
Abstract
The lack of adequate toxicity data for the vast majority of chemicals in the environment has spurred the development of high-throughput transcriptomics (HTT) to support pathway-based screening of chemicals. The main challenge is how to decipher molecular response into adverse effects from omics data. This chapter describes an adverse outcome pathway (AOP) network-based approach for chemical screening using HTT in a compendium of human cells. First, the methodology for conducting HTT, concentration-dependent modeling analysis and AOP network analysis is introduced. Two case studies are presented: (1) cross-species comparison of transcriptomic dose–response of short-chain chlorinated paraffins and (2) high-throughput transcriptomics screening of chemicals with various known modes of action using human cells, which demonstrate the ability of HTT for chemical screening, classification and tiered chemical risk assessment by HTT-based AOP network profiles. In summary, the AOP network-based chemical screening provides a rapid and efficient omics-based approach for ranking, clustering and assessment of chemical hazards.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Introduction
A major challenge for chemical risk assessment is the lack of sufficient toxicological information for thousands of chemicals. The numbers of registered chemicals are approximately 140,000, 85,000 and 45,600 in Europe (ECHA 2016), USA (EPA 2016) and China (MEE 2013), respectively, while the majority of chemicals have inadequate toxicity data. Toxicology in the twenty-first century has envisioned a shift from traditional animal-based experiments to the mechanistic understanding of biological pathways via high-throughput screening (HTS) (Collins et al. 2008; Dix et al. 2007). In the last decade, in vitro bioassays (e.g., US EPA ToxCast and Tox21 programs), were extensively applied to characterize the concentration responses of >10,000 chemicals on hundreds of molecular targets (Richard et al. 2016). However, in vitro bioassays are limited to the targeted biological endpoints within known toxicological pathways, which cannot capture the molecular signals over comprehensive biological space (Gaytán and Vulpe 2014; Huang et al. 2018; North and Vulpe 2010).
High-throughput transcriptomics (HTT) that can measure global gene expressions in cellular systems is a transformative phase of HTS to allow large-scale screening of chemicals (Dai 2018; Harrill et al. 2019; Mav et al. 2018; Zhang et al. 2018). Concentration-dependent HTT is a powerful approach to characterize chemical concentration-dependent responses of comprehensive biological pathways, which can be used to derive transcriptional point-of-departure (PODT) as potency thresholds and estimate putative molecular mechanisms (Farmahin et al. 2017; Thomas et al. 2007, 2013). Our previous works have developed and applied HTT platforms in both human cells and zebrafish embryos for the screening of environmental chemicals (Fang et al. 2020; Wang et al. 2018, 2020a, b; Xia et al. 2017, 2020b; Zhang et al. 2018, 2020). US EPA ToxCast Phase III recently demonstrated the ability of HTT to yield PODT aligned with previous ToxCast high-throughput in vitro screening assays (Harrill et al. 2021; Ramaiahgari et al. 2019). However, omics has a longstanding limitation in translating molecular perturbations into apical toxicity, which relies heavily on expert-based interpretation (Herwig et al. 2016).
The adverse outcome pathway (AOP) framework uses a modular structure to organize existing knowledge concerning the linkage between a molecular-level perturbation of a biological system and the adverse outcome(s) that the perturbation by chemicals may cause (Doering et al. 2018). An AOP describes a consecutive chain of key events (KEs) that link a molecular initiating event (MIE) to an adverse outcome (AO) across different levels of biological organization (Ankley et al. 2010). Efforts have been made to incorporate omics data into the description of KEs (Labib et al. 2015; Martens et al. 2018; Nymark et al. 2018). Genome annotations (e.g., Gene Ontology or GO terms) can be manually curated and assigned to each KE in the AOPs, and these gene-KE assignments can be used to link AOPs with omics data. Current applications of using omics data to decipher AOP events have been limited to only a few specific AOPs (Nymark et al. 2018). However, the application of deciphering omics data in the context of the entire AOP knowledge base is scarce.
