
Abstract
Immune checkpoint inhibitors that target T-cells to activate immune response against tumors have shown remarkable clinical responses emerging as new potent weapon against cancer. This therapy has led to durable responses in hard to treat tumor types with long-term remissions. The field of immune-oncology has greatly evolved in recent times primarily by our enhanced understanding of T-cell stimulation and checkpoint blockade, primarily of CTLA-4 and PD-1. Clinical responses although remarkable are, however, limited to limited pool of patients and indications. This calls for further understanding of underlying biological mechanism and function of an optimal immune response. As the immune response evolves, it is unlikely to have a single actionable biomarker to predict clinical response but rather we would need a panel of markers to guide in development of therapy. Clinically validated biomarkers would therefore be needed ultimately for optimal patient and regimen selection. Clearly, the way forward is deeper understanding of our immune system and its dynamic interaction with tumor environment. The magnitude of immune response and its regulation will have to be targeted through a combination approach to provide benefit to wide range of patients and tumor types. If done properly, there is a strong chance of turning this hope into reality.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
2.1 Introduction
Immune response as a treatment modality for response to cancerous growth has been investigated for several decades. In one of the earliest efforts, William Coley, a surgeon observed that cancer patients with post-operative infections tend to have a better clinical outcome, thus correlating immune response with cancer growth inhibition. In decades to follow, several immunotherapeutics were approved for cancer that included Bacillus Calmette-Guerin, interferon-α, and interleukin-2 (IL-2). Effects of Interleukin-2 in clinic were significant with regard to mechanism of action as it demonstrated durable effect in several tumors. These clinical studies acted as Proof of Concept (POC) that a cytokine capable of expanding T-cells demonstrated durable response in difficult to treat tumors such as advanced melanoma. In essence it also emphasized the important role of adaptive immunity in treating tumorous growth. In the ensuing decades, intense research activity was focused on developing mechanistic understanding in fields of immunology, virology, molecular biology, and cell biology. The decades of investment in basic sciences finally yielded two major breakthroughs in cancer immunotherapy: Chimeric Antigen Modified Receptor (CAR) modified T-cells and Immune activation using antibodies that act as immune checkpoint inhibitors.
2.2 Preclinical Evidence of Immune Modulation
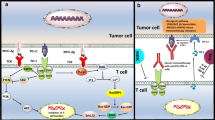
Tumor microenvironment is very heterogeneous consisting of cancerous cells with multiple genetic alterations, fibroblasts, and host of other immune cells. They include cells with lytic capacity such as NK cells, macrophages, and most importantly T-cells. As a result of this lysis, tumor specific antigens expressed on tumor cells get bound to Major Histocompatibility Complex (MHC) on cells. T-cells get activated on recognition of tumor antigens leading to proliferation, differentiation and leading to destruction of cells expressing antigens. T-cell response, however, is a complex process consisting of stimulatory and inhibitory pathways which ultimately determines its response to cancer and eradication of tumors.
