
Abstract
The hydrogenation of carbon dioxide (CO2) to formic acid (FA; HCOOH), a renewable hydrogen storage material, is a promising approach to realize to address climate change since the target product is valuable as commodity chemicals as well as a renewable hydrogen storage material. Unfortunately, the development of heterogeneous catalysts lags significantly over the homogeneous catalysts in spite of the obvious practical advantages of such materials. Herein, the state of the art in the exploitation of promising metal alloy nanoparticle catalysts designed by surface engineering with atomic precision for this targeted reaction is presented. Especially, isolated and electron-rich Pd atoms created with the aid of neighboring Ag atoms in Pd@Ag alloy NPs boost the electronegativity of the dissociated hydride species, which exhibit ten times higher activity than those provided by monometallic Pd/TiO2 based on equivalent quantities of surface Pd atoms. Moreover, the post-modification of PdAg alloy NPs supported on TiO2 was performed using metal–organic framework ZIF-8. The observed increase in catalytic activity is ascribed to the positive electronic effects on both the active metal centers and reactants, as well as an anchoring stabilization effect preventing the undesired agglomeration of NPs during the reaction. Furthermore, core–shell structured catalyst (ZIF-8@Pd1Ag2@ZIF-8) was developed by the encapsulation of PdAg NPs within the interface of ZIF-8.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
The chemical transformation of carbon dioxide (CO2) into synthetically valuable compounds is of great interest in industrial chemistry because this transformation not only reduces the emission of CO2, but also supplies valuable chemical and fuel resources [1]. The hydrogenation of CO2 to produce formic acid (FA; HCOOH), which is a liquid at room temperature and contains 4.4 wt% hydrogen, is a promising approach to establish FA as a renewable hydrogen storage material since the chemically stored H2 in the FA can be liberated in controllable fashion in the presence of appropriate catalysts even at room temperature [2,3,4,5,6]. Thus, the design of novel catalysts for CO2 hydrogenation to form FA is a crucial task in the realization of economical CO2-mediated hydrogen energy cycles. The gas phase hydrogenation of CO2 to produce formic acid has a positive free energy change [7].
However, this reaction proceeds more readily in aqueous solution (CO2 (aq) + H2 (aq) → HCOOH (aq), ΔG = −4 kJ mol−1). The reaction is typically performed with the addition of a weak base, such as a tertiary amine or alkali/alkaline earth bicarbonate, which shifts the thermodynamic equilibrium to the product side.
Significant progress has been made utilizing homogeneous complexes in basic media [8, 9]. However, the development of heterogeneous catalysts lags significantly behind that of homogeneous catalysts, in spite of their practical utility [10,11,12,13,14], and, to make matters worse, their use frequently requires a high catalyst concentration, organic solvents, and extremely high pressures.
Metal nanoparticle-based catalysts are gaining increasing attention to bridge the gap between mononuclear metal complexes and heterogeneous bulk catalysts because of their existence on borderline molecular states with discrete quantum energy levels [15]. Their large surface area-to-volume ratio allows effective utilization of expensive metals. The variation in size, composition, morphology, and supports significantly influence the catalytic activities. Additionally, the accurate control of the geometric and electronic effect of bimetallic nanoparticles, in which the architectural configuration of two metals is as random alloys, segregated or a core–shell structure, is a key technology in attaining superior catalytic performances to the monometallic counterparts [16]. The interplay of the neighboring different metals creates specific new catalytically active sites, which frequently enables the fine-tuning of the geometric and electronic properties originating from synergic alloying effects [17,18,19,20]. Moreover, the replacement of the precious noble metal nanoparticles with inexpensive metals contributes to the atomic economy [21, 22]. Thus, the successful synthesis of bimetallic nanoparticles with controllable size, shape, and composition plays a crucial role in designing highly functionalized catalysts. However, the insights into the promising design strategy as well as the additional elucidation of the catalytically active species in the supported metal nanoparticles are required.