Assemblages of AOPs that share one or more KEs can be interconnected to generate an AOP network (Knapen et al. 2018). AOP network can capture and extend the diversity of biological perturbations that may occur in different species and target organs (Villeneuve et al. 2018). For instance, multiple MIEs may contribute to the same AO within an AOP network. Importantly, incorporation of concentration or time-response data into the AOP network can help define the potency values of KEs that can be ranked to identify KEs sequentially affected across dose and time, which can quantitatively inform the most plausibly impacted pathways by particular chemicals (Pollesch et al. 2019; Song et al. 2020). To date, the use of the AOP network for chemical screening is still in its infancy.
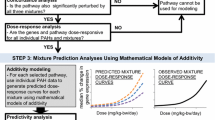
Here, an AOP network-based approach for chemical risk assessment using HTT is proposed (Fig. 13.1). Briefly, concentration-dependent HTT was conducted to profile the concentration–response of thousands of genes and pathways, followed by deriving PODT values to estimate the transcriptional potency of chemicals. The perturbed GO terms identified by HTT were matched to AOP KEs to visualize the specific patterns of perturbed AOP network by chemicals, which can be used to examine the KEs that were perturbed in a concentration-dependent manner as potential key molecular mechanisms. Lastly, the AOP network profiles of chemicals can be used for chemical classification/read-across. In the following part of this chapter, we elaborate on the details of conducting, analyzing and applying AOP network-based chemical risk assessment using high-throughput omics data.

Workflow for AOP network-based analysis of high-throughput transcriptomics (HTT) data (Note AOP, adverse outcome pathway; POD, point of departure; KE, key event; MIE, molecular initiating event; AO, adverse outcome; KER, key event relationship)
Pipeline for AOP-Network Chemical Risk Assessment by HTT
HTT technologies can be categorized into three types by the breadth of genes measured, including quantitative reverse transcription-polymerase chain reaction (qRT-PCR) arrays for dozens of genes, e.g., ToxChip array (Crump et al. 2016; Xia et al. 2020a; Zahaby et al. 2021), reduced transcriptomics using targeted RNA-Seq on customized panels of hundreds or thousands of genes, e.g., L1000 (Subramanian et al. 2017), S1500+ (Mav et al. 2018) and whole transcriptome analysis using microarrays or RNA-Seq (Yeakley et al. 2017). The reduced transcriptomics approach has been proposed as a cost-effective proxy to whole transcriptome analysis, and it is based on the principle that a small set of key genes can represent the expression of whole gene networks (Bild et al. 2006; Dai 2018). Currently, reduced transcriptomics have been primarily developed for testing chemicals on human cells (Xia et al. 2017) and zebrafish embryos (Wang et al. 2018). The following sections describe the design, experimental setup and analysis of reduced transcriptomics for chemical screening.
Design of Reduced Gene Panels
The reduced gene panel should include maximal coverage of biological pathways and toxicologically relevant genes (Mav et al. 2018; Zhang et al. 2018). To cover comprehensive biological pathways, a data-driven approach is employed by selecting genes from biological pathway databases (e.g., GO and KEGG), followed by bioinformatic network analysis to extract key genes that play central roles (e.g., biologically-connected to a majority of genes in a pathway). To select toxicologically relevant genes, a toxicological-driven approach is used by retrieving genes from existing toxicology testing databases, such as the gene-based endpoints tested in ToxCast and the genes associated with KEs in AOP Wiki. All the genes collected from data-driven and toxicological-driven approaches are merged as a reduced gene panel, followed by validation of the coverage of biological pathways (e.g., whether >95% pathways were covered by at least three genes of the reduced gene panel). Furthermore, the ability of the reduced gene panel to represent the whole transcriptomics profiles should be evaluated by using exiting whole transcriptomics data (e.g., comparing the performance for clustering different samples by using gene expressions of reduced genes or whole genome genes). Finally, the panel of reduced genes is submitted to the targeted RNA-Seq platform to design primers available for next-generation sequencing. For instance, in the case of amplicon sequencing technology, thousands of primers for the reduced genes can be synthesized, followed by mixing in one tube. The mixed primers are optimized for multiplex PCR amplification and the following transcriptomics sequencing (Li et al. 2015).
