
Abstract
Immune checkpoint blockade (ICB) has been proven to be an effective strategy for enhancing the effector activity of anti-tumor T cells, and checkpoint blockers targeting CTLA-4, PD-1, and PD-L1 have displayed strong and durable clinical responses in certain cancer patients. The new hope brought by ICB therapy has led to the boost in therapeutic development of ICBs in recent years. Nonetheless, the therapeutic efficacy of ICBs varies substantially among cancer types and patients, and only a proportion of cancer patients could benefit from ICBs. The emerging targets and molecules for enhancing anticancer immunity may bring additional therapeutic opportunities for cancer patients. The current challenges in the ICB therapy have been discussed, aimed to provide further strategies for maximizing the efficacy of ICB therapy.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
23.1 Introduction
Immunotherapy has emerged as an attractive treatment option for many kinds of cancer patients, in particular, immune checkpoint blockade (ICB) therapies that enhance the function of anti-tumor T lymphocytes have been especially promising in the clinic. Compared with other immunotherapies, ICB therapies often show higher response rates and long-lasting responses, even in patients with advanced cancer (Busato et al. 2019). Ipilimumab, a cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4) blocker, was the first ICB approved by the FDA in 2010 (Lipson and Drake 2011). It provided a new treatment option for the metastatic melanoma patients who previously lacked any effective treatments (Graziani et al. 2012). Until now, many kinds of ICB agents that targeting CTLA-4, programmed cell death-1 (PD-1), and PD-L1 have been approved for many kinds of cancers, such as metastatic melanoma (Postow et al. 2015; Deeks 2016; Rosenberg et al. 2016), non-small-cell lung cancer (NSCLC) (Wolchok et al. 2010; Rizvi et al. 2015; Dang et al. 2016), Hodgkin lymphoma (Kasamon et al. 2017), urothelial cancer (Kim et al. 2019; Burgess et al. 2019), hepatocellular carcinoma(HCC) (Hage et al. 2019; Kudo 2019), gastric cancer (Chen et al. 2019; Park et al. 2018), head and neck squamous cell carcinoma(HNSCC) (Yu et al. 2018; Sim et al. 2019), microsatellite instability-high colorectal cancer (CRC), and other MSIhigh cancers (Middha et al. 2019; Overman et al. 2017; Marginean and Melosky 2018). The common mechanism of these ICBs is through the activation of anti-tumor T-lymphocyte responses and overcoming tumor immune supervision.
However, the ICB therapy usually has many shortcomings, such as the effectiveness of ICBs varies in different kinds of cancers, and even if in melanoma, most of the patients cannot benefit from the ICB therapy (Puglisi et al. 2010; Khalil et al. 2016). Other more common cancers, such as breast cancer and MSIlow CRC patients rarely could benefit from ICB therapies (Reck et al. 2016; Kindler et al. 2012; Alexandrov et al. 2013; Polk et al. 2018; Hermel and Sigal 2019). The reason for this phenomenon is that the effectiveness of ICBs in different tumors and patients is strongly affected by the tumor’s mutation load and the local tumor microenvironment (TME) (Tumeh et al. 2014; Hamada et al. 2018).
In this chapter, we will first summarize the clinical development of the ICBs related to CTLA-4, PD-1, and PD-L1, then we will describe the emerging new ICB agents besides the CTLA-4, PD-1, and PD-L1, and finally we will explore the current challenges of immune checkpoint blockade therapy in cancer.
23.2 Therapeutic Development of CTLA4 Blockade
23.2.1 The CTLA-4 Immune Checkpoint
CTLA-4 was the first identified negative regulator of T-cell activation. It belongs to the immunoglobulin superfamily and has similar structures as T-cell surface molecule CD28 with similar functional properties (Linsley et al. 1994). CTLA-4 shares the same B7 ligands as CD28, including B7-1 (CD80) and B7-2 (CD86), but the affinity of CTLA-4 for both ligands is about 100-fold higher than that of CD28 (Sansom 2000). CD28 ligation by the B7 family ligands results in a positive co-stimulatory signal needed by the T lymphocytes for optimal cytokine secretion and proliferation (Chen et al. 2019). However, after T-cell receptor (TCR) activation, CTLA-4 is upregulated and binds CD80/CD86, resulting in reduced T-lymphocyte proliferation and lessened cytokine secretion (Engelhardt et al. 2006). More and more studies supported the idea that CTLA-4 functioned as an important negative regulator of T-lymphocyte activation. In the early stage of carcinogenesis, CTLA-4 could decrease the T lymphocyte activation by producing inhibitory signals to weaken the immune response against tumor cells (Rowshanravan et al. 2018); CTLA-4 could trigger reverse signaling through B7 ligands to induce indoleamine-2, 3-dioxygenase (IDO) and results in inhibition of T-cell proliferation (Boasso et al. 2005); recent studies also revealed that CTLA-4 could induce inhibition of PI3K/Akt pathways, cyclin-dependent kinases, and nuclear transcription factor (NF-κB) (Parry et al. 2005; Ghorpade et al. 2011); in addition, CTLA-4 inhibition may also involve in regulatory CD4+ T-cell (Treg) activation which then suppresses CTL functions by “stripping” CD80/CD86 from APCs (Qureshi et al. 2011).
However, in spite of extensive researches on CTLA-4, the mechanism of CTLA-4 interacting with its ligands or its downstream targets and the action of CTLA-4 blockade still need to be further investigated.
23.2.2 Immune Checkpoint Inhibitors Targeting CTLA-4
Based on CTLA-4’s role in the negative regulation of T-cell activation, antibodies
that block CTLA-4 and B7 ligands interaction have become attractive targets for cancer therapies. Antibodies that block CTLA-4 have demonstrated anti-tumor effect first in mouse models, then in cancer patients (Peggs et al. 2009). Based on the encouraging outcome of a pivotal clinical trial in 2010, the anti-CTLA-4 monoclonal antibody ipilimumab became the first immune checkpoint inhibitor approved for cancer therapy by the U.S. FDA in 2011. This study found that patients with unresectable stage III/IV melanoma exhibited improved survival following treatment by ipilimumab compared with glycoprotein 100 (gp100) peptide vaccine (median overall survival (OS) of 10 vs. 6.4 months, respectively) (Hodi et al. 2010). In the meanwhile and after that, more clinical trials were conducted to explore the treatment of melanoma by ipilimumab, and most of the studies support the idea that ipilimumab is effective for the treatment of unresectable melanoma (Zimmer et al. 2015; Chiarion Sileni et al. 2014; Alexander et al. 2014). A pooled analysis from 10 prospective and 2 retrospective studies including 1861 advanced melanoma patients found that the 3-year survival rate could reach 22% for patients receiving ipilimumab (Schadendorf et al. 2015). Meanwhile, the 3-year survival rate was only 12.2% for the metastatic melanoma patients treated with the FDA approved chemotherapeutic agent dacarbazine (Robert et al. 2011). Therefore, ipilimumab was recommended for the treatment of metastatic or unresectable melanoma patients by the National Comprehensive Cancer Network (NCCN). However, ipilimumab is not effective for a large amount of melanoma patients, and a recent clinical trial found that ipilimumab had no clinical activity in patients with metastatic uveal melanoma (Zimmer et al. 2015).
In the meanwhile, many clinical trials are exploring the potential clinical use of ipilimumab in many other cancers, but most of the results are not encouraging. Ipilimumab has been found to have a partial response in stage IIIB/IV non-small-cell lung cancer (Lynch et al. 2012), and a high dose of ipilimumab could result in a durable response in some subtypes of hematologic cancers after allogeneic hematopoietic stem-cell transplantation (HSCT) (Davids et al. 2016). The clinical activity of ipilimumab in prostate cancer was controversial (Slovin et al. 2013; Kwon et al. 2014; Beer et al. 2017). Furthermore, more clinical trials have suggested that ipilimumab monotherapy is not effective in some other solid tumors, such as colorectal cancer (O’Mahony et al. 2007), extensive-small-cell lung cancer (Reck et al. 2013), unresectable locally advanced or metastatic gastric or gastroesophageal junction cancer (Bang et al. 2017), gastrointestinal stromal tumor (GIST) (D’Angelo et al. 2017), metastatic sarcoma (D’Angelo et al. 2018), and pancreas cancer (Royal et al. 2010).
Although ipilimumab did not show complete response in many kinds of cancers, it did show some clinical activity in most kinds of cancers, therefore, some efforts have been made to assess the efficacy of combination therapy, and some twilight has been seen. A phase I clinical trial of a combination of ipilimumab and imatinib in patients with advanced GIST and melanoma have shown partial response and long-time disease stable property (Reilley et al. 2017), another RCT has shown that ipilimumab plus paclitaxel and carboplatin could improve progression-free survival (PFS) of stage IIIB/IV non-small-cell lung cancer patients (Lynch et al. 2012), Sakamuri’s study also revealed combination of ipilimumab and lenalidomide demonstrated preliminary signals of activity in patients with refractory Hodgkin lymphoma and other advanced cancers (Sakamuri et al. 2018), Formenti’s study also found that radiotherapy enhances responses of lung cancer to CTLA-4 blockade (Formenti et al. 2018). Some preclinical studies also found combination of CTLA-4 inhibitor and other chemotherapy agents could result in better response, Charlotte’s study has found local chemotherapy combined with CTLA-4 inhibitor results in a durable response to cancer therapy in melanoma and prostate cancer (Ariyan et al. 2018), Liu’s study revealed combination immunotherapy of the vaccine and anti-CTLA-4 monoclonal antibody could significantly enhance anti-tumor immune response for triple-negative breast cancer (TNBC) (Liu et al. 2018). More clinical trials are warranted to clarify the efficacy of combination therapy in these kinds of cancer patients.