In this section, the state of the art of the nanostructured alloy metal nanoparticle catalysts, especially core–shell-type catalyst, reported for CO2 hydrogenation to formic acid/formate is presented. The enhanced activity was demonstrated by the discussions based on kinetic and density functional theory (DFT) calculations.
2 Elucidating the Catalytically Active Species in Supported Pd@Ag Alloy Nanoparticles
The co-reduction of Pd and Ag precursors conventionally affords random PdAg alloy nanoparticles (PdAg/TiO2) because of the complete solid solubility and similar reduction potentials of Pd and Ag ions. The precise tuning of the surface composition of PdAg nanoparticles was performed to investigate the effect of surface-exposed active Pd atoms in alloy NPs. By applying a surface engineering approach via the successive reduction of metal precursors, Pd@Ag/TiO2 with a PdcoreAgshell structure and Ag@Pd/TiO2 with an AgcorePdshell structure were synthesized [23].
As shown in Fig. 9.1, high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images demonstrated highly dispersed PdAg alloy NPs with a narrow size distribution on the TiO2 support for all samples (Pd:Ag = 30:70), with mean particle diameters of ca. 3 nm. This value is similar to that obtained for the Pd/TiO2 (dave = 3.2 nm). Energy-dispersive X-ray spectroscopy (EDX) line analysis confirmed the successful surface engineering of the NPs. In the case of PdAg/TiO2, both Pd and Ag were situated on the same particles, confirming the formation of a random PdAg alloy. By contrast, Pd atoms were preferentially located in the core region, and the Ag atoms were situated in the shell region in the Pd@Ag/TiO2. Conversely, the Pd and Ag atoms had the opposite distribution for the Ag@Pd/TiO2.

TEM images of and elemental distributions along single NPs of a PdAg/TiO2, b Pd@Ag/TiO2, and c Ag@Pd/TiO2 as determined by cross-sectional EDX line profiling (Reproduced with permission from [23]. Copyright © 2018 American Chemical Society.)
In the XPS spectra, the Pd 3d peaks of all PdAg samples were shifted to lower binding energies than those of the Pd/TiO2, and this shift was decreased in the order of Pd@Ag/TiO2 > PdAg/TiO2 > Ag@Pd/TiO2. Thus, the Pd atoms in the PdAg NPs were obviously electron enriched by the charge transfer from Ag atoms owing to the net difference in ionization potential between the two metals (Pd: 8.34 eV, Ag: 7.57 eV). A similar tendency in the electronic state of the Pd species was observed in the FT-IR experiments using CO as a probe molecule. Monometallic Pd/TiO2 generated two distinct peaks assignable to the linear and bridging stretching vibrations of adsorbed CO at 2076 and 1943 cm–1, respectively. The contribution of bridging-type CO decreased as the Pd/Ag ratio was lowered, and was completely absent in the case of the Pd@Ag/TiO2, suggesting the isolation of Pd atoms. Additionally, linear-type CO was predominantly observed for all samples, and the peaks were gradually shifted to lower wavenumbers with decreases in the Pd/Ag ratio.
Figure 9.2 shows the comparison of catalytic activity in the CO2 hydrogenation. The Pd@Ag/TiO2 exhibited an elevated TON (2,496) based on the total quantities of Pd employed despite the low density of surface-exposed Pd atoms. As expected, a maximum TON value of 14839 was obtained from the Pd@Ag/TiO2 based on the quantity of surface Pd atoms, which was determined by pulsed CO adsorption measurements. This TON value is more than ten times higher than that of the monometallic Pd/TiO2. Moreover, a good correlation between the TON based on surface Pd atoms and the Pd 3d5/2 binding energy determined by XPS analysis is evidently observed (Fig. 9.2b).

a Comparison of the catalytic activities of a series of supported PdAg catalysts with different surface compositions and Pd/TiO2 during CO2 hydrogenation. b Relationship between the TON for CO2 hydrogenation based on surface-exposed Pd atoms (as determined by CO pulse adsorption) and the Pd 3d binding energy (as determined by XPS) (Reproduced with permission from [23]. Copyright © 2018 American Chemical Society.)
