
Abstract
Foxp3-expressing regulatory T cells (Tregs) perform a vital function in the maintenance of immune homeostasis. A large part of Treg suppressive function is derived from their ability to control and restrict the availability of co-signal molecules to other T cells. However, Tregs themselves also depend on many of the same co-signals for their own homeostasis, making this a complex system of feedback. In this chapter, we discuss the critical role of co-signaling in Treg cell biology.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Tregs
The first indications that some thymically derived cells had a critical suppressive function came from early work by Nishizuka and colleagues who demonstrated that day 3 thymectomy of mice led to severe autoimmunity, suggesting that cells produced after 3 days were responsible for the maintenance of immune homeostasis (Nishizuka and Sakakura 1969). However, it was not until 1995 when Tregs (Tregs), as we now know them, were definitively described on the basis of their expression of the IL2 receptor alpha chain (CD25 ) and that transfer of these cells could prevent autoimmunity (Sakaguchi et al. 1995). Shortly afterward, it was also found that in the neonatal period, Tregs develop in a slightly delayed fashion in comparison to effector T cells, explaining why thymectomy at day 3 allows the development of effector T cells but not Tregs (Asano et al. 1996).
Tregs are dependent on the master transcription factor Foxp3, and ectopic expression of Foxp3 into Foxp3 T cells induces suppressive function (Ramsdell and Ziegler 2014). Conversely, disruption of Foxp3 function results in the development of severe autoimmunity in both mice and humans as characterized by the scurfy mouse strain and immunodysregulation polyendocrinopathy enteropathy X-linked (IPEX) syndrome (Wildin et al. 2001). IPEX is characterized by severe allergy with hyper-IgE production, autoimmune disease such as type 1 diabetes mellitus, and inflammatory bowel disease (Wildin et al. 2001; Ramsdell and Ziegler 2014). While Foxp3 is critical for the function of Tregs, it is not alone responsible for all Treg-type gene expression as its transfection only causes partial reproduction of the Treg cell gene signature (Sugimoto et al. 2006). Treg gene expression is also stably maintained by epigenetic programming. Demethylation of key Treg genes such as CTLA-4 and Foxp3 itself allows their constitutive expression (Ohkura et al. 2013). Tregs are critical for prevention of autoimmunity throughout life, and depletion of Tregs in adult mice leads to catastrophic autoimmunity (Kim et al. 2007). In addition to the severe fatal inflammation associated with total loss of Treg function, more subtle defects of Treg number and function have been implicated in a wide range of autoimmune diseases such as SLE, Sjögren’s syndrome, psoriasis, autoimmune hepatitis, myasthenia gravis, and inflammatory bowel disease (Grant et al. 2015).
Tregs can be split into two primary categories, thymically derived Tregs (tTregs) and peripherally derived Tregs (pTregs). tTregs are essential for the maintenance of immune self-tolerance and make up the majority of Tregs in circulation. pTregs are formed from Foxp3-negative non-Tregs exposed to signals such as TGFβ and IL-2 in peripheral organs; they have a critical and overlapping role with tTregs, being particularly important for the control of inflammation at barrier sites such as the gastrointestinal tract and lungs (Josefowicz et al. 2012). A third group also exists, in vitro derived Tregs (iTregs ), consisting of originally Foxp3-negative conventional CD4 T cells (Tconv) that have been induced to convert to Foxp3+ cells by in vitro treatment with antigen, IL-2, and TGF-β. While iTregs have proven a valuable tool for understanding Treg function, they cannot be considered the exact equivalent of in vivo derived pTregs since they lack the proper epigenetic programming and as a result are unstable, tending to lose Foxp3 expression without its active maintenance (Ohkura et al. 2013). In the past, the terms pTregs and iTregs have been used interchangeably, while tTregs were also often described as natural Tregs (nTregs), leading to a certain amount of confusion. Recent recommendations to clarify the nomenclature aim to address this and should be observed where possible (Abbas et al. 2013). In addition to differences in their site of origin, Tregs also display a high level of functional diversity with tissue-resident Tregs in areas such as the visceral adipose tissue, tumor environment, and muscles, showing characteristic phenotypes (Burzyn et al. 2013). Further, Tregs may mirror effector T-cell subtypes gaining expression of Th17-, Th1-, Th2-, and Tfh-associated transcription factors (RORγt, T-bet, Gata3, and BCL6, respectively) to form functionally specialized subpopulations of Tregs. These cells gain expression of matching chemokine receptors allowing them to track the matching effector subtype to the site of inflammation to deliver suppression in situ (Wing and Sakaguchi 2012) (Fig. 7.1).

Diversity of Treg cells. Tregs can be broadly divided into the key subgroups of (a) thymically derived tTregs. (b) Peripheral Tregs (pTregs) converted from Foxp3-negative cells outside the thymus in vivo. (c) In vitro Tregs (iTregs) converted from Foxp3-negative T cells in vitro. (d) Tregs can also mirror effector T-cell subsets by gaining expression of key transcription factors and chemokine receptors but not inflammatory cytokines. These cells may be derived from either mainly tTregs (e.g., Tfr) or mainly pTregs (e.g., Th17-like Tregs)
While in most circumstances prevention of autoimmune responses to self-antigens is desirable, the presence of Tregs also suppresses responses to tumor antigens. As a result, Treg depletion or functional blockade leads to tumor regression in many cases. A number of therapies aimed at exploiting negative or positive co-signal molecules expressed by Tregs have been developed with the aim of enhancing antitumor immunity, forming an important part of antitumor immunotherapy (Tanaka and Sakaguchi 2017).
2 Control of Tregs by Co-signal Molecules
2.1 Two Signals Required for T-Cell Activation
During the immune response, antigens are processed and presented to Tregs by antigen-presenting cells (APCs) such as dendritic cells (DCs). Recognition of antigens by the T-cell receptor is essential for both the development and activation of T cells. However, TCR signals alone are not sufficient to fully activate T cells, often leading to anergy or cell death when presented without a second signal. The two-signal model of T-cell activation was initially described by Bretscher and Cohn in 1970 (Bretscher and Cohn 1970). In this model, T cells require both the recognition of a presented antigen via the TCR but also a second co-signal. The requirement for the co-signal prevents the activation of T cells by recognition of their antigen alone, and the availability of the second signal to be is closely regulated to prevent autoimmunity. Initially, CD28 signaling was considered to be the sole second signal, and while it remains of utmost importance, it has now become clear that a range of other co-signals have roles in T-cell activation and differentiation (Fig. 7.2). While the two-signal model provides a useful conceptual framework, it is also important to consider that the various second signals are not necessarily equivalent to one another and may have roles that vary at different stages of T-cell activation. The different roles may be sequential with CD28 required for initial signaling of naïve T cells, causing the upregulation of other co-signal molecules of the immunoglobulin family and tumor necrosis factor receptor superfamily (TNFRSF) members. These co-signal molecules then play important roles in the differentiation of the T cells into specialized subsets and their survival in the periphery (Watts 2005). Not all co-signal molecules are stimulatory; in some cases, they act to suppress the T cell expressing them in a cell-intrinsic manner and dampen their activation. In addition, some Treg co-signal molecules act in a cell-extrinsic manner to allow the Treg to exert control over the local cellular environment. The interplay between these positive and negative co-signals, received and given by the Tregs, allows a fine level of balance and homeostatic feedback that prevents both autoimmunity and the development of an overly immunosuppressed environment by unchecked expansion of Tregs.

Costimulatory molecules expressed by Tregs and their ligands. Tregs express a range of costimulatory molecules of the Ig superfamily (CD28 and ICOS) and the TNF-receptor superfamily (OX40, 4-1BB, GITR, CD27, CD30, DR3, and TNFR2)
2.2 Costimulators in Treg Biology
2.2.1 CD28
CD28 is a cell surface homodimer and a member of the Ig superfamily. It is constitutively expressed by almost all T cells and has a critical role in the initial activation of both naïve Tconvs and naïve Tregs (Sharpe and Freeman 2002). CD28’s ligands CD80 and CD86 (also known as B7-1 and B7-2) are expressed by activated APCs such as DCs and B cells. To a lesser extent, they are also expressed by activated T cells. Expression of CD28 by Tregs is essential for their development in the thymus with mice lacking CD28 or CD80 and CD86 having severe defects in the thymic development of Tregs and a resultant lack of Tregs in the periphery (Tang et al. 2003). CD28 signaling is required for the production of IL-2 by Tconvs in the thymus, which is in turn essential for conversion of Treg thymic precursors into mature Tregs (Tai et al. 2005). CD28 also has a cell-intrinsic role in Tregs themselves as CD28 signaling into a potential Treg cell is essential for their upregulation of Foxp3. This effect applies early in the Treg differentiation process as thymic development of both Treg precursor populations, i.e., CD25+Foxp3- and CD25-Foxp3+CD4 single positive thymocytes, are severely impaired in CD28-deficient mice (Lio and Hsieh 2008; Tai et al. 2005). Similar to its role in the thymus, CD28 is also essential for the formation of both in vivo pTregs and in vitro iTregs from CD4+Foxp3- T cells (Guo et al. 2008).
Apart from its role in the formation of Tregs, CD28 signaling is also required by Tregs for their full activation and proliferation in the periphery. Genetic deficiency of CD80 and CD86 or CD28 results in a severe defect in the numbers of peripheral Tregs; in part this is due to the loss of thymic production of Tregs, but experiments inhibiting CD80 and CD86 via CTLA-4Ig (a solubilized form of CTLA-4) in adult mice demonstrate that loss of peripheral CD28 signaling causes a similar reduction in Treg number even in mice with adult thymectomy (Salomon et al. 2000; Tang et al. 2003). Similar to the thymus, at least part of the loss of Treg cell numbers in the peripheral organs of mice lacking CD28 signaling is indirect due to a loss of IL-2 production by CD28-deficient Tconvs; however, a clear cell-intrinsic role for CD28 in Tregs was demonstrated by the finding that CD28 -deficient Tregs transferred into a wild-type mouse failed to proliferate (Tang et al. 2003). In addition to mouse studies, human Tregs also respond in a similar manner as Treg cell proliferation in vitro is dependent on the availability of CD28 signaling (Hombach et al. 2007). While loss of CD28 signaling affects both Tregs and Tconvs, overall the loss of CD28 shifts the balance of immune homeostasis toward autoimmunity. Genetic deficiency of CD28, CD80, and CD86 or their blockade by CTLA-4Ig in the diabetes-prone NOD mouse model results in accelerated development of diabetes and autoimmune exocrine pancreatitis due to the deficiency of functional Tregs. This can be reversed by transfer of Tregs from a CD28-sufficient mouse (Salomon et al. 2000; Meagher et al. 2008). Loss of tolerance to a self-antigen similar to that seen in Treg-depleted mice was also found to occur in CD80- and CD86-deficient mice upon reconstitution with CD80- and CD86-sufficient dendritic cells, demonstrating that a lack of Tregs was unable to control the self-reactive response once CD80 and CD86 signaling was made available to self-reactive T cells (Lohr et al. 2003).
While experiments examining the effect of blocking antibodies or CTLA-Ig on Tregs are compelling, they struggle to precisely separate the effects of these same reagents on Tconvs, which indirectly affect Treg homeostasis via IL-2 production. Recent work addressed this longstanding issue by the conditional depletion of CD28 in mature Foxp3+ Tregs, allowing the examination of the role of CD28 after its role in the initial differentiation of Tregs (Zhang et al. 2013). These mice have a normal number of Tregs in the peripheral lymphoid organs, and these Tregs have normal in vitro suppressive capacity. However, despite this the mice develop autoimmunity at multiple sites with particular foci in the skin and lungs. Additionally, CD28-deficient Tregs were found to have a defect in their proliferation and maturation and a resulting loss of expression of activation markers such as CTLA-4, PD1, and CCR6, suggesting a defect in the generation of effector Tregs. Additionally, CD28-deficient Tregs were unable to prevent colitis induced by Treg depletion; and the autoimmunity seen in these conditional knockout mice was preventable by the transfer of CD28-sufficient Tregs (Zhang et al. 2013).
Due to its critical role in Treg function and homeostasis, therapies to selectively expand Tregs by stimulation of CD28 have attracted significant interest. Initial experiments in mice demonstrated that superagonists against CD28 selectively expanded Tregs in vitro and in vivo and were expected to be a promising therapy in the treatment of autoimmune disease (Beyersdorf et al. 2006; Lin and Hunig 2003). However, this field has been set back by the unexpected results of a phase 1 trial in which the superagonistic anti-CD28 antibody TGH1412 triggered severe cytokine storms in healthy volunteers (Suntharalingam et al. 2006). In summation, it is clear that CD28 signaling plays a critical role at all points of Treg development and function.