Chemicals and Biologicals
Stock solutions of chemicals are prepared in vehicles (e.g. dimethyl sulfoxide (DMSO), methanol or water) and stored at −80 °C until used. First, the cytotoxic concentrations of chemicals need to be determined to ensure that the highest concentrations used for HTT testing do not induce secondary effects in cellular systems (e.g., apoptosis and cytotoxicity). For human cells, cell viability assays are commonly conducted on cells exposed to chemicals for 24 h. For zebrafish, embryonic toxicity assays are conducted on zebrafish embryos exposed to chemicals for 120 h.
Concentration-Dependent HTT Experiment
HTT conducted in a broad concentration-dependent manner is necessary to characterize comprehensive concentration–response of genes and pathways for quantitative estimation of the potency values of chemicals (Fig. 13.1a) (Farmahin et al. 2017; Thomas et al. 2007, 2013). Serial dilutions (e.g., 5x and 10x) of chemicals in six to ten concentrations were used to expose cells/embryos for a short time (e.g., 6 h, 12 h or 24 h). After exposure, cells were collected for transcriptomics analysis using RNA-Seq technology. Currently, a targeted RNA-Seq platform (e.g., amplicon-seq technology) (Xia et al. 2017) is the primary approach for the HTT experiment. US EPA has been applying Tempo-Seq technology that can directly measure gene expression using cell lysis without RNA extraction (Bushel et al. 2018, 2020). Targeted RNA-Seq has advantages in measuring mRNA expression with extremely low input RNA, in pg or ng level, by hybridization and sequencing with highly specific detector oligos. After sequencing, the read counts of each gene can be automatically generated during genome annotation and quality control (removing low-quality/expressed genes). A list of expressed genes in a matrix format (each column represents a sample and each row represents a gene) is used in further analysis.
Concentration–Response Modeling Analysis
Concentration–response modeling analysis is used to characterize the concentration–response curves of genes to derive gene-level POD, followed by deriving pathway-level POD and transcriptional potency of chemicals (Fig. 13.1b). To avoid confusion with the AOP pathway, in this section, the pathways are referred to as molecular pathways in the pathway databases (e.g., GO and KEGG). First, gene-level concentration–response modeling is conducted to identify the trends of genes perturbed at different toxicant concentrations. The concentration–response models of genes are fitted into two types, including monotonic and non-monotonic models (Smetanová et al. 2015). For the monotonic model, linear and non-linear curves are the two major graphs that represent the concentration–response relationships of genes. For the non-monotonic model, gaussian and log-transformed curves are commonly used for concentration–response modeling. Multiple tools can be used for concentration–response modeling, including R language-based packages, e.g., drc (Ritz et al. 2015) and DRomics (Larras et al. 2018) and benchmark dose software developed by US EPA (Yang et al. 2007). The best-fitted concentration–response model is assigned to each gene according to the pre-set criteria (e.g. the model that has the lowest Akaike’s Information Criterion (AIC) value). Then the gene-level POD values are derived from the best fitted concentration–response model by plotting toxicant concentration against the benchmark response (e.g., the mean value + 3 times the standard derivation of vehicle controls of that gene; 1.5-fold changes).
Pathway-level POD values can be calculated by matching genes to pathways (GO terms or KEGG pathways), which is important to translate gene-level changes to higher biological level information of perturbed pathways. The criterium to define a pathway as a potentially perturbed pathway is that the number of genes matched to that pathway is at least three. The three-gene cut-off has been widely used because three is the minimum number required to define the mean value and SD for a pathway (Thomas et al. 2007). The matched pathways can be ranked by pathway-level POD values to identify potentially sensitive pathways perturbed by chemical exposure. Moreover, the biological potency of chemicals can be estimated from the pathway-based profiles, such as the concentrations against the top 20% perturbed pathways, and the concentrations against the top number of perturbed pathways (Farmahin et al. 2017). The transcriptional potency of chemicals can relatively well distinguish their low and high potency. However, the accuracy of transcriptional potency needs further validation, for example, by using an in vivo experiment. In addition, the ability of transcriptional potency to estimate the potency of apical endpoints is unclear. Some studies have demonstrated the consistency between transcriptional potency and apical potency by in vivo testing (e.g., liver transcriptomics vs liver histopathology in rats) (Thomas et al. 2012). However, a limited number of studies have evaluated the ability of in vitro omics-derived potency to predict in vivo-based potency. The in vitro omics-derived potency value is usually lower than in vivo-based potency because the molecular-level responses happen earlier and at lower concentrations than apical effects. Whether there exists an uncertainty factor between in vitro omics-derived potency and in vivo-based potency remains to be established.