In this study, the enhancement of activity by alloying was well evidenced based on the DFT calculations, employing Pd22, Pd11Ag11, and Pd6Ag16 clusters as models for monometallic Pd and alloy nanoparticles (Fig. 9.3). The CO2 hydrogenation over Pd22 is initiated by the dissociation of H2 to form a metal-hydride species via TSI/II with a barrier of 13.9 kcal/mol (step 1). Next is the adsorption of HCO3– to produce intermediate III (step 2), followed by the attack of H atom to the C atom of HCO3– via TSIII/IV, with a barrier of 77.4 kcal/mol (step 3). Finally, the formate that is produced accompanied by H2O regenerates the initial active species (step 4). The activation energies for step 1 using Pd11Ag11 and Pd6Ag16 clusters, were 11.9 and 11.0 kcal/mol, respectively, which were similar to that obtained with the Pd22. On the other hand, the reduction of HCO3– via TSIII/IV occurs with a barrier of 58.7 and 46.2 kcal/mol for Pd11Ag11 and Pd6Ag16, respectively. These results show that the rate-determining step is step 3, and further demonstrate that the importance of low Pd/Ag ratio of the PdAg alloy nanoparticles in boosting the rate-determining step.

a Possible reaction mechanism for CO2 hydrogenation to formic acid. b Potential energy profiles as determined by DFT calculations for Pd22, Pd11Ag11, and Pd6Ag11 cluster models (Reproduced with permission from [23]. Copyright © 2018 American Chemical Society.)
The kinetic analysis further supported the above results. In the case of a reaction under a flow of H2 and D2 through the catalyst, the TOF for HD formation was almost independent of the surface composition. On the contrary, the effect of the HCO3– concentration was greatly dependent on the surface composition. The reaction rate decreased in the order Pd/TiO2 (0.67) > Ag@Pd/TiO2 (0.36) > PdAg/TiO2 (0.33) > Pd@Ag/TiO2 (0.22), which is consistent with the TON values based on the quantity of surface-exposed Pd atoms for the CO2 hydrogenation.
This can be well explained by considering the electronic state in reaction intermediate III (Fig. 9.4). Mulliken atomic charges of Pd atoms decrease in the order of –0.115 (Pd22) > –0.168 (Pd11Ag11) > –0.216 (Pd6Ag16), which consequently decreases the electronegativity of the dissociated hydride species on the Pd atoms. Contrastingly, the electronic charges of the C atoms of the adsorbed HCO3– are almost constant for all models, while keeping their positive charges. Thus, the hydride species on the Pd6Ag16 with more negative charge easily attack the C atoms of the adsorbed HCO3–, while a higher activation energy is necessary in the reaction between the less negative hydride species on the Pd22 and the positively charged C atoms. It can be concluded that isolated and electron-rich Pd atoms created with the aid of neighboring Ag atoms explain the enhanced activity, which provides advanced insights into the architecture of catalytically active sites for CO2 hydrogenation to FA.

Representative Mulliken atomic charges in the reaction intermediate III as determined by DFT calculations for a Pd22, b Pd11Ag11, and c Pd6Ag11 cluster models (Reproduced with permission from [23]. Copyright © 2018 American Chemical Society.)
3 Interfacial Engineering of PdAg/TiO2 with a Metal–Organic Framework
In the drive to improve catalytic performances during CO2 hydrogenation to produce FA, PdAg/TiO2 was further modified with an MOF using a facile pretreatment method. A zeolitic imidazolate framework (ZIF-8), a product of the reaction between Zn2+ and 2-methylimidazole, was chosen as a modifying agent because of its high chemical and thermal inertness in aqueous solution, suitable mechanical stability even under high-pressure conditions, and ready synthesis at room temperature. The time allowed for the growth of the ZIF-8 was varied between 10 min and 3 h, affording a series of PdAg/TiO2@ZIF-8. A schematic procedure is illustrated in Fig. 9.5 [24].

A schematic illustration of the synthesis of PdAg/TiO2@ZIF-8 (Reproduced with permission from [24]. Copyright © 2020 American Chemical Society.)