While ipilimumab has received FDA approval for the treatment of advanced melanoma, there is another CTLA-4 inhibitor, tremelimumab, a fully human IgG2 monoclonal antibody marketed by AstraZeneca that is also being investigated in many clinical trials. Unfortunately, it has not improved patient survival as monotherapy in most of the trials although tremelimumab has a comparable affinity and longer serum half-life (22 days versus 12 days) than ipilimumab. For example, Forty-four patients with non-small-cell lung cancer (NSCLC) were treated with 15 mg/kg of tremelimumab, only two out of 44 patients (4.5%) showed a partial response (PR) (Corrales et al. 2018); Seventeen patients diagnosed with hepatocellular carcinoma received 15 mg/kg of tremelimumab, only three of those patients experienced a confirmed PR (Sangro et al. 2013); Tremelimumab was also investigated as a second-line treatment for patients with gastric and esophageal adenocarcinomas, only one out of 18 patients achieved a PR > 32 months (Ralph et al. 2010); Tremelimumab did not significantly prolong overall survival compared with placebo in patients with previously treated malignant mesothelioma (Maio et al. 2017). According to melanoma, a phase II clinical trial gave a promising data, in this study, tremelimumab (15 mg/kg) was administered to 32 patients with metastatic melanoma, four patients benefitted with an overall response(OR), where the OS fluctuated between 2 months and 41 months, and seven patients survived > 2 years (Ribas 2010). This promising data led to the development of a two arm Phase III clinical trial, although the patients treated with tremelimumab had an objective response of 10.7 months, with a median OS of 12.6 months, there were no clinical differences between the tremelimumab and the temozolomide or dacarbazine arms (Ribas et al. 2013). Therefore, tremelimumab was not approved as cancer monotherapy to date. Tremelimumab is currently being investigated in combination with other regimens to assess whether it will have greater efficacy as part of combinatorial regimens (Jiang et al. 2019; Xie et al. 2019). The difference observed in clinical outcome between ipilimumab and tremelimumab may be attributed to their antibody isotypes. Ilipilumab in human IgG1 depletes immune-suppressive CTLA-4 high expressing regulatory T cells through antibody dependent cell-mediated cytotoxicity (ADCC) while tremelimumab in IgG2 isotype does not engage ADCC pathway (Borrie and Maleki 2018).
23.3 Therapeutic Development of PD-1/PD-L1 Blockade
23.3.1 The PD-1/PD-L1 Immune Checkpoint
Programmed cell death 1(PD-1) is a transmembrane protein, mainly expressed on the surface of activated T cells, B cells, and macrophages cells (Chemnitz et al. 2004). PD-L1(CD724) and PD-L2(CD723) were discovered as dual ligands for PD-1, and both were shown to inhibit T-cell effector activity following PD-1 engagement (Panjwani et al. 2018). Although the engagement between PD-1 and PD-L2 in cancer tissues could contribute to PD-1-mediated inhibition of cytotoxic T-lymphocyte(CTL) responses, there is no evidence that antibodies against PD-1 show higher clinical activity than antibodies against PD-L1, suggesting that PD-L1 is the dominant inhibitory ligand of PD-1 on T cells (Yearley et al. 2017). Binding of PD-L1 by PD-1 has been proposed to deliver survival signals to cancer cells, enhancing their resistance to proapoptotic effects of Fas, interferons, and CTLs (Gato-Canas et al. 2017; Kythreotou et al. 2018). In fact, tumor cells could escape the immune attack by abnormally expressing a series of negative co-stimulatory molecules such as PD-L1, which binds to PD-1 on the surface of immune cells, forming a unique immune escape microenvironment and inhibiting anti-tumor immunity (Topalian et al. 2015; Choueiri et al. 2014). This is the main mechanism of tumor immune escape. In view of this, the PD-1/PD-L1 immune checkpoint pathway has become an ideal target for immunotherapy that aim to restore the effector function of anti-tumor-specific T cells.
23.3.2 Immune Checkpoint Inhibitors Targeting PD-1/PD-L1
In recent years, PD-1 and PD-L1 antibodies have attracted more and more attention due to their promising efficacy compared with other immune therapy or chemotherapy agents. People have witnessed several different anti-PD-1 and anti-PD-L1 antibodies in simultaneous development in numerous cancer types. Among the agents, FDA has approved two PD-1 antibodies (Pembrolizumab and Nivolumab) and three PD-L1 antibodies (Atezolizumab, Avelumab, and Durvalumab) for cancer therapy. Currently, more research focused on Pembrolizumab, Nivolumab, and Atezolizumab in solid tumors.
Pembrolizumab
Pembrolizumab was the first PD-1 antibody approved by U.S. FDA for patients with metastatic melanoma in September 2014 based on two randomized clinical trials, PN002 and PN006. In trial PN002, 540 patients with ipilimumab-refractory metastatic melanoma were randomized (1:1:1) to pembrolizumab 2 or 10 mg/kg every 3 weeks or to chemotherapy (Weber et al. 2013). In trial PN006, 834 patients with ipilimumab-naive metastatic melanoma were randomized (1:1:1) to pembrolizumab 10 mg/kg every 2 or 3 weeks until disease progression or ipilimumab 3 mg/kg every 3 weeks for up to four doses (Barone et al. 2017). In both trials, patients receiving pembrolizumab demonstrated statistically significant improvements in PFS. In trial PN006, patients treated with pembrolizumab demonstrated a statistically significant improvement in overall survival compared with ipilimumab. In recent years, other more studies are conducted to assess the efficacy of pembrolizumab in different melanoma population and its long term efficacy. There was a phase 3 double-blind trial to evaluate pembrolizumab as adjuvant therapy in patients with resected, high-risk stage III melanoma (Eggermont et al. 2018). In this trial, 514 patients received 200 mg of pembrolizumab intravenously every 3 weeks for a total of 18 doses, other 505 patients received a placebo. After a median follow-up of 15 months, pembrolizumab was associated with significantly longer recurrence-free survival than a placebo. A retrospective analysis found that melanoma patients with pretreated brain metastasis could have durable systemic responses to pembrolizumab (Dagogo-Jack et al. 2017). The efficacy of pembrolizumab in melanoma was confirmed in different countries, including Spanish, Japan, and China. The long-term effect of pembrolizumab was confirmed by re-analyzing the PN006 and PN001 trials. In PN006 trial, 24-month overall survival rate was 55% in the 2-week pembrolizumab group, 55% in the 3-week pembrolizumab group, and 43% in the ipilimumab group, suggesting pembrolizumab continued to provide superior overall survival versus ipilimumab (Schachter et al. 2017). In PN001 trial (Hamid et al. 2019), 655 patients with previously treated or treatment-naive advanced/metastatic melanoma received pembrolizumab 2 mg/kg every 3 weeks, 10 mg/kg every 3 weeks, or 10 mg/kg every 2 weeks, median follow-up was 55 months. The estimated 5-year OS was 34% in all patients and 41% in treatment-naive patients; median OS was 23.8 months and 38.6 months, respectively. Estimated 5-year PFS rates were 21% in all patients and 29% in treatment-naive patients; median PFS was 8.3 months and 16.9 months, respectively. This trial confirmed the durable anti-tumor activity and tolerability of pembrolizumab in advanced melanoma.
In addition to melanoma, pembrolizumab was found to have good anti-tumor activity in other solid tumors, especially in non-small-cell lung cancer (NSCLC). The phase 1 KEYNOTE-001 trial initially revealed the efficacy of pembrolizumab in NSCLC (Leighl et al. 2019). After that, a large international multi-center phase 2/3 randomized trial found that pembrolizumab prolonged overall survival and had a favorable benefit-to-risk profile in patients with previously treated, PD-L1-positive, advanced NSCLC (Herbst et al. 2016). Based on this trial, FDA approved pembrolizumab for second-line and above treatment of NSCLC with PD-L1 positive (≥1%). Another KEYNOTE-024 trial compared the efficacy and safety of pembrolizumab and platinum-based chemotherapy in advanced NSCLC and found that in patients with advanced NSCLC and PD-L1 expression on at least 50% of tumor cells, pembrolizumab was associated with significantly longer progression-free and overall survival and with fewer adverse events than was platinum-based chemotherapy (Brahmer et al. 2017). This trial led to the approval of pembrolizumab for the first-line treatment of advanced NSCLC with PD-L1 high expression (≥50%). Another KEYNOTE-21 trial assessed whether the addition of pembrolizumab to platinum-doublet chemotherapy improves efficacy in patients with advanced non-squamous NSCLC (Langer et al. 2016). Result showed that 55% patients in the pembrolizumab plus chemotherapy group achieved an objective response compared with 29% in the chemotherapy alone group, the median PFS was significantly longer in the pembrolizumab plus chemotherapy group than in the chemotherapy group (13.0 months vs 8.9 months), with a 6-month progression-free survival rate of 77%. Based on this data, the FDA approved pembrolizumab in combination with pemetrexed/carboplatin chemotherapy for the first-line treatment of metastatic non-squamous NSCLC. Therefore, these results have changed the first-line management of advanced NSCLC. There are other clinical trials ongoing to assess the clinical use of pembrolizumab in lung cancer. A recent KEYNOTE-042 trial suggested that pembrolizumab monotherapy can be extended as first-line therapy to patients with locally advanced or metastatic non-small-cell lung cancer without sensitizing EGFR or ALK alterations and with low PD-L1 tumor proportion score (Mok et al. 2019).
In addition to melanoma and NSCLC, based on a series of clinical trials, pembrolizumab has also got approval for other cancers, including classical Hodgkin lymphoma (Chen et al. 2017), HNSCC (Larkins et al. 2017), urothelial carcinoma (Bellmunt et al. 2017), gastric cancer (Muro et al. 2016), and colorectal cancer (Wang et al. 2019). It is noteworthy that there is another milestone clinical trial of pembrolizumab in anti-tumor therapy. It is the first time that US FDA has granted a therapeutic treatment for any cancer types with a specific genetic biomarker. This NCT01876511 clinical trial included 11 dMMR (mismatch repair deficient) CRC patients, 9 dMMR other cancer patients, and 21 pMMR (mismatch repair proficient) CRC patients, all of the patient received pembrolizumab intravenously at a dose of 10 mg per kg of body weight every two weeks. The immune-related objective response rate and immune-related progression-free survival rate were 40% and 78%, respectively, for dMMR CRC and 0 and 11% for pMMR CRC patients. Based on this trial, FDA approved pembrolizumab for the treatment of microsatellite instability-high(MSI-H) or dMMR solid tumors (Marcus et al. 2019).
Nivolumab
Nivolumab is another PD-1 antibody that has been approved by FDA for the treatment of various types of cancer. Nivolumab was also firstly approved for the treatment of unresectable or metastatic melanoma based on CheckMate-037 trial and CheckMate066 trial. The CheckMate-037 trial revealed improved objective response rates to nivolumab versus chemotherapy in patients with unresectable or metastatic melanoma whose cancers had progressed following treatment with ipilimumab ± a BRAF inhibitor (Weber et al. 2015). CheckMate 066 trial compared the nivolumab and dacarbazine based chemotherapy in 418 previously untreated metastatic melanoma patients without BRAF mutation (Robert et al. 2015). The overall survival rate was 72.9% in the nivolumab group, as compared with 42.1% in the dacarbazine group. The median progression-free survival was 5.1 months in the nivolumab group versus 2.2 months in the dacarbazine group. The objective response rate was 40.0% in the nivolumab group versus 13.9% in the dacarbazine group. CheckMate 067 was a subsequent Phase III study that enrolled 945 untreated unresectable stage III or metastatic melanoma patients, aimed to assess the combination therapy of nivolumab and ipilimumab (Hodi et al. 2018). This trial showed that nivolumab combined with ipilimumab resulted in significantly longer progression-free survival than ipilimumab alone. This trial led to the approval of dual therapy with nivolumab and nivolumab for the first-line therapy of metastasis melanoma.
