
Abstract
Hydroxyapatite was obtained after heat treatment (600 ℃) of the powder synthetized by neutralizing a solution of calcium hydroxide Ca(OH)2 with phosphoric acid H3PO4 under atmospheric conditions and reduced time aging. The Ca/P molar ratio of the reagents was 2.0. The concentration of the solution of phosphoric acid influences the pH of the mixture and particle size distribution of solid products. When the Ca(OH)2 solution was neutralized with concentrated phosphoric acid, the average particle size obtained is about 9.13 µm, whereas when the Ca(OH)2 solution was neutralized with diluted solution of phosphoric acid, the average particle size is about 79.40 µm. Agglomerated particles were formed when phosphoric acid solution was used in diluted form. FTIR and DRX analyses have shown that the as-dried powder (at 100 ℃) was a calcium carbonate–phosphate with low crystallinity, which after sintering at temperature higher than 600 ℃ leads to the formation of crystalline hydroxyapatite (HA) with CaO second phase in minor quantities. No decomposition of hydroxyapatite single phase was observed even at high temperature treatment of the powder (1100 ℃).
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Hydroxyapatite Ca10 (PO4)6 (OH)2 (Ca/P = 1.67) is the most calcium phosphate used in biomedical field as biomaterial, adsorbent and catalytic support. As biomaterial, hydroxyapatite (HA) is bioactive, biocompatible, and osteoconductive but possesses a very low solubility in vivo. To overcome this problem, hydoxyapatite is sometimes associated with tricalcium phosphate (βTCP), which has good solubility. This biphasic material (HA/βTCP) combines the properties of HA and βTCP, and thus, its resorption properties in vivo are improved. Hydroxyapatite biomaterial can be obtained by heat treatment of bones, which are composed by carbonated hydroxyapatite or by synthetic routes. Due to its low solubility, hydroxyapatite has a great stability in water, which is an important parameter for adsorbent materials. Hydroxyapatite crystallizes in hexagonal system (P6/3 m space group), with a lattice parameters: a = 9.37 A° and c = 6.86 A°. Its structure is composed of PO43−, Ca2+, and OH− ions, which can be substituted by monovalent, divalent, and trivalent ions. This ionic substitution capacity makes these materials very efficient in the field of wastewater treatment, removal of dyes, and chromatography (Maya et al. 2020). Hydroxyapatite possesses very good retention capacities for heavy metal ions such as Pb2+, Cd2+, Cr3+…. (Haifeng et al. 2019; Michele et al. 2019). The retention capacities of heavy metals on surface of hydroxyapatite depend on the surface affinity of this material. Thus, the phosphate and hydroxyl groups in hydroxyapatite surface are hard Lewis bases and have good affinity for hard Lewis acid as lead cations. The ionic radii of the cation are another parameter on which depends the retention capacity. Cations with ionic radii bigger than Ca2+ are more likely to be incorporated in HA structure. In addition, hydroxyapatite with low crystallinity that corresponds to high surface area is more beneficial to remove heavy metal ions (Doan et al. 2012a, b; Sangeetha et al. 2018; Maya et al. 2020).
Hydroxyapatite can be prepared by many synthetic routes, which include dry methods, high-temperature processes, wet methods, etc. (Mehdi et al. 2013). Among these techniques, the most widely used one is the wet chemical route. The advantages of this method are that different particle sizes and morphologies can be obtained by varying the preparation conditions (Peipei et al. 2010; Elhammari et al. 2007). The common reagents used to synthesize hydroxyapatite are calcium nitrate and calcium chloride as calcium sources and ammonium orthophosphate salts as orthophosphate sources because of their high solubility in aqueous solutions. However, these reagents result in non-used ions as NO3−, Cl−, K+, and Na+, which require several washing in order to eliminate them. Calcium hydroxyde and orthophosphoric acid seem to be good reagents for the synthesis of hydroxyapatite by an acid–base neutralization (Florin et al. 2017). This process results in any non-used ion and the reagents are available at low cost (Doan et al. 2014). Hydroxyapatite used as adsorbent is stoichiometric (Ca/P = 1.67). According to previous work, in order to obtain a stoichiometric hydroxyapatite, the molar ratio of the reagents must be equal to 1.67, the temperature of the reaction must be high (90 ℃), and the aging time of the obtained precipitate must be long (above 24 h).
The objective of this work is to synthesize hydroxyapatite powder by acid–base neutralization at low temperature, reduced aging time, and different acid solution concentration. In these conditions and in order to obtain stoichiometric hydroxyapatite with a Ca/P = 1.67, the reagents were mixed in a molar ratio of 2 (Ca/P = 2). Based on the results of the analyses, the effect of phosphoric acid solution concentration on the pH of the suspension, the particle size distribution, and thermal stability of the Ca-HA are studied.
2 Materials and Methods
2.1 Reagents
The reagents were Ca(OH)2 and H3PO4 (85%) in water. All of them were of high purity (99%) and purchased from Prolabo.
2.2 Synthesis Methods
2.2.1 Powder n°1 (P1)
A solution of Ca(OH)2 was obtained by dissolution of 0.1 mol of Ca(OH)2 in 500 mL of distilled water. The solution was stirred with a magnetic stirrer at 400 rpm and maintained at room temperature (23 ℃). A volume of 3.2 mL of H3PO4 (85% wt., in water), at room temperature, was then added drop wise (1 mL/min) into the Ca(OH)2 solution with a graduated burette. After the total addition of H3PO4, the mixture was continuously agitated for an additional one hour at the same temperature. The pH of the acid–base solution was monitored with pH meter. The precipitate was separated, washed several time with distilled water, filtered then dried à 100 ℃ during 24 h.
2.2.2 Powder n°2 (P2)
A solution of Ca(OH)2 was obtained by dissolution of 0.1 mol of Ca(OH)2 in 250 mL of distilled water. The solution was stirred with a magnetic stirrer at 400 rpm and maintained at room temperature (23 ℃). 3.2 mL of H3PO4 solution was introduced in 250 mL of distilled water and then added drop wise (12.5 ml/min) into the Ca(OH)2solution. The total time addition of H3PO4 solution was 20 min. The obtained precipitate was continuously agitated for an additional one hour at the same temperature. The obtained suspension was washed several times with distilled water, filtered then dried à 100 ℃ during 24 h.
In order to study their thermal stability, the as-dried powders are sintered at 600, 800, 1000, and 1100 ℃ in a furnace under air atmosphere, with a heating rate of 5 °C/min, kept for 1 h, and then the sample was cooled down to room temperature.
2.3 Characterization Methods
2.3.1 Powder X-ray Diffraction (XRD)
The phase composition of the as-synthesized and calcined powders was analyzed by X-ray diffraction with Philips XPERT PRO diffractometer using CuKα radiation (λ = 1.54056 Å) at the X-ray tube voltage 45 kV and a current of 40 mA. The X-ray data were collected over the 2θ range of 20–70° at a step size of 0.0167°.The average crystallite size of the precipitates was estimated from the Debye–Scherrer equation:

