
Abstract
Helicobacter pylori (H. pylori) has urease and ureI gene to overcome gastric acid to survive on the acidic gastric mucosa. Colonization with H. pylori is a condition that affects the relative risk of developing various clinical disorders of the upper gastrointestinal tract or extragastroduodenal disorders. That is, long-term inflammation due to H. pylori infection causes progressive damage to the gastric mucosa and plays a causative role in a number of important diseases, including duodenal ulcer, gastric ulcer, gastric cancer, and gastric mucosa-associated lymphoid tissue (MALT) lymphoma in addition to atrophic gastritis and intestinal metaplasia. Two decades of intense research into H. pylori virulence factors such as the VacA and CagA proteins have revealed many aspects of the relationships between this bacterium, the gastric mucosal surface, and the induction of disease. Disease outcome is the result of the intricate, ongoing interplay between environmental, bacterial, and host factors. In the continuous interactions with the host, the bacteria are able to adapt by mutations and DNA rearrangements, rendering novel genotypes, and overcome the host immune mechanism. On the host side, variations in the host immune response to the chronic presence of H. pylori and genetic polymorphism of host directly impact H. pylori-associated gastric disease, resulting in the disease outcome. However, H. pylori colonization pattern such as antrum-predominant gastritis and corpus-predominant pangastritis is known to determine the disease outcome, and this gastritis pattern is mainly decided by the acid secretion when H. pylori starts to colonize in the gastric mucosa.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Colonization with Helicobacter pylori (H. pylori) is not a disease in itself but a condition that affects the relative risk of developing various clinical disorders of the upper gastrointestinal tract or extragastroduodenal disorders [1]. H. pylori causes progressive damage to the gastric mucosa and is now accepted as playing a causative role in a number of important diseases, including duodenal ulcer, gastric ulcer, gastric adenocarcinoma, and gastric mucosa-associated lymphoid tissue (MALT) lymphoma [2–4]. Indeed, H. pylori-induced gastritis is considered as the most important risk factor for peptic ulcer and its complications as well as for gastric cancer [4]. Testing for H. pylori therefore has no relevance by itself but should be performed to find the cause of an underlying condition, such as peptic ulcer disease, or for the purpose of disease prevention, such as in gastric cancer relatives [5, 6]. In these cases, a positive test result justifies treatment, and a negative test result may indicate the need to search for other etiologic factors or preventive measures [1]. For these reasons, a correct understanding of the clinical course of H. pylori-associated disorders and the effect of H. pylori eradication is needed. Two decades of intense research into H. pylori virulence factors such as the VacA and CagA proteins have revealed many aspects of the relationships between this bacterium, the gastric mucosal surface, and the induction of disease [1]. The reason for the increment of gastric cancer possibility can be explained by intestinal metaplasia (IM), dysplasia, and gastric cancer cascade due to atrophic gastritis (AG) from H. pylori infection. Disease outcome is the result of the intricate, ongoing interplay between environmental, bacterial, and host factors [1]. Strain-to-strain genetic variability in bacterial virulence factors such as vacA and cagA not only affects the ability of the organism to colonize and cause disease but also affects inflammation and gastric acid output [1]. The decrease gastric output due to H. pylori-associated gastritis can explain why the prevalence of reflux gastritis decreases in the H. pylori-infected host. Long-term interaction between H. pylori and host causes fundamental change to both of H. pylori and host. That is, in the continuous interactions with the host, the bacteria are able to adapt to the host condition by mutations and DNA rearrangements. On the host side, variations in the host immune response to the chronic presence of H. pylori directly impact H. pylori-associated gastric disease and affect gastric acid output and thereby the density and location of H. pylori cells [1]. In this chapter synopsis of H. pylori-associated diseases such as gastritis, peptic ulcer, non-ulcer dyspepsia, MALT lymphoma, gastric cancer, and extragastroduodenal disorders will be briefly described.
2 H. pylori-Associated Diseases
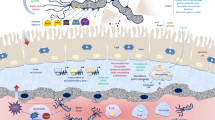
Although gastric colonization with H. pylori inevitably induces histologic gastritis in the infected individuals, 10–20 % of H. pylori-positive patients have risk of developing ulcer disease and a 1–2 % risk of developing noncardiac gastric cancer [7–9]. The risk of development of these disorders in the presence of H. pylori infection depends on a variety of bacterial, host, and environmental factors that mostly relate to the pattern and severity of gastritis [1] (Fig. 16.1).

Schematic representation of the factors contributing to gastric pathology and disease outcome in H. pylori infection. NSAID nonsteroidal anti-inflammatory drug, PPI proton pump inhibitor (Modified from Kusters et al. [1])
2.1 Acute and Chronic Gastritis
Chronic active gastritis, which occurs after colonization with H. pylori, can be observed in all H. pylori-positive subjects. The intragastric distribution and severity of this chronic inflammatory process depend on a variety of factors, such as characteristics of the colonizing strain, host genetics and immune response, diet, and the level of acid production [1]. H. pylori-induced ulcer disease, gastric cancer, and MALT lymphoma are all complications of this chronic inflammation; ulcer disease and gastric cancer in particular occur in those individuals and at those sites with the most severe inflammation [1]. Colonization with H. pylori virtually always leads to infiltration of the gastric mucosa in both the antrum and corpus with neutrophils and mononuclear cells, which consist of chronic inflammatory process.