AOP Network Analysis
AOP network analysis can integrate the above pathway-level information into a systematic and topological framework. Briefly, the identified pathways (e.g., GO terms) can be matched to KEs via GO-KE annotation database in AOP Wiki using the R package AOP (Burgoon 2015). KEs that meet the concentration-dependent ranks in a connected path in the AOP network indicate a potential key molecular mechanism of that chemical (Fig. 13.1c). The concentration-dependent KEs may be present in an existing AOP, and may also indicate a putative AOP that was not previously curated in AOP Wiki database. The identified putative AOP may support the investigation of new potential AOPs, which need further evaluation and validation such as using in vitro or in vivo bioassays. Moreover, rich network-based information can be extracted, such as the central KEs that are connected to the largest number of matched KEs, and the longest path in AOP network that has the largest number of matched KEs. For instance, the longest path between an MIE and AO may suggest the most detailed mechanistic description (Pollesch et al. 2019). Lastly, the weight of edges and nodes in the AOP network can be assigned according to AOP Wiki database. This is due to the fact that, during the development of AOPs, the KEs or key event relationships (KERs) are based on sources with different weight-of-evidence (e.g., in vivo or in vitro studies; validated or not). By assigning weight values, the identified critical paths in AOP network can be grouped into different levels of confidence.
The AOP network profiles of chemicals can be integrated into ToxPi for chemical classification. ToxPi is an interactive graphical user interface developed by the US EPA, which is a powerful tool for visual interpretation and transparent weight-of-evidence analysis (Reif et al. 2013). The AOP network profiles can be deconstructed into a matrix of all possible linear paths and each path can be scored by the mean values of POD of matched KEs (if there are no matched KEs, the score of that path is set as ‘NA’, i.e., ‘not available’). The scored AOP network profiles can be submitted to ToxPi to generate a pie plot. If multiple cell lines are used for HTT testing, the AOP network profiles from multiple cell lines can be used to generate an integrated plot (Fig. 13.1d) (Grimm et al. 2016). The AOP network-based ToxPi profiles can be used for clustering analysis. Chemicals clustered into the same group are assumed to present similar molecular modes of action, which can be used to guide future evaluations of the toxicity of these chemicals.
Examples of AOP Network-Based Chemical Screening
Cross-Species Comparison of Transcriptomic Dose–Response of Short-Chain Chlorinated Paraffins
Short-chain chlorinated paraffins (SCCPs) have attracted ever-increasing attention because of their toxicological potential in humans and wildlife at environmentally relevant doses. However, limited information is available regarding mechanistic differences across species in terms of the biological pathways that are impacted by SCCP exposure. Here, a concentration-dependent reduced human transcriptome (RHT) analysis approach was used to evaluate fifteen SCCPs in HepG2 cells and compared with our previous results using a reduced zebrafish transcriptome (RZT) analysis approach in zebrafish embryos (ZFE) (Xia et al. 2021). Generally, SCCPs induced a broader suite of biological pathways in ZFE than HepG2, while all fifteen SCCPs were more potent in HepG2 compared to ZFE. Despite these general differences, the transcriptional potency of SCCPs in both model systems showed a significant linear relationship (p = 0.0017, r2 = 0.57). C10H14Cl8 was the most potent SCCP, while C10H17Cl5 was the least potent in both ZFE and HepG2. An AOP network-based analysis demonstrated model-specific responses, such as xenobiotic metabolism that may be mediated by different nuclear receptor-mediated pathways between HepG2 (e.g., activation of the constitutive androstane receptor or CAR and the aryl hydrocarbon receptor or AhR) and ZFE (e.g., activation of the pregnane X receptor or PXR) (Fig. 13.2a). Moreover, induced transcriptional changes in ZFE associated with pathways and molecular initiating events (e.g., activation of nicotinic acetylcholine receptor or nAChR) suggest that SCCPs may disrupt neural development processes (Fig. 13.2b). This study demonstrated that the cross-model comparison of concentration-dependent transcriptomics represents a promising approach to assess and prioritize SCCPs.