Because of the lower content of ZIF-8 (0.7 wt% from CHN elemental analysis) and its high dispersity on the support, the PdAg/TiO2@ZIF-8 exhibited only peaks due to rutile TiO2, with no characteristic peaks assignable to ZIF-8 or the PdAg NPs in the XRD pattern. No significant differences were observed in the surface area (SBET = 137.5 m2 g–1 for PdAg/TiO2 and 125.9 m2 g–1 PdAg/TiO2@ZIF-8), suggesting that a very thin coating of ZIF-8 had been applied.
X-ray photoelectron spectroscopy (XPS) analysis showed the peak due to Zn 2p3/2 and Zn 2p1/2 at 1022.7 and 1045.8 eV, respectively. Peaks due to C–NH– and C=N– bonds in imidazole groups were also clearly seen in the N 1 s region at 399.7 eV. Scanning transmission electron microscopy (STEM) images confirmed that the PdAg/TiO2 was covered with a thin shell of ZIF-8 having a thickness of approximately 1.6 nm. Elemental mapping further demonstrated that Zn, C, and N atoms, which were originated from ZIF-8 layer, were highly dispersed throughout the material. From the results of XAFS, a cross-sectional EDX line profile, and high-resolution TEM image, it is evident that the PdAg alloy NPs retained their original structure even after coating with the ZIF-8 layer.
The PdAg/TiO2 without modification showed a TON of 488 at 6 h. In contrast, the PdAg/TiO2@ZIF-8 specimen synthesized using a ZIF-8 growth time of 30 min exhibited the highest TON of 913 together with >99% selectivity, which is almost twice that of the unmodified material. Additionally, the catalytic activity was greatly affected by the growth time, such that the highest TON was obtained at 30 min, with decreases in the TON when longer times were applied. This volcano-type variation in activity suggests the formation of a uniform ZIF-8 layer on the surface of the PdAg/TiO2 as well as a synergistic effect originating from integration with the ZIF-8.
Another important influence for the modification with ZIF-8 is the anchoring stabilization effect that inhibits the undesired agglomeration of NPs, which ultimately enhances the durability of the catalyst. In trials with the PdAg/TiO2, the catalytic activity gradually decreased with continued use, such that the activity was reduced by half during the second recycling experiment. Conversely, the PdAg/TiO2@ZIF-8 retained its original activity. Thus, each catalyst was isolated after catalytic reaction and subjected to TEM analysis for comparison of the average particle sizes with the initial particle sizes. The significant enlargement can be observed in the case of unmodified PdAg/TiO2, where the average diameter of the isolated catalyst was determined to be 8.9 nm, which was almost twice that of the initial particle diameter. In contrast, PdAg/TiO2@ZIF-8 catalyst suppressed the particle growth; the mean particle diameter after the reaction was determined to be 5.1 nm, which increased by only 6% compared with that of the initial catalyst particles, thus preserving their intriguing properties in catalysis applications.
To better understand the positive effect of ZIF-8 modification, adsorption energy (Ead) of HCO3– on PdAg surface and Mulliken atomic charges of selected atoms for the reaction intermediate in the rate-determining step were determined by DFT calculations (Table 9.1). Here, three representative models were considered for the calculation. These include PdAg(111) and PdAg(111) interacted with building unit of ZIF-8 framework (two 2-methylimidazole linked with Zn2+ ion) in two different configurations. As expected, the Ead of both configurations for the PdAg(111) interacted with the building unit of ZIF-8 framework was estimated to be −133.9 and −185.2 kcal/mol, respectively, which are larger than that on the pristine PdAg(111) (Fig. 9.6a vs. b and c). In the lowest-energy adsorption structure, 2-methylimidazole ring vertically interacts with surface metal atoms. According to the Brønsted–Evans–Polanyi (BEP) relationship, the larger Ead of the reaction intermediate on the metal catalyst corresponds to a lower reaction barrier. Such changes in the adsorption capacity accordingly alter the electronic charges of the C atoms of the adsorbed HCO3–, as shown in Table 9.1. In contrast, the electronic charges of the dissociated hydride species on the Pd atoms are almost constant for all calculation models. Therefore, the more positively charged C atoms of the adsorbed HCO3– will tend to undergo the attack by the dissociated hydride species. It can be concluded that the electronic effect resulting from the interplay of the neighboring ZIF-8 unit explains the enhanced activity for CO2 hydrogenation.