where Dhkl is the size (in nm), ß is the full width at half maximum of the peak at half of the maximum intensity, FWHM (in degree), λ is the diffraction wavelength of X-rays (1.5418 Å), θ is the diffraction angle for each reflection, and K the shape factor equals to 0.94.
2.3.2 Particles Size Distribution
A particle size analyzer (Malvern Mastersizer 2000) was used to determine the size distribution of the powder samples.
2.3.3 FTIR Spectroscopy
The typical groups of the synthesized and calcined powders were identified by Fourier-transform infrared spectroscopy (FTIR-4100//ATR ESPECAC). The measurements were obtained at 400–4000 cm−1 with a resolution of 0.4 cm−1.
2.3.4 Elementary Analysis
The elemental analysis was performed by fluorescence X spectroscopy (Magix PRO, PW 2540 vrc).
3 Results and Discussions
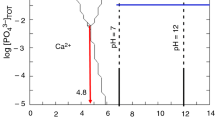
As shown in Fig. 1, the pH of the mixture varies slowly with time when the phosphoric acid solution used is in concentrated form (pH ≈ 12.3), whereas when the phosphoric acid solution used for neutralization is diluted, the pH of the mixture was slowed down to 8.8. These results indicate that the formation mechanism of the powders P1 and P2 is different. Indeed, at a pH of 12.3, the predominant species in solution are PO43− ions, while at a pH of 8.8, there are both PO43− and HPO42−.According to the results showed in Table. 1, the Ca/P molar ratio of powder P2 (Ca/P = 1.20) is higher when compared to powder P1 (Ca/P = 1.15), which can be due to a more substitution of PO43− groups by CO32−.