A demonstration of AOP networks of SCCPs covered by both reduced human transcriptome (RHT) and reduced zebrafish transcriptome (RZT) analysis (Xia et al. 2021). a 2,3,4,5,6,7,8,9-Octachlorodecane (C10H14Cl8) b 1,2,5,6,9,10-Hexachlorodecane (C10H16Cl6). Red, green and white dots represent molecular initiating events (MIE), adverse outcomes (AO) and key events (KE), respectively. Dots encircled with pink, light blue and orange represent AOP-associated events matched by only RHT, only RZT, and both RHT and RZT, respectively. Edges in yellow represent an extracted path starting from an MIE to an AO. AhR, aryl hydrocarbon receptor. nAChR, nicotinic acetylcholine receptor
High-Throughput Transcriptomics Screening of Chemicals with Various Known Modes of Action Using Human Cells
The current application of HTT is limited due to the lack of systematic evaluation of its performance for chemical screening. Concentration-dependent transcriptomics of 32 chemicals with different modes of action (i.e. genotoxicity, endocrine disruption and metabolic activity) (Table 13.1) were conducted on HepG2 and MCF7 cells using RHT approach. The pathway-based profiles identified by RHT were used to group chemicals generally consistent with their known modes of action. Comparison of the RHT and ToxCast in vitro bioassay profiles demonstrated that POD values of the pathways associated with DNA repair (i.e., GO:0000729 and GO:0006287) had a significant linear correlation (p-value < 0.05). Furthermore, the identified pathways were matched to KEs in an AOP network that arranged biological pathways into topological structures, which showed that RHT and ToxCast indicated different potentially perturbed KEs (DNA damage-associated events for RHT; hormone disruption-associated events for ToxCast). For concentration-dependently perturbed KEs, RHT and ToxCast both identified paths starting from MIE of AhR for most chemicals (Fig. 13.3), while RHT specifically identified paths involved in cellular stress processes, including suppression of constitutive androstane receptor, activation of phosphatidylinositol-3 kinase (PI3K), an increase in insulin and activation of transcription factor NRF2 (nuclear factor erythroid-2-related factor 2). The ToxPi clustering of AOP network-based profiles for chemicals tested in both HepG2 and MCF7 cells showed distinct groups of chemicals with different known modes of action (genotoxic, endocrine disrupting and metabolic activity) (Fig. 13.4). This study demonstrated that RHT can provide a novel approach for chemical screening and classification, which can be complementary to in vitro bioassays.

Plotting of an AOP network-extracted pathway starting from MIE (AhR activation-associated KE) to AO (apoptosis-associated KE) that was matched by both RHT and ToxCast for 13 chemicals. Dots encircled with pink, light blue and orange represent AOP-associated events matched by only RHT, only ToxCast and both by RHT and ToxCast, respectively. Red, green and white dots represent molecular initiating events (MIE), adverse outcomes (AO) and key events (KE), respectively. For an explanation of abbreviations see Table 13.1

ToxPi clustering plot of scores for AOP network-based profiles of chemicals tested by high-throughput transcriptomics (HTT) in HepG2 and MCF7 cells. The sectors in yellow and red colors represent the ToxPi scores of HTT pathway profiles in HepG2 and MCF7, respectively. For an explanation of abbreviations of chemicals see Table 13.1
Challenges and Perspectives
The emergent need for new approach methodologies (NAMs) has been proposed to accelerate the pace of chemical risk assessment (Harrill et al. 2019; Kavlock et al. 2018). NAMs aim to provide efficient large-scale information on chemical hazards by HTS alternatives to animal testing approaches, including a battery of high-throughput in vitro bioassays and computational models for prioritization and screening of chemicals (EPA 2018). HTT performed in in vitro test systems is considered a novel type of NAM, but its application is still in its infancy. The validity of omics-identified KEs to explain potential apical effects needs to be evaluated by comparing them to traditional in vitro bioassays or in vivo assays. Besides, multiple omics approaches (e.g., proteomics and metabolomics) are encouraged to investigate the AOP network profiles of chemicals across broad biological levels, which also requires the development of a system for biological analysis pipeline to interpret the multiple-omics data. Moreover, omics analysis at different time points is needed to identify KEs that become perturbed early. The time-dependent results can be combined with concentration-dependent omics data to obtain three-dimension patterns of AOP network profiles. The AOP Wiki database is still being updated and newly developed AOPs are being added, which will improve the quality of the currently available AOP network.