2.2.2 Inducible T-Cell Costimulator (ICOS)
ICOS (CD278) is a member of the CD28 family of the immunoglobulin superfamily. As its name suggests, ICOS expression is not found on naïve T cells but is induced by TCR and CD28 signaling and as a result expressed on activated CD8 and CD4 T cells, including Tregs (Wikenheiser and Stumhofer 2016). ICOS has a high level of sequence homology with CD28, but despite this their roles are not redundant. While CD28 is essential for initial T-cell activation, ICOS plays a more specialized function in the differentiation of effector T cells, having an important role in the formation of Th2 cells during infection and particularly a key role in antibody responses due to the dependence of T-follicular helper cells on ICOS signaling (Wikenheiser and Stumhofer 2016). ICOS also plays a role in Treg cell biology. Knockout of ICOS reveals no change to the number of thymic Tregs but a significant reduction in the number of Tregs in peripheral sites such as the spleen. This does not result in autoimmunity since the loss of Tregs is partial (from approximately 12% to 8% of CD4 T cells) and is also balanced by a loss of ICOS in effector memory T cells (Burmeister et al. 2008). Cell transfer experiments demonstrate that this is a cell-intrinsic effect dependent on a loss of proliferation and increased sensitivity to activation-induced apoptosis in ICOS-deficient Tregs (Burmeister et al. 2008). Similar to its lack of a role in the thymus, ICOS also has no clear role in the formation of pTregs from CD25-CD4+ Tconvs in vivo (Guo et al. 2008).
While loss of ICOS on Tregs does not result in the development of spontaneous autoimmunity in wild-type mice, it does increase sensitivity in already autoimmune-prone models. In two strains of mice susceptible to the induction of diabetes, NOD mice and BDC2.5 T cell receptor transgenic mice, knockout of ICOS or its blockade by antibodies results in loss of Treg protective functions and increased disease progression (Kornete et al. 2012; Herman et al. 2004). This is because Tregs resident in the pancreas express high levels of ICOS, and loss of ICOS leads to a loss of their proliferative potential and suppressive capability (Kornete et al. 2012). ICOS also plays a key role in the maintenance of mucosal tolerance. Mice given intranasal doses of myelin basic protein (MOG) peptides develop resistance to experimental autoimmune encephalomyelitis (EAE) in an ICOS-dependent and cell transferable manner (Miyamoto et al. 2005). In the lung mucosa, 50% of Tregs express ICOS, and this rises to 70–80% following exposure to inhaled antigens such as Ova. Genetic knockout of ICOS results in both lower initial levels of Tregs in the lung mucosa and a failure to expand following antigen challenge. As a result, in contrast to wild-type Tregs, ICOS-deficient Tregs are unable to induce tolerance to respiratory allergens when transferred to previously sensitized mice (Busse et al. 2012; Akbari et al. 2002). As a result, it seems that ICOS is primarily important for the maintenance and function of specialized tissue-resident Tregs.
2.2.3 Tumor Necrosis Factor Receptor Superfamily (TNFRSF)
The tumor necrosis factor receptor superfamily is a class of co-signal molecules with varied and to some extent redundant functions in Treg cell biology. Here we will focus on members of the TNFRSF with well-described roles in Treg cell biology, namely, OX40, glucocorticoid-induced tumor necrosis factor-related receptor (GITR), CD27, CD30, DR3, TNFR2, and 4-1BB (Watts 2005; Croft 2014).
2.2.3.1 GITR
Glucocorticoid-induced tumor necrosis factor-related receptor (GITR) is constitutively expressed at a high level by Tregs and can be upregulated by activated Foxp3- CD4 and CD8 T cells. Previously we found that anti-GITR antibodies could break self-tolerance, leading to the induction of autoimmune gastritis (Shimizu et al. 2002), and also overcome Treg cell-mediated suppression of the antitumor response, leading to tumor eradication (Ko et al. 2005). Furthermore, anti-GITR antibodies are also capable of abrogating Treg suppressive function in vitro (Shimizu et al. 2002). However, since GITR can also be expressed by effector T cells, it was initially unclear whether it was acting primarily to block Treg function or to stimulate effector T cells to the extent that they became resistant to Treg cell suppression (Stephens et al. 2004). A further factor is the possibility of Treg depletion by antibody-dependent cell death. While initial reports suggested that Treg depletion was not occurring, this is often a difficult factor to rule out since it is known that in some cases (such as anti-CTLA-4) an antibody that does not deplete, or even expands, Treg cell numbers in the lymphoid organs has the ability to deplete Tregs in certain microenvironments such as the colon and the tumor (Simpson et al. 2013). To address some of these issues, Shevach and colleagues used Fc-GITR-L, which mimics engagement of GITR without causing antibody-dependent cell depletion. They found that GITR stimulation induces the proliferation of both Tconvs and Tregs in naïve mice and that these Tregs retained their full suppressive capacity. However, when GITR-sufficient Tregs were co-transferred into T-cell-deficient mice with GITR-deficient effector T cells, treatment with Fc-GITR-L caused severe loss of Tregs and abrogation of the ability of the Tregs to prevent colitis. In contrast, GITR engagement by Fc-GITR-L preferentially enhances Treg proliferation in lymphoid organs (Liao et al. 2010). These findings emphasize the different roles of co-signaling pathways in different cellular contexts with the same signals resulting in either Treg expansion or loss of Tregs depending on the context. The mechanisms underlying these divergent roles are unclear but may depend on overstimulation of Tregs in highly inflammatory conditions, such as those seen in colitis induced by Treg depletion, leading to activation-induced cell death (Ephrem et al. 2013).
Genetic knockout of GITR reveals that the numbers of peripheral Tregs in the lymph nodes and spleen are reduced by around 30–50%. No spontaneous autoimmunity is seen, and the Tregs taken from these mice retain their full functional capacity, demonstrating that GITR is not essential for Treg function (Stephens et al. 2004; Ronchetti et al. 2004). GITR may also have a role in the thymic differentiation of Tregs. It is expressed by thymic Treg precursors and acts to enhance conversion of Treg precursors to mature Tregs and a resulting mild loss of thymic Tregs. Loss of GITR leads to mild defects in the production of Tregs in a competitive setting such as GITR-deficient bone marrow chimera mixed with wild-type bone marrow (Mahmud et al. 2014). In addition to quantitative changes to thymic output, competition for GITR signaling also drives the selection of Treg precursors with high-affinity TCRs. As a result, excessive GITR signaling provided by high doses of Fc-GITR-L leads to a broadening of the Treg cell TCR repertoire with the results that cells bearing lower-affinity TCRs are able to receive sufficient signaling to become Tregs (Mahmud et al. 2014).
2.2.3.2 OX40
OX40 (CD134) is a costimulatory molecule expressed on activation by Tconvs but constitutively expressed by Tregs. Its ligand OX40L is expressed by a range of APCs (DCs, B cells) and to a lesser extent by NK cells and activated T cells (Chen et al. 1999). OX40 is broadly considered to be a costimulatory molecule since blockade of OX40 signaling ameliorates autoimmunity (Redmond and Weinberg 2007). However, this overall effect masks a difference in its function on Tconvs and Tregs. OX40 signaling in Tconvs leads to an increase in cytokine production and proliferation while its role in Tregs is more complex (Webb et al. 2016). Knockout of OX40 does not lead to a severe loss of Treg cell numbers or function at most lymphoid sites; yet a mild reduction in Treg cellularity can be seen in younger mice, recovering to normal levels in older mice (Vu et al. 2007; Griseri et al. 2010; Takeda et al. 2004). OX40 also aids the proliferation of mature Tregs in vitro, suggesting that it may also have a role in the homoeostasis of mature Tregs (Takeda et al. 2004). OX40 also has a role in the conversion of Foxp3- T cells to pTregs (Vu et al. 2007).
However, despite these roles in the formation and maintenance of Tregs, OX40 also appears to suppress certain aspects of Treg cell function. Several studies have demonstrated that engagement of OX40 appears to inhibit the suppressive function of Tregs, at least partly by interfering with Foxp3 gene expression (Kinnear et al. 2013; Vu et al. 2007; Valzasina et al. 2005). More recent work demonstrates that OX40 stimulation induces initial Treg proliferation at multiple organ sites, T-cell infiltration of the lungs, and downregulation of Foxp3 expression levels in Foxp3-expressing cells, driving Tregs to an apparently exhausted phenotype. However, in the presence of additional IL-2, this can be prevented as Tregs expanded by both OX40-ligand and IL-2 in vivo proliferate well and maintain strong suppressive function. Together, these results suggest that the effect of OX40 is also dependent on the availability of cytokines since OX40 alone expands Tregs but pushes them into a relatively exhausted state unless additional supplementation with IL-2 is provided (Xiao et al. 2012).
OX40 is highly expressed by Tregs within the tumor microenvironment (Piconese et al. 2008). As a result, a number of studies have examined the effects of OX40 blockade on the antitumor response. Here, agonistic OX40 antibodies enhance immunological rejection of tumors by a combination of its stimulatory effects on Tconvs, reducing the suppressive capacity of tumor-resident Tregs, and by direct depletion of OX40 high Tregs. A number of clinical trials of anti-OX40 as an anticancer agent are now underway (Sanmamed et al. 2015). In addition to its cell-intrinsic effects, there is some evidence of a cell-extrinsic role for Treg OX40 affecting OX40 ligand-expressing cells. Tregs suppress the activation and degranulation of mast cells via engagement of Treg OX40 with OX40L expressed by mast cells (Piconese et al. 2009).
In contrast to its Treg inhibiting role in many other sites, OX40 has an important role in the ability of Tregs to maintain gut homeostasis. OX40-deficient mice have a reduction in the number of Tregs found in the lamina propria due to a cell-intrinsic defect in their ability to accumulate in the gut (Griseri et al. 2010). Upon adoptive transfer, OX40-deficient Tregs are unable to prevent colitis induced by co-transferred effector T cells. OX40-deficient mice do not spontaneously develop colitis, but this may be due to the dependence of effector T cells on OX40 for their ability to cause colitis (Griseri et al. 2010). Notably, mice expressing high levels of transgenic OX40L on T cells spontaneously develop colitis, while OX40-sufficient Tregs co-transferred alongside effector T cells into T- and B-cell-deficient RAG-KO mice expressing transgenic OX40L in many tissues under the control of chicken β-actin promoter are unable to prevent colitis (Murata et al. 2002; Takeda et al. 2004). Together these results suggest that OX40 expression on Tregs is critical for their suppression of colitis but that excessive OX40L signaling is capable of tipping the balance toward autoimmunity, either by excessive stimulation of effector T cells or by suppression of Treg cell function. As a result, the role of OX40 in Treg cells seems highly complex since it aids thymic development of Tregs and their proliferation, has an inhibitory effect on Treg suppressive activity in many situations, but also plays an important role in the maintenance and trafficking of Tregs into the gut.
2.2.3.3 4-1BB
4-1BB ( (CD137) is not constitutively expressed by human Tregs found in peripheral blood but is rapidly upregulated on their activation, and as a result, following antigen-specific stimulation in vivo, almost all 4-1BB-positive cells are Tregs at 5 hours poststimulation, while at 20 hours Tconv also upregulates 4-1BB (Schoenbrunn et al. 2012). In mouse Tregs, 4-1BB is expressed at low levels in the resting state, but similar to human Tregs, it is rapidly upregulated following stimulation (McHugh et al. 2002).
Genetic deficiency of 4-1BB does not lead to overt autoimmunity or clear abnormalities in T-cell responses (Kwon et al. 2002). 4-1BB signaling enhances the proliferation of Tregs both in vitro and in vivo (Zheng et al. 2004), and anti-4-1BBL treatment allows the large-scale expansion of Tregs ex vivo while retaining their suppressive functions and ability to prevent allogeneic pancreatic islet rejection and resulting diabetes (Elpek et al. 2007). In contrast to OX40, loss of 4-1BB on either adoptively transferred effector T cells or Tregs had no clear effect on the ability of the cells to induce or control colitis, respectively (Maerten et al. 2006).
Similar to a number of other co-signal molecules, there is significant interest in the manipulation of 4-1BB in the context of antitumor immunotherapy. Treatment with anti-4-1BB effectively enhances antitumor immunity either alone or in combination with other treatments such as anti-CTLA-4 (Kocak et al. 2006). Since 4-1BB is expressed by intratumoral Tregs and CD4 and CD8 effector T cells, it is important to understand which cells are being affected by its engagement. A more recent study found that intratumoral Treg proliferation was slightly reduced following treatment with anti-4-1BB. This was seen in conjunction with a reduction in the absolute number of Tregs infiltrating the tumor and a reduction in their expression of both PD-1 and CTLA-4, suggesting that the Tregs may have been less activated and suppressive (Curran et al. 2011). It is thus likely that anti-4-1BB enhances antitumor responses by reducing both the number and function of tumor-infiltrating Tregs.
2.2.3.4 CD27
CD27 is another member of the TNFR superfamily expressed by Tregs and activated Tconvs in the periphery and developing thymocytes. The signaling cascade resulting from CD27 stimulation is partially characterized with TRAF-mediated activation of JNK- and NIK-dependent activation of the NFκB pathway (Bullock 2017).
