
Abstract
Reducing the bandgap of TiO2 to an optimal range by doping with other atoms is a very effective method having great potential in solar energy applications. The fundamental effect of structural changes upon the electronic structure of doped semiconducting TiO2 is very much important to explore an effective doping configuration. A detailed computational study is therefore, required, to better understand the effect of different doped materials upon the structural, electronic and optical properties. We systematically study the carbon doped anatase TiO2, using first-principle density functional theory (DFT) calculations to determine the effect of carbon concentration on the structural, electronic and optical properties of C doped TiO2. We optimize the geometric structures of carbon doped anatase TiO2 using generalized gradient approximation (GGA) with Perdew–Burke–Ernzerhof (PBE) potential (GGA+PBE). Furthermore, to study optical and electronic properties, we perform the calculation with GGA+Hubbard potential (GGA+U) exchange correlational functional. The results confirm that GGA+PBE produce more accurate results for the geometric structure of undoped and carbon doped TiO2, closer to the experimental results. Moreover, GGA+U functional presents the bandgap energies of doped and undoped systems that are close to the actual values at lower computational cost. As a result of carbon doping, new impurity levels have been introduced into the bandgap region of TiO2 that leads to the decrease of bandgap energy. Narrowing the bandgap resulting in shift of the optical absorption edge to the visible region that might enhance the photocatalytic activity.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others


Keywords
1 Introduction
Energy crises and climate changes are the main reasons for researchers to direct their attention towards green renewable energy resources. Hydrogen production through solar photocatalytic water-splitting technology is assumed to be a clean renewable energy technology, in order to deal with energy crisis and environmental challenges in future [1]. Semiconductor photocatalysts has an important role as they are providing an easy way for utilization of solar energy. Since Fujishima’s report in 1972, Titanium dioxide (TiO2), has received intense attention as a promising photocatalytic material due to their excellent physical and chemical properties [1, 2] for photocatalyst applications for photocatalyst applications. But the wide bandgap of TiO2 (valued 3.2 eV for anatase) restricts their usefulness to ultraviolet radiation (< 387 nm) mainly, which is only about 3–5% of the total sunlight. The utilization of sun light is therefore playing a very minor role, to activate this metal oxide to promote oxidation and reduction surface reactions [3]. To improve the photocatalysis of TiO2, it is necessary to know that how to increase optical absorptivity coefficient of TiO2 into the visible range (43% of solar spectrum), having wavelength range (380–700 nm) therefore, it is one of the most important subjects for researchers now a days. To modify their light absorptivity and electronic structure, incorporation of the impurities into the crystal structure, is the most feasible techniques. Considerable research work is going on in order to minimize their band gap to an optimal position. Many approaches such as doping of metal cations and non-metal anions have been attempted. TiO2 doping with nonmetals including N, C, F, B and S etc. [4, 5] modify the electronic structure and hence properties by creating new impurity energy state in the host crystal structure [4, 6].
Carbon is considered an appropriate dopant element for TiO2 in order to minimize their band gap, and therefore, improving the performance of the photocatalyst by increasing the visible light absorption. Some theoretical and experimental studies have been performed for carbon doped TiO2 photocatalyst [6, 7]. Recently, Computational simulation methods, like density functional theory (DFT), is attracting more attention as a designing tool, as it is predicting many characteristics of the photocatalytic materials and to choose the suitable one for the practical application [8]. It is because, these methods provide a possibility to conduct research on complicated structures without expensive and/or dangerous experiments. Previously, most of the researchers conducted DFT calculations for the band gap of carbon doped TiO2 conventional DFT methods. These methods underestimated the value of the bandgap for strongly corelated systems like transition metal oxides. To get more effective results for the C-doping of TiO2 systems, we need a detailed information as the mechanism is not clear too much yet [6, 9].