DFT-optimized configurations for dissociated H atoms and HCO3– ions adsorbed on a PdAg (111) and b and c PdAg (111) with the ZIF-8 framework (showing two 2-methylimidazole molecules bonded to a Zn2+ ion) in two different configurations (Reproduced with permission from [24]. Copyright © 2020 American Chemical Society.)
4 Encapsulation of PdAg Nanoparticles Within ZIF-8 Framework with Core–Shell Structure
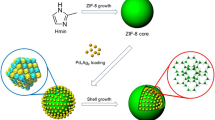
To overcome the general problems of the aggregation of supported metal NPs on the external surface of MOFs and damage of MOFs during the post-reduction process, a facile method to encapsulate metal NPs within MOFs has been developed. This “bottle around ship” approach involves the growth of ZIF-8 core at the initial stage by using 2-methylimindazole (Hmin) as an organic linker and Zn2+ as a connecting center, loading the PdAg NPs on the external surface of ZIF-8 core, and coating the PVP stabilized PdAg alloy NPs with ZIF-8. Figure 9.7 illustrates the synthetic route for the fabrication of ZIF-8@PdAg@ZIF-8 catalyst [25].

Schematic illustration for the synthesis of ZIF-8@PdAg@ZIF-8
The diffraction pattern of ZIF-8@PdAg@ZIF-8 sample is similar to that of pure ZIF-8. This suggests that the encapsulation of PdAg NPs within ZIF-8 did not change the framework structure. However, the intensities of diffraction peaks are weaker than those of pure ZIF-8, which is because the encapsulated PdAg NPs cause the disorder in MOFs crystal. In addition, the diffraction peaks assigned to Pd and Ag cannot be observed, because of their low content and small particle size. The pure ZIF-8 shows a type-I isotherm with completely reversible isothermal in the N2 sorption analysis, which is a typical feature of microporous materials. The Brunauer–Emmett–Teller (BET) surface area determined by N2 adsorption-desorption is 1110 m2 g−1. Meanwhile, ZIF-8@PdAg@ZIF-8 has a similar isotherm to pure ZIF-8 except for the slight decrease in the N2 uptake, which suggests the decrease of some micropores after the encapsulation of PdAg NPs. This resulted in a slight decrease in their surface area to 926.3 m2 g−1. Based on such results, it is concluded that the crystallinity and porosity of ZIF-8 are well preserved after PdAg encapsulation.
The morphologies of pure ZIF-8 and ZIF-8@Pd1Ag2@ZIF-8 are shown in Fig. 9.8. Pure ZIF-8 displays a rhombic dodecahedral morphology and the particle size is about 350 nm. Meanwhile, the morphology of ZIF-8@Pd1Ag2@ZIF-8 did not show obvious change. It should be noted that the external surface of ZIF-8@Pd1Ag2@ZIF-8 is very smooth, which indicates that the PdAg NPs are not loaded on the external surface of ZIF-8. From the TEM images, tiny PdAg NPs with a mean diameter of 2.8 nm can be observed, which was covered by a thin shell of ZIF-8, and the shell thickness was measured to be ca. 5 nm.

SEM images of a pure ZIF-8 and b ZIF-8@Pd1Ag2@ZIF-8, and c TEM image and d size distribution diagram of ZIF-8@Pd1Ag2@ZIF-8
The ZIF-8@Pd1Ag2@ZIF-8 samples showed the highest catalytic activity among all samples, affording 16.68 mmol g −1(catal.) after 24 h, almost two times the FA amount produced over ZIF-8@Pd3@ZIF-8 under the identical reaction condition.
