
Abstract
Organotypic skin constructs have gained significant research and commercial interest in the face of EU bans on animal-tested cosmetic products. In this project, a simplified, full-thickness organotypic skin construct was prepared using fibroblasts encapsulated in a synthetic peptide hydrogel matrix, over which keratinocytes were allowed to proliferate, differentiate and stratify. The self-assembling peptide was synthesized using solid phase chemistry. The peptide was then used to prepare hydrogels of varying concentrations and the formulation was optimized based on gelation kinetics and mechanical strength. The long-term biocompatibility of this matrix with dermal fibroblasts was also evaluated. Finally, the in vitro skin constructs were characterized using histology and electron microscopy. Potential primers to evaluate gene expression of epithelial biomarkers were also identified. In conclusion, the peptide hydrogel is an appropriate matrix for culturing organotypic skin constructs due to its stability and low cytotoxicity. Building on this model, more elaborate systems can be cultured with the addition of more cell types. These biological constructs can potentially be used to screen therapeutic candidates, as well as to evaluate the effects of compounds on skin tissue viability, permeability and cellular gene expression.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others

Keywords
1 Background and Purpose of Research
Three-dimensional (3D), multi-cellular, tissue mimetic models are powerful experimental platforms. They enable the in vitro study of mammalian tissue development, the modelling of human disease and the evaluation of novel therapeutics [1]. Organotypic 3D cultures are more biologically relevant and allow better understanding of in vivo physiology and function as integral biological processes are altered when cells are cultured in 2D versus 3D. These include immune system activation, defense response, cell adhesion and tissue development [2, 3].
Organotypic skin constructs, in particular, have gained significant research and commercial interest in the face of EU bans on animal-tested cosmetic products [4]. Human cadaver skin and excised animal skin have been traditionally used as topical and transdermal permeation models [5]. However, while human cadaver skin replicates in vivo permeation performance to some extent, there is a high sample to sample variation. Animal skin, though easily procured, is inherently different in physiology and gene expression from human skin. A simple in vitro culture of full-thickness skin can be constructed using dermal fibroblasts encapsulated in a 3D gel matrix, over which keratinocytes are seeded. Various biomaterials which mimic the extracellular matrix (ECM) have demonstrated applicability as matrices [6, 7]. Collagen is currently the most commonly used matrix for building organotypic full-thickness skin. It is a good scaffold material since it contains naturally-occurring ligands that facilitate cell attachment and proliferation [8]. However, batch-to-batch variations reduce the degree of experimental control; while potential risks of immunogenicity and pathogen transmission hinder future applications as tissue transplants [8]. Experimentally, one of the major challenges of using collagen hydrogels is remodelling by dermal fibroblasts, resulting in significant and uneven shrinkage of the tissue construct after 3 weeks of culture [9]. Hence, we propose the use of self-assembling, synthetic peptide hydrogels as a substitute supporting matrix. Short peptides are versatile building blocks for fabricating supramolecular structures. A specific motif enables ultrasmall peptides with 3–6 amino acids to self-assemble to helical supramolecular fibres [10]. This amphiphilic peptide motif consists of a tail of aliphatic nonpolar amino acids with decreasing hydrophobicity and a hydrophilic head group [10]. The peptides undergo a structural transition pathway from random coils to α-helical intermediates to β-turn structures with increasing concentration. The β-turn fibers further condense into meshed 3D nanofibrous networks, which closely resemble the ECM [10]. Due to their strong amphiphilic nature, the peptide networks entrap water, forming 3D nanofibrous hydrogels. In this study, we use a hexamer peptide, IK6, which demonstrates salt-enhanced gelation. They are highly suitable due to their outstanding biocompatibility, mild gelation conditions [10], and excellent mechanical properties. Moreover, they have been investigated as bioink candidates to facilitate bioprinting of 3D tissue constructs [11].
2 Engineering Goal
The objective of this project is to synthesize, optimize and characterize the formulation of peptide hydrogels for building full-thickness organotypic skin constructs.
3 Methods and Materials
3.1 Solid-Phase Peptide Synthesis
The ultrashort peptides were synthesized manually, using solid phase peptide chemistry (Appendix) [12]. Briefly, L-lysine residues conjugated to polystyrene beads via a Rink-amide linkage (GL Biochem, China) were swelled in dimethylformamide (JT Baker, USA), de-protected and each succeeding amino acid residue added sequentially. After the terminal amino acid had been added, the N-terminus amine group was acetylated using acetic anhydride (Sigma Aldrich, USA). The peptides were then cleaved using trifluoroacetic acid (Acros Organics, USA), precipitated and washed with diethyl ether (JT Baker, USA). After vacuum drying, the crude products were dissolved in dimethylsulfoxide (Kanto Chemical Co. Inc., Japan) and purified using reverse-phase high-performance liquid chromatography mass spectrometry. The purified peptide solution was subsequently lyophilized to obtain a dry powder.
3.2 Preparation of Peptide Hydrogels
Hydrogel samples were prepared in polydimethysiloxane moulds to obtain 8 mm diameter discs, approximately 1 mm thick. Peptide powder was first dissolved in milliQ water. 10% volume of 10× phosphate-buffered saline (PBS) was subsequently added to stimulate gelation.
3.3 Rheological Analysis
Dynamic strain and oscillatory frequency sweep experiments were carried out using the ARES-G2 Rheometer (TA Instruments, USA) with 8 mm titanium parallel plate geometry. The storage (G′) and loss (G″) moduli were recorded in response to varying strain (γ) and angular frequency (ω). The readings of 3 samples were averaged for each condition.
3.4 HDF and HDK Culture
Human dermal fibroblasts (HDFs) were obtained from Lonza (Basel, Switzerland), cultured in Dulbecco’s minimum essential media (DMEM) supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin. Human dermal keratinocytes (HDKs) were also purchased from Lonza (Basel, Switzerland), cultured in Keratinocyte Serum Free Medium (Life Technologies, USA).
3.5 Encapsulation of HDF and Cytotoxicity Assay
HDFs cultured were either cultured on tissue culture plates (24-well) or encapsulated in 6 mg/mL peptide hydrogel, and cultured for 1 or 3 weeks. Cytotoxicity was evaluated using the LIVE/DEAD assay (Thermo Fisher Scientific, USA). Constructs were incubated at 37 °C for 5 min before being viewed underneath a fluorescence microscope (Olympus IX71 Research Inverted microscope).
3.6 Organotypic Skin Co-cultures
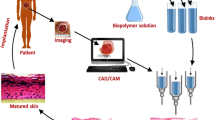
HDFs were combined with the IK6 hydrogel and NaOH and seeded onto a 24-well transwells (Corning, USA) at 4 × 106 cells/mL (Fig. 1). After a week, the HDKs seeded onto the constructs. The constructs were maintained in 50% DMEM media and 50% Keratinocyte SFM media for another week. After which, they were switched to air-liquid interphase culture in stratification media for another 2 weeks [13].