The goal of this work is to understand the effect of carbon concentration on the structural, optical and electronic properties of C doped TiO2 by using DFT calculations. For structural properties GGA+PBE and for electronic and optical properties, GGA+Hubbard potential (GGA+U) approach were applied, respectively. It is notable to highlight that GGA+U approach produces better result for band gap energy calculation in comparison with conventional DFT methods. We investigated different structural, electronic and optical properties like lattice constants, cell parameters, band structure, density of states, energy band gap and absorption of the undoped and doped model using the concept of substitutional doped model. Geometry optimization of the undoped and the different doped TiO2 model (2.08 and 4.17 at.% concentration of C), have been done with plane-wave pseudopotential approach. Geometries of the different undoped and doped structures calculated accurately and, the band gaps energies and density of states calculation also agree very well with the experimental ones.
2 Computational Methodology
In this study, all the theoretical calculations and designing of computational models have been performed with Cambridge sequential total energy package (CASTEP), one of the modules in Materials Studio program. It is based on the total energy plane-wave pseudopotential DFT approach [10]. Firstly, geometries of the structures have been optimized within the framework of GGA+PBE exchange correlational functional. The structural properties were calculated by applying GGA+PBE potential. For a system like transition metal oxides, having strongly localized orbitals (d- and f-orbitals), the bandgap energy may not be calculated too much accurately with GGA+PBE functionals [11]. Generally, the band gaps of transition metal oxides calculated with GGA+PBE functional are usually underestimated (approximately 50% smaller than the experimental value), because of the reason that it describes the strong Coulomb interactions less accurately between electrons of the d- and/or f-orbitals [12]. Therefore, in the present research work, we prefer the GGA+U scheme, for electronic and optical properties calculations, to produce more accurate results. GGA+U method produces more accurate results because of the reason that it considers the electronic interactions in d-shell, as strongly correlated interaction by introducing an on-site Coulomb’s correction term and hence describe the systems with localized d and f electrons also in a good way [12, 13]. In this method, Hubbard-potential is used in order to obtain the value of bandgap energy, which is much closer to experimental value. Different values of the Hubbard parameter (U) may be used for the valence Ti 3d electrons and the energy gap values calculated accordingly. In our present case, we choose the value of U = 8.47 eV for localized electrons and the result obtained for band gap energy is much closer to the experimental ones. This method also provides a good balance between the accuracy of the calculated results and the available computational resources.
To construct C-doped systems, we built the 48-atoms 2×2×1 anatase TiO2 supercell models. Simulation of the doped systems was conducted for the said 2×2×1 optimized supercell, containing different carbon atoms at the substituted position of the titanium atoms, as shown in Fig. 1b, c. Optimization of the atomic positions and cell parameters, were performed with residual forces below 0.03 eV/Å. To accelerate the computational runs, ultra-soft pseudopotential functional were applied to the electronic configuration of Ti; 3s2, 3p6, 3d2, 4s2, C; 2s2 2p2 and O; 2s2, 2p4. A 3× 3×2 k-point mesh was used for numerical sampling integration, over the Brillouin zone, with Monkhorst–Pack method. The cutoff energy for the valence electrons wavefunction was 300 eV. The self-consistent field (SCF) iterations convergence threshold equal to 2×10–6 eV/atom. The convergence parameters chosen with the energy values 2×10–5 eV/atom and the stress on the atom limited below 0.1 GPa. The force was set as 0.03 eV/Å and the atomic displacement less than 2×10–3 Å.

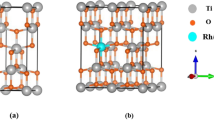
DFT simulation images for a anatase TiO2 unit cell in real apace and b in reciprocal space
3 Result and Discussion
3.1 Structural Optimization
Anatase TiO2 has a tetragonal structure with lattice parameters a = 3.776 Å, b = 3.776 Å and c = 9.486 Å. Each Titanium atom is bonded to its four nearest oxygen neighbors. Figure 1a, b shows the computationally generated DFT simulation images for anatase TiO2 unit cell in real apace and in reciprocal space.
Figure 2a shows a 2×2×1 supercell for pure anatase TiO2 including 16 titanium and 32 oxygen atoms. Figure 2b, c represents carbon-doped TiO2 supercell with 2.08 at.% (1 Ti replaced by C) and 4.17 at.% (2 Ti replaced by C), respectively. The concept of a supercell is introduced in order to effectively represent a bulk solid even if we simulate this very small block (supercell). Also, if the system contains a non-periodic entity such as a vacancy, we can apply the supercell approach by including the vacancy into the supercell [11].