The poor catalytic performance was obtained on PdAg/ZIF-8 without core–shell structure. This is due to the large particle size of PdAg NPs, whose diameter was determined to be 10.5 nm. In comparison, ZIF-8@Pd1Ag2@ZIF-8 exhibited improved catalytic activity, which can be ascribed to the high dispersibility of PdAg NPs within ZIF-8 and the positive effect of the thin shell in protecting PdAg NPs during the reaction process. The catalyst was recovered from the reaction solution by using centrifugation and wash with water. TEM image of ZIF-8@PdAg@ZIF-8 after reaction showed that the PdAg NPs are still well dispersed within ZIF-8, and no significant aggregation occurred. Moreover, the recycled ZIF-8@PdAg@ZIF-8 could be re-used at least three times without significantly loss in activity. Based on the above result, it is clear that the presented synthetic approach has advantages for overcoming the general problems of the aggregation of metal nanoparticles on the external surface of MOFs and damage of MOFs during the post-reduction process, leading to the enhanced catalytic activity for CO2 hydrogenation to produce FA.
5 Summary and Outlook
By tuning the surface-exposed Pd atoms in the alloy NPs, the isolated Pd atoms surrounded by large amount of Ag atoms in Pd@Ag/TiO2 performed as an efficient catalyst for the CO2 hydrogenation to FA even under low-pressure conditions. Kinetic and DFT calculations evidenced the enhanced electronegativity was found to facilitate the rate-determining reduction step of the adsorbed HCO3– species. Moreover, the positive effects imparted by the interfacial modification of PdAg/TiO2 with ZIF-8 and the encapsulation of PdAg NPs within the ZIF-8 Framework with core–shell structure were demonstrated. These studies not only provide advanced insights into the architecture of catalytically active centers for CO2 hydrogenation to FA based on the surface engineering approach, but also emphasizes the importance of the interfacial surface engineering of catalysts for further improvement. Nevertheless, there are still some lacking aspects that should be tackled in future investigations. One of the principal issues is their insufficient stability and long durability under reaction conditions, which should be absolutely improved to meet the practical application criteria while preserving the unique surface characteristics of NPs. Further improvement is needed to develop reliable catalysts, which meet the practical application criteria in terms of efficiency, cost, and reusability, which will open a new avenue for environmentally benign CO2-mediated hydrogen storage/release systems.
References
Wang W, Wang S, Ma X, Gong J (2011) Recent advances in catalytic hydrogenation of carbon dioxide. Chem Soc Rev 40:3703–3727
Singh AK, Singh S, Kumar A (2016) Hydrogen energy future with formic acid: a renewable chemical hydrogen storage system. Catal Sci Technol 6:12–40
Joó F (2008) Breakthroughs in hydrogen storage—formic acid as a sustainable storage material for hydrogen. Chemsuschem 1:805–808
Mori K, Tanaka H, Dojo M, Yoshizawa K, Yamashita H (2015) Synergic catalysis of PdCu alloy nanoparticles within a macroreticular basic resin for hydrogen production from formic acid. Chem Eur J 21:12085–12092
Mori K, Dojo M, Yamashita H (2013) Pd and Pd–Ag nanoparticles within a macroreticular basic resin: an efficient catalyst for hydrogen production from formic acid decomposition. ACS Catal 3:1114–1119
Masuda S, Mori K, Futamura Y, Yamashita H (2018) PdAg nanoparticles supported on functionalized mesoporous carbon: promotional effect of surface amine groups in reversible hydrogen delivery/storage mediated by formic acid/CO2. ACS Catal 8:2277–2285
Moret S, Dyson PJ, Laurenczy G (2014) Direct synthesis of formic acid from carbon dioxide by hydrogenation in acidic media. Nat Commun 5:4017