Nivolumab is also a hot topic for the treatment of NSCLC. Nivolumab was the first checkpoint inhibitor approved by FDA in 2015 for the treatment of squamous cell NSCLC based on the phase 2 CheckMate 063 trial (Rizvi et al. 2015). In this trial, 117 patients with advanced, refractory squamous NSCLC received nivolumab 3 mg/kg Q2W until progression or unacceptable toxic effects. The 6 months and 1 year PFS were 25.9 and 20.0%. Median OS was 8.2 months (95% CI, 6.1–10.9) and 1 year OS was 40.8% (31.6–49.7). Nivolumab was then approved by the FDA as a second-line therapy for patients with previously treated advanced NSCLC based on CheckMate017 (Yoo et al. 2018) and CheckMate057 trials (Horn et al. 2017). The CheckMate 017 trial evaluated the efficacy and safety of nivolumab versus docetaxel in advanced squamous cell NSCLC. The results showed that the median OS was 9.2 months with nivolumab versus 6.0 months with docetaxel, the 1 year OS rate was 42% with nivolumab versus 24% with docetaxel, the ORR was 20% with nivolumab and 9% with docetaxel. Meanwhile, the CheckMate 057 trial compared nivolumab to docetaxel in previously treated advanced non-squamous NSCLC. Median OS was 12.2 months for nivolumab and 9.4 months for docetaxel; 1-year and 18-month OS rates were 51 and 39% with nivolumab versus 39 and 23% with docetaxel; ORR was 19% for nivolumab and 12% for docetaxel; 1-year PFS was 19% for nivolumab and 8% for docetaxel. Nivolumab further improved efficacy across all endpoints compared with docetaxel.
In addition, the anti-tumor potential of nivolumab has also gained a lot of support in other tumors. The CheckMate025 trial compared nivolumab with everolimus in 821 patients with renal cell carcinoma who had received previous treatment, and found that the median OS was 25.0 months with nivolumab and 19.6 months with everolimus, and the ORR was greater with nivolumab than with everolimus (25 vs. 5%) (Motzer et al. 2015). The CheckMate 275 trial has found that nivolumab monotherapy provided meaningful clinical benefit (ORR 28.4%) irrespective of PD-L1 expression in previously treated patients with metastatic or surgically unresectable urothelial carcinoma (Sharma et al. 2017). Nivolumab monotherapy also resulted in longer overall survival than treatment with standard therapy among patients with platinum-refractory, recurrent squamous cell carcinoma of the head and neck (Ferris et al. 2016). A phase 3 trial also found survival benefits of nivolumab in the treatment of pretreated patients with advanced gastric or gastro-oesophageal junction cancer (12-month OS rates were 26·2% with nivolumab and 10·9% with placebo) (Kang et al. 2017). Nivolumab also showed promising efficacy in other tumors, such as dMMR/MSI-H metastatic colorectal cancer (Overman et al. 2017), unresectable metastatic anal cancer (Morris et al. 2017), and platinum-resistant ovarian cancer (Hamanishi et al. 2015).
There is another PD-1 blocker, cemiplimab, which has been approved for the treatment of metastatic cutaneous squamous cell carcinoma (CSCC) or locally advanced CSCC who are not candidates for curative surgery or curative radiation (Migden et al. 2018).
Atezolizumab
Atezolizumab is the first approved PD-L1 monoclonal antibody, and there are currently two approved indications as monotherapy for the progression of metastatic urothelial carcinoma, metastatic NSCLC after platinum-based chemotherapy and three indications in combination with chemotherapy in metastatic SCLC and metastatic triple-negative breast cancer as well as in combination with bevacizumab in metastatic non-squamous non-small-cell lung cancer.
FDA approved atezolizumab for the treatment of local advanced or metastatic urothelium cell cancer based on the IMvigor210 trial. This trial revealed that the ORR reached 23.5% in patients treated with atezolizumab, and the median CR time is 14.4 months (Powles et al. 2014). FDA approved atezolizumab for the treatment of NSCLC based on POPLAR and OAK clinical trials. The POPLAR trial assessed efficacy and safety of atezolizumab versus docetaxel in previously treated NSCLC, and found that OS was 12·6 months for atezolizumab versus 9·7 months for docetaxel, 16 (11%) patients in the atezolizumab group versus 52 (39%) patients in the docetaxel group had treatment-related grade 3–4 adverse events (Fehrenbacher et al. 2016). The OAK trial also found that atezolizumab treatment results in a clinically relevant improvement of overall survival versus docetaxel in previously treated non-small-cell lung cancer, with a favorable safety profile (Rittmeyer et al. 2017).
Atezolizumab was approved for the treatment of metastatic triple-negative breast cancer based on a recent clinical trial that found atezolizumab plus nab-paclitaxel could prolong the PFS among patients with metastatic triple-negative breast cancer (Schmid et al. 2018). Another trial assessed the efficacy of first-line atezolizumab treatment plus chemotherapy in extensive-stage SCLC, and found a significantly longer overall survival and progression-free survival than chemotherapy alone (Horn et al. 2018). This trial led to the approval of atezolizumab in combination with chemotherapy for the treatment of SCLC.
There are another two PD-L1 blockers, avelumab and durvalumab, which have got US FDA approval for indications for some types of cancers. A phase 2 clinical trial revealed that avelumab monotherapy was associated with durable responses, most of which are still ongoing, and was well tolerated; hence, avelumab represents a new therapeutic option for advanced Merkel cell carcinoma (Kaufman et al. 2016). Another phase one clinical trial found that avelumab showed anti-tumor activity for patients with platinum-refractory metastatic urothelial carcinoma with a manageable safety profile (6% complete responses and 11% partial responses; 29% of grade 1–2 AEs, 6% of grade 3–4 AEs) (Patel et al. 2018). Based on these trials, avelumab has been approved for the treatment of advanced Merkel cell carcinoma and platinum-refractory metastatic urothelial carcinoma. In the meanwhile, durvalumab was approved for the treatment of metastatic urothelial carcinoma and unresectable stage III NSCLC. A phase 3 clinical trial has found that durvalumab monotherapy could result in a significantly longer overall survival and prolonged PFS as compared with placebo, this led to the approval of durvalumab for the unresectable stage III NSCLC (24-month overall survival rate was 66.3% in durvalumab group as compared with 55.6% in placebo group; the median PFS was 17.2 months in durvalumab group as compared with 5.6 months in the placebo group) (Antonia et al. 2018). A phase 1/2 clinical study designed to assess the efficacy and safety of durvalumab in 191 locally advanced or metastatic urothelial carcinoma patients found that durvalumab demonstrated favorable clinical activity and an encouraging and manageable safety profile (ORR was 17.8%, one-year OS rate was 55, 6.8% grade 3/4 AEs) (Powles et al. 2017). Durvalumab also showed anti-tumor activity with acceptable safety in some other cancer types such as triple-negative breast cancer (Loibl et al. 2019) and PD-L1-high patients with recurrent/metastatic head and neck squamous cell carcinoma (Zandberg et al. 2019), as monotherapy or in combination with chemotherapy. However, these results warranted further investigation in phase 3 clinical trials.
23.4 Therapeutic Development of Combined Blockade of CTLA4 and PD-1
Although the drugs targeting PD1-/PD-L1 and CTLA4 have got great success in the treatment of many kinds of cancers, only a small percentage of patients were seen to respond to monotherapy. A combination of CTLA-4 and PD-1/PD-L1 blockers was suggested to have a synergistic effect in the treatment of cancer patients and could increase the response rates. A large amount of clinical trials have been conducted to test the efficacy and safety of the combination in different cancer types, and some of the trials have suggested combination therapy which showed a remarkable increase in response rates and median survival times, resulting in approval of the combination treatment of ipilimumab and nivolumab.
A combination of ipilimumab and nivolumab has been approved for the treatment of metastatic melanoma, metastatic renal cell carcinoma, and CRC with MSI-H and MMR aberrations. This combination has been studied extensively in metastatic melanoma patients and the efficacy and safety of the combination therapy were demonstrated in multiple clinical trials. Ipilimumab plus nivolumab combination was reported to increase the ORR to 61% in a phase 1 study (Postow et al. 2015), the combination therapy increased the 2-year OS rate to 63.8% in a phase 2 study (Hodi et al. 2016), and the combination therapy had higher ORR, longer median progression-free survival and lower incidence of disease progression or death compared to ipilimumab and nivolumab monotherapy in a phase 3 study (Larkin et al. 2015). The combination of ipilimumab and nivolumab was approved for the treatment of metastatic renal cell carcinoma based on two trials. A phase 1 study found that the ORR reached to 40.4% and 2-year OS rate reached to 69.6% in the combination group (Hammers et al. 2017), a following phase 3 study reported the 18-month OS rate was 75%, ORR was 42%, and median PFS was 11.6 months in the nivolumab 3 mg/kg plus ipilimumab 1 mg/kg combination group (Motzer et al. 2018). The combination of ipilimumab and nivolumab was approved for the treatment of CRC with MSI-H and MMR aberrations based on the results of CheckMate-142 trial. This trial revealed a ORR was 55%, PFS rate was 71%, and OS was 85% in 12 months (Overman et al. 2018).
There are also multiple studies exploring the efficacy and safety of anti-PD-1/PD-L1 plus anti-CTLA-4 antibodies in other types of cancer. A phase 1 study evaluated the safety and efficacy of durvalumab (anti-PD-L1) and tremelimumab (anti-CTLA-4) combination in patients with advanced NSCLC and reported the ORR was 23% (Antonia et al. 2016). Another phase 3 study has been conducted to test the safety and activity of nivolumab and ipilimumab combination as first-line therapy for NSCLC. The study showed that in patients with high tumor mutational burden, a combination of nivolumab and ipilimumab achieved ORR of 45.3%, 1-year PFS rate of 42.6%, and median PFS of 7.2 months (Hellmann et al. 2018). Combination of nivolumab plus ipilimumab was also tested in patients with malignant pleural mesothelioma (Scherpereel et al. 2019), locally advanced or metastatic esophagogastric cancers (Janjigian et al. 2018), metastatic prostate cancer (Boudadi et al. 2018), and metastatic sarcoma (D’Angelo et al. 2018), and have showed promising activity in these patients, therefore, the combination therapy may provide new option for these patients in the future.