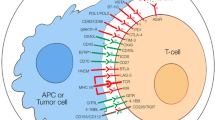
Recognition of antigen by T-cell receptor is just not sufficient to activate naïve T-cell which requires additional costimulatory signals. This costimulation is in essence a checkpoint to ensure whether T-cell activation is truly required. These costimulatory signals are provided by engagement of CD28 on T-cell surface with B7 molecules (CD80 and CD86) on APC. B7 molecules are typically expressed on subsets of hematopoietic cells such as dendritic cells that possess special ability for effective antigen presentation. Cancer cells typically do not possess B7 molecules and are thus as a result largely invisible to immune system. In an inflammatory response which leads to killing of tumor cells, APC presenting cells such as dendritic cells take up antigens and present it along with B7 molecules needed for effective activation of T-cells. Once the tumor specific T-cells are activated, they move to tumor site to mount an attack. Effective tumor site infiltration is a critical hurdle that must be overcome for effective tumor response. Tumor microenvironment can be a significant barrier put up by a host of cancer cells, stroma, regulatory T-cells, suppressor cells, and cytokines effectively blunting immune response. A major breakthrough in understanding of immune modulation occurred when a protein known as cytotoxic T Lymphocyte-associated protein 4 (CTLA-4) displayed potent inhibitory role in managing T-cell response. CTLA-4 in resting T-cells is located within the cell. Studies done by two groups, one led by James Allison and other by Jeffrey Bluestone showed that during the T-cell receptor (TCR) engagement, there is a costimulatory signal activated via CD28 binding with costimulatory molecules (CD80, CD86) (Chambers et al. 2001; Walunas et al. 1994). Following the activation, CTLA-4 moves to cell surface and outcompetes CD28 for binding with costimulatory molecules (CD80, CD86) due to much higher binding affinity. Because both CD80 and CD86 provide positive costimulatory signals through CD28, inhibition of both molecules by CTLA-4 is necessary for effectively attenuating T-cell activation. Both CD28 and CTLA-4 exhibit rapid binding kinetics with B7–1 which coupled with different binding strengths allows for swift competitive inhibition by CTLA-4. In addition to enhanced expressions, CTLA-4 contained in intracellular vesicles is rapidly transported to immunologic synapse. This movement to synapse is directly correlated with TCR signal strength. At synapse, CTLA-4 is stabilized by B7–1 binding allowing for accumulation and outcompeting with CD28. This ultimately leads to robust regulation of TCR signal amplitude and activity. Thus CTLA-4 primarily functions to regulate T-cell activity at sites of T-cell priming and in peripheral tissues. The binding of CTLA-4 with costimulatory molecules leads to inhibition of T-cell activation resulting in loss of proliferation and activation. Allison proposed that if this inhibition of T-cell activation could be blocked temporarily preferably using antibodies, this would allow the T-cell to activate and proliferate above the normal physiological levels. They further proposed to combine CTLA-4 inhibition with agents that directly kill tumor cells to release tumor specific antigens for presentation by APC which would improve anti-tumor response. To validate this hypothesis, CTLA-4 knockout animals were generated to understand the effect of CTLA-4 deficiency in a whole animal model. CTLA-4 knockout animals died due to hyperimmune activation leading to lymphocyte infiltration in several organs confirming the role of CTLA-4 as a non-redundant co-inhibitory protein.
In early POC preclinical studies to evaluate blockade of CTLA-4, effect of Anti-CTLA-4 Antibody were evaluated in xenograft tumor models. The tumors evaluated were wild type, unmanipulated established tumors. These studies conclusively showed that checkpoint inhibition using Anti-CTLA-4 Antibody led to durable regression of established tumors insyngeneic animal models (Leach et al. 1996). Thus the Anti-CTLA-4 Antibody had a dramatic curative effect on tumors. In addition, these studies suggested that CTLA-4 blockade provided enhanced immunity to secondary challenge. Animals that were cured of tumors were reinjected with tumor cells approximately two months after earlier tumors had regressed. Significant number of animals remained tumor free suggesting that tumor rejection mediated by CTLA-4 blockade results in immunologic memory. These definitive preclinical POC studies demonstrated that removing inhibitory signaling in costimulatory pathway enhances anti-tumor immunity. This is achieved by affecting T-cell response in two nonexclusive ways; non-reactive T-cells convert to active cells by lowering the threshold of activation or removing inhibitory signaling leads to sustained T-cell proliferation.
