
Abstract
Pituitary tumors are the most common neoplasm of sellar origin, comprising of 10–15% of all primary intracranial tumor. Pituitary adenoma (PA) is the commonest pituitary tumor which is further divided based on the size (microadenoma <1 cm and macroadenoma >1 cm) and secretory activity (functioning and nonfunctioning). Majority of the patients present with headache, visual disturbance, or sign and symptoms of hormonal dysfunction. Dynamic contrast enhanced magnetic resonance imaging (CEMRI) of the sella is the imaging of choice. Hormonal assessment is important and includes evaluation of thyroid stimulating hormone (TSH), free thyroxine (T4), adrenocorticotrophic hormone (ACTH), cortisol, prolactin (PRL), somatomedin C or insulin-like growth factor-1 (IGF-1), luteinising hormone (LH), follicle-stimulating hormone (FSH), and testosterone. Surgery with or without radiotherapy (RT) and medical therapy is the preferred treatment for most of the patients with PA. Radiotherapy is often given after incomplete resection of the tumor. Definitive RT alone is considered when surgery is not feasible or contraindicated.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Sellar and suprasellar regions contain several diverse and closely approximated tissues, abnormal growth of which may lead to several neoplastic and non-neoplastic lesions. Pituitary tumors are the most common cause of sellar-sprasellar mass and represent around 10–15% of primary intracranial tumors. A slight female preponderance is seen, with presentation commonly in the age group of 45–55 years. Majority of the pituitary tumors are benign. Pituitary adenoma (PA) is the commonest pituitary tumor accounting for around 10% of all intracranial tumors [1]. Pituitary carcinoma is rare. Classification of various pituitary gland tumors has been shown in Table 31.1 [2].
2 Pituitary Anatomy and Physiology
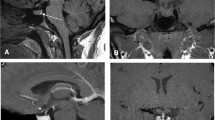
Normal pituitary gland (master gland) is a pea-sized structure, situated in the sella turcica of sphenoid bone. It has two different embryological origins. Adenohypophysis (anterior pituitary, composed of anterior and intermediate lobes) is derived from rathke’s pouch while neurohypophysis (posterior pituitary/ lobe) and pituitary stalk are derived from the diencephalon. Adenohypophysis is responsible for most of the endocrine function of pituitary gland. Anterior lobe produces prolactin (PRL), adrenocorticotropic hormone (ACTH), follicle-stimulating hormone (FSH), luteinising hormone (LH), growth hormone (GH), and thyroid-stimulating hormone (TSH) while intermediate lobe produces melanocyte-stimulating hormone (MSH). The posterior lobe stores and releases vasopressin and oxytocin produced by the hypothalamus. Pituitary stalk connects pituitary gland and hypothalamus forming hypothalamic-pituitary axis. The gland is closely related to the optic chiasma, anterior cerebral arteries, floor of the third ventricle and cavernous sinuses that contain internal carotid artery and cranial nerves III, IV, V, and VI. Tumor extension to these regions can lead to cranial nerves palsies and visual defects (Fig. 31.1).

Depicts the Pituitary gland and structures in relation to it
3 Classification of Pituitary Adenoma
PA can be broadly classified based on their secretory activity and size. Based on the size, these can be devided into microadenoma (<1 cm) and macroadenoma (>1 cm). The term Pico adenoma is used for tumors less than 3 mm in size [3]. Tumors more than 4 cm are termed as giant adenomas (6–10% of all pituitary adenoma) [4]. Based on their secretory activity, PA can be classified as functioning PA (FPA) or non-functioning PA(NFPA). Prolactinoma is the commonest PA followed by NFPA. Other functioning adenoma are GH-secreting adenoma, ACTH-secreting adenoma, and TSH-secreting adenoma.