Lastly, HTT is proposed to be incorporated into the tiered testing of chemicals (Fig. 13.5). First, concentration-dependent HTT is used to profile the disrupted biological pathways and transcriptional potency of a group of reference chemicals with well-known toxicity information, which helps to establish a reference database. Then, the effects of emerging chemicals for which there is no toxicological data are analyzed using HTT, and the results are compared to the HTT-based reference database to prioritize chemicals with similar profiles to reference chemicals. The prioritized chemicals are further evaluated by a set of in vitro bioassays that cover multiple endpoints, including hepatotoxicity, immunotoxicity, developmental toxicity, mitochondrial toxicity, and developmental neurotoxicity as proposed by the US EPA (Patlewicz et al. 2019). The chemicals that are validated to be able to induce in vitro toxicity are submitted to risk assessment by evaluating the margin of exposure (Buesen et al. 2017), which is finally applied to chemical management. In conclusion, HTT can provide a novel approach for NAM-based chemical risk assessment.

Tiered approach for HTT-based chemical risk assessment
References
Ankley GT, Bennett RS, Erickson RJ et al (2010) Adverse outcome pathways: a conceptual framework to support ecotoxicology research and risk assessment. Environ Toxicol Chem 29(3):730–741
Bild AH, Yao G, Chang JT et al (2006) Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature 439(7074):353–357
Buesen R, Chorley BN, da Silva LB et al (2017) Applying ‘omics technologies in chemicals risk assessment: report of an ECETOC workshop. Regul Toxicol Pharmacol 91:S3–S13
Burgoon LD (2015) AOP: a biocondcutor package for sufficient causal analysis in pathway-based screening of drugs and chemicals for adversity. BioRxiv 029694
Bushel PR, Ferguson SS, Ramaiahgari SC et al (2020) Comparison of normalization methods for analysis of TempO-Seq targeted RNA sequencing data. Front Genet 11:594
Bushel PR, Paules RS, Auerbach SS (2018) A comparison of the TempO-Seq S1500+ platform to RNA-Seq and microarray using rat liver mode of action samples. Front Genet 9:485
Collins FS, Gray GM, Bucher JR (2008) Transforming environmental health protection. Sci 319(5865):906
Crump D, Farhat A, Chiu S et al (2016) Use of a novel double-crested cormorant ToxChip PCR array and the EROD assay to determine effects of environmental contaminants in primary hepatocytes. Environ Sci Technol 50(6):3265–3274
Dai J (2018) Reduced transcriptomic approach for screening and prediction of chemical toxicity. Chem Res Toxicol 31(7):532–533
Dix DJ, Houck KA, Martin MT et al (2007) The ToxCast program for prioritizing toxicity testing of environmental chemicals. Toxicol Sci 95(1):5–12
Doering JA, Wiseman S, Giesy JP et al (2018) A cross-species quantitative adverse outcome pathway for activation of the aryl hydrocarbon receptor leading to early life stage mortality in birds and fishes. Environ Sci Technol 52(13):7524–7533
ECHA (2016) Guidance on information requirements and chemical safety assessment Chapter R.7b: endpoint specific guidance. Available at: https://echa.europa.eu/guidance-documents/guidance-on-information-requirements-and-chemical-safety-assessment
EPA (2016) About the TSCA chemical substance inventory. Available at: https://www.epa.gov/tsca-inventory/about-tsca-chemical-substance-inventory
EPA (2018) Strategic plan to promote the development and implementation of alternative test methods within the TSCA program. USEPA, Washington, DC
Fang W, Peng Y, Yan L et al (2020) A tiered approach for screening and assessment of environmental mixtures by omics and in vitro assays. Environ Sci Technol 54(12):7430–7439
Farmahin R, Williams A, Kuo B et al (2017) Recommended approaches in the application of toxicogenomics to derive points of departure for chemical risk assessment. Arch Toxicol 91(5):2045–2065