Schematic representation of preparation of 3D organotypic skin constructs. a Peptide hydrogels encapsulating human dermal fibroblasts are pipetted into transwells and allowed to gel at 37 °C for 30 min. The hydrogels were subsequently submerged in fibroblast media for a week. b Keratinocytes are seeded onto the hydrogel surface and the construct is maintained under submerged conditions in mixed media for another week. c Air-liquid interface culture wherein the construct is exposed to stratification media on the basolateral surface. This results in keratinocyte differentiation into a stratified epidermis
3.7 Histology
Samples were fixed in 4% paraformaldehyde. Paraffin embedding was carried out and 5 μm sections were stained with hematoxylin and eosin (H&E) for routine histological evaluation under an Olympus IX71 microscope.
3.8 Field Emission Scanning Electron Microscopy
Organotypic skin constructs were dehydrated by sequential immersion in ethanol solutions of increasing concentration, followed by crucial point drying using an Autosamdri-815 Series A Critical Point Dryer. The dried samples were then sputter-coated with platinum in a JEOL JFC 1600 High Resolution Sputter Coater. The coated sample was then examined with a JEOL JSM-7400F FESEM system using an accelerating voltage of 5 kV.
3.9 Qualitative Gene Expression Analysis Using Real Time PCR
The constructs were dissolved in 500 μL of TRIzol (Life Technologies, USA). The mRNA was extracted using chloroform extraction, followed by isopropanol-ethanol precipitation. The concentration of mRNA content was quantified via absorbance reading using NanoDrop 2000 Spectrophotometer (Thermo Fisher Scientific, USA). 500 ng mRNA was subsequently used to prepare cDNA via reverse transcription using Super Script (Thermo Fisher Scientific, USA). 4 μL of template cDNA, 5 μL SYBR Green (Kapa Biosystems, USA) and 0.2 μL ROX dye were combined to make the template mix. 9 μL of the template mix and 1 μL of diluted primer (col3A1, CRABP2, KLK5, CDSN2 and GADPH) were added to the walls of a 96 well plate. The plate was sealed with thermosensitive film and incubated in the 7500 Fast Real-time PCR System (Applied Biosystems, USA).
4 Results and Discussion
4.1 Peptide Synthesis
The peptide was synthesized by solid-phase peptide synthesis and subsequently purified using high performance liquid chromatography. The collection of the desired product is triggered by the detection of the 641 mass peak in the spectrometer. With reference to the chromatograph (Fig. 2a) and mass spectra of the collected fragment (Fig. 2b), there is little contamination of the crude product by deletion, addition or substitution peptide sequences. The collected product (Fig. 2c) is very pure.