Evolution of pH with time addition of phosphoric acid to Ca(OH)2 solution
The X-ray diffraction patterns of the as-dried powders are similar as shown in Fig. 2. All the peaks are identified and correspond to HA phase (ICDD N°96-900-2217). The broad and weak peaks diffraction indicates that the powder is a poorly crystallized apatitic phosphate with small crystallite size. This is confirmed by the crystallite sizes estimated from the Debye–Scherrer equation, which are about 8.77 and 10.20 nm for powder P1 and P2, respectively. The low crystallinity of the formed apatitic phosphate can be attributed to the low synthesis temperature used (Elhammari et al. 2007; Elena et al. 2004), which lead to fast precipitation that prevented the crystallization. Moreover, the presence of B-carbonate in anapatite lattice causes a decrease in crystallinity (Elena 2004). Souzaa et al. (2019) showed that the amorphous calcium phosphate (ACP) is very reactive, thus adsorbs atmospheric CO2 and forms a carbonated apatite.

XRD patterns of as-dried powder P1 and P2 at 100 ℃ for 24 h
The particle size distribution of the powder P1 is homogeneous (Fig. 3). 90% of particles have smaller size than 34.85 μm (d90), and 50% of them present smaller size than 9.13 μm (d50). Visually and during synthesis process, the precipitate P2 formed has spherical agglomerates compared to the precipitate P1, which has a very fine appearance. The formation of agglomerates in the case of powder P2 was confirmed by particle size analyses (Fig. 4), which showed that 90% of particles have smaller size than 350.23 μm (d90), and 50% (d50) of them present smaller than 79.40 μm. Our results are not really in agreement with those obtained by Azade and Suat (2018), who have shown that slow or rapid addition of H3PO4 to Ca(OH)2 solution has no influence on the particles size distribution.

Particle size distribution of as-dried powder P1 at 100 ℃ and 24 h

Particle size distribution of as-dried powder P2 at 100 ℃ end 24 h
The FTIR spectra of the as-dried powders P1 and P2 are shown in Fig. 5. All the bands detected for powder P1 are characteristics of functional groups of an apatitic phase (Peipei et al. 2004; Heughebaert et al. 1977; Blumenthal et al. 1972). Intense absorption bands of PO43− groups have been found at 1045 cm−1 (ν3 asymmetric stretching mode), 604 and 571 cm−1 (ν4 bending mode). The ν1 asymmetric stretching mode at 960 cm−1 and ν2 at 470 cm−1 is not observed in the sample P1. The absorption band at 1422 cm−1 is attributed to CO32− groups (ν3 stretching mode) which resulted from partial substitution of PO43− groups by carbonate ions (B-type substitution). The presence of CO32− groups in the lattice of apatitic phase formed is resulted from atmospheric CO2 contamination during the precipitation process. The reactor exposed to air, medium alkaline reaction (pH > 8), and low precipitation temperature, which increases the solubility of CO2 are the main factors that caused this contamination.