2.1.1 Acute Gastritis
When H. pylori enters the stomach of a human host and colonizes itself on gastric mucosa, heavy neutrophils infiltrate into the mucosa. Neutrophil infiltration and the severity of gastric mucosa damage are closely related, and it seems like gastric epithelial cell damage is caused by infiltrated leukocytes due to H. pylori infection [10]. Once the bacteria enter the stomach and induce infection, subjects developed variable gastrointestinal and constitutional symptoms. Dyspeptic symptoms were mild to moderate in severity and typically appeared during the first week after challenge and peaked between days 9 and 12 [11]. These symptoms start within the first week of infection, and these symptoms reach their maximum degrees on 9–12 days after the infection [11]. Then, the symptoms are attenuated by themselves, and most of the symptoms are gone after 2 weeks of the infection [11–14]. It is unclear whether this initial colonization can be followed by spontaneous clearance and resolution of gastritis and, if so, how often this occurs [1]. Follow-up studies of young children with serology or breath tests suggested that infection may spontaneously disappear in some patients in this age group [15–17]. However, it does not occur in adults except disappearance of H. pylori in case of AG and IM [18]. In addition, studies of homozygotic twins showed a concordance in their H. pylori status irrespective of whether they had grown up together or apart, but this concordance was not observed among heterozygotic twins [19]. These results suggest that some individuals are prone to H. pylori colonization, while others may be able to prevent colonization or clear an established infection [1].
2.1.2 Chronic Gastritis
When colonization does become persistent, a close correlation exists between the level of acid secretion and the distribution of gastritis, which determines the gastric hormonal responses with dynamic changes of gastric mucosa. Finally, they determine the outcomes of H. pylori infection. In subjects with intact acid secretion, H. pylori in particular colonizes the gastric antrum, where few acid secretory parietal cells are present because they like weak acid environment such as pH 5.0 instead of strong acid. This colonization pattern is associated with an antrum-predominant gastritis [1] (Fig. 16.1). Histological evaluation of gastric corpus specimens in these cases reveals limited chronic inactive inflammation and low numbers of superficially colonizing H. pylori bacteria [1]. In this state duodenal ulcer (DU) can easily develop [1] (Fig. 16.1). Subjects in whom acid secretion is impaired have a more even distribution of bacteria in the antrum and corpus, and bacteria in the corpus are in closer contact with the mucosa, leading to a corpus-predominant pangastritis [1, 20] (Fig. 16.1). In this state the acid secretion will decrease and could be a condition for gastric ulcer or gastric cancer development when the bacteria continuously interact with the host. As predisposition condition of DU and gastric cancer is contradictory, the chance of development of gastric cancer is known to be half in case of DU history. Similarly, there was a report that gastric cancers occurred only in patients with previous benign gastric ulcer (BGU) and not in patients with former DU [21]. When this chronic inflammation continues, AG defined as the loss of glands [22] occurs and slowly progresses to IM, which is defined as the replacement of the surface, foveolar, and glandular epithelium in the gastric mucosa by intestinal epithelium with the presence of Paneth cells, goblet cells, and absorptive cells [23]. H. pylori infection was the most important risk factor of AG and IM, and AG is considered to be an antecedent to IM [24]. Actually the time difference of AG and IM could be different, but Korean report suggested 10 years [24]. According to the Correa model, the pathogenesis of intestinal-type gastric cancer can be explained by a multistep process from chronic gastritis through AG, intestinal metaplasia IM, and dysplasia to cancer [22]. The risk of gastric cancer increases with greater extent and higher degree of gastric mucosal atrophy [23] and more than tenfold by IM [25]. Several studies suggest that AG and IM are not related with gastroduodenal symptoms [26], but they are major precursor lesions of gastric cancer [25, 27, 28].
2.2 Non-ulcer Dyspepsia
Non-ulcer or functional dyspepsia (FD) is defined as the presence of symptoms of upper gastrointestinal distress without any identifiable structural abnormality during diagnostic workup, in particular including upper gastrointestinal endoscopy [29]. Uninvestigated dyspepsia is defined as the presence of dyspeptic symptoms for which no further diagnostic evaluation has been performed. These symptoms are frequently experienced by 20–30 % of the adult population of the Western world [29]. FD was divided into four symptoms based on the Rome III criteria as follows: epigastric pain, epigastric burning, bothersome postprandial fullness, and early satiation (prevents finishing regular-sized meals). Rome III criteria classified FD into two subcategories of FD based on cohort and population-based studies: postprandial distress syndrome (PDS) and epigastric pain syndrome (EPS) [29]. A recent meta-analysis reported that the odds ratio (OR) for the occurrence of post-infectious FD was 2.54 (95 % confidence interval [CI], 1.76–3.65) more than 6 months after acute gastroenteritis compared with the control [30]. Another meta-analysis found an OR of 2.18 (95 % CI, 1.70–2.81) for FD risk following acute gastroenteritis [31]. These results suggest that gastrointestinal infection is associated with an increased risk of FD, supporting an inflammatory and immunological mechanism in the pathogenesis of FD [32]. H. pylori infection is the main cause of gastroduodenal inflammation [33] and provokes activation of a complex cytokine and chemokine response in the gastric mucosa [34], which may induce dyspepsia [35]. However, 30–60 % of patients with functional dyspepsia carry H. pylori, but this prevalence is not much different from that in the unaffected population [28, 36]. This result could be explained by that FD is a multifactorial disease rather than H. pylori-dependent disease. Thus, the effect of eradication on FD could be the only method whether H. pylori infection is related with FD or not. A Cochrane meta-analysis was performed on 17 randomized controlled trials and identified an association between H. pylori eradication and improvement in FD symptoms [37]. A small but significant benefit of H. pylori eradication therapy was observed with a number needed to treat of 14 (95 % CI, 10–25) [37]. In another recent study, the number needed to treat was 8 [38]. In addition, a systemic review and meta-analysis from China reported that the summary OR for improvement in FD patients after H. pylori eradication was 3.61 (95 % CI, 2.62–4.98) [39]. Similarly, a recent meta-analysis of randomized controlled trials with 12 months follow-up revealed that dyspeptic symptoms were significantly improved in the eradication group (OR 1.38; 95 % CI, 1.18–1.62) [40]. Based on these considerations, the Kyoto global consensus proposed that patients who remain symptomatic after successful H. pylori eradication should be regarded as having H. pylori-associated dyspepsia separated from FD [41]. In addition, H. pylori eradication has been recommended as first-line treatment for dyspeptic patients with H. pylori infection, because eradication therapy for dyspeptic symptoms is better than placebo in H. pylori-infected dyspeptic patients [41]. However, as symptom resolution takes months after completion of eradication therapy, this time delay makes some scholars to not agree with this proposal.