CD70, the ligand for CD27, is constitutively expressed by medullary thymic epithelial cells and thymic DCs, suggesting a role in the thymic development of T cells (Coquet et al. 2013). Mice lacking either CD27 or CD70 expression have a partial loss of Treg cell numbers in both the thymus and the periphery but no loss of suppressive function by the remaining Tregs. Mechanistically, it appears that CD27 co-signals in developing Tregs suppress their apoptosis by suppressing the expression of proapoptotic BCL-2 family members such as Puma and Bak, allowing them to survive positive selection (Coquet et al. 2013). Expression of CD27 by tumor-infiltrating Tregs may also have a role on their survival and function. CD27-deficient Tregs fail to accumulate in the tumor environment, and as a result, antitumor responses are enhanced. Similar to the situation in the thymus, this appears to primarily be a defect in the survival of Tregs (Claus et al. 2012).
Tregs may also possess the cell-extrinsic ability to regulate CD70 expression on DCs via CD27. CD27-expressing Tregs cause the loss of surface CD70 from interacting DCs in a contact-dependent manner. In a mirror image of the transendocytosis central to the function of CTLA-4, in this case the DC internalizes both CD70 and bound CD27 from the Treg cell (Dhainaut et al. 2015).
2.2.3.5 CD30, DR3, and TNFR2
CD30 is expressed by activated effector T cells and Tregs, while CD30L (CD154) is expressed by DCs, thymic epithelia cells, B cells, eosinophils, and neutrophils. The role of CD30 in Treg biology is not well characterized, but several groups have reported that CD30 expression by Tregs plays a role in transplantation tolerance. CD30-deficient Tregs are not capable of suppressing CD8 T-cell memory responses responsible for the rejection of allogenic skin grafts. In this case, CD30 expressed by Tregs is required for their contact-dependent suppressive function. Whether this is by direct cell-extrinsic function of CD30 or indirectly via a requirement for CD30 signaling to activate the full suppressive function of the Tregs is unclear, although the latter seems more credible (Dai et al. 2004). Tregs lacking CD30 expression also have a significantly weakened ability to prevent graft versus host disease (GvHD), while antibody blockade of CD30L in the early but not late stages of GvHD induction prevents Treg control of the disease (Zeiser et al. 2007).
DR3 (TNFRSF25) is selectively expressed on Tregs in mice with little expression seen in Foxp3- Tconvs. Agonistic antibodies to DR3 preferentially cause the proliferation of Tregs in a TCR- and IL-2-dependent manner. Interestingly due to its more Treg-specific expression profile, anti-DR3 seems better able to specifically expand Tregs than other TNFRSF molecules such as GITR, CD27, OX40, and 4-1BB, making it a good candidate immunotherapeutic agent (Schreiber et al. 2010). Accordingly, mice treated with anti-DR3 are resistant to allergic lung inflammation and have delayed rejection of allogenic heart transplantation; and T cells from anti-DR3-treated hosts mediate reduced GvHD (Kim et al. 2015; Schreiber et al. 2010).
TNFR2 is expressed by both resting and activated Tregs. TNFR2 signaling induced by its ligand TNF-α causes the expansion of both mouse and human Tregs (Okubo et al. 2013; Chen et al. 2007). Murine Tregs expanded in this manner have enhanced in vitro suppressive function; however, contrasting results have been reported by different groups as to the effect of TNF on human Treg suppressive function, although the majority suggest that Tregs treated with TNF-α have reduced suppressive function (Nie et al. 2016). Genetic deficiency of TNFR2 results in a reduction in the number of Tregs in both the thymus and the spleen, a phenotype also seen in mice with triple knockout of the TNFR2 ligands: TNF, lymphotoxin-α, and lymphotoxin-β (Chen et al. 2013). Further to this, addition of TNF, to cultures containing pre-Tregs, enhances their conversion to mature Tregs in comparison to IL-2 alone (Mahmud et al. 2014). A role for TNFR2 in the in vivo suppressive function of Tregs was demonstrated as Tregs lacking TNFR2 are unable to prevent colitis due to a deficiency in their stability and competitive fitness (Chen et al. 2013).
2.2.3.6 Redundancy of TNFRSF Members in Treg Cell Function
Some redundancy between different members of the TNFR family is seen. While the magnitude of the reductions of Treg numbers in GITR-, OX40-, or TNFR2-deficient mice is relatively mild, combining OX40 genetic depletion with tailless dominant negative forms of GITR or TNFR reveals a much more severe defect in Treg cell numbers with all the combination of all three leading to a near total loss of thymic Treg production. This role for TNFR family members is in turn dependent on CD28 signaling for the induction of their initial expression (Mahmud et al. 2014).
Many of the TNFRSF members have signaling cascades that terminate in activation of the transcription factor NF-κB family of transcription factors. In particular, cRel and RelA have vital roles in the thymic production, maintenance, and suppressive function of Tregs (Li and Jacks 2017). Engagement of GITR, OX40, and 4-1BB induces the phosphorylation of RelA, which anti-CD3 and anti-CD28 fail to do, suggesting they are acting via distinct signaling pathways. While individual knockouts of GITR, OX40, and 4-1BB result in relatively mild defects in Treg formation and function, conditional knockout of RelA in mature Tregs results in serious autoimmune pathology, resulting in the death of the mice at around 100 days of age. The Treg cell defect was cell intrinsic and seems to primarily result from a loss of the ability of naïve/central Tregs to differentiate into highly suppressive effector Tregs (Vasanthakumar et al. 2017). As a result, it seems that loss of one TNFRSF member expressed by Tregs can be compensated for by the action of others, but loss of multiple receptors or of the downstream signaling molecules they share leads to severe defects in either Treg formation in the thymus or the differentiation and function of Tregs in the periphery, demonstrating that TNFRSF members, as a group, have an essential role in Treg cell function.
2.3 Coinhibitors in Treg Biology
2.3.1 Coinhibitory Molecules
Coinhibitory receptors are integral to the synchrony of immune responses. They provide a means for the immune system to coordinate its defense mechanisms to achieve sustainable immunity without development of autoimmunity. Their expression generally coincides with the activation of immune cells; however, each is highly dependent on the availability of their respective ligands to elicit immunomodulatory signals. From a therapeutic viewpoint, coinhibitory receptors and ligands present avenues for the development of antagonists to counter their effects in chronic viral infections and cancer, diseases that capitalize on the immune checkpoint receptors as Achilles heels of viral- and tumor antigen-specific T cells to restrain their responses. This strategy has produced promising results in recent years with certain limitations that are potentially due to the lack of understanding of coinhibitory molecules in immune cells other than conventional T cells. In this section, we discuss the roles of coinhibitory receptors in Tregs, which are known to express them in substantial amounts (Fig. 7.3).

Coinhibitory molecules expressed by Tregs and their ligands on antigen-presenting cells. Suppressive function may be mainly either cell intrinsic (TIM3, PD1, Lag-3), cell extrinsic (ligand capture by CTLA-4), or a mixture of both (cell-intrinsic signaling by TIGIT and TIGIT triggered release of the suppressive molecule Fgl2)
2.3.2 PD-1
Programmed cell death-1 (PD-1 or CD279 )) is a coinhibitory surface receptor belonging to the CD28 superfamily. It is a 50–55 kDa type 1 transmembrane glycoprotein with a single IgV domain in its extracellular region that shares 21–33% sequence homology with CTLA-4, CD28, and ICOS. One key feature of PD-1 that distinguishes it from other CD28 family members is its inability to homodimerize due to a lack of membrane cysteine (Zhang et al. 2004). The intracellular cytoplasmic region of PD-1 consists of two tyrosine-based residues – tyrosine-based inhibitory motif (ITIM) and immunoreceptor tyrosine-based switch motif (ITSM); the latter is responsible for transducing negative signaling (Long 1999; Sidorenko and Clark 2003). PD-1 is expressed on activated T cells, B cells, NK cells, NKT cells, dendritic cells, and monocytes. The level of PD-1 expression increases with constant antigen exposure and stimulation. High PD-1 expression is a characteristic feature of exhausted unresponsive T cells (Freeman et al. 2006); however, it is also expressed by highly activated T cells, making PD-1 alone unable to differentiate between these contrasting cell types. Upon activation in T cells, the nuclear factor of activated T cells c1 (NFATc1) translocates to the promoter region of PD-1 to drive PD-1 expression (Oestreich et al. 2008). Interestingly, a recent study suggested that this may be epigenetically repressed by the chromatin organizer special AT-rich sequence-binding protein-1 (Satb1) through its recruitment of nucleosome remodeling deacetylase (NURD) complex to the PD-1 promoter, a process that is partly counterbalanced by Smad proteins competing for the same promoter-binding region (Stephen et al. 2017). The exact mechanism which dictates PD-1 expression remains to be determined.
PD-1 binds to two ligands, namely, PD-L1 and PD-L2 , with stronger binding to PD-L2 (Freeman et al. 2000; Latchman et al. 2001; Tseng et al. 2001; Karunarathne et al. 2016). In mice, while PD-L1 is constitutively expressed on a wide variety of immune cells including T and B cells, dendritic cells, macrophages, and bone-marrow-derived mast cells, PD-L2 expression is mainly limited to primed professional antigen-presenting cells such as DCs, macrophages, peritoneal B1 B cells, and germinal center B cells (Freeman et al. 2000; Yamazaki et al. 2002; Zhong et al. 2007). Additionally, PD-L1 can be induced on cells of nonhematopoietic origin (e.g., vascular endothelial cells) (Eppihimer et al. 2002). In humans, PD-L2 is also present on T cells and vascular endothelial cells (Tseng et al. 2001; Messal et al. 2011). PD-L1 expression is preferentially enhanced by IFNγ, whereas PD-L2 is upregulated by IL-4 and GM-CSF (Yamazaki et al. 2002; Loke and Allison 2003). Besides PD-1, PD-L1 serves as a ligand for CD80/B7-1 for which the effect of this interaction leans toward inhibition of T-cell responses (Butte et al. 2007). An alternative binding partner for PD-L2 has also been proposed with contrasting properties to PD-1 (Shin et al. 2003). This is supported by the increased killing of PD-L2-expressing tumor cells compared to non-PD-L2-expressing tumor cells by PD-1-/- cytotoxic T cells (Liu et al. 2003). Identifying the receptor is paramount to make further inroads into this pathway.
Upon ligation to PD-Ls, ITSM of PD-1 becomes phosphorylated and recruits SHP-2 (SH2 domain-containing tyrosine phosphatase 2) which in turn dephosphorylates other signaling molecules in the TCR complex, for example, CD3, ZAP70 (ζ-associated protein of 70 kDa), and PI3K (phosphatidylinositol-3-kinase) (Parry et al. 2005). Consequently, T cells have reduced capacity to proliferate and produce cytokines as well as the transcription factors – T-bet, Eomes, and GATA3 – required for specialized Th1/Th2 functions (Nurieva et al. 2006). In the event of strong TCR and CD28 stimulation, PD-1-mediated inhibitory effects can be overcome (Freeman et al. 2000). This can also occur through the STAT5-dependent cytokine pathways of IL-2, IL-7, and IL-15 (Bennett et al. 2003). Mice rendered genetically deficient in PD-1 are susceptible to strain-specific autoimmune diseases. PD-1KO C57Bl6 mice sustain mild lupus-like nephritis and arthritis with late onset, while PD-1KO BALB/c and NOD mice develop cardiomyopathy and type 1 diabetes, respectively (Nishimura et al. 1999; Nishimura et al. 2001; Wang et al. 2005). The strain- and tissue-specific nature of the observed autoimmunity suggests a loss of peripheral tolerance rather than central tolerance in PD-1KO mice. Nevertheless, it is not known whether autoimmunity in PD-1KO mice results from increased activation of T cells per se or whether DCs and macrophages, which have been shown to secrete larger amounts of cytokines when deficient in PD-1 (Rui et al. 2013), also contribute through the promotion of inflammation.
Alternatively, the breakdown in peripheral versus central tolerance in PD-1KO mice may be explained by the role that PD-1 plays in T-cell development. Given that PD-1 is first expressed in early CD4-CD8- thymocytes and is upregulated upon TCR recognition in CD4+CD8+ thymocytes, PD-1 can affect the maturation of T cells by calibrating the TCR signal threshold for positive and negative selection (Nishimura et al. 1996; Blank et al. 2003). Hence, it is likely that thymic emigrants in mice lacking inhibitory PD-1 contain less self-recognizing Tconvs and perhaps more Tregs. Indeed, the percentage and number of thymic CD4+Foxp3+ cells in young PD-1KO mice are higher compared to wild-type controls. The authors also demonstrated reduced total CD4 single positive PD-1KO thymic T cells specific for MJ23, a native prostate antigen, in chimeric mice containing a mixture of MJ23 TCR transgenic Rag1-/- PD-1-deficient and PD-1-sufficient bone marrow cells, indicative of possible increased negative selection of PD-1KO immature CD4 T cells. On the other hand, there was increased differentiation of PD-1KO MJ23-specific thymic T cells into Tregs. Nevertheless, studies have yet to be performed to determine whether the TCR repertoire of mature Tconvs has decreased affinity for self-antigens and that of Tregs has increased affinity in PD-1KO mice.