The FDA approved indications of ICBs targeting CTLA-4, PD-1, and PD-L1 were summarized in Table 23.1.
23.5 Therapeutic Development of Next Generation Immune Checkpoint Blockade
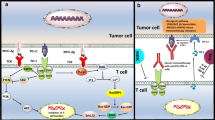
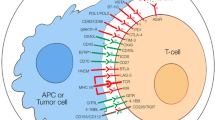
CTLA-4 and PD-1/PD-L1 blockade only confers clinical benefits in a limited proportion of cancer patients, therefore, therapeutic agents that target immune checkpoints other than CTLA-4 and PD-1/PD-L1 are currently under clinical investigations. Here, we summarized the therapeutic development of new targets in immune checkpoint blockade, including TIM-3, LAG-3, TIGIT, VISTA, CD39, CD73, A2AR, and NKG2A.
TIM-3
T-cell immunoglobulin mucin-3 (TIM-3), also known as HAVCR2, is a member of the TIM gene family. As a negative regulatory immune checkpoint, TIM-3 is detected in different types of immune cells, including T cells, Tregs, DCs, B cells, macrophages, NK cells, and mast cells. It has four ligands including galectin-9 (Gal-9), high-mobility group protein B1 (HMGB1), carcinoembryonic antigen cell adhesion molecule 1 (CEACAM-1), and phosphatidylserine (PS) (Anderson et al. 2016). By binding to these ligands, TIM-3 could inhibit cancer immunity by negatively regulating T-cell immunity.
TIM-3 expression has several roles in cancer. Firstly, TIM-3 expression is associated with severe T-cell dysfunction in several types of cancers including NSCLC, hepatocellular carcinoma (HCC), CRC, cervical cancer, ovarian cancer, gastric cancer, RCC, head and neck cancer, and so on. TIM-3 could inhibit anti-tumor immunity by mediating T-cell exhaustion in these cancers (Zhu et al. 2015). For example, TIM-3+ CD8+ T cells could impair the functioning of CD8+ T cells in gastric cancer (Wang et al. 2015); in CRC, upregulation of TIM-3 could restrict T-cell responses and might participate in tumorigenesis (Xu et al. 2015); in RCC, TIM-3 expressed on cancer cells and in myeloid cells could inhibit cancer immunity (Komohara et al. 2015); in ovarian cancer, TIM-3 could negatively regulate various T-cell subsets (Fucikova et al. 2019). Secondly, TIM-3 expression on tumor-infiltrating T cells has been suggested to have a role in resistance to PD-1/PD-L1 blockade. It was reported that PD-1 blockade may lead to an increased expression of TIM-3 in a mouse model of lung cancer, and additional TIM-3 blockade conferred survival benefits (Koyama et al. 2016). PD-1 and TIM-3 inhibitors could enhance T cells’ response to tumor antigens, and had a synergistic function, therefore, the combined use of TIM-3 blockade and PD-1 blockade could be more effective than the TIM-3 or PD-1 blockade alone. It was reported that Dual TIM-3 and PD-1 blockade synergistically restored the function of tumor-infiltrating T cells from HCC patients (Zhou et al. 2017), melanoma patients (Fourcade et al. 2014), and gastric cancer patients (Lu et al. 2017).
Currently, several clinical trials are focusing on TIM-3 alone or combined with PD-1 as a new approach for the treatment of cancer. Three anti-TIM-3 antibodies, MBG453 (Novartis Pharmaceuticals), LY3321367(Eli Lilly and Company), and TSR-022 (Tesaro, Inc.) are under clinical evaluation in combination with PD-1 blockade for patients with advanced solid tumors, and the clinical benefits are worth looking forward to (He et al. 2018).
LAG-3
Lymphocyte activation gene-3 (LAG-3) is a member of the immunoglobulin superfamily mainly expressed on activated T cells, NK cells, Tregs, B cells, and dendritic cells (DCs). LAG-3 could bind to MHC class II and LSECtin, however, recently fibrinogen-like protein 1 (FGL-1) has been identified as a major inhibitory ligand for LAG-3 (Wang et al. 2019). By binding to these ligands, LAG-3 could suppress T-cells activation and cytokines secretion, and could exert differential inhibitory impacts on various types of lymphocytes (Goldberg and Drake 2011).
Importantly, over-expression of LAG-3 is detected on various TILs and exhibits significant immune regulatory impacts. For example, expression of LAG-3 on tumor-specific CD8+ T cells was first described in ovarian cancer and co-expression of LAG-3 and PD-1 was linked to a more severe T-cell dysfunction (Matsuzaki et al. 2010); LAG-3 is also expressed at a high level on Treg cells, and LAG-3+ Treg cells have a more activated phenotype and confer higher suppressive effect (Chew et al. 2017); LAG-3 blockade can potentially affect CD4+ T-cell populations, lead to a relative skewing from a Treg phenotype, and modulate the function of CD4+ T cells to be suppressed (Durham et al. 2014); In melanoma patient samples, LAG-3 is highly expressed on tumor-infiltrating pDCs, contributing to directing an immune-suppressive environment (Camisaschi et al. 2014). Therefore, LAG-3 may be a promising therapeutic target in cancer immunotherapy.
Interestingly, LAG-3 has remarkable interactions with other immune checkpoints especially PD-1. Increasing evidence has elucidated that LAG-3 has remarkable cooperation with PD-1/PD-L1, which can conjointly mediate immune homeostasis, and enhance tumor-induced tolerance (Okazaki et al. 2011). In animal studies, the striking synergy between LAG-3 and PD-1 has been reported in melanoma, fibrosarcoma, and CRC models, the combinational blockade against LAG-3 and PD-1 could effectively eradicate most established tumors resistant to single agent treatment (Woo et al. 2012). In tumor samples from patients, co-expression of LAG-3 and PD-1 can modulate T-cells exhaustion state (Matsuzaki et al. 2010). A recent study in human NSCLC revealed that over-expression of LAG-3 on TILs significantly correlates with PD-1/PD-L1 expression (Deng et al. 2016). Overall, these preclinical data suggest an apparent synergy between LAG-3 and PD-1/PD-L1, providing the foundation for combinational treatment strategy (Dempke et al. 2017).
Currently, several anti-LAG-3 antibodies, such as BMS-986016, LAG525, MGD013, REGN3767, TSR-033, and INCAGN022385 are under clinical evaluation mostly in combination with PD-1 blockade for cancer patients (Long et al. 2018). Among these agents, BMS-986016 is actively being evaluated in various phase I or II clinical trials in hematological and solid tumors. Notably, the combination of BMS-986016 and nivolumab exhibited exciting preliminary efficacy in melanoma patients who were refractory to anti–PD-1/PD-L1 therapy (Ascierto and McArthur 2017). These promising results support the ongoing more extensive exploration of LAG-3 as an alternative immunotherapy target.
TIGIT
T-cell immunoglobulin and ITIM domain (TIGIT) is a member of the immunoglobulin superfamily that is expressed on T cells and NK cells and functions as an inhibitory checkpoint receptor (Dougall et al. 2017). TIGIT has two ligands, CD115 and CD112, and has a much higher affinity to CD115 (Zhang et al. 2014). Interaction of TIGIT with CD112 and CD155 can be happened in trans or in cis. TIGIT competes with immunoactivator receptor DNAX accessory molecule-1 (DNAM-1) for the same set of ligands CD155 (Sanchez-Correa et al. 2019). It is also reported that TIGIT could inhibit immunosurveillance through direct inhibition of DNAM-1.
TIGIT appears to have an important role in the suppression of CD8+ TILs. It is reported that TIGIT expressed at a higher level on CD8+ TILs than on other immune checkpoint receptors, and its expression was also correlated with impaired effector function of CD8+ TILs in acute myeloid leukemia (Wang et al. 2018), multiple myeloma (Guillerey et al. 2018), and gastric cancer (He et al. 2017). TIGIT also has an important role in the suppressive activity of tumor-infiltrating Treg cells. It was proposed that TIGIT primarily suppresses anti-tumor T-cell responses via Tregs rather than CD8+ T cells in mouse models (Kurtulus et al. 2015). Zhang’s study also suggested that TIGIT was highly expressed on exhausted tumor-infiltrating NK cells, and TIGIT blockade could reverse NK-cell exhaustion and restore NK cell cytotoxic activity (Zhang et al. 2018). TIGIT’s role in the tumor microenvironment may also be intertwined with the microbiome. It was suggested that Fusobacterium nucleatum could directly interact with TIGIT, and cause inhibition of NK cell cytotoxicity (Gur et al. 2015). Furthermore, TIGIT and PD-1 were found to be co-expressed in multiple tumor-associated T cells, and this was seen in colon, endometroid, breast, and renal clear cell carcinoma (Chauvin et al. 2015). These findings suggested that both TIGIT and PD-1 are partners in inducing T-cell exhaustion.
Preclinical trials have revealed the anti-tumor activity of anti-TIGIT agents alone or combined with anti-PD-1 antibodies (Solomon and Garrido-Laguna 2018). Currently, several phase 1 clinical trials evaluating the therapeutic efficacy of anti-TIGIT monoclonal antibodies BMS-9862, OMP-313M32, MTIG7192A, MK-7684, AB154, CGEN-15137, and CASC-TIGIT alone or in combination with anti-PD-1 therapy are ongoing (Dixon et al. 2018).
VISTA
V-domain Ig-containing Suppressor of T-cell Activation (VISTA, also known as PD-1H) is a type I transmembrane protein of the B7 family, and shares similarities with PD-1, CD28, and CTLA-4, with the highest identity with PD-1 (Wang et al. 2011). However, analysis of the IgV domain of VISTA shows the greatest homology with PD-L1, suggesting that VISTA may act as both a ligand and receptor in regulating immune responses (Lines et al. 2014). Unlike other immune checkpoints, VISTA is primarily, if not exclusively, found in hematopoietic tissue cells, including macrophages, dendritic cells, myeloid-derived suppressor cells (MDSCs), and neutrophils. VSIG3, VSIG8, and PSGL-1 have been reported to interact with VISTA and mediate the suppressive effect of VISTA (Wang et al. 2019). In vitro binding study demonstrated that multimeric form of VISTA was bound to activated T cells at acidic pH but not at physiological pH7.0. Co-immunoprecipitation analysis has identified that PSGL-1 interacted with VISTA at acidic pH (Johnston et al. 2019).