4 Etiology and Pathogenesis
Pathogenesis of pituitary tumors is not well understood. Most pituitary tumors are believed to be sporadic in origin; however, syndromic association is seen in approximately 5% of the patients. The genes responsible for syndromic association include (1) multiple endocrine neoplasia I (MEN I), (2) cyclin dependent kinase inhibitor 1 B (CDKN), (3) protein kinase, cAMP dependent regulatory 1A (PRKAR)—Carney complex (CNC)—characterised by endocrine overactivity, schwannomas, skin pigmentation, and myxoma, (4) aryl hydrocarbon receptor-interacting protein (AIP) [5]. No causal relationship has been found with smoking, alcoholism, or trauma.
5 Clinical Presentation
Clinical features depend on tumor extent and secretory status. FPA are usually detected on evaluation for features of hyperpituitarism (acromegaly, cushing’s syndrome, galactorrhea) and are usually microadenomas. NFPA are either detected incidentally during imaging for unrelated reasons or present with pressure symptoms over normal pituitary or over visual pathway manifesting as vision loss that can include bitemporal or homonymous hemianopia, supero-inferior field effects and central scotoma, and uncommonly cranial nerve dysfunction and seizures. Signs and symptoms of hypopituitarism such as hypogonadism, adrenal insufficiency and rarely diabetes insipidus may also be present. Rarely patients can present with pituitary apoplexy.
6 Evaluation
Detailed physical examination, laboratory assessment and imaging should be done to assess visual, endocrinological and neurological abnormalities. Laboratory investigation includes complete blood count, blood chemistry and urine examination. Hormonal analysis depends on patient’s age, gender and symptoms and may include TSH, free thyroxine (T4), ACTH, cortisol, PRL, somatomedin C or insulin-like growth factor-1 (IGF-1), LH, FSH, and testosterone. Dynamic contrast enhanced T1 weighted magnetic resonance imaging (MRI) of the brain or sella is the imaging modality of choice. Thin slices (1 mm) should be obtained in the coronal/axial and sagittal planes to define tumor extent and its relation to surrounding structures (optic pathway, cavernous sinuses, and anterior cerebral vessels). PA is generally hypointense on T1W MRI and hyperintense on T2W MRI. Contrast enhanced computed tomography (CT) scan of the brain can be done when there is a suspicion of bone destruction or MRI is not feasible. Role of functional imaging, i.e., somatostatin receptor scintigraphy or fluorodeoxyglucose (FDG) and 11 C-methionine (11 C-Met) positron emission tomography (PET) has been explored by some groups but its utility remains unknown [6]. Susceptibility weighted imaging (SWI) can be used to detect any hemorrhagic component.
7 Classification
Grade | Criteria |
|---|---|
A. Hardy’s classification of pituitary adenoma [7] | |
Grade I | Microadenoma (<10 mm diameter) |
Grade II | Macroadenoma (>10 mm diameter), within Sella |
Grade III | >10 mm, focal sellar erosions, outside the Sella |
Grade IV | Infiltrates sphenoid and cavernous sinuses, compresses optic nerves, cranial nerves and/or invades adjacent brain parenchyma |
B. Knosp’s grading of parasellar invasion [8] | |
Grade 0 | Adenoma does not extend beyond the medial carotid line |
Grade 1 | Adenoma extends beyond the medial line, but doesn’t reach the median line, or intercarotid line |
Grade 2 | Adenoma extends to the lateral aspects of internal carotid artery (ICA) |
Grade 3a | Adenoma extends beyond lateral aspects of ICA and into the superior cavermons sinus compartment |
Grade 3b | Adenoma extends beyond lateral aspects of ICA and into the inferior cavenous sinus compartment |
Grade 4 | Adenoma totally wraps around the intracavernous carotid artery |
8 Treatment
Goals of the treatment are to control over-secretion of hormones, to correct hormone deficiencies and to reduce tumor size while minimising potential morbidity such as hypopituitarism and vision changes. Three main treatment modalities are surgery, medical management, and radiotherapy (RT). Observation is an option for selected group of patients.
Observation
NFPA patients without hypopituitarism, mass effect or visual abnormalities can be considered for observation. Follow-up clinical examination and MRI should be done to rule out progression as approximately 10% of microadenoma and 23% of macroadenoma may progress without treatment.