FTIR spectra of as-dried powder P1 and P2 at 100 ℃ during 24 h
The reaction of atmospheric CO2 dissolution in an alkaline medium is:
The weak band at 874 cm−1 is attributed to CO32− (A-type substitution) or HPO42− groups (Moghimian et al. 2012). The high intensity bands at 3441 and 1641 cm−1 are attributed to absorbed water molecules. The absence of the bands at 3750 and 630 cm−1 characteristic of OH− groups of hydroxyapatite is assigned to the substitution of these groups by carbonate ions. Although, Gema et al. (2018) have found the same result and indicate that the presence of strong carbonate bands in the range of 1400–1460 cm−1 and the band at 875 cm−1 implies the presence of AB type carbonated substituted apatites (Ca10−x (PO4)6−x (CO3)x (OH)2−x-2y (CO3)y).
The FTIR spectrum of the as-dried powder P2 is similar to that of powder P1. However, two supplementary bands have been identified at 960 cm−1 and 470 cm−1, which are, respectively, attributed to ν1symmetric stretching mode and ν2 bending mode of PO43−. The broad band at 1045 cm−1 corresponding to PO43− groups indicates that the synthetized apatite is poorly crystalline, which confirms the XRD results.
Infrared spectra of the powder P1 calcined at 600, 800, 1000, and 1100 ℃ are reported in Fig. 6. The absorption bands of OH groups of hydroxyapatite appeared at about 600 ℃ and all other peaks in the spectra were characteristics of hydroxyapatite. The sharp peaks of PO43− groups, when the calcination temperature increases, indicate that the crystallinity of hydroxyapatite increases. No decomposition of hydroxyapatite was observed even at 1100 ℃, which confirms their stability (presence of OH groups at 3570 cm−1) (Kang et al. 2019).The same result was obtained with the calcined powder P2 (Fig. 7).The diminution of the band at 1450 cm−1 is assigned to the decomposition of carbonate ions to form CO2 (g).

FTIR spectra of the powder P1: as-dried and calcined at 600, 800, 1000, 1100 ℃ for 1 h at 5 ℃/min

FTIR spectra of the powder P2: as-dried and calcined at 600, 800, 1100 ℃ for 1 h at 5 ℃/min
The XRD spectra of the calcined powder P1 at 600, 800, 1000, and 1100 ℃ are reported in Fig. 8. In comparison with the as-dried powder, the crystallinity of the powders calcined at 800, 1000, and 1100 ℃ was raised and all the diffractions peaks correspond to the hydroxyapatite phase (ICDD N°96-900-2217). Low-intensity peaks at 37.44° and 54.55° in the DRX paterns of powder P1 calcined at 800, 1000 and 1100 ℃ are attibuted to CaO phase, which corresponds to the remained Ca (OH)2 of the reaction or to the no total decomposition of carbonate ions present in the lattice of hydroxyapatite powder (Landi et al. 2004).