Although PD-1 is highly expressed by activated Tregs, there is limited knowledge on its exact function in Tregs. In comparison to PD-1- Tregs, PD-1hi-expressing Tregs have reduced demethylation in the Foxp3 locus, are less proliferative in vitro, exert weaker suppression on Tconvs, and have shorter telomeres, traits consistent with terminal differentiation and exhaustion (Lowther et al. 2016). This observation also raises two important questions pertaining to dormancy and memory of Tregs since the graded manner which PD-1 is expressed could be associated with activation status. On this note, the effect of blocking PD-1 signaling during activation of Tregs needs to be carefully assessed in future studies. According to current evidence, it is likely that PD-1 blockade on Tregs would bring about increased immunosuppression. This stems from a study that found T-follicular regulatory cells, a subtype of Tregs residing within lymphoid follicular zones, derived from PD-1KO mice are more efficient in preventing Tconv proliferation in vitro (Sage et al. 2013). A second study made a similar finding in vivo by transferring CD4+Foxp3+ Tregs from either wild-type or PD-1KO mice into mice that were rendered susceptible to pancreatitis by partial impairment of Foxp3 expression (Zhang et al. 2016). Not only did the transfer of PD-1KO Tregs result in decreased conventional CD4 and CD8 T-cell activation; there were also less cellular infiltrates and pathological damage in the pancreas. Nonetheless, one caveat with assessing Tregs from PD-1KO mice is the possibility that whole Treg activity is enhanced as a compensatory response to the global increase in T-cell activation and not directly due to PD-1 deficiency itself.
Another area of interest in PD-1 that is related to Tregs is its role in the conversion of Tconvs to peripheral Tregs (pTregs). PD-L1 expression on antigen-presenting cells has been shown to be critical for TGFβ-induced development of pTregs from Tconvs (Francisco et al. 2009). Furthermore, transfer of naïve CD4 Tconvs into RagKO mice deficient in both PD-L1 and PD-L2 produced less pTregs and caused more severe inflammatory disease. Downregulation of Akt/mTOR signaling is believed to facilitate PD-L1-mediated generation of pTregs. However, it should be noted that these data cannot definitively link the PD-L1:PD-1 axis to pTreg development as PD-L1, in addition to PD-1, binds to CD80 which is expressed on Tconvs as well. This could be inferred by an apparent increase in number, but not frequency which remained unchanged, of pTregs converted from PD-1KO Tconvs in RagKO mice (Ellestad et al. 2014). Here, due consideration must also be given to the analysis and interpretation of pTreg frequency, and absolute number in the various organs (i.e., blood and lymph nodes) for PD-1KO Tconvs is inherently more proliferative and readily mobilized. Therefore, an increase in pTregs may merely be a consequence of increased expansion and activation of PD-1KO Tconvs rather than an absence of PD-1-mediated regulation of pTreg conversion. Despite this, the results clearly suggest that PD-1 may not be absolutely essential for Tconvs to differentiate into pTregs. More stringent experimental designs are required to investigate the effect of PD-1 on this aspect of immune regulation.
Due to its high expression in Tconv, Treg, and CD8 cells in the tumor environment, targeting the PD-1 pathway has been considered a key candidate for antitumor immunotherapy (Iwai et al. 2017). Early experiments in mice demonstrated that either anti-PD-1 or anti-PD-L1 is effective at inducing antitumor immune responses and anti-PD-1 agents such as Nivolumab have proven a relative success in the clinic either alone or in combination with other immunotherapeutic agents such as anti-CTLA-4 (Callahan et al. 2014).
2.3.3 CTLA-4
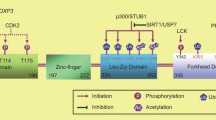
Cytotoxic T-cell lymphocyte antigen-4 (CTLA-4 , CD152 )) is a close relative of CD28 and shares the same ligands, CD80 and CD86. However, while CD28 is a critical costimulatory molecule, CTLA-4 is known for its suppressive function. CTLA-4 is constitutively expressed by Tregs and can also be expressed by activated Tconvs. Genetic knockout of CTLA-4 results in severe fatal autoimmunity and lymphoproliferation in a manner similar to that seen in Foxp3-deficient scurfy mice (Tivol et al. 1995). Correspondingly genetic knockout of both CD80 and CD86 or CD28 prevents the autoimmunity seen in CTLA-4-deficient mice demonstrating the opposing roles of CTLA-4 inhibition and CD28 stimulation (Mandelbrot et al. 1999; Tai et al. 2007). Conditional knockout of CTLA-4 specifically on Tregs results in severe inflammation with similar consequences but slightly delayed fatality to fully CTLA-4-deficient mice (Wing et al. 2008). Surprisingly, in contrast to germline or Treg conditional knockout, CTLA-4 depletion in adult mice is not fatal and even leads to resistance to EAE. Despite this, CTLA-4 depletion in adult mice does result in severe autoimmunity (pneumonitis, gastritis, insulitis, and sialadenitis) (Klocke et al. 2016). These findings suggest that while CTLA-4 is critical for Treg cell suppressive function during the neonatal period, if depleted in the mature immune system, other Treg suppressive mechanisms are able to partially compensate for this to prevent fatal autoimmunity. This partial redundancy of other suppressive molecules replacing CTLA-4 function is also demonstrated by the finding that highly activated CTLA-4 KO Tregs taken from CTLA-4-deficient mice are suppressive in vitro while CTLA-4 KO Tregs taken from bone marrow chimera or mosaic mice are not. This is because strong activation of Tregs, which occurs in a mouse with a total lack of CTLA-4, allows upregulation of other suppressive molecules. This does not occur in the marrow chimera where immune homeostasis is retained (Wing et al. 2008). The key role of CTLA-4 in the maintenance of immune homeostasis in humans has recently been demonstrated by the discovery of a rare patient group suffering from severe autoimmunity due to heterozygous loss of function mutations of the CTLA-4 gene (Schubert et al. 2014; Kuehn et al. 2014). In contrast, heterozygous loss of CTLA-4 in mice has little measurable effect, possibly due to the unchallenging clean conditions that specific pathogen-free mice are kept in.
CTLA-4 has been demonstrated to have both cell-extrinsic and cell-intrinsic functions (Wing et al. 2011). The key cell-extrinsic function of CTLA-4 is to control availability of its ligands CD80 and CD86 on antigen-presenting cells in a contact-dependent manner (Onishi et al. 2008). This ability to deplete CD80 and CD86 can be blocked by anti-CTLA-4 antibodies and is also lost in Tregs with genetic deficiency of CTLA-4 (Wing et al. 2008). CTLA-4 rapidly cycles on and off the cell membrane into intracellular pools, where the majority of CTLA-4 is located. As it engages with CD80 and CD86, it is able to pull them off the surface of APCs, sequestering them into the Treg where they are then degraded; as a result, Tregs rapidly remove CD80 and CD86 from APCs in a contact-dependent manner (Walker and Sansom 2015).
CTLA-4 has also been reported to have cell-intrinsic functions. Recent work suggests that CTLA-4 may affect the motility of Tregs via interaction of its cytoplasmic tail with the protein kinase C isoform PKC-η. This complex recruits a GIT2–αPIX–PAK complex that controls the disassembly of focal adhesion points between the Treg and APCs and as a result affects the ability of the Treg to disengage from an APC in order to seek new targets. This results in a partial loss of suppressive function in Tregs lacking PKC-η, causing a loss of ability to prevent antitumor responses, but still retaining the capacity to prevent colitis following cell transfer into RAG-deficient mice (Kong et al. 2014). Several other reports have also suggested that CTLA-4 may play a role in the arrest or enhancement of T cell and Treg mobility on contact with APCs; however, this has proven controversial with different studies providing conflicting evidence (Walker and Sansom 2015). While cell-intrinsic mechanisms may play a role in the fine-tuning of CTLA-4 function, they appear dispensable for the main role of CTLA-4 in the control of immune homeostasis since bone marrow chimera experiments and use of conditional knockout mice where half of Tregs express CTLA-4 demonstrate that CTLA-4-deficient Tconvs and Tregs do not have a clear phenotype as long as they are in the presence of CTLA-4-sufficient Tregs (Bachmann et al. 1999; Wing et al. 2008).
CTLA-4 expression by Tregs is also critical for their ability to prevent antitumor responses, and as a result, CTLA-4 cKO mice rapidly clear tumors (Wing et al. 2008). Treatment of tumor-bearing mice with anti-CTLA-4 leads to tumor regression. In the clinic, anti-CTLA-4 is already in use with compounds such as ipilimumab , already showing efficacy in the treatment of human melanoma. Treatment attempts at other cancers such as prostate and non-small cell lung cancer have not been initially successful, but combination therapies with Nivolumab (anti-PD-1) have yielded promising initial results (Callahan et al. 2014). Due to its importance in regulation of the immune system, other reagents targeting the CTLA-4 pathway have been developed. CTLA-4Ig, in which CTLA-4 has been fused with the Fc region of IgG1 to create a solubilized form of CTLA-4 that blocks CD80 and CD86, is a currently licensed drug (abatacept ) for the treatment of rheumatoid arthritis and has been shown to have significant benefit to this patient group. However, trials in other autoimmune/inflammatory conditions such as asthma, lupus, and ulcerative colitis have been less successful (Adams et al. 2016).
2.3.4 Lag-3
Lymphocyte activation gene-3 (Lag-3 ) is a transmembrane protein with a conformation reminiscent of the CD4 coreceptor. It mainly binds to major histocompatibility complex II (MHCII) and has greater affinity compared to the binding of CD4 to MHCII (Huard et al. 1995). In spite of this, Lag-3 can also regulate MHCI-restricted CD8 T cells intrinsically (Grosso et al. 2007), suggesting a role of other Lag-3 ligands such as LSECtin, a member of the C-type lectin receptor superfamily, found predominantly in dendritic cells in blockade of T-effector responses (Xu et al. 2014). It is notable that deficiency in Lag-3 leads to spontaneous autoimmunity only in autoimmune-prone mice stains such as the NOD background (Bettini et al. 2011). During T-cell activation, Lag-3 cross-links with CD3 and modulates T-cell activity through a pathway that has yet to be uncovered (Hannier et al. 1998). To date, it is only known that Lag-3 depends on the KIEELE motif of its intracellular domain to transduce inhibitory signals in CD4 Tcons (Workman et al. 2002). Whether Lag-3 requires the other two motifs, one containing serine-phosphorylation sites and the other containing glutamic acid-proline repeats, for its function in other T-cell subsets remains to be determined (Workman et al. 2002). This is worth consideration particularly for Tregs where Lag-3 may either act as a cell-intrinsic inhibitory coreceptor or serve as a cell-extrinsic immune suppressive arm as discussed below.
Within the activated population of whole CD4 T cells, Lag-3 is preferentially expressed and maintained on pTregs. Using a Lag-3 blocking antibody, it was found that Lag-3 was crucial for pTregs to mount efficient suppression on Tconvs and protect mice from death in a model of lethal pneumonitis (Huang et al. 2004). A similar finding was obtained for tTregs although this was debatable due to the ambiguous nature of identifying tTregs as CD4+CD25+ cells in earlier studies (Huang et al. 2004). This controversy is compounded by another study that showed wild-type and Lag-3KO CD4+CD25hi Tregs did not differ in their ability to prevent allogeneic GvHD (Sega et al. 2014). As GvHD is only preventable by the transfer of CD62L+ naïve Tregs, which are essentially tTregs, into host mice (Ermann et al. 2005), the initial claim that Lag-3 is indispensable for tTreg function may not be completely true. On a similar note, one must account for differences, if any, in the proportion of CD62L+ naïve Tregs within the CD4+CD25hi cells in wild-type and Lag-3KO mice. A recent study addressed some of these issues by generating NOD mice with a conditional knockout of Lag-3 specifically in Foxp3+ Tregs. It was found that such mice had a lower incidence of diabetes, which may be attributed to Lag-3 restricting the maintenance and proliferation of islet-infiltrating Tregs through downregulation of Eos and IL-2-Stat5 signaling (Zhang et al. 2017). Importantly, the inhibition of Eos by Lag-3 likely subjects Tregs to become unstable and disposed to reprogramming, a typical feature of pTregs (Sharma et al. 2013).