Surgery
Surgery with or without RT remains the first line of treatment for most of the PA including apoplexy and prolactinoma resistant to medical treatment. Debulking of the tumor provides histological diagnosis, rapidly reduces mass effect and hormone hypersecretion, arrests progression of neurological deficits and vision loss. Surgical management yields a long-term control of 80–90% in microadenoma and 25–50% in macroadenomas. Transsphenoidal surgery (TSS) approach is appropiate for majority of the patients; however, if the tumor extends to frontal or temporal lobe, transcranial route is preferred. Complications of TSS approach such as perioperative mortality, central diabetes insipidus, cerebrospinal fluid (CSF) rhinorrhoea, and meningitis occur only in 1–3% of the patients [9].
Medical Therapy
Medical management alone, or in combination with surgery or RT, is important as hormonal excess or deficiency both can have a detrimental effect on quality of life of a patient.
Type of hormonal therapy depends on tumor type, laboratory investigations and clinical condition of the patient. Often, hydrocortisone and cortisone acetate are used for glucocorticoid replacement. For correction of hypothyroidism, L-thyroxine is used. Gonadal steroids oestrogen and progestin are used for women and testosterone for men, if deficient. For prolactinoma, medical management is the first treatment of choice that includes therapy with a dopamine agonist, such as bromocriptine or cabergoline. These drugs rapidly normalise PRL levels and reduce tumor size in 80–90% of patients [10]. For GH-secreting adenoma or acromegaly, medical treatment can be used as an adjunct if patient is not a candidate for surgery or has not responded to surgery or RT. Most commonly used drugs are first generation somatostatin analogues (octreotide and lanreotide) administered once a month which inhibit GH production or antagonise its action [11]. Normalisation of IGF-1 and GH at 1 year can be achieved in 25% and 38% patients, respectively [12]. Cabergoline can be added if patients do not show adequate response to somatostatin analogues. Pasireotide, a multisomatostatin receptor (SSTR)-ligand has a longer half-life than first generation somatostatin analogues and is used in resistant cases [13]. GH receptor antagonist pegvisomant, in combination with somatostatin analogue has also been tested in placebo controlled trial in resistant cases which has shown to normalise IGF-1 levels in 90–97% of patients [14]. Pegvisomant monotherapy can increase the tumor size due to lack of negative feedback as it does not inhibit GH secretion. Other limitations of pegvisomant therapy are high cost and need for lifelong therapy.
For ACTH-secreting adenoma or cushing’s disease, medical treatment is considered for those who do not respond to surgery or RT. Steroidogenesis inhibitors, i.e., ketoconazole, aminoglutethimide, metyrapone, mitotane, and etomidate are used for hormonal normalisation. Ketoconazole is a well tolerated which results in control of cortisol excess in up to 70% of the patients [15]. Pasireotide has also shown biochemical control in approximately 25% of the patients [16]. Glucocorticoid receptor inhibitor mifepristone may reduce the systemic side effects of cortisol [17]. Bilateral adrenalectomy may be considered in severe refractory disease [18].
TSH-secreting adenomas are exceedingly rare, and thus, treatment paradigm is not well defined. Surgery with or without RT is typically the first line of the treatment and medical treatment is considered only in resistant cases. Somatostatin analogues such as octreotide have been shown to normalise TSH levels in approximately 79% of patients [19]. A more detailed description of medical management is provided in the chapter on neuro-endocrinology.
Radiotherapy (RT)
Typical indications of RT include incomplete resection of a large NFPA, cavernous sinus invasion, recurrent or progressive tumors, inoperable symptomatic or potentially symptomatic tumors, or refractory secreting tumors. RT techniques for PA have evolved from conventional two-dimensional (2D) to three-dimensional (3D) treatment, intensity modulated radiotherapy and stereotactic treatments, with either fractionated stereotactic radiotherapy (SRT) or stereotactic radiosurgery (SRS). Sophisticated RT techniques can provide a steeper dose gradient thus delivering higher dose to the tumor and minimising the dose to surrounding critical structures, thereby allowing for maximum tumor control and lesser long-term RT-induced side effects. Figure 31.2 depicts IMRT dose color wash for a prescription dose of 54 Gy in 30 fractions planned for large residual functioning pituitary adenoma. Figure 31.3 depicts dose volume histogram (DVH) of optic chiasm for the same patient.