2.3 Gastric or Duodenal Ulcers
Gastric or duodenal ulcers are defined as mucosal defects with a diameter of at least 0.5 cm penetrating through the muscularis mucosa [1], but the guideline could be different to 0.3 cm. BGU mostly occurs along the lesser curvature of the stomach, in particular, at the transition from the corpus to antrum mucosa [42]. DU usually occurs in the duodenal bulb, which is the area most exposed to gastric acid. In Western countries, DU is approximately fourfold more common than BGU; elsewhere, BGU is more common, especially, when aged population increases [1]. For instance, the ratio of BGU (n = 265) to DU (n = 210) was 1.26:1 in Korea [43]. In terms of age, DU frequently occurs between 20 and 50 years of age, while BGU predominantly arises in subjects over 40 years old. Recently H. pylori prevalence rapidly decreases, the mean age of DU also increases, but still there is age difference between DU and BGU [43]. Both gastric and duodenal ulcer diseases are strongly related to H. pylori. In the first decade after the discovery of H. pylori, approximately 95 % of DU and 85 % of BGU occurred in the presence of H. pylori infection [9]. Several cohort studies estimated that the lifetime risk for ulcer disease in H. pylori-positive subjects is three to ten times higher than in H. pylori-negative subjects [44]. Furthermore, H. pylori eradication provided strong evidence for a causal relationship between H. pylori and ulcer disease by showing that eradication of this bacterium strongly reduced the risk of recurrent ulcer disease [45–50]. Ulcer development in the presence of H. pylori is influenced by a variety of host and bacterial factors [51]. If acid output is decreased, the gastric transitional zone between the corpus and antrum is the place where gastric ulcer can occur [1]. If acid production is normal to high, the most severe inflammation usually is found in the distal stomach and proximal duodenum, giving rise to juxtapyloric and duodenal ulcer disease [1]. Recently the incidence and prevalence of peptic ulcer have been decreasing [52]. Hygiene improvement and low prevalence of H. pylori infection are suspected as main reasons, but the increase of aging population and the use of nonsteroidal anti-inflammatory drug (NSAID) become the major cause of peptic ulcer diseases instead of H. pylori infection. In addition, idiopathic peptic ulcer rapidly increases [53].
2.4 Gastric MALT Lymphoma
The gastric mucosa does not normally contain lymphoid tissue, but MALT nearly always appears in response to colonization with H. pylori. In rare cases, a monoclonal population of B cells may arise from this tissue and slowly proliferate to form a MALT lymphoma [1]. The histological criteria for the diagnosis of gastric MALT lymphoma and the differentiation from polyclonal reactive infiltrates remain controversial. In particular diagnosis is based on histological appearance during routine microscopy and on demonstration of clonality by immunohistochemistry or molecular techniques, such as PCR. Nearly all MALT lymphoma patients are H. pylori positive [54], and H. pylori-positive subjects have a significantly increased risk for the development of gastric MALT lymphoma [55]. Because of the diagnostic controversies and the relative rarity of this disorder, the exact incidence in H. pylori-positive subjects is unknown, but MALT lymphomas occur in less than 1 % of H. pylori-positive subjects [56]. H. pylori eradication can lead to complete remission in approximately 60–80 % of patients with stage IE MALT lymphoma confined to the stomach, but some 10 % continue to have signs of minimal residual disease, and the remainder shows no response or disease progression [57–61]. Ten to 35 % of those who initially reach complete remission after H. pylori eradication show recurrent disease during further follow-up. A major predictor for response appears to be the presence of a t(11;18) (q21;q21) translocation. This translocation is associated with API2-MALT1 fusion, the former being involved in regulation of apoptosis, the latter resembling a caspase-like protein, but with as-yet-unknown biological function. Together, the fusion leads to suppression of apoptosis. Several studies have reported that MALT lymphomas with this translocation do not at all or only rarely respond to H. pylori eradication [61–63].