In multiple mouse models, VISTA plays a critical role in shaping anti-tumor immunity. Wang’s study initially demonstrated that over-expression of VISTA in fibrosarcoma tumor cells significantly increased tumor growth due to an impact of the ligand activity of VISTA on suppressing T-cell immunity (Wang et al. 2011). Le Mercier ‘s study showed that anti-VISTA monotherapy significantly reduced growth in many different solid tumor models regardless of their immunogenic status or origin (Le Mercier et al. 2014). Taking together, the preclinical studies suggested that anti-VISTA monotherapy reshapes the suppressive nature of the TME by reducing the number of MDSCs and tumor-specific Tregs, and increasing the proliferation of TIL and promoting T-cell effector function. It is also reported that a combined blockade of VISTA and PD-1 achieved optimal synergistic anti-tumor activity in a mouse model (Liu et al. 2015). Currently, the therapeutic efficacy of CA170, a selective inhibitor of VISTA,is under evaluation (Nowak et al. 2017).
NKG2A
NKG2A is another promising inhibitory checkpoint receptor in cancer immunotherapy. NKG2A is mainly expressed on the surface of T cells and NK cells in a heterodimeric form with CD94, and the main ligand is HLA-E (Manser and Uhrberg 2016). NKG2A has important roles in tumor-infiltrating NK cells. As to know, NK cells play a major role in the anti-tumor immune response by controlling both tumor progression and metastases. However, tumor cells have the ability to escape from NK cell-mediated immune surveillance within the tumor microenvironment (Pahl and Cerwenka 2017). It is reported that cancer cells could inhibit the effector functions of tumor-infiltrating NK cells via the upregulation CD94/NKG2A heterodimer on NK cells (Schleypen et al. 2003). NK cells from AML patients also show an increased expression of NKG2A and impaired effector functions. The increased expression of NKG2A in tumor-infiltrating NK cells is also emerging as a contributor in determining the poor prognosis of cancer, such as hepatocellular carcinoma, lung carcinoma, and invasive breast cancer. Therefore, NKG2A blockade could restore the cytotoxic capacity of NK cells and targeting NKG2A represents a promising cancer immunotherapy.
Monalizumab is a humanized NKG2A blocking antibody. The impact of monalizumab had been first investigated in in vitro and in vivo studies, and the success of preliminary investigations made it possible to develop clinical trials in human cancer patients. Monalizumab was first used after haplo-HSCT because it is demonstrated that the in vitro blockade of CD94/NKG2A early after haplo-HSCT is able to promote NK cell alloreactivity (Roberto et al. 2018). The potential clinical utility of monalizumab in the Chronic Lymphocytic Leukemia (CLL) is also investigated in combination with irutinib, a tyrosine kinase inhibitor already used in the treatment of CLL (McWilliams et al. 2016). Other clinical trials are ongoing for the treatment of different solid tumors including head and neck cancer, ovarian and endometrial cancer, and metastatic colon cancer (Zandberg et al. 2019).
CD39/CD73/A2AR pathway
CD39/CD73/A2AR/adenosine pathway has recently drawn lots of attention in cancer immunotherapy field. Adenosine is involved in many pathophysiological processes particularly it supports development of immunosuppressive cells like regulatory T cells and myeloid-derived suppressor cells (MDSC) through the binding and activation of A2AR. Ectoenzyme CD39 hydrolyzes extracellular ATP to ADP and AMP, where CD73 converts AMP to adenosine. Adenosine exerts its biological functions through binding to adenosine receptors (Perrot et al. 2019). Expression of CD39 and CD73 have been shown to be upregulated in tumor microenvironment that promotes the development of immune-suppressive cells like regulatory T cells, myeloid-derived suppressor cells M2 macrophage, at the same time, inhibits T-cell functions (through upregulation of CTLA-4, PD-L1, and LAG-3) (Zarek et al. 2008), dendritic cells activation, reduction of NK cell cytotoxic capability, neutrophils attachment. Oleclumab (<EDI9447), a fully human anti-CD73 antibody from Medimmune/AstraZeneca is currently in Ph I and II clinical studies as a single agent or in combination with anti-PDL-1 or chemotherapy across various solid tumors including advanced NSCLC (Vigano et al. 2019), metastatic TNBC, and pancreatic cancer as well as PD-1/PD-L1 resistant NSCLC. Many anti-CD39 and anti-CD73 therapeutics antibodies are in preclinical stage.
23.6 Current Challenges of Immune Checkpoint Blockade Therapy in Cancer
In order to maximize the efficacy of immune checkpoint blockade therapy for cancer patients, some major challenges in this field must be addressed.
One of the major challenges is the toxicities associated with immune checkpoint blockade therapy for cancer. The immune checkpoint inhibitors are not directed solely to tumor-specific T cells, therefore, these drugs may lead to activation of non-tumor-specific immune responses that target self antigens expressed on healthy tissue. This can result in immune-related adverse events (irAEs) due to enhanced T-cell responsiveness, and the activation of self-reactive T cells. The most common irAEs include pruritis and mucositis, vitiligo, diarrhea, and immune-mediated colitis. Less common irAEs include hepatotoxicity, endocrinopathies, and pneumonitis, and rare irAEs include renal toxicity, neurotoxicity, pancreatitis, cardiovascular toxicity, and hematological abnormalities (Kumar et al. 2017). A recent systematic review concluded that grade 3/4 treatment-related adverse events occurred in 14% of patients treated with PD-1/PD-L1 inhibitors, in contrast to 34% of patients treated with CTLA-4 blockade, increasing to 55% during PD-1/PD-L1 and CTLA-4 combination therapy (Arnaud-Coffin et al. 2019). The majority of the irAEs could be treated with corticosteroids and other immunosuppressive drugs. Such drugs might be expected to counteract the action of ICBs, although some studies have reported no obvious therapeutic disadvantage to patients treated with ICBs when corticosteroids were used to alleviate the symptoms of irAEs (Garant et al. 2017). Therefore, it is urgent to explore the methods to retain the efficacy and alleviate the side effects. Ishihara’s study has shown that the safety of anti-PD-L1 antibody in mouse models can be improved by fusing it to the collagen-binding domain of von Willebrand factor, thereby allowing it to bind to the tumor stroma and exert its effects locally (Ishihara et al. 2019). Optimization of dosing regimens could also reduce irAEs in some studies (Lebbe et al. 2019). More efforts are needed to alleviate the side effects of ICBs in future studies.
Another challenge is to gain insight into factors that influence response outcomes to ICB therapy and to better understand and overcome tumor resistance to ICB therapy. It is a fact that most cancer patients do not respond or do not show long-lasting remission after ICB treatment. Despite the clinical benefits of ICBs, the response rates to date have rarely exceeded 40% (Pitt et al. 2016). Patients that do not respond to ICB are said to have “innate resistance”, while those responding transiently before disease progresses have “acquired resistance” (Park et al. 2019). There is urgent to clarify the mechanisms underpinning innate and acquired resistance, and develop accurate ways of predicting which patients will benefit from ICB therapy.
Apart from “innate resistance”, another major factor that contributes to low response rate for ICB is the lack of tumor T-cell infiltration or so-called “cold tumor”. The lack of T-cell infiltration includes lack of tumor-specific antigens, defect in antigen presentation by antigen presenting cells, inhibition of T-cell activation, or homing to the tumor sites. Conversion of “cold tumor” to “hot tumor” have been a focus in improving the overall response rate of ICB. Various approaches or therapeutic combinations are being tested both in patients or animal models. Combination of ICB with chemotherapy or radiation, oncolytic viruses, tumor antigen vaccination as well as DC activation agents (Toll like receptor agonist or CD40 agonist) is being explored to enhance T-cell activation or priming. Anti-TGF, anti-angiogenic agents or IL-2/IL-15 have been used to improve T-cell trafficking and infiltration into tumor microenvironment. Bispecific antibody such as T-cell engager or NK cell engager is another approach being investigated for recruiting T cells or NK cells to tumor sites. Bispecific antibodies comprising antibodies against ICI and innate immunity targets such as anti-PD-L1/anti-TGF and anti-PD-L1/anti-CD47 are under extensive investigation.
Researchers have proposed some possible mechanisms that may be responsible for this resistance. One of the mechanism is the tumor mutational burden. It is reported that high mutational burden is usually associated with a positive outcome for patients treated with ICB. This is because they contain more potential neo-Ags, therefore increasing the chance of anti-tumor T cells becoming activated (Gandara et al. 2018; Samstein et al. 2019; Hellmann et al. 2019). The second potential mechanism is T-cell priming and infiltration of the TME. Responses to ICB therapy depend on the number and diversity of previously activated tumor-specific T cells present in the tumor patient. Tumors with extensive effector T-cell infiltrates will respond best to the ICB therapy. In other words, tumors will be resistance to ICB therapy if there are insufficient tumor-specific T cells, or if these cells are unable to enter the TME to exert anti-tumor activities (Gide et al. 2019; Smith et al. 2019). Another mechanism is the accumulation of additional metabolic and immunosuppressive factors in the TME may limit the efficacy of T-cell responses elicited by ICB therapy. It is reported that tumor cells can outcompete T cells for glucose to reduce glycolytic activity and IFN-γ production by T cells, ICB therapy could restore T-cell glycolysis (Chang et al. 2015). Genetic defects in IFNγ pathway-related genes are also involved in the resistance to ICB therapy (Gao et al. 2016). It is also emerging that the microbiome could influence responses to ICB therapy. Studies on patients with melanoma or NSCLC have revealed that the certain bacterial species in the oral or gut microbiome could influence the responses to PD-1/PD-L1 blockade, and antibiotics can reduce the clinical benefit of PD-1/PD-L1 blockade therapy in cancer patients and mice (Matson et al. 2018; Routy et al. 2018). In-depth study and understanding these mechanisms could develop effective strategies to overcome the resistance to ICB therapy.
23.7 Conclusion
Immune checkpoint inhibitor therapy, especially anti-PD-1/PD-L1 therapy has demonstrated clinical efficacy in multiple types of solid and hematologic tumors, thus FDA has approved six ICB drugs for the treatment of various tumors in recent years, and more promising clinical trials are ongoing to explore the potential anti-tumor activity in more kinds of cancer. However, the irAEs and resistance to ICB therapy are the current major challenges, and more efforts are warranted to develop more effective strategies to overcome the resistance to ICB therapy.