Dose color wash of radiotherapy planning in a 30 years old patient with large residual FPA. CT/MR fusion was done for RT planning. Gross target volume (GTV) was delineated as seen on imaging and a planning target volume (PTV) margin of 3 mm was given. No margin was given for clinical target volume (CTV). She received 54 Gy/30 fractions by IMRT technique. Figure 31.2 is showing 95% dose color wash of PTV

Dose volume histogram (DVH) of optic chiasm. Dmax-54 Gy, 0.02 cc of optic chiasm received 54 Gy
Normalisation of excess hormone production is slow after RT as compared to surgery and medical therapy.
Fractionated RT vs SRS
As the dose gradient is sharper in SRS, the likelihood of radiation injury to surrounding non-contiguous normal structures may be lesser. Response to RT depends on the type of the tumor. In general, either form of RT controls the tumor in approximately 90% of the patients at 5 years. Evidence so far does not distinguish between these two approaches based on efficacy or safety. A meta-analysis of eight studies has shown comparable results with fractionated RT and SRS [20]. Few retrospective studies have suggested that compared to fractionated RT, SRS is more effective in functioning adenoma, while both are equi-efficacious in NFPA [21]. SRS, for now, only offers the time advantage of treatment in a single session against a longer 5 weeks course of fractionated RT, and may be considered as treatment of choice in tumors situated atleast 2 mm away from optic structures, or cavernous sinus involvement or persistent high serum hormone levels postoperatively. Fractionated RT is preferred over SRS when optic pathway is involved or closely apposed, or for large lesions (diameter >3 cm) where risk of radio necrosis with SRS is high.
SRS
Optic apparatus is the most important dose-limiting structure in RT planning of PA. To minimise the risk of radiation related optic neuropathy, a minimum gap of 2 mm between the tumor and optic apparatus is crucial. SRS which is generally delivered as a single fraction of high dose, may result in more rapid biochemical control. However, 2–5 fractions of SRS can be used if there is a higher probability of radiation damage to nearby structures. In functioning PA, where the treatment goal is to achieve radiological control as well as to reduce endocrine oversecretion, a higher dose of SRS (16–25 Gy) is generally required as compared to NFPA where a SRS dose of 13–14 Gy is adequate for long-term tumor control and acceptable neurological toxicity. (20) In a multicenter retrospective analysis of NFPA, SRS dose of <12 Gy was associated with poor outcome while a dose of >20 Gy did not provide additional tumor control as compared to doses between 12 and 20 Gy [22]. SRS is effective in achieving 5-year tumor control rate of 90–100% in NFPA.
Hypopituitarism is the most commonly reported toxicity after SRS with a 5-year incidence of increasing pituitary deficits ranging from 10% to 40% in different studies [23]. Risk of radiation related optic neuropathy is less than 3% if the maximum point dose to optic apparatus is 8–10 Gy, but significantly increases after 12 Gy [24]. Other cranial nerve deficits resulting from RT are rarely reported.
Fractionated RT
Adaquete RT doses for NFPA and functioning PA are 45-50.4 Gy and 50.4-54 Gy respectively at 1.8 Gy/fraction [25]. Studies have shown that local control with conventional RT can vary from 80% to 94% and 75% to 90% at 10 and 20 years respectively for both functioning and nonfunctioning adenoma [26]. Normalisation of GH/IGF-1 occurs in approximately 50% of the patients after 5–10 years of RT for acromegaly [27]. Biochemical control in cushing disease can be seen in 50–80% patients within 3 years of fractionated RT [28]. RT in prolactinomas is indicated in those who do not respond to medical therapy. Around one third of the patients with prolactinomas respond to RT with normalisation of PRL after 5–10 years [7].