2.3 Early Clinical Evidence of Immune Modulation
After it was conclusively shown in animal models that immune checkpoint inhibition leads to regression of tumors, the next step was clinical validation of these preclinical results. Early Phase-I trials with Anti-CTLA-4 antibody, Ipilumumab (MDX-010) with multiple tumor types including melanoma showed remarkable results (Ribas et al. 2005; Hodi et al. 2003). In hard to treat tumors such as melanoma, a remarkable >10% objective response (OR) was observed. This regression of tumor was quite durable (>10 years) even after stopping therapy. In contrast to a typical chemotherapy response where the responses are observed early, there is a delayed response in case of immune checkpoint inhibition where there is initial progression or new tumorous growth appearing followed by regression. This atypical response, however, led to challenges in the manner efficacy could be evaluated for such treatment where the typical regulatory assessment involved calculating an objective response (OR) or progression free survival (PFS). In case of immune checkpoint inhibition, a more long-term efficacy assessment such as Overall Survival (OS) as a primary end point would be more appropriate. Following an impressive early phase trials that clearly demonstrated efficacy of Ipilumumab in hard to treat tumors such as melanoma, large scale phase three trials were conducted to confirm these early phase findings. Ipilumumab clearly extended PFS compared to peptide vaccine or standard dacarbazine chemotherapy in late stage melanoma patients becoming the first immunotherapeutic agent to be approved by FDA in 2011.
2.4 PD-1 Another Important Immune Checkpoint Player
Another important immune checkpoint that has a dominant impact on downregulating T-cell activation is programmed cell death (PD-1) receptor. Initially discovered role of PD-1 was cell death inducer as name suggest. However, further work shed light on another important role an as immune checkpoint. PD-1 receptor has two ligands, PD-L1 which is broadly expressed by somatic cells in response to proinflammatory cytokines, and PD-L2 which is more restricted to antigen presenting cells (APC) (Ishida et al. 1992).
CTLA-4 as discussed above is a downregulator of T-cell activation upon engagement with tumor specific antigen on presentation by APC which primarily happens at lymph nodes. Once the T-cell is activated by blocking CTLA-4, they circulate inside body to locate cognate antigen presented by cancer cells. Upon engagement with antigen, TCR activation also leads to expression of PD-1 which acts as negative regulatory receptor (Keir et al. 2006). There are two ways by which tumor cells can express PD-L1 providing them immunity from immune response; “innate immune resistance” and “adaptive immune resistance.” Innate immune resistance refers to constitutive expression of PD-L1 by tumor cells due to gene amplification or aberrant oncogenic pathway activation. In contrast to innate immune resistance, adaptive immune resistance refers to PD-L1 expression in response to proinflammatory cytokines released by tumor/immune cells in response to immune response. While INF-g is primary cytokine thought to be responsible for PD-L1 expression, other cytokines which are resident of tumor microenvironment such as IL-1, IL-10, IL-27, and IL-32 can upregulate PD-L1 in tumor/immune cells. This deactivation of T-cells can be overcome by blocking PD-L1/PD-1 pathway.
2.5 Impressive Anti-Tumor Effect on PD-1 Blockade
Blocking of PD-1 or PD-L1 interaction becomes relevant to cancer cells as it leads to deactivation of immune response via preferential blockade of activated T-cells. The clinical effect of blockade of activated T-cells was tested in an early Phase-I trial using a fully human monoclonal Antibody, Nivolumab as PD-1 inhibitor. Nivolumab is a fully human IgG4 antibody which binds to PD-1 receptor with nanomolar affinity along with high specificity for PD-1. Impressive Objective responses (OR) in the range of 40% were observed in a wide range of tumors that included melanoma, renal cell carcinoma, and non-small cell lung cancer with low incidences of toxicity (Joseph et al. 2013). These impressive clinical responses with PD-1 blockade in heterogeneous tumor population led to several clinical trials with PD-1 and PD-L1 Antibodies. First FDA approvals for PD-1 inhibitors were granted in 2014 in refractory melanoma and non-small cell lung cancer in 2015 under accelerated and breakthrough pathways. Subsequently, in 2016 first PD-L1 antibody approved was atezolizumab for urothelial cancers followed by avelumab for Merkel cell carcinoma in 2017. The next immediate question is to identify the type of tumors that would respond to PD-1/PD-L1 blockade. As the body of evidence grows, responding tumors appear to be either carcinogen induced or viral infections driven. Common variants of Melanoma which are carcinogen induced have shown high response rates in range of 35–40% with PD-1 blockade. Another series of cancers associated with carcinogenic effect of cigarette smoking such as NSCLC have also shown impressive response rates of 20% with PD-1 blockade.