2.3.5 TIGIT
The coinhibitory T-cell immunoreceptor with Ig and ITIM domains (TIGIT )) and its partner costimulatory receptor CD226 can be deemed to regulate T-cell responses in ways similar to CD28/CTLA-4 (Joller et al. 2011). Both TIGIT and CD226 bind to CD155 and CD112 with the former exhibiting stronger affinity (Anderson et al. 2016). The intracellular domain of TIGIT consists of an ITIM motif and an immunoglobulin tail tyrosine (ITT)-like motif (Yu et al. 2009). The cell-intrinsic inhibitory function of TIGIT hinges on its ITT-like motif which becomes phosphorylated and recruits SHIP1 to dampen NF-κB signaling upon ligation of TIGIT to its ligand (Li et al. 2014). Besides, TIGIT has been shown to exert cell-extrinsic immunoregulatory effects by stimulating IL-10 and blocking IL-12 production by DCs through its interaction with CD155, hence repressing Th1 immunity (Yu et al. 2009). This can also be achieved by TIGIT-expressing Tregs (Joller et al. 2014). In humans, TIGIT+ T cells are particularly enriched in the Foxp3+Helios+ thymic Treg fraction. Moreover, while level of TIGIT expression is markedly higher, CD226 expression is lower in Helios+ compared to Helios- Tregs and Tconvs (Joller et al. 2014). It is currently postulated that TIGIT is a late activation marker and limits the proliferative capacity of tTregs. In contrast, TIGIT-CD226+ Tregs harbor properties that are synonymous to iTregs such as higher IFNγ and IL-10 production and reduced Foxp3-TSDR demethylation (Fuhrman et al. 2015; Joller et al. 2014). Intriguingly, TIGIT was found to be highly necessary for the conversion of murine Tconvs into iTregs in vitro.
Due to the novel identification of TIGIT+ Tregs, the function of this particular Treg subset is still unclear. It was earlier shown that upregulation of TIGIT during Treg activation was hindered in an IL-12-mediated Th1 environment, suggesting a possible role in counterbalancing T-helper effector responses (Fuhrman et al. 2015). This concept has been validated in mice where, as in humans, tTregs are the major source of TIGIT+ Tregs which coexpress neuropilin-1 and Helios and are distinctly more activated and suppressive in comparison to TIGIT- Tregs (Joller et al. 2014). Elevated expression of costimulatory (e.g., ICOS) and coinhibitory molecules (e.g., PD-1, CTLA-4, Tim3, Lag3) and Treg signature genes (e.g., Foxp3, CD25, GITR, IL-10) bear testament to the highly immunosuppressive phenotype of TIGIT+ Tregs (Joller et al. 2014). This was similarly observed in tumor-infiltrating TIGIT+ Tregs, which were behind the suppression of antitumor CD8 T-cell responses (Kurtulus et al. 2015). Perhaps, one peculiar trait of TIGIT+ Tregs is their preferential expression of Th1- and Th17-specific genes over those of Th2 (Joller et al. 2014). This includes the respective chemokine receptors. In line with their gene expression profile, TIGIT+ Tregs suppress Th1 and Th17 but not Th2 responses. Mechanistically, this particular function of TIGIT+ Tregs owes to their increased propensity to secrete soluble fibrinogen-like protein 2 (Fgl2) in addition to IL-10 (Joller et al. 2014). It is conceivable that the combined effect of Fgl2 and IL-10 from TIGIT+ Tregs tilts the T-helper balance from Th1/Th17 to Th2. Interestingly, in comparison to other activated Tregs, TIGIT has twofold greater expression by highly differentiated CD25-negative T-follicular regulatory cells located in germinal centers, suggesting that it may have a role in the regulation of antibody responses (Wing et al. 2017).
2.3.6 Tim-3
T-cell immunoglobulin-3 (Tim-3) is the first among the Tim family of proteins that was discovered (Anderson et al. 2016). As a coinhibitory molecule, any compromise on Tim-3 function unleashes activated T cells, particularly Th1, to instigate immune-mediated diseases. This can be brought about by modifications to the Tim-3 gene or blocking Tim-3 with anti-Tim3 antibody (Monney et al. 2002; Koguchi et al. 2006). The spontaneity of autoimmune development from antagonizing Tim-3 firmly underlines Tim-3 as a major immune regulator. Reversion of Tim-3+ T cells from a dysfunctional state to a highly autoreactive state has been shown to account for the effect of Tim-3 blockade (Koguchi et al. 2006), though it is necessary to recognize that Tim-3 is expressed in multiple other cells such as Tregs, NK cells, NKT cells, and APCs (Anderson et al. 2016).
Several ligands have been identified for Tim-3, one of which is galactin-9 (GAL-9). Engagement of GAL-9 leads to cell death in Tim-3+ Th1 cells and protects mice from experimental autoimmune encephalomyelitis. Other ligands include phosphatidylserine (PS), high mobility group protein B1 (HGMB1), and carcinoembryonic antigen cell adhesion molecule-1 (Ceacam-1) (Anderson et al. 2016). Since T cells do not possess any ability to phagocytose apoptotic cell function, the PS:Tim-3 pathway has not been examined in T cells. As for HMGB-1 whose role is to chaperone DNA from apoptotic cells to dendritic cells and macrophages, it is proposed that Tim-3 sequesters it to keep inflammatory responses under control (Chiba et al. 2012). Lately, Ceacam-1 has emerged as an interesting candidate that is coexpressed with Tim-3 in activated T cells. Both Ceacam-1 and Tim-3 interact with each other in cis and trans through their N-terminal domains to mediate Tim-3-dependent inhibition. Heterodimerization of the molecules in cis is especially vital for Tim-3 maturation and maintenance on the cell surface. This is evidently manifested in Ceacam-1-deficient T cells which are low in Tim-3 expression and are highly pathogenic.
Clearly, Tim-3 is important to immune tolerance, and the degree of redundancy it shares with Tregs ought to be ascertained. Thus far, it is known that Tim-3+ Tregs belong to a specific PD-1-expressing activated Treg subset that is almost exclusively found within inflamed tissues (Sakuishi et al. 2013). The same can be assumed for human Tim-3+ Tregs which are not immediately detectable ex vivo but only after stimulation with anti-CD3 and anti-CD28 (Gautron et al. 2014). The in vitro activated Tim-3+ Tregs are more suppressive against Th1 and Th17 responses compared to their Tim-3- counterparts which are barely effective in Th17 suppression. In mice, Tim-3+PD-1+ Tregs were reported to have high expression of the classic regulatory genes (e.g., CD25, CTLA-4, and IL-10) and display strong in vitro immunosuppression (Sakuishi et al. 2013). Notwithstanding these traits, the short-term survival of Tim-3+PD-1+ Tregs allows them to only delay but not prevent allograft rejection (Gupta et al. 2012). On a more positive note, future cancer treatment can leverage combined blockade of Tim-3 and PD-1, which abrogates much of the enhanced immunoregulatory functions of Tim-3+ Tregs, in particular IL-10 production (Sakuishi et al. 2013). This would facilitate a two-pronged approach to relive antitumor T cells from checkpoint blockade as well as suppression by Tregs (Fig. 7.4).

The role of diverse suppressive and coinhibitory signals in key checkpoints in Treg development and function. Plus or minus signs denote a positive or negative contribution to the noted function
3 Conclusion
One notable similarity between a range of the co-signal molecules discussed here is the strongly context-dependent nature of their function. Many positive co-signal molecules are capable of enhancing Treg proliferation when given in the context of an environment with relatively low inflammatory milieu. On the other hand, these same signals cause the death or loss of function of the Tregs in more activated environments, notably inside tumors. The exact mechanisms underlying this phenomenon are not fully clear but may be the result of either overstimulation leading to activation-induced cell death or alterations to intracellular conditions of the Tregs leading to different co-factors becoming involved in the downstream signaling events following engagement of the co-signal molecule. For example, the TNFRSF members CD27, GITR, OX40, and 4-1BB can bind to TRAF2, which leads to activation of canonical NF-κB signaling and proliferation and activation of Tregs. Alternatively, the proapoptotic molecule Siva can also interact with the signaling domains of CD27 and GITR, resulting in increased apoptosis of Tregs via the TRAF pathway (Spinicelli et al. 2002; Nocentini and Riccardi 2005). The choice of these opposing pathways may be the result of the environment and signaling through other ligands or cytokines resulting in changes to the availability of downstream adaptor molecules.
Tregs are critical for the prevention of autoimmunity while also being capable of preventing beneficial responses such as antitumor immunity. As a result, their function must be tightly regulated by a range of co-signals either enhancing or suppressing their proliferation and function dependent on context. Further Tregs themselves control the availability of co-signals to other T cells, most notably by their ability to regulate CD80 and CD86 expression by CTLA-4 and thus restrict the availably of CD28 signals to Tconv. These different costimulatory and coinhibitory factors intertwine to result in a fine-tuned system to control immune homeostasis with multiple levels of feedback and redundancy and balance the need for protective immune responses with reducing the likelihood of autoimmunity.
References
Abbas AK, Benoist C, Bluestone JA, Campbell DJ, Ghosh S, Hori S, Jiang S, Kuchroo VK, Mathis D, Roncarolo MG, Rudensky A, Sakaguchi S, Shevach EM, Vignali DA, Ziegler SF (2013) Regulatory T cells: recommendations to simplify the nomenclature. Nat Immunol 14(4):307–308. https://doi.org/10.1038/ni.2554
Adams AB, Ford ML, Larsen CP (2016) Costimulation blockade in autoimmunity and transplantation: the CD28 pathway. J Immunol 197(6):2045–2050. https://doi.org/10.4049/jimmunol.1601135
Akbari O, Freeman GJ, Meyer EH, Greenfield EA, Chang TT, Sharpe AH, Berry G, DeKruyff RH, Umetsu DT (2002) Antigen-specific regulatory T cells develop via the ICOS-ICOS-ligand pathway and inhibit allergen-induced airway hyperreactivity. Nat Med 8(9):1024–1032. https://doi.org/10.1038/nm745
Anderson AC, Joller N, Kuchroo VK (2016) Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity 44(5):989–1004. https://doi.org/10.1016/j.immuni.2016.05.001
Asano M, Toda M, Sakaguchi N, Sakaguchi S (1996) Autoimmune disease as a consequence of developmental abnormality of a T cell subpopulation. J Exp Med 184(2):387–396
Bachmann MF, Kohler G, Ecabert B, Mak TW, Kopf M (1999) Cutting edge: lymphoproliferative disease in the absence of CTLA-4 is not T cell autonomous. J Immunol 163(3):1128–1131
Bennett F, Luxenberg D, Ling V, Wang IM, Marquette K, Lowe D, Khan N, Veldman G, Jacobs KA, Valge-Archer VE, Collins M, Carreno BM (2003) Program death-1 engagement upon TCR activation has distinct effects on costimulation and cytokine-driven proliferation: attenuation of ICOS, IL-4, and IL-21, but not CD28, IL-7, and IL-15 responses. J Immunol 170(2):711–718
Bettini M, Szymczak-Workman AL, Forbes K, Castellaw AH, Selby M, Pan X, Drake CG, Korman AJ, Vignali DA (2011) Cutting edge: accelerated autoimmune diabetes in the absence of LAG-3. J Immunol 187(7):3493–3498. https://doi.org/10.4049/jimmunol.1100714