Gaytán BD, Vulpe CD (2014) Functional toxicology: tools to advance the future of toxicity testing. Front Genet 5:110
Grimm FA, Iwata Y, Sirenko O et al (2016) A chemical–biological similarity-based grouping of complex substances as a prototype approach for evaluating chemical alternatives. Green Chem 18(16):4407–4419
Harrill JA, Everett LJ, Haggard DE et al (2021) High-throughput transcriptomics platform for screening environmental chemicals. Toxicol Sci 181(1):68–89
Harrill JA, Shah I, Setzer RW et al (2019) Considerations for strategic use of high-throughput transcriptomics chemical screening data in regulatory decisions. Curr Opin Toxicol 15:64–75
Herwig R, Hardt C, Lienhard M et al (2016) Analyzing and interpreting genome data at the network level with ConsensusPathDB. Nat Protoc 11(10):1889
Huang R, Xia M, Sakamuru S et al (2018) Expanding biological space coverage enhances the prediction of drug adverse effects in human using in vitro activity profiles. Sci Rep 8(1):1–12
Kavlock RJ, Bahadori T, Barton-Maclaren TS et al (2018) Accelerating the pace of chemical risk assessment. Chem Res Toxicol 31(5):287–290
Knapen D, Angrish MM, Fortin MC et al (2018) Adverse outcome pathway networks I: development and applications. Environ Toxicol Chem 37(6):1723–1733
Labib S, Williams A, Yauk CL et al (2015) Nano-risk science: application of toxicogenomics in an adverse outcome pathway framework for risk assessment of multi-walled carbon nanotubes. Part Fibre Toxicol 13(1):15
Larras F, Billoir E, Baillard V et al (2018) DRomics: a turnkey tool to support the use of the dose–response framework for Omics data in ecological risk assessment. Environ Sci Technol 52(24):14461–14468
Li W, Turner A, Aggarwal P et al (2015) Comprehensive evaluation of AmpliSeq transcriptome, a novel targeted whole transcriptome RNA sequencing methodology for global gene expression analysis. BMC Genom 16(1):1–13
Martens M, Verbruggen T, Nymark P et al (2018) Introducing WikiPathways as a data-source to support adverse outcome pathways for regulatory risk assessment of chemicals and nanomaterials. Front Genet 9:661
Mav D, Shah RR, Howard BE et al (2018) A hybrid gene selection approach to create the S1500+ targeted gene sets for use in high-throughput transcriptomics. PLoS ONE 13(2):e0191105
MEE (2013) List of existing chemical substances in China. Available at: https://www.mee.gov.cn/ywgz/gtfwyhxpgl/hxphjgl/wzml/
North M, Vulpe CD (2010) Functional toxicogenomics: mechanism-centered toxicology. Int J Mol Sci 11(12):4796–4813
Nymark P, Rieswijk L, Ehrhart F et al (2018) A data fusion pipeline for generating and enriching adverse outcome pathway descriptions. Toxicol Sci 162(1):264–275
Patlewicz G, Richard AM, Williams AJ et al (2019) A chemical category-based prioritization approach for selecting 75 per-and polyfluoroalkyl substances (PFAS) for tiered toxicity and toxicokinetic testing. Environ Health Perspect 127(01):014501
Pollesch NL, Villeneuve DL, O’Brien JM (2019) Extracting and benchmarking emerging adverse outcome pathway knowledge. Toxicol Sci 168(2):349–364
Ramaiahgari SC, Auerbach SS, Saddler TO et al (2019) The power of resolution: contextualized understanding of biological responses to liver injury chemicals using high-throughput transcriptomics and benchmark concentration modeling. Toxicol Sci 169(2):553–566
Reif DM, Sypa M, Lock EF et al (2013) ToxPi GUI: an interactive visualization tool for transparent integration of data from diverse sources of evidence. Bioinformatics 29(3):402–403
Richard AM, Judson RS, Houck KA et al (2016) ToxCast chemical landscape: paving the road to 21st century toxicology. Chem Res Toxicol 29(8):1225–1251
Ritz C, Baty F, Streibig JC et al (2015) Dose-response analysis using R. PLoS ONE 10(12):e0146021
Smetanová S, Riedl J, Zitzkat D et al (2015) High-throughput concentration–response analysis for omics datasets. Environ Toxicol Chem 34(9):2167–2180