2.6 Predictive Biomarkers of Immune Response
There is a compelling need to identify biomarkers that would aid in patient and regimen selection to ultimately predict clinical outcome to immunotherapy. Although there are several strong candidates, a definitive single predictive biomarker is still lacking. Several candidate biomarkers are largely based on mechanistic understanding of anti-tumor immune response. Simplistically, T-cell should be able to infiltrate tumor microenvironment, get activated by immune modulating agents and recognize tumor derived antigens. Intuitively, extent of T-cell infiltration along with PD-1/PDL-1 expression in a tumor microenvironment would be a good predictor of immune response. Indeed, patients responding to immunotherapy had a higher degree of existing activated T-cells (CD8+) at tumor margins. Another range of biomarkers would be immune checkpoint expression along with their respective ligands: PD-1, PD-L1 or CTLA-4. Higher expression of inhibitory proteins such as PD-1 or CTLA-4 on circulating T-cells was associated with better clinical outcomes (PFS) after treatment with Anti-CTLA-4 therapy. Melanoma patients who had higher expression of CTLA-4 and PD-2 responded favorably to Anti-CTLA-4 antibody Ipilumumab compared to those with lower expression. A similar result was observed in lung cancer patients where patients with >50% PDL-1 positive showed an improved response (Daud et al. 2016). Although these results have tempted oncologists to use PD-L1 as a marker for selection of patients, the data is yet not conclusive. Interestingly, there are significant numbers of patients that have benefitted with Anti-PD-1 therapy in spite of lower expressions of PD-L1. This could be due to limitations of single biopsy sample not able to capture the dynamic expression of PD-L1 or heterogeneity of expression. Ability of cells to express PD-L1 is directly correlated with expression of Interferon Regulatory Factor (IRF-1). In clinical trials of melanoma treatment by different immune modulators (Anti-PD-1, Anti-CTLA-4), IRF-1 expression was found to be correlated with good response rate.
Cancerous cells are often product genetic mutations that result in novel proteins which can act as antigens. The uniqueness of antigen presentation can lead to effective immune response upon activation of T-cells. As the number of mutations in a cancerous cell increases, so is the possibility of having a unique antigen capable of invoking immune response. In melanoma which is known to harbor high rate of mutations, patients treated with Anti-CTLA-4 was significantly associated with clinical outcome. However, high mutational burden is not an exclusive predictor of clinical outcome. In the final step of immune response, Cytotoxic killer T-cells mount cell killing response involving perforin and granzyme. It was observed retrospectively that Ipilumumab responders had enriched perforin and granzyme transcripts compared to non-responders.
Several peripheral blood markers have shown promise as a prognostic marker for response. As such these markers are appealing because of ease of assessment and offering longitudinal evaluation. One such marker is serum lactate dehydrogenase (LDH) where elevated levels have indicated worst prognosis in case of melanoma. In one of the trials, melanoma patients with lower levels of baseline LDH had a better overall survival. Several other markers that have shown promise in clinical setting are absolute monocyte count, CD14+ monocytes, and absolute eosinophil count.
Clinical features can also be useful indicator of immune response resulting from immunotherapy. Analysis of patient subset that have responded to immunotherapy also tend to have immune related adverse event (irAEs). The nature of adverse event suggests immune activation by immunotherapy. In melanoma trials with PD-1 inhibitor Nivolumab, those patients that had experienced any irAEs had a significantly longer overall survival compared to those who did not (Freeman-Keller et al. 2016). Among the notable, cutaneous irAEs such as rash and vitiligo were strongly correlated to longer overall survival. irAEs can thus provide valuable prognostic information in a minimally non-invasive manner.