Beyersdorf N, Hanke T, Kerkau T, Hunig T (2006) CD28 superagonists put a break on autoimmunity by preferentially activating CD4+CD25+ regulatory T cells. Autoimmun Rev 5(1):40–45. https://doi.org/10.1016/j.autrev.2005.06.001
Blank C, Brown I, Marks R, Nishimura H, Honjo T, Gajewski TF (2003) Absence of programmed death receptor 1 alters thymic development and enhances generation of CD4/CD8 double-negative TCR-transgenic T cells. J Immunol 171(9):4574–4581
Bretscher P, Cohn M (1970) A theory of self-nonself discrimination. Science 169(3950):1042–1049
Bullock TN (2017) Stimulating CD27 to quantitatively and qualitatively shape adaptive immunity to cancer. Curr Opin Immunol 45:82–88. https://doi.org/10.1016/j.coi.2017.02.001
Burmeister Y, Lischke T, Dahler AC, Mages HW, Lam KP, Coyle AJ, Kroczek RA, Hutloff A (2008) ICOS controls the pool size of effector-memory and regulatory T cells. J Immunol 180(2):774–782
Burzyn D, Benoist C, Mathis D (2013) Regulatory T cells in nonlymphoid tissues. Nat Immunol 14(10):1007–1013. https://doi.org/10.1038/ni.2683
Busse M, Krech M, Meyer-Bahlburg A, Hennig C, Hansen G (2012) ICOS mediates the generation and function of CD4+CD25+Foxp3+ regulatory T cells conveying respiratory tolerance. J Immunol 189(4):1975–1982. https://doi.org/10.4049/jimmunol.1103581
Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ (2007) Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity 27(1):111–122. https://doi.org/10.1016/j.immuni.2007.05.016
Callahan MK, Postow MA, Wolchok JD (2014) CTLA-4 and PD-1 pathway blockade: combinations in the clinic. Front Oncol 4:385. https://doi.org/10.3389/fonc.2014.00385
Chen AI, McAdam AJ, Buhlmann JE, Scott S, Lupher ML Jr, Greenfield EA, Baum PR, Fanslow WC, Calderhead DM, Freeman GJ, Sharpe AH (1999) Ox40-ligand has a critical costimulatory role in dendritic cell: T cell interactions. Immunity 11(6):689–698
Chen X, Baumel M, Mannel DN, Howard OM, Oppenheim JJ (2007) Interaction of TNF with TNF receptor type 2 promotes expansion and function of mouse CD4+CD25+ T regulatory cells. J Immunol 179(1):154–161
Chen X, Wu X, Zhou Q, Howard OM, Netea MG, Oppenheim JJ (2013) TNFR2 is critical for the stabilization of the CD4+Foxp3+ regulatory T. cell phenotype in the inflammatory environment. J Immunol 190(3):1076–1084. https://doi.org/10.4049/jimmunol.1202659
Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, Fujioka Y, Ohba Y, Gorman JV, Colgan JD, Hirashima M, Uede T, Takaoka A, Yagita H, Jinushi M (2012) Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol 13(9):832–842. https://doi.org/10.1038/ni.2376
Claus C, Riether C, Schurch C, Matter MS, Hilmenyuk T, Ochsenbein AF (2012) CD27 signaling increases the frequency of regulatory T cells and promotes tumor growth. Cancer Res 72(14):3664–3676. https://doi.org/10.1158/0008-5472.CAN-11-2791
Coquet JM, Ribot JC, Babala N, Middendorp S, van der Horst G, Xiao Y, Neves JF, Fonseca-Pereira D, Jacobs H, Pennington DJ, Silva-Santos B, Borst J (2013) Epithelial and dendritic cells in the thymic medulla promote CD4+Foxp3+ regulatory T cell development via the CD27-CD70 pathway. J Exp Med 210(4):715–728. https://doi.org/10.1084/jem.20112061
Croft M (2014) The TNF family in T cell differentiation and function – unanswered questions and future directions. Semin Immunol 26(3):183–190. https://doi.org/10.1016/j.smim.2014.02.005
Curran MA, Kim M, Montalvo W, Al-Shamkhani A, Allison JP (2011) Combination CTLA-4 blockade and 4-1BB activation enhances tumor rejection by increasing T-cell infiltration, proliferation, and cytokine production. PLoS One 6(4):e19499. https://doi.org/10.1371/journal.pone.0019499
Dai Z, Li Q, Wang Y, Gao G, Diggs LS, Tellides G, Lakkis FG (2004) CD4+CD25+ regulatory T cells suppress allograft rejection mediated by memory CD8+ T cells via a CD30-dependent mechanism. J Clin Invest 113(2):310–317. https://doi.org/10.1172/JCI19727
Dhainaut M, Coquerelle C, Uzureau S, Denoeud J, Acolty V, Oldenhove G, Galuppo A, Sparwasser T, Thielemans K, Pays E, Yagita H, Borst J, Moser M (2015) Thymus-derived regulatory T cells restrain pro-inflammatory Th1 responses by downregulating CD70 on dendritic cells. EMBO J 34(10):1336–1348. https://doi.org/10.15252/embj.201490312
Ellestad KK, Thangavelu G, Ewen CL, Boon L, Anderson CC (2014) PD-1 is not required for natural or peripherally induced regulatory T cells: severe autoimmunity despite normal production of regulatory T cells. Eur J Immunol 44(12):3560–3572. https://doi.org/10.1002/eji.201444688
Elpek KG, Yolcu ES, Franke DD, Lacelle C, Schabowsky RH, Shirwan H (2007) Ex vivo expansion of CD4+CD25+FoxP3+ T regulatory cells based on synergy between IL-2 and 4-1BB signaling. J Immunol 179(11):7295–7304
Ephrem A, Epstein AL, Stephens GL, Thornton AM, Glass D, Shevach EM (2013) Modulation of Treg cells/T effector function by GITR signaling is context-dependent. Eur J Immunol 43(9):2421–2429. https://doi.org/10.1002/eji.201343451
Eppihimer MJ, Gunn J, Freeman GJ, Greenfield EA, Chernova T, Erickson J, Leonard JP (2002) Expression and regulation of the PD-L1 immunoinhibitory molecule on microvascular endothelial cells. Microcirculation 9(2):133–145. https://doi.org/10.1038/sj/mn/7800123
Ermann J, Hoffmann P, Edinger M, Dutt S, Blankenberg FG, Higgins JP, Negrin RS, Fathman CG, Strober S (2005) Only the CD62L+ subpopulation of CD4+CD25+ regulatory T cells protects from lethal acute GVHD. Blood 105(5):2220–2226. https://doi.org/10.1182/blood-2004-05-2044
Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, Sharpe AH (2009) PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med 206(13):3015–3029. https://doi.org/10.1084/jem.20090847
Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, Horton HF, Fouser L, Carter L, Ling V, Bowman MR, Carreno BM, Collins M, Wood CR, Honjo T (2000) Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med 192(7):1027–1034
Freeman GJ, Wherry EJ, Ahmed R, Sharpe AH (2006) Reinvigorating exhausted HIV-specific T cells via PD-1-PD-1 ligand blockade. J Exp Med 203(10):2223–2227. https://doi.org/10.1084/jem.20061800
Fuhrman CA, Yeh WI, Seay HR, Saikumar Lakshmi P, Chopra G, Zhang L, Perry DJ, McClymont SA, Yadav M, Lopez MC, Baker HV, Zhang Y, Li Y, Whitley M, von Schack D, Atkinson MA, Bluestone JA, Brusko TM (2015) Divergent phenotypes of human regulatory T cells expressing the receptors TIGIT and CD226. J Immunol 195(1):145–155. https://doi.org/10.4049/jimmunol.1402381
Gautron AS, Dominguez-Villar M, de Marcken M, Hafler DA (2014) Enhanced suppressor function of TIM-3+ FoxP3+ regulatory T cells. Eur J Immunol 44(9):2703–2711. https://doi.org/10.1002/eji.201344392
Grant CR, Liberal R, Mieli-Vergani G, Vergani D, Longhi MS (2015) Regulatory T-cells in autoimmune diseases: challenges, controversies and--yet--unanswered questions. Autoimmun Rev 14(2):105–116. https://doi.org/10.1016/j.autrev.2014.10.012
Griseri T, Asquith M, Thompson C, Powrie F (2010) OX40 is required for regulatory T cell-mediated control of colitis. J Exp Med 207(4):699–709. https://doi.org/10.1084/jem.20091618
Grosso JF, Kelleher CC, Harris TJ, Maris CH, Hipkiss EL, De Marzo A, Anders R, Netto G, Getnet D, Bruno TC, Goldberg MV, Pardoll DM, Drake CG (2007) LAG-3 regulates CD8+ T cell accumulation and effector function in murine self- and tumor-tolerance systems. J Clin Invest 117(11):3383–3392. https://doi.org/10.1172/JCI31184
Guo F, Iclozan C, Suh WK, Anasetti C, Yu XZ (2008) CD28 controls differentiation of regulatory T cells from naive CD4 T cells. J Immunol 181(4):2285–2291
Gupta S, Thornley TB, Gao W, Larocca R, Turka LA, Kuchroo VK, Strom TB (2012) Allograft rejection is restrained by short-lived TIM-3+PD-1+Foxp3+ Tregs. J Clin Invest 122(7):2395–2404. https://doi.org/10.1172/JCI45138
Hannier S, Tournier M, Bismuth G, Triebel F (1998) CD3/TCR complex-associated lymphocyte activation gene-3 molecules inhibit CD3/TCR signaling. J Immunol 161(8):4058–4065
Herman AE, Freeman GJ, Mathis D, Benoist C (2004) CD4+CD25+ T regulatory cells dependent on ICOS promote regulation of effector cells in the prediabetic lesion. J Exp Med 199(11):1479–1489. https://doi.org/10.1084/jem.20040179
Hombach AA, Kofler D, Hombach A, Rappl G, Abken H (2007) Effective proliferation of human regulatory T cells requires a strong costimulatory CD28 signal that cannot be substituted by IL-2. J Immunol 179(11):7924–7931
Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, Hipkiss EL, Ravi S, Kowalski J, Levitsky HI, Powell JD, Pardoll DM, Drake CG, Vignali DA (2004) Role of LAG-3 in regulatory T cells. Immunity 21(4):503–513. https://doi.org/10.1016/j.immuni.2004.08.010
Huard B, Prigent P, Tournier M, Bruniquel D, Triebel F (1995) CD4/major histocompatibility complex class II interaction analyzed with CD4- and lymphocyte activation gene-3 (LAG-3)-Ig fusion proteins. Eur J Immunol 25(9):2718–2721. https://doi.org/10.1002/eji.1830250949
Iwai Y, Hamanishi J, Chamoto K, Honjo T (2017) Cancer immunotherapies targeting the PD-1 signaling pathway. J Biomed Sci 24(1):26. https://doi.org/10.1186/s12929-017-0329-9
Joller N, Hafler JP, Brynedal B, Kassam N, Spoerl S, Levin SD, Sharpe AH, Kuchroo VK (2011) Cutting edge: TIGIT has T cell-intrinsic inhibitory functions. J Immunol 186(3):1338–1342. https://doi.org/10.4049/jimmunol.1003081
Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, Xia J, Tan TG, Sefik E, Yajnik V, Sharpe AH, Quintana FJ, Mathis D, Benoist C, Hafler DA, Kuchroo VK (2014) Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity 40(4):569–581. https://doi.org/10.1016/j.immuni.2014.02.012
Josefowicz SZ, Niec RE, Kim HY, Treuting P, Chinen T, Zheng Y, Umetsu DT, Rudensky AY (2012) Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature 482(7385):395–399. https://doi.org/10.1038/nature10772
Karunarathne DS, Horne-Debets JM, Huang JX, Faleiro R, Leow CY, Amante F, Watkins TS, Miles JJ, Dwyer PJ, Stacey KJ, Yarski M, Poh CM, Lee JS, Cooper MA, Rénia L, Richard D, McCarthy JS, Sharpe AH, Wykes MN (2016) Programmed death-1 ligand 2-mediated regulation of the PD-L1 to PD-1 axis is essential for establishing CD4(+) T cell immunity. Immunity 45(2):333–345. https://doi.org/10.1016/j.immuni.2016.07.017
Kim JM, Rasmussen JP, Rudensky AY (2007) Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat Immunol 8(2):191–197. https://doi.org/10.1038/ni1428
Kim BS, Nishikii H, Baker J, Pierini A, Schneidawind D, Pan Y, Beilhack A, Park CG, Negrin RS (2015) Treatment with agonistic DR3 antibody results in expansion of donor Tregs and reduced graft-versus-host disease. Blood 126(4):546–557. https://doi.org/10.1182/blood-2015-04-637587
Kinnear G, Wood KJ, Fallah-Arani F, Jones ND (2013) A diametric role for OX40 in the response of effector/memory CD4+ T cells and regulatory T cells to alloantigen. J Immunol 191(3):1465–1475. https://doi.org/10.4049/jimmunol.1300553
Klocke K, Sakaguchi S, Holmdahl R, Wing K (2016) Induction of autoimmune disease by deletion of CTLA-4 in mice in adulthood. Proc Natl Acad Sci U S A 113(17):E2383–E2392. https://doi.org/10.1073/pnas.1603892113
Ko K, Yamazaki S, Nakamura K, Nishioka T, Hirota K, Yamaguchi T, Shimizu J, Nomura T, Chiba T, Sakaguchi S (2005) Treatment of advanced tumors with agonistic anti-GITR mAb and its effects on tumor-infiltrating Foxp3+CD25+CD4+ regulatory T cells. J Exp Med 202(7):885–891. https://doi.org/10.1084/jem.20050940