2.5 Gastric Cancer
Despite the global decrease, gastric cancer is still a burdensome disease internationally. It is the fifth most common cancer and the third leading cause of cancer mortality worldwide [64]. The prognosis of gastric cancer patients depends on the tumor stage at the time of initial diagnosis that the 5-year survival rates of early gastric cancer exceeding 90 % make a striking contrast to those of advanced gastric cancer reaching below 50 % [65–68]. Therefore, the effort to detect gastric cancer when the tumor is in early stage can reduce the overall socioeconomic burden of gastric cancer. Traditionally, gastric cancer was thought to develop due to dietary carcinogens produced by old ways of food preservation such as smoking and salting. However, in recent decades, H. pylori has been marked as the main culprit of gastric cancer development [69]. According to the Lauren’s classification system [70], gastric cancer can be classified into two major histological variants: an intestinal type and a diffuse type. It is generally accepted that intestinal-type gastric adenocarcinoma arises through a multistep process from chronic gastritis that progresses through stages of atrophy, IM, and dysplasia and finally intestinal-type cancer [22]. On the other hand, diffuse-type gastric cancer is thought to be primarily genetically determined and to be less associated with environmental factors than the intestinal type and not to progress through severe AG [71]. In a meta-analysis of 19 cohort or case-control studies, a summary odds ratio for gastric cancer was estimated to be 1.92 (95 % CI, 1.32–2.78) in H. pylori-infected subjects compared to uninfected subjects regardless of histological type of gastric cancer such as intestinal type or diffuse type [72]. Furthermore, several randomized control studies have shown that H. pylori eradication reduces the risk of gastric cancer [73]. Among those, the most recent study reported that H. pylori eradication reduced gastric cancer incidence by 25 % [74], confirming the importance of H. pylori eradication for the prevention of gastric cancer. In addition to H. pylori infection, low socioeconomic status, smoking, and family history of gastric cancer were established as risk factors of gastric cancer [5, 6, 75–78]. Factors such as alcohol, fruits, vegetables, and salty and spicy food intake have been subjects of controversy [79–82]. Regarding ABO blood type’s association with gastric cancer, blood group A was associated with increased risk of gastric cancer [83, 84], and B allele was associated with decreased risk of gastric cancer in a Japanese study [85]. These findings suggest that gastric cancer is a multifactorial disease with H. pylori being the primary cause, and H. pylori’s effect on carcinogenesis is modulated by microbial, environmental, and host factors [86].
2.6 Gastroesophageal Reflux Disease
Many studies suggested that H. pylori might protect against the development of gastroesophageal reflux disease (GERD) and as such also be of benefit to their hosts [1]. This slowly emerging concept came from repeated observations of a low prevalence of H. pylori among GERD patients, particularly of more virulent strains [87], opposing time and geographical trends for H. pylori prevalence compared with the incidence of GERD and its complications, a potentially increased incidence of GERD after H. pylori eradication [88], and the recognition that H. pylori-induced corpus gastritis reduced acid secretion. The hypothesis was that H. pylori-induced inflammation of the gastric corpus had an acid-suppressive effect, thus preventing patients from contracting GERD. However, there was no evidence that H. pylori eradication has a considerable impact on either the new development of GERD [89, 90]. These data show that although epidemiologic data suggest that there may be an inverse relation between H. pylori and GERD, the risk for new development or worsening of preexistent GERD is not an issue in the decision of whether or not to treat H. pylori.
2.7 Extraintestinal Manifestations of H. pylori Infection
H. pylori has been linked to a variety of extraintestinal disorders. These include coronary heart disease, asthma, dermatological disorders such as rosacea and idiopathic urticaria, autoimmune thyroid disease and thrombocytopenic purpura, iron deficiency anemia, Raynaud’s phenomenon, scleroderma, migraine, and Guillain-Barré syndrome [1]. The underlying hypothetical mechanisms include chronic low-grade activation of the coagulation cascade, accelerating atherosclerosis, and antigenic mimicry between H. pylori and host epitopes leading to autoimmune disorders [91]. This has led to large numbers of case studies of patients with these disorders. Several groups in particular have studied patients with idiopathic thrombocytopenic purpura and showed that when these patients are colonized with H. pylori, eradication therapy has a significant effect over placebo for improvement of thrombocyte counts [92–94] and iron deficiency anemia [95, 96]. Currently, according to American College of Gastroenterology Guideline and the Maastricht IV/Florence Consensus Report, H. pylori eradication is recommended in unexplained iron deficiency anemia and idiopathic thrombocytopenic purpura [4, 97]. In patients with other conditions mentioned above, there is as yet no role for H. pylori eradication, and further adequate, randomized trials are needed.
3 Conclusions
H. pylori causes progressive damage to the gastric mucosa and is now accepted as playing a causative role in a number of important diseases, including gastritis, duodenal ulcer, gastric ulcer, gastric cancer, gastric MALT lymphoma, and extraintestinal disorders. This disease outcome is the result of the intricate, ongoing interplay between environmental, bacterial, and host factors. Long-term interaction between H. pylori and host causes fundamental change to both H. pylori and host. That is, in the continuous interactions with the host, the bacteria are able to adapt to the host condition by mutations and DNA rearrangements. On the host side, variations in the host immune response to the chronic presence of H. pylori directly impact H. pylori-associated gastric disease and affect gastric acid output and thereby the density and location of H. pylori cells. It is well known that H. pylori colonization pattern such as antrum-predominant gastritis and corpus-predominant pangastritis is very important for the disease outcome, and gastritis pattern is mainly decided by the acid secretion when H. pylori starts to colonize in the gastric mucosa because H. pylori likes the weak acid condition instead of strong acid.
