
Abstract
The myopathies are inherited or acquired neuromuscular disorders characterized primarily by muscle weakness due to dysfunction of muscle fiber. Sleep disturbances are frequent in patients with myopathy but are often ignored or missed. Timely identification and treatment of these disorders can improve the quality of life of such patients. Therefore, patients with myopathy should routinely be assessed for features of sleep disturbances because of their treatable nature in an otherwise progressive disease process. This chapter covers various sleep disorders in myopathies, their clinical presentations, role of various investigations, and available treatment options. Before discussing that, it is important to have a clear concept of the functional anatomy and physiology of sleep and breathing. The first section contains an overview of the anatomy and physiology of sleep, clinical presentations of various myopathies, and control of breathing during sleep. The second section focuses on myopathy-associated sleep abnormalities and their management.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Myopathy
- Sleep apnea
- Sleep-related breathing disorders
- Parasomnias
- Circadian rhythm sleep-wake disorders
- Sleep-related movement disorders
1 Introduction
Sleep and breathing are controlled by anatomic structures situated in both central as well as peripheral nervous system. Thus, neurologic illnesses can adversely affect sleep and breathing and vice versa [1, 2]. The effect of acute and chronic muscle disorders on sleep and the resulting interactions with breathing has not received wider attention yet. This understanding may prove essential for deciding the treatment as well as prognosis in various muscle disorders. Varied presentations of sleep disturbances in myopathies include the following:
-
1.
Breathing abnormalities: Sleep-related breathing disorders (SBD), resulting in hypopnea, apnea, alveolar hypoventilation.
-
2.
Duration abnormalities: Insomnia, hyposomnia, or hypersomnia.
-
3.
Behavior abnormalities: Parasomnias (sleep terrors, sleep talking, somnambulism, nightmares).
-
4.
Tone abnormality: Sleep-related movement disorders (rapid eye movement [REM] sleep behavior disorders), periodic limb movements of sleep (PLMS), restless leg syndrome (RLS).
-
5.
Rhythm disorders: Circadian rhythm sleep-wake disorders.
-
6.
Excessive day time sleepiness.
2 Functional Anatomy of Sleep and Awake State
The neuroanatomic structures involved in maintenance of wakefulness, rapid eye movement (REM) sleep, and non-rapid eye movement (NREM) sleep are located in different parts of the central nervous system (CNS) [3, 4]. There are no separate sleep and wake-promoting centers in brain; rather, these states are generated by changes in the balance between the interconnecting neuronal circuits modulated by neurotransmitters and neuromodulators. The parts of CNS and concerned neurotransmitters involved in sleep physiology are illustrated in Fig. 35.1.

The neurobiology & physiology of sleep and wakefulness. (a) Ascending reticular activating system (ARAS): Two major pathways are shown in figure a. One (in yellow) depicting upper brain stem input to be delivered to the thalamic-relay nuclei as well as reticular nucleus of the thalamus. This input is coming from acetylcholine (ACh)-producing neuronal groups, which are located in the Pedunculopontine and laterodorsal tegmental (PPT/LDT) nuclei of brainstem. The second major group of neurons (in red) are located in noradrenergic (NA)- locus coeruleus (LC), serotoninergic (5-HT)- dorsal and median raphe nuclei, dopaminergic (DA)- periaqueductal gray matter (vPAG), and histaminergic (His)- tuberomamillary neurons (TMN). Additional cortical input also originates from the GABA or Ach neurotransmitter containing basal forebrain (BF) neurons as well as from lateral hypothalamic (LH) peptidergic neurons that contain melanin-concentrating hormone (MCH) or orexin (hypocretin) (ORX). (b) Projections from the ventrolateral preoptic nucleus (VLPO) to the components of the ascending wakefulness system. PPT pedunculopontine tegmental area, LDT laterodorsal tegmental area, NA noradrenergic, LC locus coeruleus, 5HT serotoninergic, DA dopaminergic, vPAG periaqueductal gray matter, His histaminergic, TMN tuberomamillary neurons, BF basal forebrain, LH lateral hypothalamus, MCH melanin-concentrating hormone, ORX orexin, VLPO ventrolateral preoptic nucleus. Reproduced from van Someren E., Cluydts R. (2015) Sleep Regulation and Insomnia. In: Pfaff D., Volkow N. (eds) Neuroscience in the twenty-first Century. Springer, New York, NY [5]
3 Myopathies
They are inherited or acquired disorders, characterized by muscle weakness, muscle cramps, stiffness, and spasm and can be grouped into multiple types (Table 35.1) [12].