Most reported late toxicity of RT is hypopituitarism. Following conventional RT at doses of 45–50.4 Gy at 1.8 Gy per fraction, hypopituitarism can be seen in 30–50% of the patients after 5 years [28]. The reported incidence of vision loss due to radiation-induced optic neuropathy varies from 0 to 6% (<2% at RT dose of 45 Gy in 1.8 Gy daily fraction) [28]. Increased incidence of cerebrovascular accidents (CVA) has also been reported. Conventional RT for PA can increase the risk of CVA by four fold [8]. Radiation induced second cancer is another long-term complication of RT.
Target delieation in fractionated RT- Postoperative T1w MRI images are extremely helpful in target delineation. T2w MRI which is used to assess brain parenchyma infiltration and perilesion edema is generally not used. Any lesion which is visible on MRI is demarketed as GTV. Margin for CTV is generally not given except in the intracavernous portion of aggressive adenoma to encompass potential microscopic spread. A 3-5 mm margin is added for PTV for setup uncertainities. Reference-Minniti, G., Osti, M.F. & Niyazi, M. Target delineation and optimal radiosurgical dose for pituitary tumors. Radiat Oncol 11, 135 (2016) Particle radiation has been also been successfully used in the treatment of pituitary adenomas [29]. However, the potential advantage of proton therapy over photon remains unclear.
Pituitary Incidentaloma (PI)
It is defined as an unexpected pituitary lesion (adenoma and cystic lesion), identified on brain imaging for an unrelated reason. Although most of the pituitary incidentalomas are <1 cm in size and nonfunctioning, hormonal deficiencies or excess hormone secretion can also seen in these patients [30]. Studies have shown that the prevalence of microincidentaloma on MRI for an unrelated reason varies from 10% to 38% while that for macroincidentaloma is far less and varies from 0.16% to 0.3% [31]. Series of untreated PRL secreting microadenoma has shown that conversion rate to PRL macroadenoma was less than 5%. Endocrine society recommends that all patients of incidentaloma should be investigated for the presence of signs and symptoms of pituitary hormone dysfunction irrespective of their size [32]. All patients with macroincidentalomas or large solid microincidentalomas with a diameter of >6 mm should undergo laboratory investigation for the presence of hypopituitarism. For microincidentalomas of size >6 mm, assessment of PRL and IGF-I alone is sufficient [32]. Hormonal assessment is not required for cystic microincidentalomas and solid lesions <5 mm in diameter. Visual campimetry is necessary when the tumor extends till optic chiasm [33]. Treatment is always indicated for functioning macroadenomas depending on the type of tumor.
Pituitary Carcinoma
It is a rare entity comprising of <0.5% of all pituitary tumors. Most pituitary carcinomas originate from adenohypophysis and secrete PRL and ACTH. Clinical presentation resembles that of PA. These tumors have a high propensity for CSF and systemic metastases. Mitotic count/Ki-67 index indicates aggressiveness of the tumor. Despite aggressive treatment with surgery and RT, overall prognosis remains poor with mean survival of 1.9 years. Studies have suggested that temozolomide (TMZ) is effective when used as a first-line chemotherapeutic agent after surgery [34]. Concurrent RT with TMZ may further improve the outcome.
Pituicytoma is a rare low-grade glial neoplasm which originates from neurohypophysis, and can be misdiagnosed preoperatively as a pituitary adenoma [35].
Pituitary Blastoma
It is a rare aggressive malignant neoplasm of the pituitary gland mostly seen in children [36]. Pituitary blastomas are generally seen as a part of pleuropulmonary blastoma (PPB)-familial tumor, which is caused by heterozygous germline mutations in the DICER1gene.