Purification of the peptide product. The a chromatograph and b mass spectra of the collected fraction reflect the purity of the product. The desired product has a molecular weight of 640. It is detected in the mass spectrometer as a singly charged species as denoted by the 641 peak. The 642 and 643 peaks contain deuterium isotopes, while the 321 peak is indicative of the double charged species. c The purified, lyophilized peptide product is a fluffy white powder
4.2 Gelation Behavior and Mechanical Properties
Rigid hydrogels with storage moduli (G′) in the range of 10 kPa were obtained, using PBS as the buffer. From the frequency sweep study (Fig. 3a), as frequency increases, the gel remains fairly stable. Increasing peptide concentration increases both storage and loss (G″) moduli. Variation between samples is very small (n = 3). From the amplitude sweep study (Fig. 3b), G′ and G″ drops sharply when the strain increases past a certain point, indicating that the hydrogel’s microstructure is breaking down. In both studies, G′ remains higher than G″, demonstrating that the samples remain as gels throughout the measurement. When peptide concentration increases from 6 to 7.5 mg/mL, G′ increases from 104 to 1.5 × 104 Pa. When peptide concentration increases from 7.5 to 9 mg/mL, G′ increases from 1.5 × 104 to 6 × 104 Pa.

The peptide hydrogels demonstrate good mechanical strength, as reflected by high storage moduli in the range of 10 kPa. a Frequency and b amplitude sweep studies show that hydrogel rigidity increases with peptide concentration
The 6 mg/mL hydrogel formulation was chosen for preparation of tissue constructs as it sufficiently stable (above 104 Pa). The lower concentration hydrogel conserves material and reduces cost, resulting in significant savings when producing tissue constructs on a large scale. Moreover, the gelation time of the 6 mg/mL hydrogel was about 30 min, which is comparable to that observed in literature. During this window, the gel is very soft and fluid. It can be mixed to distribute the cells uniformly, and consistently dispensed into the transwells before solidification.
4.3 HDF Encapsulation
The cell viability of the encapsulated HDFs is high following encapsulation at the one- and three-week time points (Fig. 4), demonstrating that the hydrogel has low cytotoxicity. These results suggest that the peptide hydrogel can support the long-term culture of fibroblasts in organotypic skin constructs.

Encapsulated fibroblasts maintain their viability after a one and b three weeks of culture, as evident from calcein (green) staining
4.4 Organotypic Skin Culture
The gel retained its shape and size after 3 weeks of culture; no shrinkage was observed (Fig. 5a). The surface of the gel at the air-liquid interphase was smooth.

Organotypic skin constructs after culturing for 3 weeks. Comparison of the collagen control (left) and peptide hydrogels (right) reflect the dramatic shrinkage of the former while the latter remains largely unchanged in both the a top view and b side view
From the histology images (Fig. 6), the stratified layer of the organotypic skin construct can be observed. Similar to the keratinized stratified squamous epithelium of human skin, the upper most layer of the construct consists of tightly packed, flattened keratinocytes, layered on top of more cuboidal cells near the hydrogel.