Most of the muscle diseases share common features of diaphragmatic weakness, reduced strength of upper airway dilators, as well as cardiomyopathy, leading to sleep disorders including sleep-disordered breathing (SDB).
4 Pathophysiology of Sleep Disorders in Myopathies
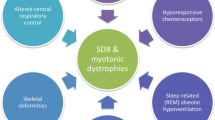
During sleep, arterial partial pressure of carbon dioxide (PaCO2) usually increases by 2–4 mmHg due to reduction of alveolar ventilation (because of a fall in central respiratory drive), blunted arousal thresholds, and reduced respiratory muscle activity. Disproportionate loss of upper airway muscle tone and diaphragm weakness may contribute additional loads, particularly during rapid eye movement (REM) sleep, the time of maximal muscle hypotonia. GABA- and glycine-mediated muscle atonia in REM sleep reduces the contribution of intercostal muscles toward tidal volume by up to 19% as compared to 40% occurring in awake state as well as during early sleep [13,14,15,16]. In normal state, an intact activity of diaphragm and accessory respiratory muscles compensate for these type of sleep alterations [17]. But these sleep changes may precipitate respiratory dysfunction in muscle disorders. Imbalance between respiratory load, drive, and muscular capacity predisposes to SDB with desaturations, hyperpnea, obstructive and central apneas, and hypercapnic hypoventilation which occur preferentially during REM sleep. Various myopathies and their factors leading to breathing disorders during sleep are shown in Fig. 35.2, while pathophysiological process is depicted in Fig. 35.3.

Common myopathies and their Factors promoting sleep-disordered breathing [18]

Pathophysiological mechanisms responsible for sleep disordered breathing in patients with myopathy and muscular dystrophy [19].
5 Patterns of Sleep Disorders in Myopathies
In the early stage, SDB may manifest as REM-related alveolar hypoventilation. Later as the disease progresses, there will be gradual worsening of respiration in NREM sleep followed by disordered breathing in the awake state. In addition, weakness of upper airway dilator muscles may cause superimposed OSA. Diaphragmatic weakness usually develops late in the course of disease. In myopathies not affecting diaphragmatic function sufficiently enough to cause alveolar hypoventilation, REM-related respiratory abnormalities such as hypopneas, apneas, and paradoxical breathing may occur.
6 Various Sleep Disorders in Specific Myopathies Are as Follows
-
1.
Duchene muscular dystrophy (DMD), an X-linked recessive disease characterized by aggressive progression of skeletal muscle weakness along with development of pseudohypertrophy of the calves and skeletal deformities (scoliosis). Alveolar hypoventilation (caused by respiratory muscle weakness with scoliosis) results in severe nocturnal sleep hypoxemia and hypercapnia. Obstructive apnea and hypopnea are also common (resulting from upper airway muscle weakness). The utility of noninvasive positive pressure ventilation (NIPPV) in DMD is debated and its use for preventive purposes should be avoided in patients with FVC between 20% and 50% of predicted values [20].
-
2.
Congenital myopathies can also present with sleep abnormalities, respiratory insufficiency, and SDB. Reduced total sleep time along with decreased REM sleep duration and presence of signs of upper airway resistance has also been reported [21, 22].
-
3.