Song Y, Xie L, Lee Y et al (2020) De novo development of a quantitative adverse outcome pathway (qAOP) network for ultraviolet B (UVB) radiation using targeted laboratory tests and automated data mining. Environ Sci Technol 54(20):13147–13156
Subramanian A, Narayan R, Corsello SM et al (2017) A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell 171(6):1437–1452
Thomas RS, Allen BC, Nong A et al (2007) A method to integrate benchmark dose estimates with genomic data to assess the functional effects of chemical exposure. Toxicol Sci 98(1):240–248
Thomas RS, Clewell HJ III, Allen BC et al (2012) Integrating pathway-based transcriptomic data into quantitative chemical risk assessment: a five chemical case study. Mutat Res 746(2):135–143
Thomas RS, Wesselkamper SC, Wang NCY et al (2013) Temporal concordance between apical and transcriptional points of departure for chemical risk assessment. Toxicol Sci 134(1):180–194
Villeneuve DL, Angrish MM, Fortin MC et al (2018) Adverse outcome pathway networks II: network analytics. Environ Toxicol Chem 37(6):1734–1748
Wang P, Wang Z, Xia P et al (2020a) Concentration-dependent transcriptome of zebrafish embryo for environmental chemical assessment. Chemosphere 245:125632
Wang P, Xia P, Wang Z et al (2020b) Evidence-based assessment on environmental mixture using a concentration-dependent transcriptomics approach. Environ Pollut 265:114839
Wang P, Xia P, Yang J et al (2018) A reduced transcriptome approach to assess environmental toxicants using zebrafish embryo test. Environ Sci Technol 52(2):821–830
Xia P, Crump D, Chiu S et al (2020a) Toxicogenomic assessment of complex chemical signatures in double-crested cormorant embryos from variably contaminated great lakes sites. Environ Sci Technol 54(12):7504–7512
Xia P, Peng Y, Fang W et al (2021) Cross-model comparison of transcriptomic dose-response of short-chain chlorinated paraffins. Environ Sci Technol 55(12):8149–8158
Xia P, Zhang H, Peng Y et al (2020b) Pathway-based assessment of single chemicals and mixtures by a high-throughput transcriptomics approach. Environ Int 136:105455
Xia P, Zhang X, Zhang H et al (2017) Benchmarking water quality from wastewater to drinking waters using reduced transcriptome of human cells. Environ Sci Technol 51(16):9318–9326
Yang L, Allen BC, Thomas RS (2007) BMDExpress: a software tool for the benchmark dose analyses of genomic data. BMC Genomics 8(1):1–8
Yeakley JM, Shepard PJ, Goyena DE et al (2017) A trichostatin A expression signature identified by TempO-Seq targeted whole transcriptome profiling. PLoS ONE 12(5):e0178302
Zahaby Y, Xia P, Crump D et al (2021) ToxChip PCR arrays for two arctic-breeding seabirds: applications for regional environmental assessments. Environ Sci Technol 55(11):7521–7530
Zhang X, Wang P, Xia P (2020) Dose-dependent transcriptomic approach for mechanistic screening in chemical risk assessment. In: Jiang G, Li X (eds) A new paradigm for environmental chemistry and toxicology. Springer, Singapore, pp 33–56
Zhang X, Xia P, Wang P et al (2018) Omics advances in ecotoxicology. Environ Sci Technol 52(7):3842–3851
Acknowledgements
This work is supported by the National Natural Science Foundation of China (Grant No. 41977206), Jiangsu Environmental Protection Research Fund (2018001) and National Key Research and Development Program of China (2019YFC1804004).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Xia, P., Wang, P., Fang, W., Zhang, X. (2022). Adverse Outcome Pathway Network-Based Chemical Risk Assessment Using High-Throughput Transcriptomics. In: Guo, LH., Mortimer, M. (eds) Advances in Toxicology and Risk Assessment of Nanomaterials and Emerging Contaminants. Springer, Singapore. https://doi.org/10.1007/978-981-16-9116-4_13
Download citation
DOI: https://doi.org/10.1007/978-981-16-9116-4_13
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-16-9115-7
Online ISBN: 978-981-16-9116-4
eBook Packages: Earth and Environmental ScienceEarth and Environmental Science (R0)