Pituitary Apoplexy
This is an uncommon clinical condition that occurs because of acute hemorrhage or infarction in the pituitary gland, often in PA patients. It affects 0.2–0.6% of the general population and 2–12% of patients with pituitary adenomas [37]. Headache is the most common symptom followed by vision loss, hypopituitarism, nausea, diplopia and altered consciousness [37].
Patients may present in acute (within 24–72 h of symptom onset) or subacute setting. The classic triad of acute pituitary apoplexy constitutes (1) severe, sudden-onset headache (“Worst headache of life!”), (2) acute hypopituitarism (addisonian crisis, hyponatremia), and (3) visual disturbances. If imaging shows acute hemorrhage or infarction a diagnosis of pituitary apoplexy should be made. Acute pituitary apoplexy is a medical emergency. A rapid treatment with hydrocortisone is often lifesaving. Once patient is medically stable, urgent resection of the hemorrhagic pituitary tumor should be done.
Several studies have compared conservative approach with surgical management in pituitary apoplexy. A systematic review has demonstrated a significantly higher recovery rate for ocular palsy and visual field in surgically treated patients, but no significant difference was seen in visual acuity or pituitary function recovery [38]. A large, multi-institution data registry (PASTOR—Pituitary Apoplexy Surgical Timing & Outcomes Registry) is prospectively analysing observation versus surgery in acute or sub-acute onset pituitary apoplexy. At present, urgency of surgical treatment depends on symptom severity and duration. Urgent surgery is the mainstay for the treatment of acute pituitary apoplexy, especially in those with vision loss. However, in milder cases, a conservative approach can be preferred.
9 Follow-up
Patients are followed up with clinical evaluation to see any change in pretreatment visual or hormonal symptoms. Hormonal evaluation is done 3 months after treatment and then at least once in 6 months. Ophthalmology evaluation (visual acuity, fields, fundus, oculomotor evaluation) should be conducted every 6 months for those with baseline abnormalities and every year in those who had normal vision at baseline. First follow-up MRI is generally recommended at 3–6 months after treatment and then every year for 5 years, and subsequently every 2–3 years in absence of progression.
10 Conclusions
Pituitary tumors are a common cause of morbidity and often require multidisciplinary management. Observation, surgery, medical management, and radiotherapy are treatment options that are tailored according to the clinical circumstances. With suitable therapy, long-term control with good quality of life is feasible for over 90% of the patients. Long-term follow-up with periodic imaging, hormonal and vision assessments is required to assess disease control and treatment sequelae.
References
Elston MS, McDonald KL, Clifton-Bligh RJ, Robinson BG. Familial pituitary tumor syndromes. Nat Rev Endocrinol. 2009;5(8):453–61.
Mete O, Lopes MB. Overview of the 2017 WHO classification of pituitary tumors. Endocr Pathol. 2017;28(3):228–43.
Bonneville J-F, Bonneville F, Cattin F. Magnetic resonance imaging of pituitary adenomas. Eur Radiol. 2005;15(3):543–8.
Iglesias P, Rodríguez Berrocal V, Díez JJ. Giant pituitary adenoma: histological types, clinical features and therapeutic approaches. Endocrine. 2018;61(3):407–21.
Crabtree JS, Scacheri PC, Ward JM, Garrett-Beal L, Emmert-Buck MR, Edgemon KA, et al. A mouse model of multiple endocrine neoplasia, type 1, develops multiple endocrine tumors. Proc Natl Acad Sci U S A. 2001;98(3):1118–23.
Bashari WA, Senanayake R, Fernández-Pombo A, Gillett D, Koulouri O, Powlson AS, et al. Modern imaging of pituitary adenomas. Best Pract Res Clin Endocrinol Metab. 2019;33(2):101278.
Tsagarakis S, Grossman A, Plowman PN, Jones AE, Touzel R, Rees LH, et al. Megavoltage pituitary irradiation in the management of prolactinomas: long-term follow-up. Clin Endocrinol. 1991;34(5):399–406.
van Varsseveld NC, van Bunderen CC, Ubachs DHH, Franken AAM, Koppeschaar HPF, van der Lely AJ, et al. Cerebrovascular events, secondary intracranial tumors, and mortality after radiotherapy for nonfunctioning pituitary adenomas: a subanalysis from the Dutch National Registry of Growth Hormone Treatment in Adults. J Clin Endocrinol Metab. 2015;100(3):1104–12.