Myotonic dystrophy type 1 (DM1), an autosomal dominant multisystem disorder characterized by muscle weakness and extra-skeletal features. More than 50% patients suffer excessive daytime somnolence and show presence of sleep-onset REM periods during the multiple sleep latency test (MSLT), while up to two-thirds manifest fatigue. A combination of OSA, central apneas, periodic breathing, and nocturnal hypoventilation commonly predispose these patients to SDB, which may not be detected by daytime pulmonary function test. Developmental orofacial structural abnormalities due to poor muscle tone may further predispose them to OSA [23]. Such patients often report longer sleep periods, less restorative sleep, more difficulty falling asleep, being less alert in the morning, and having more trouble staying awake after meals. The excessive daytime sleepiness is often out of proportion to their degree of SDB and persists despite adequate treatment of SDB. They have short sleep latencies on multiple sleep latency test (MSLT) as well as short sleep-onset REM periods (SOREMPs), suggesting a narcoleptic phenotype. However, no correlation has been reported between the CSF hypocretin level and the degree of hypersomnolence. Nevertheless, this along with the occurrence of periodic breathing suggests a central mechanism, responsible for sleep dysfunction in DM1, and a correlation has been found between the degree of corpus callosum atrophy and severity of hypersomnolence detected on MSLT [24].
-
4.
Myotonic dystrophy type 2 (DM2 or proximal myotonic myopathy, PROMM) is clinically and genetically distinct from DM1. Such patients seem to be less hypersomnolent, but more fatigued than patients with DM1. PSG-based analysis suggested increased arousals, decreased sleep efficiency, alpha-delta sleep, obstructive apneas, paradoxical breathing in REM sleep, excessive daytime sleepiness, snoring, or insomnia. An abnormality of central control of breathing and sleep-wakefulness related to a common generalized membrane abnormality of the brainstem may be responsible for sleep-wake and respiratory dysfunction [25].
-
5.
Mitochondrial myopathies: They are a heterogeneous group of disorders caused by heritable or spontaneous mutations in nuclear or mitochondrial DNA and share the common feature of mitochondrial abnormalities in muscle fibers on histochemical (ragged-red fibers on modified Gomori’s trichrome stain) and electron-microscopic examination. Both nuclear and mitochondrial DNA mutations can lead to cellular energy failure, resulting in both central as well as peripheral neuromuscular alteration. All these can cause abnormal sleep patterns, commonly manifesting as central sleep apnea and poor ventilatory response to hypercapnia, which predispose them to develop sleep-related respiratory disorders. This activates a vicious cycle of progressive daytime fatigue, hypotonia, and exercise intolerance, leading to poor quality of life [26].
-
6.
Acid maltase deficiency: Acid maltase deficiency, also called Pompe’s disease is a glycogen storage disease primarily involving cardiac and skeletal muscle. The clinical features depend on residual acid maltase enzyme activity. The disease may have either a childhood or an adulthood presentation. SDB and respiratory failure are reported in this disorder. Respiratory muscle weakness resulting in respiratory failure is the most common cause of early mortality. Sleep disturbances resulting from diaphragmatic weakness and obstructive sleep apnea are common factors predisposing to sleep disturbances [27]. Diaphragmatic weakness in the early stage of the disease may result in hypoventilation. It is initially seen during REM sleep but may cause daytime respiratory failure as the disease progresses. Macroglossia and weakness involving tongue muscles may result in upper airway dysfunction causing OSA [28].
7 Clinical Manifestations
The clinical features can be grouped into specific and general features. Specific manifestations depend on the nature of the neurologic deficit. General features include excessive daytime sleepiness, fatigue, early morning headache, unexplained pedal edema, disturbed nocturnal sleep, intellectual deterioration, personality changes, and in men, impotence, plus nocturnal restlessness, frequent unexplained awakenings, and loud snoring [21]. Difficulty waking in the morning with prolonged sleep inertia may interfere with morning activities. During the day, patients may manifest with somnolence, fatigue, and inappropriate napping that underlie failure to thrive in the very young and declining school grades or poor work performance at later ages. More ominously, some patients develop nocturnal cyanosis, severe insomnia, morning lethargy, headaches, vomiting, and leg edema that indicate the insidious but relentless occurrence of acute respiratory failure and cor pulmonale.
8 General Approach and Suggested Management
-
1.