References
Kusters JG, van Vliet AH, Kuipers EJ. Pathogenesis of Helicobacter pylori infection. Clin Microbiol Rev. 2006;19:449–90.
Malfertheiner P, Chan FK, McColl KE. Peptic ulcer disease. Lancet. 2009;374:1449–61.
Fock KM, Graham DY, Malfertheiner P. Helicobacter pylori research: historical insights and future directions. Nat Rev Gastroenterol Hepatol. 2013;10:495–500.
Malfertheiner P, Mégraud F, O’Morain CA, Atherton J, Axon AT, Bazzoli F, et al. Management of Helicobacter pylori infection—the Maastricht IV Florence Consensus Report. Gut. 2012;61:646–64.
Shin CM, Kim N, Yang HJ, Cho SI, Lee HS, Kim JS, et al. Stomach cancer risk in gastric cancer relatives: interaction between H. pylori infection and family history of gastric cancer for the risk of stomach cancer. J Clin Gastroenterol. 2010;44:e34–9.
Shin CM, Kim N, Lee HS, Lee DH, Kim JS, Jung HC, et al. Intrafamilial aggregation of gastric cancer: a comprehensive approach including environmental factors, Helicobacter pylori virulence and genetic susceptibility. Eur J Gastroenterol Hepatol. 2011;23:411–7.
Ernst PB, Gold BD. The disease spectrum of Helicobacter pylori: the immunopathogenesis of gastroduodenal ulcer and gastric cancer. Annu Rev Microbiol. 2000;54:615–40.
Kuipers EJ. Review article: exploring the link between Helicobacterpylori and gastric cancer. Aliment Pharmacol Ther. 1999;13 Suppl 1:3–11.
Kuipers EJ, Thijs JC, Festen HP. The prevalence of Helicobacter pylori in peptic ulcer disease. Aliment Pharmacol Ther. 1995;9 Suppl 2:59–69.
Dundon WG, Nishioka H, Polenghi A, Papinutto E, Zanotti G, Montemurro P, et al. The neutrophil-activating protein of Helicobacter pylori. Int J Med Microbiol. 2002;291:545–50.
Graham DY, Opekun AR, Osato MS, El-Zimaity HM, Lee CK, Yamaoka Y, et al. Challenge model for Helicobacter pylori infection in human volunteers. Gut. 2004;53:1235–43.
Marshall BJ, Armstrong JA, McGechie DB, Glancy RJ. Attempt to fulfill Koch’s postulates for pyloric Campylobacter. Med J Aust. 1985;142:436–9.
Morris A, Nicholson G. Ingestion of Campylobacter pyloridis causes gastritis and raised fasting gastric pH. Am J Gastroenterol. 1987;82:192–9.
Morris AJ, Ali MR, Nicholson GI, Perez-Perez GI, Blaser MJ. Long-term follow-up of voluntary ingestion of Helicobacter pylori. Ann Intern Med. 1991;114:662–3.
Granstrom M, Tindberg Y, Blennow M. Seroepidemiology of Helicobacter pylori infection in a cohort of children monitored from 6 months to 11 years of age. J Clin Microbiol. 1997;35:468–70.
Malaty HM, Graham DY, Wattigney WA, Srinivasan SR, Osato M, Berenson GS. Natural history of Helicobacter pylori infection in childhood: 12-year follow-up cohort study in a biracial community. Clin Infect Dis. 1999;28:279–82.
Pérez-Pérez GI, Sack RB, Reid R, Santosham M, Croll J, Blaser MJ. Transient and persistent Helicobacter pylori colonization in Native American children. J Clin Microbiol. 2003;41:2401–7.
Kang HY, Kim N, Park YS, Hwang JH, Kim JW, Jeong SH, et al. Progression of atrophic gastritis and intestinal metaplasia driving Helicobacter pylori out of the gastric mucosa. Dig Dis Sci. 2006;51:2310–5.
Malaty HM, Engstrand L, Pedersen NL, Graham DY. Helicobacter pylori infection: genetic and environmental influences. A study of twins. Ann Intern Med. 1994;120:982–6.
Kuipers EJ, Uyterlinde AM, Peña AS, Hazenberg HJ, Bloemena E, Lindeman J, et al. Increase of Helicobacter pylori-associated corpus gastritis during acid suppressive therapy: implications for long-term safety. Am J Gastroenterol. 1995;90:1401–6.
Take S, Mizuno M, Ishiki K, Nagahara Y, Yoshida T, Yokota K, et al. The effect of eradicating Helicobacter pylori on the development of gastric cancer in patients with peptic ulcer disease. Am J Gastroenterol. 2005;100:1037–42.
Correa P. Chronic gastritis: a clinico-pathological classification. Am J Gastroenterol. 1988;83:504–9.
de Vries AC, Kuipers EJ. Epidemiology of premalignant gastric lesions: implications for the development of screening and surveillance strategies. Helicobacter. 2007;12 Suppl 2:22–31.
Kim N, Park YS, Cho SI, Lee HS, Choe G, Kim IW, et al. Prevalence and risk factors of atrophic gastritis and intestinal metaplasia in a Korean population without significant gastroduodenal disease. Helicobacter. 2008;13:245–55.