Histology of the tissue constructs after 2 weeks of stratification culture. a At low magnification (4×), it is observed that the keratinocytes formed a continuous layer on the hydrogel. b At higher (10×) magnification, the organization of differentiated keratinocytes into a pluristratified epidermis was seen
The surface topology of the organotypic skin constructs was smooth (Fig. 7a). From the SEM image of the underside, the hydrogel is visibly present and the nanofibrous microarchitecture is preserved even after 3 weeks of culture (Fig. 7b).

Field emission scanning microscopy images of the a apical and b basolateral surfaces of the tissue constructs. a The stratified keratinocytes have a smooth, continuous surface. b The basolateral surface reflects the nanofibrous microarchitecture of the peptide hydrogel
4.5 Gene Expression
After exploring different primer sequences, the following primers are found to be appropriate for detecting gene expression in these organotypic skin constructs. Col3A1 codes for type 3 collagen, while CRABP2 codes for the Cellular Retinoic Acid Binding Protein, both of which are likely expressed by the fibroblasts. CDSN2 codes for corneodesmosin, which is involved in corneocyte maturation, and KLK5 codes for Kallikrein-related peptidase 5, which is suggested to regulate desquamation, which are likely expressed by the keratinocytes (Fig. 8).

Relative expression of genes
5 Conclusions, Challenges Encountered and Recommendations for Future Work
During the course of this project, the specific aims to synthesize a suitable peptide candidate, optimize the formulation of peptide hydrogels and characterise a simple organotypic skin construct were achieved. These biological constructs can subsequently be used to screen therapeutic candidates, as well as to evaluate the effects of compounds on skin tissue viability, permeability and cellular gene expression.
Due to the limited time frame of this project, the evaluation of a model test compound—retinoic acid, was limited in scope. The results are presented in Appendix. Briefly, two concentrations of retinoic acid (RA) (0.05 and 0.25%) were applied to the constructs. The preliminary data suggests that KLK5 and CDSN2 were upregulated with increasing concentration of RA. However, the small sample set and limited data points did not allow reliable conclusions to be drawn. One of the major challenges encountered was designing the dosages for the in vitro model to correlate well with published studies on human samples. Majority of the published literature utilize human patient participants who applied the test compound as a cream to parts of their skin, which was subsequently either biopsied or evaluated non-invasively. To eliminate potentially confusing results from possible permeation enhancers or toxic additives in the cream, attempts were made to dissolve RA in dimethylsulfoxide and dilute in PBS for application to the in vitro constructs. Due to differences in application medium, as well as differences in surface area of application, there were discrepancies between our results and literature published date. The RA applied in our study is likely to be of a higher concentration than what is applied to skin in vivo per unit area. Therefore, in the future, determining concentrations of RA more similar to that of what is applied topically can be explored in order for more reliable comparisons between organotypic skin constructs and in vivo studies can be made. Additionally, a wider variety of model compounds, such as salicylic acid or benzoic acid, across a wide range of concentrations should be tested, to validate the model for evaluating novel therapeutic candidates.
In this project, a simplified organotypic skin construct was prepared using fibroblasts encapsulated in a synthetic peptide hydrogel matrix, over which keratinocytes were allowed to proliferate, differentiate and stratify. Building on this model, more elaborate systems can be cultured with the addition of more cell types such as immune cells and adipocytes into the dermal compartment, as well as melanocytes into the epidermis. For example, the addition of normal versus malignant melanocytes can be used to study the mechanism of epidermal pigmentation and melanoma progression [14].