DRX patterns of powder P1: as-dried and calcined at 600, 800, 1000, 1100 ℃ for 1 h at 5 ℃/min
4 Conclusion and Future Perspective
Acid–base neutralization is an easy method to prepare hydroxyapatite powder by using Ca(OH)2 and H3PO4 as precursors. The results of this work have shown that using rich calcium solution allows preparing hydroxyapatite at room temperature (23 ℃) at reduced aging time (1 h) by the calcination of the obtained precipitate at 600 ℃ for 1 h. The particle size of the obtained powder depends on the concentration of the acidic solution. The evolution of pH of synthesis with time depends on the concentration of the acid solution as well. The obtained hydroxyapatite powder is stable even at high temperature of treatment. Depending on the application field of the synthesized powder, other properties are required to study in the future as the morphology of the particles, surface area, and porosity of the powder.
References
Azade Y, Suat Y (2018) Wet chemical precipitation synthesis of hydroxyapatite (HA) powders. Ceram Int 44:9703–9710
Blumenthal C, Posner AS, Holms JM (1972) Effect of preparation conditions on the propperties and transformation of amorphous calcium phosphate. Mat Res Bull 7:1181–1190
Doan PM, Haroun S, Ange N, Patrick S (2012a) Apatitic calcium phosphate: Synthesis, characterization and reactivity in the removal of lead (II) from aqueos solution. Chem Eng J 180–190
Doan PM, Nathalie L, Haroun S, Ange N, Sharrock P (2012b) Synthesis of calcium hydroxyapatite from calcium carbonate and different orthophosphate sources: a comparative study. Mater Sci Eng B 177:1080–1089
Doan PM, Sébastien R, Patrick S, Haroun S, Nathalie L, Ngoc DT, Mohamed R, Ange N (2014) Hydroxyapatite starting from calcium carbonate and orthophosphoric acid: synthesis, characterization, and applications. J Mater Sci 49(12):4261–4269
Elena L, Anna T, Giancarlo C, Lucia V, Monica S (2004) Influence of synthesis andsintering parameters on the characteristics of carbonate apatite. Biomaterials 25:1763–1770
Elhammari L, Merroun H, Coradin T, Cassaignon S, Laghzizil A, Saoiabi A (2007) Mesoporous hydroxyapatites prepared in ethanol–water media: structure and surface properties. Mater Chem Phys 104:448–453
Florin M, Aura-Catalina M, Catalina AD, Andreea M, Dan B, Andrei B, Stefan IV, Marian M, Vijay KT, Lucian TC (2017) Facile synthesis and characterization of hydroxyapatite particles for high value nanocomposites and biomaterials. Vacuum 146:614–622
Gema G, Cesar CV, Luis JB, Damarys S, Lisbeth L, Jose IC, Camilo JD, Luis L (2018) Effect of carbonates on hydroxyapatite self-activated photoluminescence response. J Lumin 195:385–395
Haifeng G, Changhua J, Zhenxuan X, Pingying L, Zhaofu F, Jia Z (2019) Synthesis of bitter gourd-shaped nanoscaled hydroxyapatite and its adsorption property for heavy metal ions. Mater Lett 241:176–179
Heughebaert J-C, Montel G (1977) Etude de l’évolution de l’orthophosphate tricalcique non cristalin en phosphate apatitique à la faveur d’une réaction chimique, à température ordinaire. Revue de physique appliquée. Tome 12
Kang S, Seo JT, Park S-H, Jung Y, Lee CY, Park J-W (2019) Qualitative analysis on crystal growth of synthetic hydroxyapatite influenced by fluoride concentration. Arch Oral Biol 104:52–59
Landi E, Tampieri A, Celotti G, Vichi L, Sandri M (2004) Influence of synthesis and sintering parameters on the characteristics of carbonate apatite. Biomaterials 25:1763–1770
Maya I, Madona L, Jean-M G, Jean-François L (2020) Hydroxyapatite, a multifunctional material for air, water and soil pollution control: a review materials. J Hazard 383:121–139
Mehdi SS, Mohammad-TK, Ehsan DK, Ahmad (2013) J Synthesis methods for nanosized hydroxyapatite with diverse structures. Acta Biomaterialia 9:7591–7621
Michele F, Sebastiano C, Marco S, Claudio E, Paolo C, Antonella G (2019) In-depth study of the mechanism of heavy metal trapping on the surface of hydroxyapatite. Appl Surf Sci 475:397–409
Moghimian P, Najafi A, Afshar S, Javadpour CJ (2012) Effect of low temperature on formation mechanism of calcium phosphate nano powder via precipitation method. Adv Powder Technol 23:744–751
Peipei W, Caihong L, Haiyan G, Xuerong J, Hongqiang W, Kaixing L (2010) Effects of synthesis conditions on the morphology of hydroxyapatite nanoparticles produced by wet chemical process. Powder Technol 203(2):315–321
Sangeetha K, Vidhya G, Vasugi G, Girija EK (2018) Lead and cademium removal from single and binary metal ion solution by novel hydroxyapatite/alginate/gelatin nanocomposites. J Environ Chem Eng 6(1):1118–1126
Souzaa FS, Matosb MJS, Galvãoa BRL, Arapiracaa AFC, Silvaa SN, Pinheiroa IP (2019) Adsorption of CO2 on biphasic and amorphous calcium phosphates: an experimental and theoretical analysis. Chem Phys Lett714:143–148
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Zamoume, O., Mammeri, R. (2021). Synthesis of Hydroxyapatite by Wet Method: Effect of Acid Solution Concentration on Powder Granulometry. In: Trache, D., Benaliouche, F., Mekki, A. (eds) Materials Research and Applications. Materials Horizons: From Nature to Nanomaterials. Springer, Singapore. https://doi.org/10.1007/978-981-15-9223-2_9
Download citation
DOI: https://doi.org/10.1007/978-981-15-9223-2_9
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-9222-5
Online ISBN: 978-981-15-9223-2
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)