Enthaler S, von Langermann J, Schmidt T (2010) Carbon dioxide and formic acid-the couple for environmental-friendly hydrogen storage? Energy Environ Sci 3:1207–1217
Mellmann D, Sponholz P, Junge H, Beller M (2016) Formic acid as a hydrogen storage material—development of homogeneous catalysts for selective hydrogen release. Chem Soc Rev 45:3954–3988
Preti D, Resta C, Squarcialupi S, Fachinetti G (2011) Carbon dioxide hydrogenation to formic acid by using a heterogeneous gold catalyst. Angew Chem Int Ed 50:12551–12554
Xu Z, McNamara ND, Neumann GT, Schneider WF, Hicks JC (2013) Catalytic hydrogenation of CO2 to formic acid with silica-tethered iridium catalysts. ChemCatChem 5:1769–1771
Filonenko GA, Vrijburg WL, Hensen EJM, Pidko EA (2016) On the activity of supported Au catalysts in the liquid phase hydrogenation of CO2 to formates. J Catal 343:97–105
Lee JH et al (2014) Carbon dioxide mediated, reversible chemical hydrogen storage using a Pd nanocatalyst supported on mesoporous graphitic carbon nitride. J Mater Chem A 2:9490–9495
Mori K, Taga T, Yamashita H (2017) Isolated single-atomic Ru catalyst bound on a layered double hydroxide for hydrogenation of CO2 to formic acid. ACS Catal 7:3147–3151
Mori K, Yamashita H (2010) Progress in design and architecture of metal nanoparticles for catalytic applications. Phys Chem Chem Phys 12:14420–14432
Gao F, Goodman DW (2012) Pd-Au bimetallic catalysts: understanding alloy effects from planar models and (supported) nanoparticles. Chem Soc Rev 41:8009–8020
Tedsree K et al (2011) Hydrogen production from formic acid decomposition at room temperature using a Ag-Pd core-shell nanocatalyst. Nat Nanotechnol 6:302–307
Mori K, Naka K, Masuda S, Miyawaki K, Yamashita H (2017) Palladium copper chromium ternary nanoparticles constructed in situ within a basic resin: enhanced activity in the dehydrogenation of formic acid. ChemCatChem 9:3456–3462
Masuda S et al (2018) Simple route for the synthesis of highly active bimetallic nanoparticle catalysts with immiscible Ru and Ni combination by utilizing a TiO2 support. ChemCatChem 10:3526–3531
Masuda S, Shun K, Mori K, Kuwahara Y, Yamashita H (2020) Synthesis of a binary alloy nanoparticle catalyst with an immiscible combination of Rh and Cu assisted by hydrogen spillover on a TiO2 support. Chem Sci 11:4194–4203
Mori K, Kondo Y, Yamashita H (2009) Synthesis and characterization of FePd magnetic nanoparticles modified with chiral BINAP ligand as a recoverable catalyst vehicle for the asymmetric coupling reaction. Phys Chem Chem Phys 11:8949–8954
Mori K, Yoshioka N, Kondo Y, Takeuchi T, Yamashita H (2009) Catalytically active, magnetically separable, and water-soluble FePt nanoparticles modified with cyclodextrin for aqueous hydrogenation reactions. Green Chem 11:1337–1342
Mori K, Sano T, Kobayashi H, Yamashita H (2018) Surface engineering of a supported PdAg catalyst for hydrogenation of CO2 to formic acid: elucidating the active pd atoms in alloy nanoparticles. J Am Chem Soc 140:8902–8909
Mori K, Konishi A, Yamashita H (2020) Interfacial engineering of PdAg/TiO2 with a metal-organic framework to promote the hydrogenation of CO2 to formic acid. J Phys Chem C 124:11499–11505
Wen M et al (2019) PdAg nanoparticles within core-shell structured zeolitic imidazolate framework as a dual catalyst for formic acid-based hydrogen storage/production. Sci Rep 9:15675
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Mori, K., Yamashita, H. (2021). Supported Core–Shell Alloy Nanoparticle Catalysts for the Carbon Dioxide Hydrogenation to Formic Acid. In: Yamashita, H., Li, H. (eds) Core-Shell and Yolk-Shell Nanocatalysts. Nanostructure Science and Technology. Springer, Singapore. https://doi.org/10.1007/978-981-16-0463-8_9
Download citation
DOI: https://doi.org/10.1007/978-981-16-0463-8_9
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-16-0462-1
Online ISBN: 978-981-16-0463-8
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)