(2×2×1) anatase TiO2 supercell models with different concentrations of C a 0 at.%, b 2.08 at.%, c 4.17 at.%
To analyze the structural modifications as a result of carbon dopants incorporation, the optimized lattice parameters of the undoped and C-doped TiO2 structures is then compared. For undoped system, the optimized bulk lattice parameters are a = b = 3.797 A˚ and c = 9.549 Å, that agrees well with the experimental values of the anatase having values, a = b = 3.785 Å and c = 9.502 Å [12]. It also indicating that our calculation method is more reliable. For carbon doped system having 2.08% doping concentration, the optimized bulk lattice parameters are a = b = 3.786 Å and c = 9.497 Å, while for 4.17% doped system, a = b = 3.772 Å, and c = 9.585 Å. The bulk lattice parameters ‘a’ and ‘b’ of the doped systems decreases, whereas the value of ‘c’ tends to increase after carbon doping. In doped models, the average bond length of Ti–O is shorter, then the C–O bond lengths, while volume of the doped system reduces, as the carbon concentration increases [13]. The contraction in the overall volume is due to the differences in the radii of the host and dopant atoms. Table 1 summarized the different structure optimization parameters, obtained from of simulation of undoped and doped systems.
3.2 Electronic Structure/Properties
Addition of the dopant carbon atoms to the TiO2 lattice causes the structural distortions and as well as modifying the electronic structures of semiconducting materials also. New impurity states introduced in the bandgap causing changes in the electronic structure [14]. The electronic structure and their related properties have great impact on the performance of TiO2. In the present study, the DFT+U (Hubbard coefficient) approach has been adopted to find out the electronic and optical properties. DFT+U approach improves the bandgap energy calculation partially more reliable as discussed earlier [11]. Firstly, the bandgap energy for pure anatase TiO2, has been found out with simple GGA functionals, which is 1.91 eV and much smaller than the experimental value of 3.20 eV. We then calculated the bandgap with GGA+U functionals. With U value equal to 8.47 eV, for Titanium 3d states, the calculated bandgap is 3.13 eV and is closer to the experimental value [16]. Generally, valence band (VB) for pure anatase TiO2, has more O 2p states and less Ti 3d states, whereas the conduction band (CB) has more Ti 3d states with smaller number of O 2p states, as previously reported by many researchers [15, 16].
Figure 3 indicates the band structure of the undoped and carbon doped models. The zero-point energy is the Fermi energy level and the bandgap energy (Eg) for undoped anatase TiO2 is 3.13 eV, as shown in the band structure plot in Fig. 3a. At 2.08% C concentration, the bandgap decreases to 3.02 eV, due to the carbon-doped impurity states in the bandgap region. At 4.17 at.% as shown in Fig. 3b, the bandgap of C-doped TiO2 narrows to 2.52 eV as the conduction band minima (CBM) shifts toward the Fermi level continuously, which results in an effective reduction of the bandgap of TiO2. The result also indicates that the decrease in bandgap energy from 2.08 at.% to 4.17 at.% is more obvious than that from 0 to 2.08 at.%. By narrowing the bandgap, the excitation energy of the electrons in the valence band decreases as the electrons in the valence band excited to impurity states easily, and subsequently to the conduction band also [17]. As electron transition energy decreases in C-doped TiO2, the material will absorb more visible light as the redshift of the optical absorption edge occurs.

Electronic band structure of anatase TiO2 a 0 at.% (undoped) and b 4.17 at.%. Red-dashed line corresponds to the Fermi energy level
The density of states (DOS) graphs also confirming the reduction in the bandgap energy as shown in Fig. 4. The DOS graphs also showing small peaks appearing in the valence band (VB) region for carbon-doped models, that were not present in the undoped TiO2, and is due to the induction of carbon impurity states. Thus, the valence band was found to be more broaden as the concentration of incorporated carbon increases into TiO2. An increase in the bandwidth of the valence band (WVB) might benefit the hole mobility in VB [18]. In this way, the effect of both i.e. narrowing the bandgap and increase in the hole mobility can improve the photocatalytic performance of the system.