2.7 Resistance to Immune Checkpoint Inhibition
Although the overall response to immunotherapy in broad range of tumors has been quite remarkable, there are still certain type of tumors that do not respond (Primary Resistance) and in some cases those that respond initially develop an acquired resistance to intervention. As a result these non-responder patients endure toxicities and treatment cost with no clinical benefit. Interestingly, different responses have been observed for different tumors even within the same patient. Functioning of Immune system is dependent on host of different factors such as environment, genetic factors as well as interventions such as chemotherapy and radiation. Accordingly, the anti-tumor response within a patient is also dynamic and is affected by host of different factors leading to either primary or acquired resistance. Both of these types of resistance could be attributed directly to Tumor cell or Non-Tumor cell related factors. Multiple Tumor cell related factors have been identified such as (1) Activated MAP pathways, (2) expression of WNT/b-catenin pathway, (3) loss of IFN-y pathway, and (4) lack of T-cell response due to poor antigen presentation. Activation of oncogenic MAP pathway is known to inhibit T-cell recruitment and function. The interferon-gamma pathway appears to have both positive and a negative effect on anti-tumor immune response. Interferon-gamma expression upon T-cell activation has shown to recruit other immune cells, improved antigen presentation and have a direct pro-apoptotic effect on tumor cells. On the other hand, continuous Interferon-gamma signaling can lead to immunoediting of cancer cells.
Outside of tumor, several mechanisms can contribute to immunotherapy resistance. Tumor microenvironment contains a host of immune related components such as regulatory T-cells (T-regs), myeloid derived suppressor cells (MDSC) all of them can contribute to anti-tumor response. It is well known that T-regs can down regulate effector T-cells response by secreting inhibitory cytokines. Depletion of T-regs in tumor microenvironment animal model has shown to enhance or restore anti-tumor immunity. MDSC have lately shown to play a major role in regulating immune response. MDSC have been implicated in tumor metastasis and invasion. The mere presence of MDSC of tumor microenvironment is correlated with decreased efficacy of immunotherapy agents. Macrophages especially those associated with tumor (TAM) are known to provide anti-tumor immunity along with pro-tumorigenic effect.
In spite of remarkable anti-tumor response especially in hard to treat tumor such as melanoma, close to one third patients who had previously shown improvement with anti-CTLA-4 or Anti-PD-1 tend to progress. This acquired resistance to immunotherapy is in spite of continued drug therapy. Several mechanisms can contribute to this resistance that includes loss of T-cell function (Exhaustion), lack of antigen recognition, and development of mutant forms of tumor. Activated T-cells can undergo phenotypic changes leading to loss in their cytotoxic activity as a result patient develops tumor relapse. T-cell activation occurs after tumor antigen presentation and recognition. Any changes in these neo-antigens can lead to non-recognition by T-cells. Any genetic deletions, mutations or epigenetic changes can lead to loss of expression of neo-antigens making the tumor non-responsive to immunotherapy. Continued research in this area is shedding light on several other inhibitory checkpoints in tumor microenvironment such as LAG-3, TIGIT, and VISTA. Classical approach of combining therapies with different mechanism has shown promise with immunotherapy agents as well. Combination of anti-CTLA-4 plus anti-PD-1 has shown improved overall survival compared to monotherapy alone for patients with metastatic melanoma.
2.8 Clinical Strategies to Overcome Resistance
As we develop a better mechanistic understanding of drug resistance, combination strategies using multiple targeted approach is evolving and is being currently tested in clinic. The hope behind this approach is by of targeting different immune escape pathways is that it leads to improved patient outcomes.
One of the initial strategies was to combine anti-CTLA-4 along with anti-PD-1 which is now already approved in multiple tumor types. Long-term survival data with combination therapy is descriptively superior with dual blockade. Complimentary mechanism of dual blockade is a likely reason for beneficial effect. In addition, anti-CTLA-4 leads to depletion of T-regs along with broader antigen recognition. Combination with chemotherapy leads to tumor cell death leading to increased release of antigens. Similar is the effect of combination with radiation therapy leading to inflamed tumor microenvironment.