Jane JA, Catalino MP, Laws ER. Surgical treatment of pituitary adenomas. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, Dungan K, Grossman A, et al., editors. Endotext [internet]. South Dartmouth, MA: MDText.com, Inc.; 2000. Available from: http://www.ncbi.nlm.nih.gov/books/NBK278983/.
Gillam MP, Molitch ME, Lombardi G, Colao A. Advances in the treatment of prolactinomas. Endocr Rev. 2006;27(5):485–534.
Mehta GU, Lonser RR. Management of hormone-secreting pituitary adenomas. Neuro Oncol. 2017;19(6):762–73.
Melmed S, Cook D, Schopohl J, Goth MI, Lam KSL, Marek J. Rapid and sustained reduction of serum growth hormone and insulin-like growth factor-1 in patients with acromegaly receiving lanreotide Autogel therapy: a randomized, placebo-controlled, multicenter study with a 52 week open extension. Pituitary. 2010;13(1):18–28.
Grasso LFS, Auriemma RS, Pivonello R, Colao A. Somatostatin analogs, cabergoline and pegvisomant: comparing the efficacy of medical treatment for acromegaly. Expert Rev Endocrinol Metab. 2017;12(1):73–85.
Buchfelder M, van der Lely A-J, Biller BMK, Webb SM, Brue T, Strasburger CJ, et al. Long-term treatment with pegvisomant: observations from 2090 acromegaly patients in ACROSTUDY. Eur J Endocrinol. 2018;179(6):419–27.
Schteingart DE. Drugs in the medical treatment of Cushing’s syndrome. Expert Opin Emerg Drugs. 2009;14(4):661–71.
Colao A, Petersenn S, Newell-Price J, Findling JW, Gu F, Maldonado M, et al. A 12-month phase 3 study of pasireotide in Cushing’s disease. N Engl J Med. 2012;366(10):914–24.
Fleseriu M, Findling JW, Koch CA, Schlaffer S-M, Buchfelder M, Gross C. Changes in plasma ACTH levels and corticotroph tumor size in patients with Cushing’s disease during long-term treatment with the glucocorticoid receptor antagonist mifepristone. J Clin Endocrinol Metab. 2014;99(10):3718–27.
Nagesser SK, van Seters AP, Kievit J, Hermans J, Krans HM, van de Velde CJ. Long-term results of total adrenalectomy for Cushing’s disease. World J Surg. 2000;24(1):108–13.
Beck-Peccoz P, Brucker-Davis F, Persani L, Smallridge RC, Weintraub BD. Thyrotropin-secreting pituitary tumors. Endocr Rev. 1996;17(6):610–38.
Li X, Li Y, Cao Y, Li P, Liang B, Sun J, et al. Safety and efficacy of fractionated stereotactic radiotherapy and stereotactic radiosurgery for treatment of pituitary adenomas: a systematic review and meta-analysis. J Neurol Sci. 2017;372:110–6.
Scheick S, Amdur RJ, Kirwan JM, Morris CG, Mendenhall WM, Roper S, et al. Long-term outcome after fractionated radiotherapy for pituitary adenoma: the curse of the secretory tumor. Am J Clin Oncol. 2016;39(1):49–54.
Sheehan JP, Starke RM, Mathieu D, Young B, Sneed PK, Chiang VL, et al. Gamma Knife radiosurgery for the management of nonfunctioning pituitary adenomas: a multicenter study. J Neurosurg. 2013;119(2):446–56.
Minniti G, Flickinger J. The risk/benefit ratio of radiotherapy in pituitary tumors. Best Pract Res Clin Endocrinol Metab. 2020;33(2):101269. Available from: https://pubmed.ncbi.nlm.nih.gov/31053487/?from_term=The+risk%2Fbenefit+ratio+of+radiotherapy+in+pituitary+tumors&from_pos=1.