References
Alexander M, Mellor JD, McArthur G, Kee D (2014) Ipilimumab in pretreated patients with unresectable or metastatic cutaneous, uveal and mucosal melanoma. Med J Aust 201(1):49–53
Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV et al (2013) Signatures of mutational processes in human cancer. Nature 500(7463):415–421
Anderson AC, Joller N, Kuchroo VK (2016) Lag-3, Tim-3, and TIGIT: co-inhibitory receptors with specialized functions in immune regulation. Immunity 44(5):989–1004
Antonia S, Goldberg SB, Balmanoukian A, Chaft JE, Sanborn RE, Gupta A et al (2016) Safety and antitumour activity of durvalumab plus tremelimumab in non-small cell lung cancer: a multicentre, phase 1b study. Lancet Oncol 17(3):299–308
Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R et al (2018) Overall survival with durvalumab after chemoradiotherapy in stage III NSCLC. N Engl J Med 379(24):2342–2350
Ariyan CE, Brady MS, Siegelbaum RH, Hu J, Bello DM, Rand J et al (2018) Robust antitumor responses result from local chemotherapy and CTLA-4 blockade. Cancer Immunol Res 6(2):189–200
Arnaud-Coffin P, Maillet D, Gan HK, Stelmes JJ, You B, Dalle S et al (2019) A systematic review of adverse events in randomized trials assessing immune checkpoint inhibitors. Int J Cancer 145(3):639–648
Ascierto PA, McArthur GA (2017) Checkpoint inhibitors in melanoma and early phase development in solid tumors: what’s the future? J Transl Med 15(1):173
Bang YJ, Cho JY, Kim YH, Kim JW, Di Bartolomeo M, Ajani JA et al (2017) Efficacy of sequential ipilimumab monotherapy versus best supportive care for unresectable locally advanced/metastatic gastric or gastroesophageal junction cancer. Clin Cancer Res 23(19):5671–5678 An Official Journal of the American Association for Cancer Research
Barone A, Hazarika M, Theoret MR, Mishra-Kalyani P, Chen H, He K et al (2017) FDA approval summary: pembrolizumab for the treatment of patients with unresectable or metastatic melanoma. Clin Cancer Res 23(19):5661–5665 An Official Journal of the American Association for Cancer Research
Beer TM, Kwon ED, Drake CG, Fizazi K, Logothetis C, Gravis G et al (2017) Randomized, double-blind, phase III trial of ipilimumab versus placebo in asymptomatic or minimally symptomatic patients with metastatic chemotherapy-naive castration-resistant prostate cancer. J Clin Oncol 35(1):40–47 Official Journal of the American Society of Clinical Oncology
Bellmunt J, de Wit R, Vaughn DJ, Fradet Y, Lee JL, Fong L et al (2017) Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N Engl J Med 376(11):1015–1026
Boasso A, Herbeuval JP, Hardy AW, Winkler C, Shearer GM (2005) Regulation of indoleamine 2,3-dioxygenase and tryptophanyl-tRNA-synthetase by CTLA-4-Fc in human CD4 + T cells. Blood 105(4):1574–1581
Borrie AE, Maleki Vareki S (2018) T lymphocyte-based cancer immunotherapeutics. Int Rev Cell Mol Biol 341:201–276
Boudadi K, Suzman DL, Anagnostou V, Fu W, Luber B, Wang H et al (2018) Ipilimumab plus nivolumab and DNA-repair defects in AR-V7-expressing metastatic prostate cancer. Oncotarget 9(47):28561–28571
Brahmer JR, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A et al (2017) Health-related quality-of-life results for pembrolizumab versus chemotherapy in advanced, PD-L1-positive NSCLC (KEYNOTE-024): a multicentre, international, randomised, open-label phase 3 trial. Lancet Oncol 18(12):1600–1609
Burgess EF, Livasy C, Hartman A, Robinson MM, Symanowski J, Naso C, et al (2019) Discordance of high PD-L1 expression in primary and metastatic urothelial carcinoma lesions. Urol Oncol 37(5):299 e19–e25
Busato D, Mossenta M, Baboci L, Di Cintio F, Toffoli G, Dal Bo M (2019) Novel immunotherapeutic approaches for hepatocellular carcinoma treatment. Expert Rev Clin Pharmacol 12(5):453–470
Camisaschi C, De Filippo A, Beretta V, Vergani B, Villa A, Vergani E et al (2014) Alternative activation of human plasmacytoid DCs in vitro and in melanoma lesions: involvement of LAG-3. J Investig Dermatol 134(7):1893–1902
Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD et al (2015) Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 162(6):1229–1241
Chauvin JM, Pagliano O, Fourcade J, Sun Z, Wang H, Sander C et al (2015) TIGIT and PD-1 impair tumor antigen-specific CD8(+) T cells in melanoma patients. J Clin Investig 125(5):2046–2058
Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL (2004) SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol 173(2):945–954
Chen R, Zinzani PL, Fanale MA, Armand P, Johnson NA, Brice P et al (2017) Phase II study of the efficacy and safety of pembrolizumab for relapsed/refractory classic hodgkin lymphoma. J Clin Oncol 35(19):2125–2132 Official Journal of the American Society of Clinical Oncology
Chen C, Zhang F, Zhou N, Gu YM, Zhang YT, He YD et al (2019a) Efficacy and safety of immune checkpoint inhibitors in advanced gastric or gastroesophageal junction cancer: a systematic review and meta-analysis. Oncoimmunology 8(5):e1581547
Chen Q, Wang J, Chen W, Zhang Q, Wei T, Zhou Y et al (2019b) B7-H5/CD28H is a co-stimulatory pathway and correlates with improved prognosis in pancreatic ductal adenocarcinoma. Cancer Sci 110(2):530–539
Chew V, Lai L, Pan L, Lim CJ, Li J, Ong R et al (2017) Delineation of an immunosuppressive gradient in hepatocellular carcinoma using high-dimensional proteomic and transcriptomic analyses. Proc Natl Acad Sci U S A 114(29):E5900–E5909
Chiarion Sileni V, Pigozzo J, Ascierto PA, Grimaldi AM, Maio M, Di Guardo L et al (2014) Efficacy and safety of ipilimumab in elderly patients with pretreated advanced melanoma treated at Italian centres through the expanded access programme. J Exper Clin Cancer Res CR 33:30
Choueiri TK, Fay AP, Gray KP, Callea M, Ho TH, Albiges L et al (2014) PD-L1 expression in nonclear-cell renal cell carcinoma. Ann Oncol 25(11):2178–2184 Official Journal of the European Society for Medical Oncology
Corrales L, Scilla K, Caglevic C, Miller K, Oliveira J, Rolfo C (2018) Immunotherapy in lung cancer: a new age in cancer treatment. Adv Exper Med Biol 995:65–95
Dagogo-Jack I, Lanfranchi M, Gainor JF, Giobbie-Hurder A, Lawrence DP, Shaw AT et al (2017) A retrospective analysis of the efficacy of pembrolizumab in melanoma patients with brain metastasis. J Immunother 40(3):108–113
Dang TO, Ogunniyi A, Barbee MS, Drilon A (2016) Pembrolizumab for the treatment of PD-L1 positive advanced or metastatic non-small cell lung cancer. Expert Rev Anticancer Ther 16(1):13–20
D’Angelo SP, Shoushtari AN, Keohan ML, Dickson MA, Gounder MM, Chi P et al (2017) Combined KIT and CTLA-4 blockade in patients with refractory GIST and other advanced sarcomas: a phase Ib study of dasatinib plus ipilimumab. Clin Cancer Res 23(12):2972–2980 An Official Journal of the American Association for Cancer Research
D’Angelo SP, Mahoney MR, Van Tine BA, Atkins J, Milhem MM, Jahagirdar BN et al (2018) Nivolumab with or without ipilimumab treatment for metastatic sarcoma (Alliance A091401): two open-label, non-comparative, randomised, phase 2 trials. Lancet Oncol 19(3):416–426
Davids MS, Kim HT, Bachireddy P, Costello C, Liguori R, Savell A et al (2016) Ipilimumab for patients with relapse after allogeneic transplantation. N Engl J Med 375(2):143–153
Deeks ED (2016) Pembrolizumab: a review in advanced melanoma. Drugs 76(3):375–386
Dempke WCM, Fenchel K, Uciechowski P, Dale SP (2017) Second- and third-generation drugs for immuno-oncology treatment-the more the better? Eur J Cancer 74:55–72
Deng WW, Mao L, Yu GT, Bu LL, Ma SR, Liu B et al (2016) LAG-3 confers poor prognosis and its blockade reshapes antitumor response in head and neck squamous cell carcinoma. Oncoimmunology 5(11):e1239005
Dixon KO, Schorer M, Nevin J, Etminan Y, Amoozgar Z, Kondo T et al (2018) Functional anti-TIGIT antibodies regulate development of autoimmunity and antitumor immunity. J Immunol 200(8):3000–3007
Dougall WC, Kurtulus S, Smyth MJ, Anderson AC (2017) TIGIT and CD96: new checkpoint receptor targets for cancer immunotherapy. Immunol Rev 276(1):112–120
Durham NM, Nirschl CJ, Jackson CM, Elias J, Kochel CM, Anders RA et al (2014) Lymphocyte Activation Gene 3 (LAG-3) modulates the ability of CD4 T-cells to be suppressed in vivo. PLoS ONE 9(11):e109080
Eggermont AMM, Blank CU, Mandala M, Long GV, Atkinson V, Dalle S et al (2018) Adjuvant pembrolizumab versus placebo in resected stage III melanoma. N Engl J Med 378(19):1789–1801
Engelhardt JJ, Sullivan TJ, Allison JP (2006) CTLA-4 overexpression inhibits T cell responses through a CD28-B7-dependent mechanism. J Immunol 177(2):1052–1061
Fehrenbacher L, Spira A, Ballinger M, Kowanetz M, Vansteenkiste J, Mazieres J et al (2016) Atezolizumab versus docetaxel for patients with previously treated non-small-cell lung cancer (POPLAR): a multicentre, open-label, phase 2 randomised controlled trial. Lancet 387(10030):1837–1846
Ferris RL, Blumenschein G Jr, Fayette J, Guigay J, Colevas AD, Licitra L et al (2016) Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med 375(19):1856–1867
Formenti SC, Rudqvist NP, Golden E, Cooper B, Wennerberg E, Lhuillier C et al (2018) Radiotherapy induces responses of lung cancer to CTLA-4 blockade. Nat Med 24(12):1845–1851
Fourcade J, Sun Z, Pagliano O, Chauvin JM, Sander C, Janjic B et al (2014) PD-1 and Tim-3 regulate the expansion of tumor antigen-specific CD8(+) T cells induced by melanoma vaccines. Cancer Res 74(4):1045–1055
Fucikova J, Rakova J, Hensler M, Kasikova L, Belicova L, Hladikova K, et al (2019) TIM-3 dictates functional orientation of the immune infiltrate in ovarian cancer. Clin Cancer Res. An Official journal of the American Association for Cancer Research
Gandara DR, Paul SM, Kowanetz M, Schleifman E, Zou W, Li Y et al (2018) Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat Med 24(9):1441–1448
Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al (2016) Loss of IFN-γ pathway genes in tumor cells as a mechanism of resistance to Anti-CTLA-4 therapy. Cell 167(2):397–404 e9
Garant A, Guilbault C, Ekmekjian T, Greenwald Z, Murgoi P, Vuong T (2017) Concomitant use of corticosteroids and immune checkpoint inhibitors in patients with hematologic or solid neoplasms: a systematic review. Crit Rev Oncology/Hematology 120:86–92
Gato-Canas M, Zuazo M, Arasanz H, Ibanez-Vea M, Lorenzo L, Fernandez-Hinojal G et al (2017) PDL1 signals through conserved sequence motifs to overcome interferon-mediated cytotoxicity. Cell Rep 20(8):1818–1829
Ghorpade DS, Kaveri SV, Bayry J, Balaji KN (2011) Cooperative regulation of NOTCH1 protein-phosphatidylinositol 3-kinase (PI3K) signaling by NOD1, NOD2, and TLR2 receptors renders enhanced refractoriness to transforming growth factor-beta (TGF-beta)- or cytotoxic T-lymphocyte antigen 4 (CTLA-4)-mediated impairment of human dendritic cell maturation. J Biol Chem 286(36):31347–31360
Gide TN, Quek C, Menzies AM, Tasker AT, Shang P, Holst J, et al (2019) Distinct immune cell populations define Response to Anti-PD-1 monotherapy and anti-PD-1/anti-CTLA-4 combined therapy. Cancer Cell 35(2):238–55 e6
Goldberg MV, Drake CG (2011) LAG-3 in Cancer Immunotherapy. Curr Top Microbiol Immunol 344:269–278
Graziani G, Tentori L, Navarra P (2012) Ipilimumab: a novel immunostimulatory monoclonal antibody for the treatment of cancer. Pharmacol Res 65(1):9–22
Guillerey C, Harjunpaa H, Carrie N, Kassem S, Teo T, Miles K et al (2018) TIGIT immune checkpoint blockade restores CD8(+) T-cell immunity against multiple myeloma. Blood 132(16):1689–1694
Gur C, Ibrahim Y, Isaacson B, Yamin R, Abed J, Gamliel M et al (2015) Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity 42(2):344–355
Hage C, Hoves S, Strauss L, Bissinger S, Prinz Y, Poschinger T, et al (2019) Sorafenib induces pyroptosis in macrophages and triggers natural killer cell-mediated cytotoxicity against hepatocellular carcinoma. Hepatology
Hamada T, Soong TR, Masugi Y, Kosumi K, Nowak JA, da Silva A et al (2018) TIME (Tumor Immunity in the MicroEnvironment) classification based on tumor CD274 (PD-L1) expression status and tumor-infiltrating lymphocytes in colorectal carcinomas. Oncoimmunology 7(7):e1442999
Hamanishi J, Mandai M, Ikeda T, Minami M, Kawaguchi A, Murayama T et al (2015) Safety and antitumor activity of anti-PD-1 antibody, nivolumab, in patients with platinum-resistant ovarian cancer. J Clin Oncol 33(34):4015–4022 Official Journal of the American Society of Clinical Oncology
Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R et al (2019) Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann Oncol 30(4):582–588 Official Journal of the European Society for Medical Oncology
Hammers HJ, Plimack ER, Infante JR, Rini BI, McDermott DF, Lewis LD et al (2017) Safety and efficacy of nivolumab in combination with ipilimumab in metastatic renal cell carcinoma: the CheckMate 016 study. J Clin Oncol 35(34):3851–3858 Official Journal of the American Society of Clinical Oncology
He W, Zhang H, Han F, Chen X, Lin R, Wang W et al (2017) CD155T/TIGIT signaling regulates CD8(+) T-cell metabolism and promotes tumor progression in human gastric cancer. Cancer Res 77(22):6375–6388
He Y, Cao J, Zhao C, Li X, Zhou C, Hirsch FR (2018) TIM-3, a promising target for cancer immunotherapy. OncoTargets Ther 11:7005–7009
Hellmann MD, Ciuleanu TE, Pluzanski A, Lee JS, Otterson GA, Audigier-Valette C et al (2018) Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med 378(22):2093–2104
Hellmann MD, Callahan MK, Awad MM, Calvo E, Ascierto PA, Atmaca A et al (2019) Tumor mutational burden and efficacy of nivolumab monotherapy and in combination with ipilimumab in small-cell lung cancer. Cancer Cell 35(2):329
Herbst RS, Baas P, Kim DW, Felip E, Perez-Gracia JL, Han JY et al (2016) Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet 387(10027):1540–1550
Hermel DJ, Sigal D, The emerging role of checkpoint inhibition in microsatellite stable colorectal cancer. J Personal Med 9(1) (2019)
Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB et al (2010) Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med 363(8):711–723
Hodi FS, Chesney J, Pavlick AC, Robert C, Grossmann KF, McDermott DF et al (2016) Combined nivolumab and ipilimumab versus ipilimumab alone in patients with advanced melanoma: 2-year overall survival outcomes in a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol 17(11):1558–1568
Hodi FS, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Cowey CL et al (2018) Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol 19(11):1480–1492
Horn L, Spigel DR, Vokes EE, Holgado E, Ready N, Steins M et al (2017) Nivolumab versus docetaxel in previously treated patients with advanced non-small-cell lung cancer: two-year outcomes from two randomized, open-label, phase III trials (CheckMate 017 and CheckMate 057). J Clin Oncol 35(35):3924–3933 Official Journal of the American Society of Clinical Oncology
Horn L, Mansfield AS, Szczesna A, Havel L, Krzakowski M, Hochmair MJ et al (2018) First-line atezolizumab plus chemotherapy in extensive-stage small-cell lung cancer. N Engl J Med 379(23):2220–2229
Ishihara J, Ishihara A, Sasaki K, Lee SS, Williford JM, Yasui M, et al (2019) Targeted antibody and cytokine cancer immunotherapies through collagen affinity. Sci Transl Med 11(487)
Janjigian YY, Bendell J, Calvo E, Kim JW, Ascierto PA, Sharma P et al (2018) CheckMate-032 study: efficacy and safety of nivolumab and nivolumab plus ipilimumab in patients with metastatic esophagogastric cancer. J Clin Oncol 36(28):2836–2844 Official Journal of the American Society of Clinical Oncology
Jiang DM, Fyles A, Nguyen LT, Neel BG, Sacher A, Rottapel R et al (2019) Phase I study of local radiation and tremelimumab in patients with inoperable locally recurrent or metastatic breast cancer. Oncotarget 10(31):2947–2958
Johnston RJ, Su LJ, Pinckney J, Critton D, Boyer E, Krishnakumar A et al (2019) VISTA is an acidic pH-selective ligand for PSGL-1. Nature 574(7779):565–570
Kang YK, Boku N, Satoh T, Ryu MH, Chao Y, Kato K et al (2017) Nivolumab in patients with advanced gastric or gastro-oesophageal junction cancer refractory to, or intolerant of, at least two previous chemotherapy regimens (ONO-4538-12, ATTRACTION-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 390(10111):2461–2471
Kasamon YL, de Claro RA, Wang Y, Shen YL, Farrell AT, Pazdur R (2017) FDA approval summary: nivolumab for the treatment of relapsed or progressive classical Hodgkin lymphoma. Oncologist 22(5):585–591
Kaufman HL, Russell J, Hamid O, Bhatia S, Terheyden P, D’Angelo SP et al (2016) Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. Lancet Oncol 17(10):1374–1385
Khalil DN, Smith EL, Brentjens RJ, Wolchok JD (2016) The future of cancer treatment: immunomodulation, CARs and combination immunotherapy. Nat Rev Clin Oncol 13(5):273–290
Kim J, Kwiatkowski D, McConkey DJ, Meeks JJ, Freeman SS, Bellmunt J et al (2019) The cancer genome atlas expression subtypes stratify response to checkpoint inhibition in advanced urothelial cancer and identify a subset of patients with high survival probability. Eur Urol 75(6):961–964
Kindler HL, Karrison TG, Gandara DR, Lu C, Krug LM, Stevenson JP et al (2012) Multicenter, double-blind, placebo-controlled, randomized phase II trial of gemcitabine/cisplatin plus bevacizumab or placebo in patients with malignant mesothelioma. J Clin Oncol 30(20):2509–2515 Official Journal of the American Society of Clinical Oncology
Komohara Y, Morita T, Annan DA, Horlad H, Ohnishi K, Yamada S et al (2015) The coordinated actions of TIM-3 on cancer and myeloid cells in the regulation of tumorigenicity and clinical prognosis in clear cell renal cell carcinomas. Cancer Immunol Res 3(9):999–1007
Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG et al (2016) Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun 7:10501
Kudo M (2019) Targeted and immune therapies for hepatocellular carcinoma: predictions for 2019 and beyond. World J Gastroenterol 25(7):789–807
Kumar V, Chaudhary N, Garg M, Floudas CS, Soni P, Chandra AB (2017) Current diagnosis and management of immune related adverse events (irAEs) induced by immune checkpoint inhibitor therapy. Front Pharmacol 8:49
Kurtulus S, Sakuishi K, Ngiow SF, Joller N, Tan DJ, Teng MW et al (2015) TIGIT predominantly regulates the immune response via regulatory T cells. J Clin Investig 125(11):4053–4062
Kwon ED, Drake CG, Scher HI, Fizazi K, Bossi A, van den Eertwegh AJ et al (2014) Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol 15(7):700–712
Kythreotou A, Siddique A, Mauri FA, Bower M, Pinato DJ (2018) Pd-L1. J Clin Pathol 71(3):189–194
Langer CJ, Gadgeel SM, Borghaei H, Papadimitrakopoulou VA, Patnaik A, Powell SF et al (2016) Carboplatin and pemetrexed with or without pembrolizumab for advanced, non-squamous non-small-cell lung cancer: a randomised, phase 2 cohort of the open-label KEYNOTE-021 study. Lancet Oncol 17(11):1497–1508
Larkin J, Hodi FS, Wolchok JD (2015) Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 373(13):1270–1271
Larkins E, Blumenthal GM, Yuan W, He K, Sridhara R, Subramaniam S et al (2017) FDA approval summary: pembrolizumab for the treatment of recurrent or metastatic head and neck squamous cell carcinoma with disease progression on or after platinum-containing chemotherapy. Oncologist 22(7):873–878
Le Mercier I, Chen W, Lines JL, Day M, Li J, Sergent P et al (2014) VISTA regulates the development of protective antitumor immunity. Cancer Res 74(7):1933–1944
Lebbe C, Meyer N, Mortier L, Marquez-Rodas I, Robert C, Rutkowski P et al (2019) Evaluation of two dosing regimens for Nivolumab in combination with Ipilimumab in patients with advanced melanoma: results from the phase IIIb/IV CheckMate 511 trial. J Clin Oncol 37(11):867–875 Official Journal of the American Society of Clinical Oncology