History and examination: To make a clinical diagnosis of sleep disorders, a careful history from the patient and their caregivers along with a detailed physical examination is essential (Fig. 35.4). Diaphragm weakness is the most important determinant of sleep-related respiratory insufficiency. Chest wall weakness and restrictive lung diseases resulting from chest wall deformities and kyphoscoliosis also contribute to hypoventilation in REM and NREM sleep. Weakness of the pharyngeal wall muscles, compounded with obesity of sedentary origin and craniofacial maldevelopment, may facilitate the appearance of obstructive sleep apneas. Some patients exhibit nocturnal hypoventilation in excess of muscle weakness or diaphragmatic failure, suggesting an alteration of central respiratory drive.
-
2.
Polysomnographic evaluation: It is a gold standard test used to distinguish different causes of sleep disturbance and to assess the severity of the disorder (Fig. 35.4). A sleep apnea protocol is recommended. The study may show obstructive, central, and mixed sleep apneas, hypoventilation with oxygen saturations under 89%, or profound REM sleep-related desaturation of oxygen events indicating diaphragmatic failure. The sleep architecture may reveal fragmentation of sleep with many arousals and awakenings, many of them associated with episodes of respiratory interruption. Various polysomnographic patterns of myopathy are shown in Fig. 35.5. Since PSG may be expensive and may not be available at all centers; the simplest way to assess SDB is overnight oximetry monitoring.
-
3.
Hematological investigations: Analysis of arterial blood gases as well as PaO2 and PaCO2 (partial pressure of oxygen and carbon dioxide) to look for presence of hypoxia (Pa o 2 < 60 mm Hg) and hypercapnia (Pa co 2 > 45 mm Hg) [21].
-
4.
Pulmonary function test (PFT): It is used to diagnose and classify ventilatory disorders, whether they are of intrinsic pulmonary, neurologic, or musculoskeletal etiology. Various tests including lung volumes, forced vital capacity (FVC) in sitting and supine positions, forced expiratory volume in 1 s (FEV1), FEV1/FVC ratio, maximal peak inspiratory force (PImax or negative inspiratory force, NIF), maximal peak expiratory force (PEmax) should be done to check for presence of any restrictive lung disease related to myopathy and its impact on sleep architecture.
-
5.
Other tests: Maximal sniff maneuvers, esophageal, transdiaphragmatic, nasal pressures (Pnsn), overnight pulse oximetry, and capnography to check for presence of dynamic obstructions of upper airways due to upper airway muscle.

Algorithm for management of sleep disorders in myopathies [29]

Polysomnographic patterns in normal respiratory effort and sleep apnea. Airflow_Th: signal from the nasal thermistor, Airflow_Pr: signal from the nasal pressure cannula, RespEffort3D: Respiratory effort signal derived from the depth signal of the 3D camera, RIP Sum thorax/abdomen respiratory inductance plethysmography belts’ sign. Coronel, C., Wiesmeyr, C., Garn, H. et al. Somnologie; 2019; 23: 86, Springer Medizin
In short, no single diurnal test has been found to be uniformly predictive of nocturnal hypoventilation, and overnight oximetry plus capnography have their own limitations. A combination of maximal inspiratory pressure and nasal sniff pressure testing may be the most sensitive test in this regard, but in-laboratory polysomnography is the recommended gold standard diagnostic tool for SDB in patients with myopathies.
9 Consequences of Sleep Disorders
Persistent nocturnal hypoxemia resulting from SDB from any cause results in cardiovascular and pulmonary morbidity as well as mortality from various causes such as lethal cardiac arrhythmias, pulmonary hypertension, right heart failure (cor pulmonale), and propensity to develop myocardial infarction as well as stroke. In addition, SDB causes fragmentation of sleep and excessive daytime sleepiness and fatigue, decreasing the quality of life and affecting mood and cognition.
10 General Principles in the Treatment of Sleep Dysfunction in Myopathies
Most myopathies have no specific treatment, and management is largely supportive. In addition to optimization of treatment and alleviation of discomfort from symptoms attributable to the neurologic disease itself, the early diagnosis and treatment of SDB associated with myopathies is crucial. The goal of treatment of SDB is to support the weakened ventilatory muscles, thereby improving alveolar ventilation and gas exchange. In the case of syndromes related to increased nocturnal upper airway resistance or decreased drive to breathe (OSA and central sleep apnea, respectively), treatment is aimed at overcoming these issues, primarily through the use of devices meant to supply upper airway pressurization. In turn, this leads to improved sleep quality.