Leung WK, Sung JJ. Review article: intestinal metaplasia and gastric carcinogenesis. Aliment Pharmacol Ther. 2002;16:1209–16.
Kim SE, Park HK, Kim N, Joo YE, Baik GH, Shin JE, et al. Prevalence and risk factors of functional dyspepsia: a nationwide multicenter prospective study in Korea. J Clin Gastroenterol. 2014;48:e12–8.
Uemura N, Okamoto S, Yamamoto S, Matsumura N, Yamaguchi S, Yamakido M, et al. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med. 2001;345:784–9.
Ohata H, Kitauchi S, Yoshimura N, Mugitani K, Iwane M, Nakamura H, et al. Progression of chronic atrophic gastritis associated with Helicobacter pylori infection increases risk of gastric cancer. Int J Cancer. 2004;109:138–43.
Tack J, Talley NJ, Camilleri M, Holtmann G, Hu P, Malagelada JR, et al. Functional gastroduodenal disorders. Gastroenterology. 2006;130:1466–79.
Futagami S, Itoh T, Sakamoto C. Systematic review with meta-analysis: post-infectious functional dyspepsia. Aliment Pharmacol Ther. 2015;41:177–88.
Pike BL, Porter CK, Sorrell TJ, Riddle MS. Acute gastroenteritis and the risk of functional dyspepsia: a systematic review and meta-analysis. Am J Gastroenterol. 2013;108:1558–63.
Gwee KA. Post-infectious irritable bowel syndrome, an inflammation-immunological model with relevance for other IBS and functional dyspepsia. J Neurogastroenterol Motil. 2010;16:30–4.
Suzuki H, Hibi T, Marshall BJ. Helicobacter pylori: present status and future prospects in Japan. J Gastroenterol. 2007;42:1–15.
D’Elios MM, Andersen LP. Inflammation, immunity, and vaccines for Helicobacter pylori. Helicobacter. 2009;14 Suppl 1:21–8.
Suzuki H, Matsuzaki J, Hibi T. What is the difference between Helicobacter pylori-associated dyspepsia and functional dyspepsia? J Neurogastroenterol Motil. 2011;17:124–30.
Talley NJ, Hunt RH. What role does Helicobacter pylori play in dyspepsia and nonulcer dyspepsia? Arguments for and against H. pylori being associated with dyspeptic symptoms. Gastroenterology. 1997;113:S67–77.
Moayyedi P, Soo S, Deeks J, Delaney B, Harris A, Innes M, et al. Eradication of Helicobacter pylori for non-ulcer dyspepsia. Cochrane Database Syst Rev. 2006;(2):CD002096.
Mazzoleni LE, Sander GB, Francesconi CF, Mazzoleni F, Uchoa DM, De Bona LR, et al. Helicobacter pylori eradication in functional dyspepsia: HEROES trial. Arch Intern Med. 2011;171:1929–36.
Jin X, Li YM. Systematic review and meta-analysis from Chinese literature: the association between Helicobacter pylori eradication and improvement of functional dyspepsia. Helicobacter. 2007;12:541–6.
Zhao B, Zhao J, Cheng WF, Shi WJ, Liu W, Pan XL, et al. Efficacy of Helicobacter pylori eradication therapy on functional dyspepsia: a meta-analysis of randomized controlled studies with 12-month follow-up. J Clin Gastroenterol. 2014;48:241–7.
Sugano K, Tack J, Kuipers EJ, Graham DY, El-Omar EM, Miura S, et al. Kyoto global consensus report on Helicobacter pylori gastritis. Gut. 2015;64:1353–67.
Van Zanten SJ, Dixon MF, Lee A. The gastric transitional zones: neglected links between gastroduodenal pathology and Helicobacter ecology. Gastroenterology. 1999;116:1217–29.
Kim JJ, Kim N, Lee BH, Kang JM, Seo P, Lim MK, et al. Risk factors for development and recurrence of peptic ulcer disease. Korean J Gastroenterol. 2010;56:220–8.
Nomura A, Stemmermann GN, Chyou PH, Perez-Perez GI, Blaser MJ. Helicobacter pylori infection and the risk for duodenal and gastric ulceration. Ann Intern Med. 1994;120:977–81.
Rauws EA, Tytgat GN. Cure of duodenal ulcer associated with eradication of Helicobacter pylori. Lancet. 1990;335:1233–5.
Kim N, Lim SH, Lee KH, Choi SE. Long-term effect of Helicobacter pylori eradication on gastric metaplasia in patients with duodenal ulcer. J Clin Gstroenterol. 1998;27:246–52.
Kim N, Oh JH, Lee CG, Lim C, Won KH, Choi WR, et al. Effect of H. pylori eradication on the benign gastric ulcer recurrence – a 24 month follow-up study. Korean J Intern Med. 1999;14:9–14.
Hentschell E, Brandstätter G, Dragosics B, Hirschl AM, Nemec H, Schütze K, et al. Effect of ranitidine and amoxicillin plus metronidazole on the eradication of Helicobacter pylori and the recurrence of duodenal ulcer. N Engl J Med. 1993;328:308–12.
Treiber G, Lambert JR. The impact of Helicobacter pylori eradication on peptic ulcer healing. Am J Gastroenterol. 1998;93:1080–4.
van der Hulst RW, Rauws EA, Köycü B, Keller JJ, ten Kate FJ, Dankert J, et al. Helicobacter pylori reinfection is virtually absent after successful eradication. J Infect Dis. 1997;176:196–200.