Density of states of TiO2 doped with 0 at.%, 2.08 at.% and 4.17 at.% of C
3.3 Optical Properties
Pure anatase TiO2 only absorbs ultraviolet light radiation due to wide bandgap energy, due to which, their application range is very limited. To shift the absorption edge to the longer wavelength range, red shift could be attributed to the bandgap reduction and is due to the introduction of impurity states in the bandgap of TiO2 [19, 20]. We plotted the optical absorption (UV–visible) spectra for undoped and C-doped models, as shown in Fig. 5. The absorption spectra show that undoped TiO2 mainly absorbs the UV radiation in the wavelength region between 200 and 360 nm, which agrees very well with previous theoretical calculations [21, 22]. After the C-doping of TiO2, the changes in electronic structure extend the solar absorption edge from the UV towards the longer wavelength visible region, resulting in the red-shift of the optical absorption edge. Compared with undoped TiO2, the doped model with 4.17 at.% C-doping concentration, the absorption band edge shifted up to 450 nm and thus resulting in red-shift of the absorption band edge. This redshift of the absorption edge is due to the bandgap narrowing, which reduces the energy required for electron excitation. It is to be noted that the C-doping not only resulting in red-shift of the optical spectra, but also improving the absorption visible light significantly.

The absorption plot for undoped and carbon doped anatase TiO2
4 Conclusions
In this study, we investigate the structural properties of undoped and carbon doped TiO2, through DFT calculations using GGA+PBE exchange correlational functional. A simple and efficient GGA+U technique is also used to find out the electronic and optical properties of anatase TiO2 by investigating the influence of different carbon doping concentrations. The calculated results imply that carbon concentration induces new impurity energy states in the band structure of TiO2 and higher concentrations of carbon effectively narrowing the bandgap of anatase TiO2 to absorb more visible light. The bandgap energy value for pure anatase TiO2, with the GGA+U technique (U = 8.47 eV), is around 3.13 eV and is in very close agreement with experimental value (3.20 eV). An increase in carbon impurities decreases the bandgap energy from 3.13 eV, for undoped TiO2 (0 at.% C), to 3.01 and 2.52 eV for 2.08 and 4.17% of carbon doping respectively. As a result of band gap reduction, the redshift in the optical absorption edge occurs, that improving the visible light absorption also. Therefore, we conclude that the carbon doping of TiO2 with a proper mechanism is a feasible method to improve the photocatalytic performance of TiO2 under solar light absorption.
References
Liu, Y., Liang, W., Zhang, W., Zhang, J., Han, P.: First principle study of Cu and N-doped anatase TiO2. Solid State Commun. 164, 27–31 (2013)
Fujishima, A., Honda, K.: Electrochemical photolysis of water at a semiconductor electrode. Nature 238(5358), 37–38 (1972)
Bashiri, R., Mohamed, N.M., Kait, C.F., Sufian, S.: Hydrogen production from water photosplitting using Cu/TiO2 nanoparticles: effect of hydrolysis rate and reaction medium. Int. J. Hydrogen Energy 40(18), 6021–6037 (2015)
Giannakas, A., Antonopoulou, M., Daikopoulos, C., Deligiannakis, Y., Konstantinou, I.: Characterization and catalytic performance of B-doped, B-N co-doped and B–N–F tri-doped TiO2 towards simultaneous Cr (VI) reduction and benzoic acid oxidation. Appl. Catal. B 184, 44–54 (2016)
Chen, H., Li, X., Wan, R., Kao-Walter, S., Lei, Y., Leng, C.: A DFT study on modification mechanism of (N, S) interstitial co-doped rutile TiO2. Chem. Phys. Lett. 695, 8–18 (2018)
Li, X., et al.: A DFT study on the modification mechanism of (Cr, C) co-doping for the electronic and optical properties of anatase TiO2. Comput. Mater. Sci. 129, 295–303 (2017)