In conclusion, IK6 is an appropriate matrix for culturing organotypic skin constructs due to its stability and low cytotoxicity. With great advancements in biomedical engineering in the last decade, it can be hoped that such constructs will be reliable and accurate solution to the complex problems of animal testing, including inherent differences in skin physiology and gene expression between human and animal samples.
References
Shamir, E., & Ewald, A. (2014). Three-dimensional organotypic culture: Experimental models of mammalian biology and disease. Nature Reviews Molecular Cell Biology, 15(10), 647–664. https://doi.org/10.1038/nrm3873.
Weigelt, B., Lo, A. T., Park, C. C., Gray, J. W., & Bissell, M. J. (2010). HER2 signaling pathway activation and response of breast cancer cells to HER2-targeting agents is dependent strongly on the 3D microenvironment. Breast Cancer Research and Treatment, 122, 35–43. https://doi.org/10.1007/s10549-009-0502-2.
Kenny, P. A., Lee, G. Y., Myers, C. A., Neve, R. M., Semeiks, J. R., Spellman, P. T., et al. (2007). The morphologies of breast cancer cell lines in three-dimensional assays correlate with their profiles of gene expression. Molecular Oncology, 1, 84–96. https://doi.org/10.1016/j.molonc.2007.02.004.
EUR-Lex—32009R1223—EN—EUR-Lex. (2017). Eur-lex.europa.eu. Retrieved December 28, 2017, from http://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:32009R1223.
Panchagnula, R., Stemmer, K., & Ritschel, W. A. (1997). Animal models for transdermal drug delivery. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/9379782.
Andersen, T., Auk-Emblem, P., & Dornish, M. (2015). 3D cell culture in alginate hydrogels. Microarrays, 4(2), 133–161. https://doi.org/10.3390/microarrays4020133.
Lee, K., & Mooney, D. (2001). Hydrogels for tissue engineering. Chemical Reviews, 101(7), 1869–1880. https://doi.org/10.1021/cr000108x.
Rosso, F., Giordano, A., Barbarisi, M., & Barbarisi, A. (2004). From cell-ECM interactions to tissue engineering. Journal of Cellular Physiology, 199(2), 174–180. https://doi.org/10.1002/jcp.10471.
Timpson, P., Mcghee, E., Erami, Z., Nobis, M., Quinn, J., Edward, M., et al. (2011). Organotypic collagen I assay: A malleable platform to assess cell behaviour in a 3-dimensional context. Journal of Visualized Experiments, (56). http://dx.doi.org/10.3791/3089.
Hauser, C., Deng, R., Mishra, A., Loo, Y., Khoe, U., Zhuang, F., et al. (2011). Natural tri- to hexapeptides self-assemble in water to amyloid β-type fiber aggregates by unexpected α-helical intermediate structures. Proceedings of the National Academy of Sciences, 108(4), 1361–1366. https://doi.org/10.1073/pnas.1014796108.
Loo, Y., Lakshmanan, A., Ni, M., Toh, L., Wang, S., & Hauser, C. (2015). Peptide bioink: Self-assembling nanofibrous scaffolds for three-dimensional organotypic cultures. Nano Letters, 15(10), 6919–6925. https://doi.org/10.1021/acs.nanolett.5b02859.
Kirin, S., Noor, F., Metzler-Nolte, N., & Mier, W. (2017). Manual solid–phase peptide synthesis of metallocene–peptide bioconjugates.
Sriram, G., Bigliardi, P., & Bigliardi-Qi, M. (2015). Fibroblast heterogeneity and its implications for engineering organotypic skin models in vitro. European Journal of Cell Biology, 94(11), 483–512. https://doi.org/10.1016/j.ejcb.2015.08.00.
Eves, P., Haycock, J., Layton, C., Wagner, M., Kemp, H., Szabo, M., et al. (2003). Anti-inflammatory and anti-invasive effects of α-melanocyte-stimulating hormone in human melanoma cells. British Journal of Cancer, 89(10), 2004–2015. https://doi.org/10.1038/sj.bjc.6601349.
Acknowledgements
This work was supported by the Youth Research Program (YRP) at the Institute of Bioengineering and Nanotechnology (Biomedical Research Council, Agency for Science, Technology and Research, Singapore).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Appendix
Appendix

Solid phase peptide synthesis scheme.

Primers used in real time PCR

Relative expression of genes under 0.05 and 0.25% retinoic acid
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this paper

Cite this paper
Lee, A.C., Loo, Y., Wan, A. (2019). Biofabrication of Organotypic Full-Thickness Skin Constructs. In: Guo, H., Ren, H., Bandla, A. (eds) IRC-SET 2018. Springer, Singapore. https://doi.org/10.1007/978-981-32-9828-6_21
Download citation
DOI: https://doi.org/10.1007/978-981-32-9828-6_21
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-32-9827-9
Online ISBN: 978-981-32-9828-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)