Kocak E, Lute K, Chang X, May KF Jr, Exten KR, Zhang H, Abdessalam SF, Lehman AM, Jarjoura D, Zheng P, Liu Y (2006) Combination therapy with anti-CTL antigen-4 and anti-4-1BB antibodies enhances cancer immunity and reduces autoimmunity. Cancer Res 66(14):7276–7284. https://doi.org/10.1158/0008-5472.CAN-05-2128
Koguchi K, Anderson DE, Yang L, O’Connor KC, Kuchroo VK, Hafler DA (2006) Dysregulated T cell expression of TIM3 in multiple sclerosis. J Exp Med 203(6):1413–1418. https://doi.org/10.1084/jem.20060210
Kong K-F, Fu G, Zhang Y, Yokosuka T, Casas J, Canonigo-Balancio AJ, Becart S, Kim G, Yates JR, Kronenberg M, Saito T, Gascoigne NRJ, Altman A (2014) Protein kinase C-η controls CTLA-4-mediated regulatory T cell function. Nat Immunol 15(5):465–472. https://doi.org/10.1038/ni.2866
Kornete M, Sgouroudis E, Piccirillo CA (2012) ICOS-dependent homeostasis and function of Foxp3+ regulatory T cells in islets of nonobese diabetic mice. J Immunol 188(3):1064–1074. https://doi.org/10.4049/jimmunol.1101303
Kuehn HS, Ouyang W, Lo B, Deenick EK, Niemela JE, Avery DT, Schickel JN, Tran DQ, Stoddard J, Zhang Y, Frucht DM, Dumitriu B, Scheinberg P, Folio LR, Frein CA, Price S, Koh C, Heller T, Seroogy CM, Huttenlocher A, Rao VK, Su HC, Kleiner D, Notarangelo LD, Rampertaap Y, Olivier KN, McElwee J, Hughes J, Pittaluga S, Oliveira JB, Meffre E, Fleisher TA, Holland SM, Lenardo MJ, Tangye SG, Uzel G (2014) Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science 345(6204):1623–1627. https://doi.org/10.1126/science.1255904
Kurtulus S, Sakuishi K, Ngiow SF, Joller N, Tan DJ, Teng MW, Smyth MJ, Kuchroo VK, Anderson AC (2015) TIGIT predominantly regulates the immune response via regulatory T cells. J Clin Invest 125(11):4053–4062. https://doi.org/10.1172/JCI81187
Kwon BS, Hurtado JC, Lee ZH, Kwack KB, Seo SK, Choi BK, Koller BH, Wolisi G, Broxmeyer HE, Vinay DS (2002) Immune responses in 4-1BB (CD137)-deficient mice. J Immunol 168(11):5483–5490
Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, Iwai Y, Long AJ, Brown JA, Nunes R, Greenfield EA, Bourque K, Boussiotis VA, Carter LL, Carreno BM, Malenkovich N, Nishimura H, Okazaki T, Honjo T, Sharpe AH, Freeman GJ (2001) PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol 2(3):261–268. https://doi.org/10.1038/85330
Li A, Jacks T (2017) Driving Rel-iant Tregs toward an identity crisis. Immunity 47(3):391–393. https://doi.org/10.1016/j.immuni.2017.08.014
Li M, Xia P, Du Y, Liu S, Huang G, Chen J, Zhang H, Hou N, Cheng X, Zhou L, Li P, Yang X, Fan Z (2014) T-cell immunoglobulin and ITIM domain (TIGIT) receptor/poliovirus receptor (PVR) ligand engagement suppresses interferon-γ production of natural killer cells via β-arrestin 2-mediated negative signaling. J Biol Chem 289(25):17647–17657. https://doi.org/10.1074/jbc.M114.572420
Liao G, Nayak S, Regueiro JR, Berger SB, Detre C, Romero X, de Waal Malefyt R, Chatila TA, Herzog RW, Terhorst C (2010) GITR engagement preferentially enhances proliferation of functionally competent CD4+CD25+FoxP3+ regulatory T cells. Int Immunol 22(4):259–270. https://doi.org/10.1093/intimm/dxq001
Lin CH, Hunig T (2003) Efficient expansion of regulatory T cells in vitro and in vivo with a CD28 superagonist. Eur J Immunol 33(3):626–638. https://doi.org/10.1002/eji.200323570
Lio CW, Hsieh CS (2008) A two-step process for thymic regulatory T cell development. Immunity 28(1):100–111. https://doi.org/10.1016/j.immuni.2007.11.021
Liu X, Gao JX, Wen J, Yin L, Li O, Zuo T, Gajewski TF, Fu YX, Zheng P, Liu Y (2003) B7DC/PDL2 promotes tumor immunity by a PD-1-independent mechanism. J Exp Med 197(12):1721–1730. https://doi.org/10.1084/jem.20022089
Lohr J, Knoechel B, Jiang S, Sharpe AH, Abbas AK (2003) The inhibitory function of B7 costimulators in T cell responses to foreign and self-antigens. Nat Immunol 4(7):664–669. https://doi.org/10.1038/ni939
Loke P, Allison JP (2003) PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc Natl Acad Sci U S A 100(9):5336–5341. https://doi.org/10.1073/pnas.0931259100
Long EO (1999) Regulation of immune responses through inhibitory receptors. Annu Rev Immunol 17:875–904. https://doi.org/10.1146/annurev.immunol.17.1.875
Lowther DE, Goods BA, Lucca LE, Lerner BA, Raddassi K, van Dijk D, Hernandez AL, Duan X, Gunel M, Coric V, Krishnaswamy S, Love JC, Hafler DA (2016) PD-1 marks dysfunctional regulatory T cells in malignant gliomas. JCI Insight 1(5). https://doi.org/10.1172/jci.insight.85935
Maerten P, Kwon BS, Shen C, De Hertogh G, Cadot P, Bullens DM, Overbergh L, Mathieu C, Van Assche G, Geboes K, Rutgeerts P, Ceuppens JL (2006) Involvement of 4-1BB (CD137)-4-1BBligand interaction in the modulation of CD4 T cell-mediated inflammatory colitis. Clin Exp Immunol 143(2):228–236. https://doi.org/10.1111/j.1365-2249.2005.02991.x
Mahmud SA, Manlove LS, Schmitz HM, Xing Y, Wang Y, Owen DL, Schenkel JM, Boomer JS, Green JM, Yagita H, Chi H, Ka H, Ma F (2014) Costimulation via the tumor-necrosis factor receptor superfamily couples TCR signal strength to the thymic differentiation of regulatory T cells. Nat Immunol 15(5):473–481. https://doi.org/10.1038/ni.2849
Mandelbrot DA, McAdam AJ, Sharpe AH (1999) B7-1 or B7-2 is required to produce the lymphoproliferative phenotype in mice lacking cytotoxic T lymphocyte-associated antigen 4 (CTLA-4). J Exp Med 189(2):435–440
McHugh RS, Whitters MJ, Piccirillo CA, Young DA, Shevach EM, Collins M, Byrne MC (2002) CD4(+)CD25(+) immunoregulatory T cells: gene expression analysis reveals a functional role for the glucocorticoid-induced TNF receptor. Immunity 16(2):311–323
Meagher C, Tang Q, Fife BT, Bour-Jordan H, Wu J, Pardoux C, Bi M, Melli K, Bluestone JA (2008) Spontaneous development of a pancreatic exocrine disease in CD28-deficient NOD mice. J Immunol 180(12):7793–7803
Messal N, Serriari NE, Pastor S, Nunès JA, Olive D (2011) PD-L2 is expressed on activated human T cells and regulates their function. Mol Immunol 48(15-16):2214–2219. https://doi.org/10.1016/j.molimm.2011.06.436
Miyamoto K, Kingsley CI, Zhang X, Jabs C, Izikson L, Sobel RA, Weiner HL, Kuchroo VK, Sharpe AH (2005) The ICOS molecule plays a crucial role in the development of mucosal tolerance. J Immunol 175(11):7341–7347
Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, Manning S, Greenfield EA, Coyle AJ, Sobel RA, Freeman GJ, Kuchroo VK (2002) Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature 415(6871):536–541. https://doi.org/10.1038/415536a
Murata K, Nose M, Ndhlovu LC, Sato T, Sugamura K, Ishii N (2002) Constitutive OX40/OX40 ligand interaction induces autoimmune-like diseases. J Immunol 169(8):4628–4636
Nie H, Zheng Y, Li R, Zhang J (2016) Reply to Suppressive activity of human regulatory T cells is maintained in the presence of TNF. Nat Med 22(1):18–19. https://doi.org/10.1038/nm.4018
Nishimura H, Agata Y, Kawasaki A, Sato M, Imamura S, Minato N, Yagita H, Nakano T, Honjo T (1996) Developmentally regulated expression of the PD-1 protein on the surface of double-negative (CD4-CD8-) thymocytes. Int Immunol 8(5):773–780
Nishimura H, Nose M, Hiai H, Minato N, Honjo T (1999) Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity 11(2):141–151
Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, Sasayama S, Mizoguchi A, Hiai H, Minato N, Honjo T (2001) Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science 291(5502):319–322. https://doi.org/10.1126/science.291.5502.319
Nishizuka Y, Sakakura T (1969) Thymus and reproduction: sex-linked dysgenesia of the gonad after neonatal thymectomy in mice. Science 166(3906):753–755
Nocentini G, Riccardi C (2005) GITR: a multifaceted regulator of immunity belonging to the tumor necrosis factor receptor superfamily. Eur J Immunol 35(4):1016–1022. https://doi.org/10.1002/eji.200425818
Nurieva R, Thomas S, Nguyen T, Martin-Orozco N, Wang Y, Kaja MK, Yu XZ, Dong C (2006) T-cell tolerance or function is determined by combinatorial costimulatory signals. EMBO J 25(11):2623–2633. https://doi.org/10.1038/sj.emboj.7601146
Oestreich KJ, Yoon H, Ahmed R, Boss JM (2008) NFATc1 regulates PD-1 expression upon T cell activation. J Immunol 181(7):4832–4839
Ohkura N, Kitagawa Y, Sakaguchi S (2013) Development and maintenance of regulatory T cells. Immunity 38(3):414–423. https://doi.org/10.1016/j.immuni.2013.03.002
Okubo Y, Mera T, Wang L, Faustman DL (2013) Homogeneous expansion of human T-regulatory cells via tumor necrosis factor receptor 2. Sci Rep 3:3153. https://doi.org/10.1038/srep03153
Onishi Y, Fehervari Z, Yamaguchi T, Sakaguchi S (2008) Foxp3(+) natural regulatory T cells preferentially form aggregates on dendritic cells in vitro and actively inhibit their maturation. Proc Natl Acad Sci U S A 105(29):10113–10118. https://doi.org/10.1073/Pnas.0711106105
Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, Linsley PS, Thompson CB, Riley JL (2005) CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol 25(21):9543–9553. https://doi.org/10.1128/MCB.25.21.9543-9553.2005
Piconese S, Valzasina B, Colombo MP (2008) OX40 triggering blocks suppression by regulatory T cells and facilitates tumor rejection. J Exp Med 205(4):825–839. https://doi.org/10.1084/jem.20071341
Piconese S, Gri G, Tripodo C, Musio S, Gorzanelli A, Frossi B, Pedotti R, Pucillo CE, Colombo MP (2009) Mast cells counteract regulatory T-cell suppression through interleukin-6 and OX40/OX40L axis toward Th17-cell differentiation. Blood 114(13):2639–2648. https://doi.org/10.1182/blood-2009-05-220004
Ramsdell F, Ziegler SF (2014) FOXP3 and scurfy: how it all began. Nat Rev Immunol 14(5):343–349. https://doi.org/10.1038/nri3650
Redmond WL, Weinberg AD (2007) Targeting OX40 and OX40L for the treatment of autoimmunity and cancer. Crit Rev Immunol 27(5):415–436
Ronchetti S, Zollo O, Bruscoli S, Agostini M, Bianchini R, Nocentini G, Ayroldi E, Riccardi C (2004) GITR, a member of the TNF receptor superfamily, is costimulatory to mouse T lymphocyte subpopulations. Eur J Immunol 34(3):613–622. https://doi.org/10.1002/eji.200324804
Rui Y, Honjo T, Chikuma S (2013) Programmed cell death 1 inhibits inflammatory helper T-cell development through controlling the innate immune response. Proc Natl Acad Sci U S A 110(40):16073–16078. https://doi.org/10.1073/pnas.1315828110
Sage PT, Francisco LM, Carman CV, Sharpe AH (2013) The receptor PD-1 controls follicular regulatory T cells in the lymph nodes and blood. Nat Immunol 14(2):152–161. https://doi.org/10.1038/ni.2496
Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M (1995) Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol 155(3):1151–1164
Sakuishi K, Ngiow SF, Sullivan JM, Teng MW, Kuchroo VK, Smyth MJ, Anderson AC (2013) TIM3(+)FOXP3(+) regulatory T cells are tissue-specific promoters of T-cell dysfunction in cancer. Oncoimmunology 2(4):e23849. https://doi.org/10.4161/onci.23849
Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, Bluestone JA (2000) B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity 12(4):431–440
Sanmamed MF, Pastor F, Rodriguez A, Perez-Gracia JL, Rodriguez-Ruiz ME, Jure-Kunkel M, Melero I (2015) Agonists of Co-stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin Oncol 42(4):640–655. https://doi.org/10.1053/j.seminoncol.2015.05.014
Schoenbrunn A, Frentsch M, Kohler S, Keye J, Dooms H, Moewes B, Dong J, Loddenkemper C, Sieper J, Wu P, Romagnani C, Matzmohr N, Thiel A (2012) A converse 4-1BB and CD40 ligand expression pattern delineates activated regulatory T cells (Treg) and conventional T cells enabling direct isolation of alloantigen-reactive natural Foxp3+ Treg. J Immunol 189(12):5985–5994. https://doi.org/10.4049/jimmunol.1201090