Mohamed, M.A., et al.: Revealing the role of kapok fibre as bio-template for In-situ construction of C-doped g-C3N4@ C, N co-doped TiO2 core-shell heterojunction photocatalyst and its photocatalytic hydrogen production performance. Appl. Surf. Sci. 476, 205–220 (2019)
Würger, T., et al.: Adsorption of acetone on rutile TiO2: a DFT and FTIRS study. J. Phys. Chem. C 122(34), 19481–19490 (2018)
Shao, J., et al.: In situ synthesis of carbon-doped TiO2 single-crystal nanorods with a remarkably photocatalytic efficiency. Appl. Catal. B 209, 311–319 (2017)
Clark, S.J., et al.: First principles methods using CASTEP. Zeitschrift für Kristallographie-Crystalline Mater. 220(5/6), 567–570 (2005)
Lee, J.G.: Computational Materials Science: An Introduction. CRC Press (2016)
Li, X., Gao, H., Liu, G.: A LDA+ U study of the hybrid graphene/anatase TiO2 nanocomposites: Interfacial properties and visible light response. Comput. Theor. Chem. 1025, 30–34 (2013)
Zhang, H.-X., Zhu, Y.F., Zhao, M.: Interface charge transfer and enhanced visible light response of graphene/anatase TiO2 (110) systems with and without oxygen vacancy: a DFT+ U calculation. Appl. Surf. Sci. 420, 105–109 (2017)
Zou, Y., Shi, J.-W., Ma, D., Fan, Z., Lu, L., Niu, C.: In situ synthesis of C-doped TiO2@ g-C3N4 core-shell hollow nanospheres with enhanced visible-light photocatalytic activity for H2 evolution. Chem. Eng. J. 322, 435–444 (2017)
German, E., Faccio, R., Mombru, A.W.: Comparison of standard DFT and Hubbard-DFT methods in structural and electronic properties of TiO2 polymorphs and H-titanate ultrathin sheets for DSSC application. Appl. Surf. Sci. 428, 118–123 (2018)
Long, R., English, N.J.: Density functional theory studies of doping in titania. Mol. Simul. 36(7–8), 618–632 (2010)
Zhu, Z., Chen, J.-Y., Su, K.-Y., Wu, R.-J.: Efficient hydrogen production by water-splitting over Pt-deposited C-HS–TiO2 hollow spheres under visible light. J. Taiwan Inst. Chem. Eng. 60, 222–228 (2016)
Wu, H.-C., Li, S.-H., Lin, S.-W.: Effect of Fe concentration on Fe-doped anatase TiO2 from GGA. Int. J. Photoenergy 2012 (2012)
Zhang, J., et al.: Engineering the absorption and field enhancement properties of Au–TiO2 nanohybrids via whispering gallery mode resonances for photocatalytic water splitting. ACS Nano 10(4), 4496–4503 (2016)
Basu, K., et al.: 4.3 Highly Stable Photoelectrochemical Cells for Hydrogen Production Using, I dedicate this thesis to my family, p. 111 (2018)
Yan, J., et al.: Heterostructured CoO/3D-TiO2 nanorod arrays for photoelectrochemical water splitting hydrogen production. J. Solid-State Electrochem. 21(2), 455–461 (2017)
Kandiel, T.A., Takanabe, K.: Solvent-induced deposition of Cu–Ga–In–S nanocrystals onto a titanium dioxide surface for visible-light-driven photocatalytic hydrogen production. Appl. Catal. B 184, 264–269 (2016)
Acknowledgements
This work is supported financially by Universiti Teknologi PETRONAS (UTP) and ministry of higher education (MOHE) Malaysia, Fundamental Research Grant Scheme (FRGS) (Grant No. FRGS/1/2019/STG07/UTP/01/1).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this paper

Cite this paper
Mohamed, N.M., Ullah, F., Bashiri, R., Kait, C.F., Saheed, M.S.M., Shahid, M.U. (2021). Hubbard’s Modified Density Functional Theory Calculations for the Electronic Structure and Optical Properties of Carbon Doped Anatase TiO2. In: Abdul Karim, S.A., Abd Shukur, M.F., Fai Kait, C., Soleimani, H., Sakidin, H. (eds) Proceedings of the 6th International Conference on Fundamental and Applied Sciences. Springer Proceedings in Complexity. Springer, Singapore. https://doi.org/10.1007/978-981-16-4513-6_32
Download citation
DOI: https://doi.org/10.1007/978-981-16-4513-6_32
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-16-4512-9
Online ISBN: 978-981-16-4513-6
eBook Packages: Physics and AstronomyPhysics and Astronomy (R0)