bond Functionalization Org. Process Res. Dev. 17 273
Mitchell D, Cole K P, Pollock P M, Coppert D M, Burkholder T P and Clayton J R 2012 Development and a Practical Synthesis of the JAK2 Inhibitor LY2784544 Org. Process Res. Dev. 16 70
(a) Fürstner A, Domostoj M M and Scheiper B 2005 Total Synthesis of Dictyodendrin B J. Am. Chem. Soc. 127 11620; (b) Hirao S, Sugiyama Y, Iwao M and Ishibashi F 2009 Synthetic Approach to Telomerase Inhibitor Dictyodendrin B: Synthesis of the Pyrrolo[2,3-c]carbazole Core Biosci. Biotechnol. Biochem. 73 1764; (c) Okano K, Fujiwara H, Noji T, Fukuyama T and Tokuyama H 2010 Total Synthesis of Dictyodendrin A and B Angew. Chem. Int. Edit. 49 5925; (d) Liang J, Hu W, Tao P and Jia Y 2013 Total Synthesis of Dictyodendrins B and E J. Org. Chem. 78 5810
Pitts A K, O’Hara F, Snell R H and Gaunt M J 2015 A Concise and Scalable Strategy for the Total Synthesis of Dictyodendrin B Based on Sequential CH Functionalization Angew. Chem. 127 5541
Acknowledgements
We thank the management of Dr. Reddy’s Laboratories Ltd for supporting this work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Basu, D., Kumar, S., V, S.S. et al. Transition metal catalyzed C-H activation for the synthesis of medicinally relevant molecules: A Review. J Chem Sci 130, 71 (2018). https://doi.org/10.1007/s12039-018-1468-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12039-018-1468-6