Kim JY, Kim N, Nam RH, Suh JH, Chang H, Lee JW, et al. The association of polymorphisms in virulence factor of Helicobacter pylori and gastroduodenal diseases in South Korea. J Gastroenterol Hepatol. 2014;29:984–91.
Sung JJ, Kuipers EJ, El-Serag HB. Systematic review: the global incidence and prevalence of peptic ulcer disease. Aliment Pharmacol Ther. 2009;29:938–46.
Kim JJ, Kim N, Park HK, Jo HJ, Shin CM, Lee SH, et al. Clinical characteristics of patients diagnosed as peptic ulcer disease in the third referral center in 2007. Korean J Gastroenterol. 2012;59:338–46.
Eidt S, Stolte M, Fischer R. Helicobacter pylori gastritis and primary gastric non-Hodgkin’s lymphomas. J Clin Pathol. 1994;47:436–9.
Parsonnet J, Hansen S, Rodriguez L, Gelb AB, Warnke RA, Jellum E, et al. Helicobacter pylori infection and gastric lymphoma. N Engl J Med. 1994;330:1267–71.
Berg DE, Hoffman PS, Appelmelk BJ, Kusters JG. The Helicobacter pylori genome sequence: genetic factors for long life in the gastric mucosa. Trends Microbiol. 1997;5:468–74.
de Mascarel A, Ruskone-Fourmestraux A, Lavergne-Slove A, Megraud F, Dubus P, Merlio JP. Clinical, histological and molecular follow-up of 60 patients with gastric marginal zone lymphoma of mucosa-associated lymphoid tissue. Virchows Arch. 2005;446:219–24.
Fischbach W, Goebeler-Kolve ME, Dragosics B, Greiner A, Stolte M. Long term outcome of patients with gastric marginal zone B cell lymphoma of mucosa associated lymphoid tissue (MALT) following exclusive Helicobacter pylori eradication therapy: experience from a large prospective series. Gut. 2004;53:34–7.
Nakamura S, Matsumoto T, Suekane H, Nakamura S, Matsumoto H, Esaki M, et al. Long-term clinical outcome of Helicobacter pylori eradication for gastric mucosa-associated lymphoid tissue lymphoma with a reference to second-line treatment. Cancer. 2005;104:532–40.
Wundisch T, Thiede C, Morgner A, Dempfle A, Günther A, Liu H, et al. Long-term follow-up of gastric MALT lymphoma after Helicobacter pylori eradication. J Clin Oncol. 2005;23:8018–24.
Choi YJ, Kim N, Paik JH, Kim JM, Lee SH, Park YS, et al. Characteristics of Helicobacter pylori-positive and Helicobacter pylori-negative gastric mucosa-associated lymphoid tissue lymphoma and their influence on clinical outcome. Helicobacter. 2013;18:197–205.
Inagaki H, Nakamura T, Li C, Sugiyama T, Asaka M, Kodaira J, et al. Gastric MALT lymphomas are divided into three groups based on responsiveness to Helicobacter pylori eradication and detection of API2-MALT1 fusion. Am J Surg Pathol. 2004;28:1560–7.
Liu H, Ye H, Ruskone-Fourmestraux A, De Jong D, Pileri S, Thiede C, et al. T(11;18) is a marker for all stage gastric MALT lymphomas that will not respond to H. pylori eradication. Gastroenterology. 2002;122:1286–94.
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:E359–86.
Everett SM, Axon AT. Early gastric cancer in Europe. Gut. 1997;41:142–50.
Leung WK, Wu MS, Kakugawa Y, Kim JJ, Yeoh KG, Goh KL, et al. Screening for gastric cancer in Asia: current evidence and practice. Lancet Oncol. 2008;9:279–87.
Bang YJ, Van Cutsem E, Feyereislova A, Chung HC, Shen L, Sawaki A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376:687–97.
Lello E, Furnes B, Edna TH. Short and long-term survival from gastric cancer. A population-based study from a county hospital during 25 years. Acta Oncol. 2007;46:308–15.
Schistosomes, liver flukes and Helicobacter pylori. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Lyon, 7–14 June 1994. IARC Monogr Eval Carcinog Risks Hum. 1994;61:1–241.
Lauren P. The two histological main types of gastric carcinoma: diffuse and so-called intestinal-type carcinoma. An attempt at a histo-clinical classification. Acta Pathol Microbiol Scand. 1965;64:31–49.
Nardone G. Review article: molecular basis of gastric carcinogenesis. Aliment Pharmacol Ther. 2003;17 Suppl 2:75–81.
Huang JQ, Sridhar S, Chen Y, Hunt RH. Meta-analysis of the relationship between Helicobacter pylori seropositivity and gastric cancer. Gastroenterology. 1998;114:1169–79.
Tan VP, Wong BC. Gastric cancer chemoprevention: the current evidence. Gastroenterol Clin North Am. 2013;42:299–316.
Lee YC, Chen TH, Chiu HM, Shun CT, Chiang H, Liu TY, et al. The benefit of mass eradication of Helicobacter pylori infection: a community-based study of gastric cancer prevention. Gut. 2013;62:676–82.
Barker DJ, Coggon D, Osmond C, Wickham C. Poor housing in childhood and high rates of stomach cancer in England and Wales. Br J Cancer. 1990;61:575–8.