Overall immune response after T-cell activation largely depends upon extent of costimulation and coinhibition. T-cells can be activated using several costimulatory agonists including OX40, CD40, GITR, and ICOS. There appears a strong rationale for combining these with immune checkpoint inhibitors. Targeting suppressive signaling in tumor microenvironment (TME) along with depleting T-regs may improve response to immunotherapy. Colony stimulating factor-1 receptor (CSF1R) inhibition along with simultaneous anti-CTLA-4 or anti-PD-1 has shown promise in tumor inhibition in a preclinical pancreatic tumor model. CSF1R inhibition also leads to reprogramming of macrophages for better antigen presentation. Chemokines/cytokines can also modify TME via recruitment of inhibitory cells leading to drug resistance. They bind to their respective receptors on immune-suppressive cells including CXCR2 and CXCR4. Inhibition of these pathways along with anti-PD1 has shown to prevent immune evasion in preclinical models.
Combination with small molecule or large molecule targeted therapies has shown mixed results. Different growth factors and angiogenic factors are known to affect immune response leading to immune suppression. Immune therapy is often associated with reduced recruitment of blood vessels and activated cytotoxic T-cells, anti-angiogenic therapy with similar effects could potentially act in synergistic manner. VEGF inhibitors in combination with immune checkpoint inhibitors have led to normalization of immune-suppressive TME and thereby reversing resistance. Based on positive clinical outcome, Bevacizumab in combination with atezolizumab and chemotherapy recently gained US-FDA approval in patients with metastatic NSCLC. Combining BRAF/MEK inhibitors with anti-CTLA4 led to increased toxicity while combination with anti-PD1 has shown promise with enhanced anti-tumor immunity and tolerability. Early preclinical data with inhibitors of PI3K, CDK4/6 in combination with checkpoint inhibitors have also shown promising results suggesting a potential treatment option.
2.9 Future Path
Cancer treatment modalities have shown a remarkable improvement with the introduction of immune checkpoint inhibitors. The development of immune checkpoint inhibitors has brought hope to patients with hard to treat tumors and opened new avenues in understanding of cancer immunology. As a consequence, there are several hundred clinical trials exploring anti-tumor effect in various tumor types. Despite these remarkable results, for the majority of those enrolled patients the benefits are short lived if at all it occurs followed by rapid resistance. High incidences of immune related adverse reactions make it ethically challenging to assign trials without a strong biological rationale. In some cases the curative effect though has been restricted to specific tumors due to tumor intrinsic or extrinsic resistance mechanism.
There are a several emerging checkpoint inhibitors that could act as monotherapy targets or as a part of combination therapy. Although the focus currently is on anti-CTLA-4 and anti-PD-1 inhibitors, some of these emerging checkpoint inhibitors are much more potent and could possibly offer a better patient outcome. Some of these next generation targets are VISTA, LAG-3, TIGIT, and TIM-3. One of the possible scenarios could be anti-CTLA-4 and anti-PD-1 inhibitors forming the primary activation pathway while the newer generation inhibitors which have overlapping effects could act in synergistic manner.
Overall, Immune checkpoint inhibitors are a powerful modulator of cell immunity forming backbone for all future cancer treatment modalities. However, to realize this would require an understanding of genomic, epigenomic, and cellular features that drive both tumor response and resistance. Having a clear understanding of immune pathway to outline whether a sequential or a combination approach would be best to prevent immune evasion. Although progress is made in identifying inflamed or mutated tumors that are likely to respond, ability to counteract cold and excluded tumors is still lacking. Further, current treatment modalities such as chemotherapy, targeted therapy, radiation that directly regulate TME, timing of immune checkpoint inhibitors to maximize immune response is critical. Patient selection in clinical trials is also critical since majority of those are heavily pretreated suggesting that early immune inhibition might be more beneficial. Further understanding of tumor biology and early induction of immune response might lead to early tumor effect followed by long-term remission. Overall, it is clear that Immune checkpoint inhibitors will be cornerstone for cancer therapy and in future could offer cure for devastating disease.