Schreiber TH, Wolf D, Tsai MS, Chirinos J, Deyev VV, Gonzalez L, Malek TR, Levy RB, Podack ER (2010) Therapeutic Treg expansion in mice by TNFRSF25 prevents allergic lung inflammation. J Clin Invest 120(10):3629–3640. https://doi.org/10.1172/JCI42933
Schubert D, Bode C, Kenefeck R, Hou TZ, Wing JB, Kennedy A, Bulashevska A, Petersen BS, Schaffer AA, Gruning BA, Unger S, Frede N, Baumann U, Witte T, Schmidt RE, Dueckers G, Niehues T, Seneviratne S, Kanariou M, Speckmann C, Ehl S, Rensing-Ehl A, Warnatz K, Rakhmanov M, Thimme R, Hasselblatt P, Emmerich F, Cathomen T, Backofen R, Fisch P, Seidl M, May A, Schmitt-Graeff A, Ikemizu S, Salzer U, Franke A, Sakaguchi S, Walker LS, Sansom DM, Grimbacher B (2014) Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat Med 20(12):1410–1416. https://doi.org/10.1038/nm.3746
Sega EI, Leveson-Gower DB, Florek M, Schneidawind D, Luong RH, Negrin RS (2014) Role of lymphocyte activation gene-3 (Lag-3) in conventional and regulatory T cell function in allogeneic transplantation. PLoS One 9(1):e86551. https://doi.org/10.1371/journal.pone.0086551
Sharma MD, Huang L, Choi JH, Lee EJ, Wilson JM, Lemos H, Pan F, Blazar BR, Pardoll DM, Mellor AL, Shi H, Munn DH (2013) An inherently bifunctional subset of Foxp3+ T helper cells is controlled by the transcription factor eos. Immunity 38(5):998–1012. https://doi.org/10.1016/j.immuni.2013.01.013
Sharpe AH, Freeman GJ (2002) The B7-CD28 superfamily. Nat Rev Immunol 2(2):116–126. https://doi.org/10.1038/nri727
Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S (2002) Stimulation of CD25(+)CD4(+) regulatory T cells through GITR breaks immunological self-tolerance. Nat Immunol 3(2):135–142. https://doi.org/10.1038/ni759
Shin T, Kennedy G, Gorski K, Tsuchiya H, Koseki H, Azuma M, Yagita H, Chen L, Powell J, Pardoll D, Housseau F (2003) Cooperative B7-1/2 (CD80/CD86) and B7-DC costimulation of CD4+ T cells independent of the PD-1 receptor. J Exp Med 198(1):31–38. https://doi.org/10.1084/jem.20030242
Sidorenko SP, Clark EA (2003) The dual-function CD150 receptor subfamily: the viral attraction. Nat Immunol 4(1):19–24. https://doi.org/10.1038/ni0103-19
Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, Roddie C, Henry JY, Yagita H, Wolchok JD, Peggs KS, Ravetch JV, Allison JP, Quezada SA (2013) Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med 210(9):1695–1710. https://doi.org/10.1084/jem.20130579
Spinicelli S, Nocentini G, Ronchetti S, Krausz LT, Bianchini R, Riccardi C (2002) GITR interacts with the pro-apoptotic protein Siva and induces apoptosis. Cell Death Differ 9(12):1382–1384. https://doi.org/10.1038/sj.cdd.4401140
Stephen TL, Payne KK, Chaurio RA, Allegrezza MJ, Zhu H, Perez-Sanz J, Perales-Puchalt A, Nguyen JM, Vara-Ailor AE, Eruslanov EB, Borowsky ME, Zhang R, Laufer TM, Conejo-Garcia JR (2017) SATB1 expression governs epigenetic repression of PD-1 in tumor-reactive T cells. Immunity 46(1):51–64. https://doi.org/10.1016/j.immuni.2016.12.015
Stephens GL, McHugh RS, Whitters MJ, Young DA, Luxenberg D, Carreno BM, Collins M, Shevach EM (2004) Engagement of glucocorticoid-induced TNFR family-related receptor on effector T cells by its ligand mediates resistance to suppression by CD4+CD25+ T cells. J Immunol 173(8):5008–5020
Sugimoto N, Oida T, Hirota K, Nakamura K, Nomura T, Uchiyama T, Sakaguchi S (2006) Foxp3-dependent and -independent molecules specific for CD25+CD4+ natural regulatory T cells revealed by DNA microarray analysis. Int Immunol 18(8):1197–1209. https://doi.org/10.1093/intimm/dxl060
Suntharalingam G, Perry MR, Ward S, Brett SJ, Castello-Cortes A, Brunner MD, Panoskaltsis N (2006) Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med 355(10):1018–1028. https://doi.org/10.1056/NEJMoa063842
Tai X, Cowan M, Feigenbaum L, Singer A (2005) CD28 costimulation of developing thymocytes induces Foxp3 expression and regulatory T cell differentiation independently of interleukin 2. Nat Immunol 6(2):152–162. https://doi.org/10.1038/ni1160
Tai X, Van Laethem F, Sharpe AH, Singer A (2007) Induction of autoimmune disease in CTLA-4-/- mice depends on a specific CD28 motif that is required for in vivo costimulation. Proc Natl Acad Sci U S A 104(34):13756–13761. https://doi.org/10.1073/pnas.0706509104
Takeda I, Ine S, Killeen N, Ndhlovu LC, Murata K, Satomi S, Sugamura K, Ishii N (2004) Distinct roles for the OX40-OX40 ligand interaction in regulatory and nonregulatory T cells. J Immunol 172(6):3580–3589
Tanaka A, Sakaguchi S (2017) Regulatory T cells in cancer immunotherapy. Cell Res 27(1):109–118. https://doi.org/10.1038/cr.2016.151
Tang Q, Henriksen KJ, Boden EK, Tooley AJ, Ye J, Subudhi SK, Zheng XX, Strom TB, Bluestone JA (2003) Cutting edge: CD28 controls peripheral homeostasis of CD4+CD25+ regulatory T cells. J Immunol 171(7):3348–3352
Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH (1995) Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 3(5):541–547
Tseng SY, Otsuji M, Gorski K, Huang X, Slansky JE, Pai SI, Shalabi A, Shin T, Pardoll DM, Tsuchiya H (2001) B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J Exp Med 193(7):839–846
Valzasina B, Guiducci C, Dislich H, Killeen N, Weinberg AD, Colombo MP (2005) Triggering of OX40 (CD134) on CD4(+)CD25+ T cells blocks their inhibitory activity: a novel regulatory role for OX40 and its comparison with GITR. Blood 105(7):2845–2851. https://doi.org/10.1182/blood-2004-07-2959
Vasanthakumar A, Liao Y, Teh P, Pascutti MF, Oja AE, Garnham AL, Gloury R, Tempany JC, Sidwell T, Cuadrado E, Tuijnenburg P, Kuijpers TW, Lalaoui N, Mielke LA, Bryant VL, Hodgkin PD, Silke J, Smyth GK, Nolte MA, Shi W, Kallies A (2017) The TNF receptor superfamily-NF-kappaB axis is critical to maintain effector regulatory T cells in lymphoid and non-lymphoid tissues. Cell Rep 20(12):2906–2920. https://doi.org/10.1016/j.celrep.2017.08.068
Vu MD, Xiao X, Gao W, Degauque N, Chen M, Kroemer A, Killeen N, Ishii N, Li XC (2007) OX40 costimulation turns off Foxp3+ Tregs. Blood 110(7):2501–2510. https://doi.org/10.1182/blood-2007-01-070748
Walker LSK, Sansom DM (2015) Confusing signals: recent progress in CTLA-4 biology. Trends Immunol:1–8. https://doi.org/10.1016/j.it.2014.12.001
Wang J, Yoshida T, Nakaki F, Hiai H, Okazaki T, Honjo T (2005) Establishment of NOD-Pdcd1-/- mice as an efficient animal model of type I diabetes. Proc Natl Acad Sci U S A 102(33):11823–11828. https://doi.org/10.1073/pnas.0505497102
Watts TH (2005) TNF/TNFR family members in costimulation of T cell responses. Annu Rev Immunol 23:23–68. https://doi.org/10.1146/annurev.immunol.23.021704.115839
Webb GJ, Hirschfield GM, Lane PJ (2016) OX40, OX40L and autoimmunity: a comprehensive review. Clin Rev Allergy Immunol 50(3):312–332. https://doi.org/10.1007/s12016-015-8498-3
Wikenheiser DJ, Stumhofer JS (2016) ICOS co-stimulation: friend or foe? Front Immunol 7:304. https://doi.org/10.3389/fimmu.2016.00304
Wildin RS, Ramsdell F, Peake J, Faravelli F, Casanova JL, Buist N, Levy-Lahad E, Mazzella M, Goulet O, Perroni L, Bricarelli FD, Byrne G, McEuen M, Proll S, Appleby M, Brunkow ME (2001) X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet 27(1):18–20. https://doi.org/10.1038/83707
Wing JB, Sakaguchi S (2012) Multiple treg suppressive modules and their adaptability. Front Immunol 3:178. https://doi.org/10.3389/fimmu.2012.00178
Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, Nomura T, Sakaguchi S (2008) CTLA-4 control over Foxp3+ regulatory T cell function. Science 322(5899):271–275. https://doi.org/10.1126/science.1160062
Wing K, Yamaguchi T, Sakaguchi S (2011) Cell-autonomous and -non-autonomous roles of CTLA-4 in immune regulation. Trends Immunol 32(9):428–433. https://doi.org/10.1016/j.it.2011.06.002
Wing JB, Kitagawa Y, Locci M, Hume H, Tay C, Morita T, Kidani Y, Matsuda K, Inoue T, Kurosaki T, Crotty S, Coban C, Ohkura N, Sakaguchi S (2017) A distinct subpopulation of CD25- T-follicular regulatory cells localizes in the germinal centers. Proc Natl Acad Sci U S A 114(31):E6400–E6409. https://doi.org/10.1073/pnas.1705551114
Workman CJ, Dugger KJ, Vignali DA (2002) Cutting edge: molecular analysis of the negative regulatory function of lymphocyte activation gene-3. J Immunol 169(10):5392–5395
Xiao X, Gong W, Demirci G, Liu W, Spoerl S, Chu X, Bishop DK, Turka LA, Li XC (2012) New insights on OX40 in the control of T cell immunity and immune tolerance in vivo. J Immunol 188(2):892–901. https://doi.org/10.4049/jimmunol.1101373
Xu F, Liu J, Liu D, Liu B, Wang M, Hu Z, Du X, Tang L, He F (2014) LSECtin expressed on melanoma cells promotes tumor progression by inhibiting antitumor T-cell responses. Cancer Res 74(13):3418–3428. https://doi.org/10.1158/0008-5472.CAN-13-2690
Yamazaki T, Akiba H, Iwai H, Matsuda H, Aoki M, Tanno Y, Shin T, Tsuchiya H, Pardoll DM, Okumura K, Azuma M, Yagita H (2002) Expression of programmed death 1 ligands by murine T cells and APC. J Immunol 169(10):5538–5545
Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, Tom I, Ivelja S, Refino CJ, Clark H, Eaton D, Grogan JL (2009) The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol 10(1):48–57. https://doi.org/10.1038/ni.1674
Zeiser R, Nguyen VH, Hou JZ, Beilhack A, Zambricki E, Buess M, Contag CH, Negrin RS (2007) Early CD30 signaling is critical for adoptively transferred CD4+CD25+ regulatory T cells in prevention of acute graft-versus-host disease. Blood 109(5):2225–2233. https://doi.org/10.1182/blood-2006-07-038455
Zhang X, Schwartz JC, Guo X, Bhatia S, Cao E, Lorenz M, Cammer M, Chen L, Zhang ZY, Edidin MA, Nathenson SG, Almo SC (2004) Structural and functional analysis of the costimulatory receptor programmed death-1. Immunity 20(3):337–347
Zhang R, Huynh A, Whitcher G, Chang J, Maltzman JS, Turka LA (2013) An obligate cell-intrinsic function for CD28 in Tregs. J Clin Invest 123(2):580–593. https://doi.org/10.1172/JCI65013
Zhang B, Chikuma S, Hori S, Fagarasan S, Honjo T (2016) Nonoverlapping roles of PD-1 and FoxP3 in maintaining immune tolerance in a novel autoimmune pancreatitis mouse model. Proc Natl Acad Sci U S A 113(30):8490–8495. https://doi.org/10.1073/pnas.1608873113
Zhang Q, Chikina M, Szymczak-Workman AL, Horne W, Kolls JK, Vignali KM, Normolle D, Bettini M, Workman CJ, Vignali DAA (2017) LAG3 limits regulatory T cell proliferation and function in autoimmune diabetes. Sci Immunol 2(9). https://doi.org/10.1126/sciimmunol.aah4569
Zheng G, Wang B, Chen A (2004) The 4-1BB costimulation augments the proliferation of CD4+CD25+ regulatory T cells. J Immunol 173(4):2428–2434
Zhong X, Tumang JR, Gao W, Bai C, Rothstein TL (2007) PD-L2 expression extends beyond dendritic cells/macrophages to B1 cells enriched for V(H)11/V(H)12 and phosphatidylcholine binding. Eur J Immunol 37(9):2405–2410. https://doi.org/10.1002/eji.200737461
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Wing, J.B., Tay, C., Sakaguchi, S. (2019). Control of Regulatory T Cells by Co-signal Molecules. In: Azuma, M., Yagita, H. (eds) Co-signal Molecules in T Cell Activation. Advances in Experimental Medicine and Biology, vol 1189. Springer, Singapore. https://doi.org/10.1007/978-981-32-9717-3_7
Download citation
DOI: https://doi.org/10.1007/978-981-32-9717-3_7
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-32-9716-6
Online ISBN: 978-981-32-9717-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)