11 Treatment Options
Assisted mechanical ventilation at night improves the symptoms and protects patient from fatal apnea during disease. Furthermore, such treatment may prevent the development of serious complications resulting from episodic or prolonged hypoxemia, hypercapnia, and respiratory acidosis in sleep. Various treatment modalities available for sleep dysfunction in muscle disorders include the following:
-
Upper airway pressurization.
-
Continuous positive airway pressure (CPAP).
-
NIPPV (noninvasive positive pressure ventilation) [30].
-
Supplemental oxygen therapy.
-
Surgical treatment like cricoidotomy.
-
Tracheostomy.
-
Diaphragmatic pacing.
-
Pharmacotherapy like melatonin, non-benzodiazepine sedatives.
-
For excessive daytime sleepiness.
-
Wakefulness-promoting agents (modafinil, armodafinil).
-
Stimulants (methylphenidate and amphetamines).
-
-
For restless legs syndrome: Dopamine agonists such as pramipexole, ropinirole, and rotigotine; gabapentin and pregabalin; serum ferritin in case of iron deficiency state.
Specific therapeutic options for the management of sleep disorders in myopathy are summarized in Table 35.2.
12 Summary
Sleep dysfunction, particularly SDB, is common in many neuromuscular diseases, including myopathies. A high index of suspicion is crucial because patients are often unaware of the specific symptoms of SDB, attributing them to the underlying neurologic illness. A variety of diurnal and nocturnal tests, including blood gas analysis, pulmonary and sniff function testing, oximetry, and capnography are available to aid in making the diagnosis. However, there is still lack of consensus available about the optimal daytime indicator in neuromuscular patients with suspected nocturnal hypoventilation. In laboratory, PSG and upper airway titration studies remain the gold standard for diagnosis and treatment. NIPPV is the mainstay of treatment of nocturnal respiratory failure in neuromuscular diseases, although the precise timing and indications for this therapy, as well as the long-term outcomes, especially on symptoms related to sleep dysfunction, remain to be elucidated with further research.
References
Steriade M, McCarley RW. Brain control of wakefulness and sleep. 2nd ed. New York: Kluwer Academic/Plenum Publishers; 2005.
Phillipson EA, Bowes G. Control of breathing during sleep. In: Cherniack NS, Widdicombe JG, editors. Handbook of physiology, section 3, the respiratory system, part II. Bethesda, MD: American Physiological Society; 1986. p. 649–89.
Coronel C, Wiesmeyr C, Garn H, et al. Measurement of respiratory effort in sleep by 3D camera and respiratory inductance plethysmography. Somnologie. 2019;23:86–92.
Chokroverty S. An overview of normal sleep. In: Chokroverty S, Allen RP, Walters AS, Montagna P, editors. Sleep and movement disorders. 2nd ed. New York: Oxford University Press; 2013. p. 22–45.
Van Someren E, Cluydts R. Sleep regulation and insomnia. In: Pfaff D, Volkow N, editors. Neuroscience in the 21st century. New York, NY: Springer; 2015.
Khadilkar SV, Yadav RS, Patel BA. Congenital myopathies. Neuromuscul Disord. 2017:173–82.
Mukherjee M, Mittal B. Muscular dystrophies. Indian J Pediatr. 2004;71:161–8.
Ahmed ST, Craven L, Russell OM, et al. Diagnosis and treatment of mitochondrial myopathies. Neurotherapeutics. 2018;15:943–53.
Laforêt P, Weinstein DA, Peter A, Smit G. The glycogen storage diseases and related disorders. In: Saudubray JM, van den Berghe G, Walter JH, editors. Inborn metabolic diseases. Berlin: Springer; 2012.
Kansagra S, Austin S, DeArmey S, Kishnani PS, Kravitz RM. Polysomnographic findings in infantile Pompe disease. Am J Med Genet A. 2013;161(12):3196–200.
Khadilkar SV, Yadav RS, Patel BA. Idiopathic inflammatory myopathies. In: Neuromuscular disorders. Singapore: Springer; 2018.