Pollock BE, Link MJ, Leavitt JA, Stafford SL. Dose-volume analysis of radiation-induced optic neuropathy after single-fraction stereotactic radiosurgery. Neurosurgery. 2014;75(4):456–60. discussion 460
Loeffler JS, Shih HA. Radiation therapy in the management of pituitary adenomas. J Clin Endocrinol Metab. 2011;96(7):1992–2003.
Sebastian P, Balakrishnan R, Yadav B, John S. Outcome of radiotherapy for pituitary adenomas. Rep Pract Oncol Radiother J Gt Cancer Cent Poznan Pol Soc Radiat Oncol. 2016;21(5):466–72.
Biermasz NR, Dulken HV, Roelfsema F. Postoperative radiotherapy in acromegaly is effective in reducing GH concentration to safe levels. Clin Endocrinol. 2000;53(3):321–7.
Minniti G, Osti M, Jaffrain-Rea ML, Esposito V, Cantore G, Maurizi Enrici R. Long-term follow-up results of postoperative radiation therapy for Cushing’s disease. J Neuro-Oncol. 2007;84(1):79–84.
Wattson DA, Tanguturi SK, Spiegel DY, Niemierko A, Biller BMK, Nachtigall LB, et al. Outcomes of proton therapy for patients with functional pituitary adenomas. Int J Radiat Oncol Biol Phys. 2014;90(3):532–9.
Orija IB, Weil RJ, Hamrahian AH. Pituitary incidentaloma. Best Pract Res Clin Endocrinol Metab. 2012;26(1):47–68.
Vasilev V, Rostomyan L, Daly AF, Potorac I, Zacharieva S, Bonneville J-F, et al. Management Of Endocrine Disease: pituitary “incidentaloma”: neuroradiological assessment and differential diagnosis. Eur J Endocrinol. 2016;175(4):R171–84.
Boguszewski CL, de Castro Musolino NR, Kasuki L. Management of pituitary incidentaloma. Best Pract Res Clin Endocrinol Metab. 2019;33(2):101268.
Chanson P, Raverot G, Castinetti F, Cortet-Rudelli C, Galland F, Salenave S, et al. Management of clinically non-functioning pituitary adenoma. Ann Endocrinol. 2015;76(3):239–47.
McCormack A, Dekkers OM, Petersenn S, Popovic V, Trouillas J, Raverot G, et al. Treatment of aggressive pituitary tumours and carcinomas: results of a European Society of Endocrinology (ESE) survey 2016. Eur J Endocrinol. 2018;178(3):265–76.
El Hussein S, Vincentelli C. Pituicytoma: review of commonalities and distinguishing features among TTF-1 positive tumors of the central nervous system. Ann Diagn Pathol. 2017;29:57–61.
Scheithauer BW, Horvath E, Abel TW, Robital Y, Park S-H, Osamura RY, et al. Pituitary blastoma: a unique embryonal tumor. Pituitary. 2012;15(3):365–73.
Briet C, Salenave S, Bonneville J-F, Laws ER, Chanson P. Pituitary apoplexy. Endocr Rev. 2015;36(6):622–45.
Tu M, Lu Q, Zhu P, Zheng W. Surgical versus non-surgical treatment for pituitary apoplexy: a systematic review and meta-analysis. J Neurol Sci. 2016;370:258–62.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Madan, R., Yadav, A., Goyal, S. (2021). Pituitary Tumors: Diagnosis and Management. In: Mallick, S., Giridhar, P., Rath, G.K. (eds) Evidence based practice in Neuro-oncology. Springer, Singapore. https://doi.org/10.1007/978-981-16-2659-3_31
Download citation
DOI: https://doi.org/10.1007/978-981-16-2659-3_31
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-16-2658-6
Online ISBN: 978-981-16-2659-3
eBook Packages: MedicineMedicine (R0)