Roder DM. The epidemiology of gastric cancer. Gastric Cancer. 2002;5 Suppl 1:5–11.
Ladeiras-Lopes R, Pereira AK, Nogueira A, Pinheiro-Torres T, Pinto I, Santos-Pereira R, et al. Smoking and gastric cancer: systematic review and meta-analysis of cohort studies. Cancer Causes Control. 2008;19:689–701.
La Torre G, Chiaradia G, Gianfagna F, De Lauretis A, Boccia S, Mannocci A, et al. Smoking status and gastric cancer risk: an updated meta-analysis of case-control studies published in the past ten years. Tumori. 2009;95:13–22.
de Martel C, Forman D, Plummer M. Gastric cancer: epidemiology and risk factors. Gastroenterol Clin North Am. 2013;42:219–40.
Yan S, Li B, Bai ZZ, Wu JQ, Xie DW, Ma YC, et al. Clinical epidemiology of gastric cancer in Hehuang valley of China: a 10-year epidemiological study of gastric cancer. World J Gastroenterol. 2014;20:10486–94.
Dikshit RP, Mathur G, Mhatre S, Yeole BB. Epidemiological review of gastric cancer in India. Indian J Med Paediatr Oncol. 2011;32:3–11.
Yoon H, Kim N. Diagnosis and management of high risk group for gastric cancer. Gut Liver. 2015;9:5–17.
Edgren G, Hjalgrim H, Rostgaard K, Norda R, Wikman A, Melbye M, et al. Risk of gastric cancer and peptic ulcers in relation to ABO blood type: a cohort study. Am J Epidemiol. 2010;172:1280–5.
Wang Z, Liu L, Ji J, Zhang J, Yan M, Zhang J, et al. ABO blood group system and gastric cancer: a case-control study and meta-analysis. Int J Mol Sci. 2012;13:13308–21.
Nakao M, Matsuo K, Ito H, Shitara K, Hosono S, Watanabe M, et al. ABO genotype and the risk of gastric cancer, atrophic gastritis, and Helicobacter pylori infection. Cancer Epidemiol Biomarkers Prev. 2011;20:1665–72.
Correa P. Gastric cancer: overview. Gastroenterol Clin North Am. 2013;42:211–7.
Fallone CA, Barkun AN, Göttke MU, Best LM, Loo VG, Veldhuyzen van Zanten S, et al. Association of Helicobacter pylori genotype with gastroesophageal reflux disease and other upper gastrointestinal diseases. Am J Gastroenterol. 2000;95:659–69.
Labenz J, Blum AL, Bayerdörffer E, Meining A, Stolte M, Börsch G. Curing Helicobacter pylori infection in patients with duodenal ulcer may provoke reflux esophagitis. Gastroenterology. 1997;112:1442–7.
Vakil N, Hahn B, McSorley D. Recurrent symptoms and gastro-oesophageal reflux disease patients with duodenal ulcer treated for Helicobacter pylori infection. Aliment Pharmacol Ther. 2000;14:45–51.
Kim N, Lee SW, Kim JI, Baik GH, Kim SJ, Seo GS, et al. Effect of Helicobacter pylori eradication on development of reflux esophagitis and gastroesophageal reflux symptoms: a nationwide multicenter prospective study. Gut Liver. 2011;5:437–46.
Gasbarrini A, Carloni E, Gasbarrini G, Chisholm SA. Helicobacter pylori and extragastric diseases—other Helicobacters. Helicobacter. 2004;9 Suppl 1:57–66.
Gasbarrini A, Franceschi F. Does H. pylori infection play a role in idiopathic thrombocytopenic purpura and in other autoimmune diseases? Am J Gastroenterol. 2005;100:1271–3.
Jackson S, Beck PL, Pineo GF, Poon MC. Helicobacter pylori eradication: novel therapy for immune thrombocytopenic purpura? A review of the literature. Am J Hematol. 2005;78:142–50.
Suzuki T, Matsushima M, Masui A, Watanabe K, Takagi A, Ogawa Y, et al. Effect of Helicobacter pylori eradication in patients with chronic idiopathic thrombocytopenic purpura—a randomized controlled trial. Am J Gastroenterol. 2005;100:1265–70.
Choe YH, Kim SK, Son BK, Lee DH, Hong YC, Pai SH. Randomized placebo-controlled trial of Helicobacter pylori eradication for iron-deficiency anemia in preadolescent children and adolescents. Helicobacter. 1999;4:135–9.
Choe YH, Kim SK, Hong YC. Helicobacter pylori infection with iron deficiency anaemia and subnormal growth at puberty. Arch Dis Child. 2000;82:136–40.
Chey WD, Wong BC, Practice Parameters Committee of the American College of Gastroenterology. American College of Gastroenterology Guideline on the management of Helicobacter pylori infection. Am J Gastroenterol. 2007;102:1808–25.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media Singapore
About this chapter
Cite this chapter
Kim, N. (2016). Synopsis of H. pylori-Associated Diseases. In: Kim, N. (eds) Helicobacter pylori. Springer, Singapore. https://doi.org/10.1007/978-981-287-706-2_16
Download citation
DOI: https://doi.org/10.1007/978-981-287-706-2_16
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-287-705-5
Online ISBN: 978-981-287-706-2
eBook Packages: MedicineMedicine (R0)