Amato AA. Disorders of skeletal muscle. In: Daroff RB, Jankovic J, Mazziotta JC, Pomeroy SL, editors. Neurology in clinical practice. 7th ed; 2015. p. 1915–55.
Douglas NJ, White DP, Pickett CK, Weil JV, Zwillich CW. Respiration during sleep in normal man. Thorax. 1982;37:840–4.
Gould GA, Gugger M, Molloy J, Tsara V, Shapiro CM, Douglas NJ. Breathing pattern and eye movement density during REM sleep in humans. Am Rev Respir Dis. 1988;138:874–7.
Tabachnik E, Muller NL, Bryan AC, Levison H. Changes in ventilation and chest wall mechanics during sleep in normal adolescents. J Appl Physiol. 1981;51(3):557–64.
Wiegand L, Zwillich CW, White DP. Collapsibility of the human upper airway during normal sleep. J Appl Physiol. 1989;66:1800–8.
Aboussouan LS. Sleep-disordered breathing in neuromuscular disease. Am J Respir Crit Care Med. 2015;191:979–89.
Chokroverty S. Sleep and breathing in neuromuscular disorders. Handb Clin Neurol. 2011;99:1087–108.
Joseph V, Devasahayam J, Goyal M. Sleep issues in myopathic disorders and muscular dystrophies. In: Govindarajan R, Bollu PC, editors. Sleep issues in neuromuscular disorders: a clinical guide. Cham: Springer; 2018.
LoMauro A, D’Angelo MG, Aliverti A. Sleep disordered breathing in Duchenne muscular dystrophy. Curr Neurol Neurosci Rep. 2017;17(5):44.
Bhat S, Gupta D, Chokroverty S. Sleep disorders. Neurol Clin. 2012;30(4):1359–87.
Ragette R, Mellies U, et al. Patterns and predictors of sleep disordered breathing in primary myopathies. Thorax. 2002;57:724–8.
Laberge L, Gagnon C, Dauvilliers Y. Daytime sleepiness and myotonic dystrophy. Curr Neurol Neurosci Rep. 2013;13(4):340.
Martínez-Rodríguez JE, et al. Decreased hypocretin-1 (Orexin-A) levels in the cerebrospinal fluid of patients with myotonic dystrophy and excessive daytime sleepiness. Sleep. 2003;26(3)
Tieleman AA, Knoop H, van de Logt AE, et al. Poor sleep quality and fatigue but no excessive daytime sleepiness in myotonic dystrophy type 2. J Neurol Neurosurg Psychiatry. 2010;81(9):963–7.
Ramezani RJ, Stacpoole PW. Sleep disorders associated with primary mitochondrial diseases. J Clin Sleep Med. 2014;10(11):1233–9.
Mellies U, Ragette R, et al. Sleep-disordered breathing and respiratory failure in acid maltase deficiency. Neurology. 2001;57:1290–5.
Nabatame S, Taniike M, Sakai N, Kato-Nishimura K, Mohri I, Kagitani-Shimono K, et al. Sleep disordered breathing in childhood-onset acid maltase deficiency. Brain Dev. 2009;31(3):234–9.
Mayer G, Arzt M, Braumann B, et al. German S3 guideline nonrestorative sleep/sleep disorders, chapter “sleep-related breathing disorders in adults,” short version. Somnologie. 2017;21(4):290–301.
Aboussouan LS, Mireles-Cabodevila E. Sleep-disordered breathing in neuromuscular disease: diagnostic and therapeutic challenges. Chest. 2017;152(4):880–92.
Lie JD, et al. Pharmacological treatment of insomnia. P & T: a peer-reviewed. Journal for Formulary Management. 2015;40:759–71.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2022 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Desai, I., Kumar, N. (2022). Sleep Disorders in Myopathies. In: Gupta, R., Neubauer, D.N., Pandi-Perumal, S.R. (eds) Sleep and Neuropsychiatric Disorders. Springer, Singapore. https://doi.org/10.1007/978-981-16-0123-1_35
Download citation
DOI: https://doi.org/10.1007/978-981-16-0123-1_35
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-16-0122-4
Online ISBN: 978-981-16-0123-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)