References
Chambers CA, Kuhns MS, Egen JG, Allison JP (2001) C TLA -4-mediated inhibition in regulation of T cell responses: mechanisms and manipulation in tumor immunotherapy. Annu Rev Immunol 19:565–594. https://doi.org/10.1146/annurev.immunol.19.1.565
Daud AI, Loo K, Pauli ML, Sanchez-Rodriguez R, Sandoval PM, Taravati K, Tsai K, Nosrati A, Nardo L, Alvarado MD, Algazi AP, Pampaloni MH, Lobach IV, Hwang J, Pierce RH, Gratz IK, Krummel MF, Rosenblum MD (2016) Tumor immune profiling predicts response to anti-PD-1 therapy in human melanoma. J Clin Investig 126(9):3447–3452. https://doi.org/10.1172/JCI87324
Freeman-Keller M, Kim Y, Cronin H, Richards A, Gibney G, Weber JS (2016) Nivolumab in resected and unresectable metastatic melanoma: Characteristics of immune-related adverse events and association with outcomes. Clin Cancer Res 22(4):886–894. https://doi.org/10.1158/1078-0432.CCR-15-1136
Hodi FS, Mihm MC, Soiffer RJ, Haluska FG, Butler M, Seiden MV, Davis T, Henry-Spires R, MacRae S, Willman A, Padera R, Jaklitsch MT, Shankar S, Chen TC, Korman A, Allison JP, Dranoff G (2003) Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Proc Natl Acad Sci USA 100(8):4712–4717. https://doi.org/10.1073/pnas.0830997100
Ishida Y, Agata Y, Shibahara K, Honjo T (1992) Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J 11(11):3887–3895. https://doi.org/10.1002/j.1460-2075.1992.tb05481.x
Joseph G, Horak CE, Inzunza D, Cardona DM, Simon JS, Gupta AK, Sankar V, Park J-S, Kollia G, Taube JM, Anders R, Jure-Kunkel M, Novotny J Jr, Taylor CR, Zhang X, The JC (2013) Association of tumor PD-L1 expression and immune biomarkers with clinical activity in patients with non-small cell lung cancer (NSCLC) treated with nivolumab (Anti-PD-1; BMS-936558; ONO-4538). J Clin Oncol 31:2013. (Suppl; Abstr 3016)
Keir ME, Liang SC, Guleria I, Latchman YE, Qipo A, Albacker LA, Koulmanda M, Freeman GJ, Sayegh MH, Sharpe AH (2006) Tissue expression of PD-L1 mediates peripheral T cell tolerance. J Exp Med 203(4):883–895. https://doi.org/10.1084/jem.20051776
Leach DR, Krummel MF, Allison JP (1996) Enhancement of antitumor immunity by CTLA-4 blockade. Science 271:1734–1736. https://doi.org/10.1126/science.271.5256.1734
Ribas A, Camacho LH, Lopez-Berestein G, Pavlov D, Bulanhagui CA, Millham R, Comin-Anduix B, Reuben JM, Seja E, Parker CA, Sharma A, Glaspy JA, Gomez-Navarro J (2005) Antitumor activity in melanoma and anti-self responses in a phase I trial with the anti-cytotoxic T lymphocyte-associated antigen 4 monoclonal antibody CP-675,206. J Clin Oncol 23:8968–8977. https://doi.org/10.1200/JCO.2005.01.109
Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, Thompson CB, Bluestone JA (1994) CTLA-4 can function as a negative regulator of T cell activation. Immunity 1(5):405–413. https://doi.org/10.1016/1074-7613(94)90071-X
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Syed, S. (2021). Immunotherapy in Cancer: Immune Checkpoint Inhibitors; Changing Oncology Treatment Paradigm. In: Sawarkar, S.P., Nikam, V.S., Syed, S. (eds) Immunotherapy – A Novel Facet of Modern Therapeutics. Springer, Singapore. https://doi.org/10.1007/978-981-15-9038-2_2
Download citation
DOI: https://doi.org/10.1007/978-981-15-9038-2_2
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-9037-5
Online ISBN: 978-981-15-9038-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)