Leighl NB, Hellmann MD, Hui R, Carcereny E, Felip E, Ahn MJ et al (2019) Pembrolizumab in patients with advanced non-small-cell lung cancer (KEYNOTE-001): 3-year results from an open-label, phase 1 study. Lancet Respir Med 7(4):347–357
Lines JL, Pantazi E, Mak J, Sempere LF, Wang L, O’Connell S et al (2014) VISTA is an immune checkpoint molecule for human T cells. Cancer Res 74(7):1924–1932
Linsley PS, Greene JL, Brady W, Bajorath J, Ledbetter JA, Peach R (1994) Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity 1(9):793–801
Lipson EJ, Drake CG (2011) Ipilimumab: an anti-CTLA-4 antibody for metastatic melanoma. Clin Cancer Res 17(22):6958–6962 An Official Journal of the American Association for Cancer Research
Liu J, Yuan Y, Chen W, Putra J, Suriawinata AA, Schenk AD et al (2015) Immune-checkpoint proteins VISTA and PD-1 nonredundantly regulate murine T-cell responses. Proc Natl Acad Sci U S A 112(21):6682–6687
Liu L, Wang Y, Miao L, Liu Q, Musetti S, Li J et al (2018) Combination Immunotherapy of MUC1 mRNA nano-vaccine and CTLA-4 blockade effectively inhibits growth of triple negative breast cancer. Mol Ther J Am Soc Gene Ther 26(1):45–55
Loibl S, Untch M, Burchardi N, Huober J, Sinn BV, Blohmer JU, et al (2019) A randomised phase II study investigating durvalumab in addition to an anthracycline taxane-based neoadjuvant therapy in early triple negative breast cancer—clinical results and biomarker analysis of GeparNuevo study. Ann Oncol. Official Journal of the European Society for Medical Oncology
Long L, Zhang X, Chen F, Pan Q, Phiphatwatchara P, Zeng Y et al (2018) The promising immune checkpoint LAG-3: from tumor microenvironment to cancer immunotherapy. Genes Cancer 9(5–6):176–189
Lu X, Yang L, Yao D, Wu X, Li J, Liu X et al (2017) Tumor antigen-specific CD8(+) T cells are negatively regulated by PD-1 and Tim-3 in human gastric cancer. Cell Immunol 313:43–51
Lynch TJ, Bondarenko I, Luft A, Serwatowski P, Barlesi F, Chacko R et al (2012) Ipilimumab in combination with paclitaxel and carboplatin as first-line treatment in stage IIIB/IV non-small-cell lung cancer: results from a randomized, double-blind, multicenter phase II study. J Clin Oncol 30(17):2046–2054 Official Journal of the American Society of Clinical Oncology
Maio M, Scherpereel A, Calabro L, Aerts J, Cedres Perez S, Bearz A et al (2017) Tremelimumab as second-line or third-line treatment in relapsed malignant mesothelioma (DETERMINE): a multicentre, international, randomised, double-blind, placebo-controlled phase 2b trial. Lancet Oncol 18(9):1261–1273
Manser AR, Uhrberg M (2016) Age-related changes in natural killer cell repertoires: impact on NK cell function and immune surveillance. Cancer Immunol Immunother CII 65(4):417–426
Marcus L, Lemery SJ, Keegan P, Pazdur R (2019) FDA approval summary: pembrolizumab for the treatment of microsatellite instability-high solid tumors. Clin Cancer Res 25(13):3753–3758 An Official Journal of the American Association for Cancer Research
Marginean EC, Melosky B (2018) Is there a role for programmed death ligand-1 testing and immunotherapy in colorectal cancer with microsatellite instability? Part I-colorectal cancer: microsatellite instability, testing, and clinical implications. Arch Pathol Lab Med 142(1):17–25
Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre ML et al (2018) The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science 359(6371):104–108
Matsuzaki J, Gnjatic S, Mhawech-Fauceglia P, Beck A, Miller A, Tsuji T et al (2010) Tumor-infiltrating NY-ESO-1-specific CD8 + T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc Natl Acad Sci U S A 107(17):7875–7880
McWilliams EM, Mele JM, Cheney C, Timmerman EA, Fiazuddin F, Strattan EJ et al (2016) Therapeutic CD94/NKG2A blockade improves natural killer cell dysfunction in chronic lymphocytic leukemia. Oncoimmunology 5(10):e1226720
Middha S, Yaeger R, Shia J, Stadler ZK, King S, Guercio S, et al (2019) Majority of B2M-mutant and -deficient colorectal carcinomas achieve clinical benefit from immune checkpoint inhibitor therapy and are microsatellite instability-high. JCO Precis Oncol 3
Migden MR, Rischin D, Schmults CD, Guminski A, Hauschild A, Lewis KD et al (2018) PD-1 blockade with cemiplimab in advanced cutaneous squamous-cell carcinoma. N Engl J Med 379(4):341–351
Mok TSK, Wu YL, Kudaba I, Kowalski DM, Cho BC, Turna HZ et al (2019) Pembrolizumab versus chemotherapy for previously untreated, PD-L1-expressing, locally advanced or metastatic non-small-cell lung cancer (KEYNOTE-042): a randomised, open-label, controlled, phase 3 trial. Lancet 393(10183):1819–1830
Morris VK, Salem ME, Nimeiri H, Iqbal S, Singh P, Ciombor K et al (2017) Nivolumab for previously treated unresectable metastatic anal cancer (NCI9673): a multicentre, single-arm, phase 2 study. Lancet Oncol 18(4):446–453
Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S et al (2015) Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med 373(19):1803–1813
Motzer RJ, Tannir NM, McDermott DF, Aren Frontera O, Melichar B, Choueiri TK et al (2018) Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N Engl J Med 378(14):1277–1290
Muro K, Chung HC, Shankaran V, Geva R, Catenacci D, Gupta S et al (2016) Pembrolizumab for patients with PD-L1-positive advanced gastric cancer (KEYNOTE-012): a multicentre, open-label, phase 1b trial. Lancet Oncol 17(6):717–726
Nowak EC, Lines JL, Varn FS, Deng J, Sarde A, Mabaera R et al (2017) Immunoregulatory functions of VISTA. Immunol Rev 276(1):66–79
Okazaki T, Okazaki IM, Wang J, Sugiura D, Nakaki F, Yoshida T et al (2011) PD-1 and LAG-3 inhibitory co-receptors act synergistically to prevent autoimmunity in mice. J Exper Med 208(2):395–407
O’Mahony D, Morris JC, Quinn C, Gao W, Wilson WH, Gause B et al (2007) A pilot study of CTLA-4 blockade after cancer vaccine failure in patients with advanced malignancy. Clin Cancer Res 13(3):958–964 An Official Journal of the American Association for Cancer Research
Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA et al (2017) Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol 18(9):1182–1191
Overman MJ, Lonardi S, Wong KYM, Lenz HJ, Gelsomino F, Aglietta M et al (2018) Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J Clin Oncol 36(8):773–779 Official Journal of the American Society of Clinical Oncology
Pahl J, Cerwenka A (2017) Tricking the balance: NK cells in anti-cancer immunity. Immunobiology 222(1):11–20
Panjwani PK, Charu V, DeLisser M, Molina-Kirsch H, Natkunam Y, Zhao S (2018) Programmed death-1 ligands PD-L1 and PD-L2 show distinctive and restricted patterns of expression in lymphoma subtypes. Hum Pathol 71:91–99
Park R, Williamson S, Kasi A, Saeed A (2018) Immune therapeutics in the treatment of advanced gastric and esophageal cancer. Anticancer Res 38(10):5569–5580
Park R, Winnicki M, Liu E, Chu WM (2019) Immune checkpoints and cancer in the immunogenomics era. Brief Funct Genomics 18(2):133–139
Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV et al (2005) CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol 25(21):9543–9553
Patel MR, Ellerton J, Infante JR, Agrawal M, Gordon M, Aljumaily R et al (2018) Avelumab in metastatic urothelial carcinoma after platinum failure (JAVELIN Solid Tumor): pooled results from two expansion cohorts of an open-label, phase 1 trial. Lancet Oncol 19(1):51–64
Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP (2009) Blockade of CTLA-4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti-CTLA-4 antibodies. J Exper Med 206(8):1717–1725
Perrot I, Michaud HA, Giraudon-Paoli M, Augier S, Docquier A, Gros L, et al (2019) Blocking antibodies targeting the CD39/CD73 immunosuppressive pathway unleash immune responses in combination cancer therapies. Cell Rep 27(8):2411–2425 e9
Pitt JM, Vetizou M, Daillere R, Roberti MP, Yamazaki T, Routy B et al (2016) Resistance mechanisms to immune-checkpoint blockade in cancer: tumor-intrinsic and -extrinsic factors. Immunity 44(6):1255–1269
Polk A, Svane IM, Andersson M, Nielsen D (2018) Checkpoint inhibitors in breast cancer—current status. Cancer Treat Rev 63:122–134
Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D et al (2015) Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med 372(21):2006–2017
Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C et al (2014) MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature 515(7528):558–562
Powles T, O’Donnell PH, Massard C, Arkenau HT, Friedlander TW, Hoimes CJ et al (2017) Efficacy and Safety of durvalumab in locally advanced or metastatic urothelial carcinoma: updated results from a phase 1/2 open-label study. JAMA Oncol 3(9):e172411
Puglisi M, Dolly S, Faria A, Myerson JS, Popat S, O’Brien ME (2010) Treatment options for small cell lung cancer—do we have more choice? Br J Cancer 102(4):629–638
Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM et al (2011) Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science 332(6029):600–603
Ralph C, Elkord E, Burt DJ, O’Dwyer JF, Austin EB, Stern PL et al (2010) Modulation of lymphocyte regulation for cancer therapy: a phase II trial of tremelimumab in advanced gastric and esophageal adenocarcinoma. Clin Cancer Res 16(5):1662–1672 An Official Journal of the American Association for Cancer Research
Reck M, Bondarenko I, Luft A, Serwatowski P, Barlesi F, Chacko R et al (2013) Ipilimumab in combination with paclitaxel and carboplatin as first-line therapy in extensive-disease-small-cell lung cancer: results from a randomized, double-blind, multicenter phase 2 trial. Ann Oncol 24(1):75–83 Official Journal of the European Society for Medical Oncology
Reck M, Luft A, Szczesna A, Havel L, Kim SW, Akerley W et al (2016) Phase III randomized trial of ipilimumab plus etoposide and platinum versus placebo plus etoposide and platinum in extensive-stage small-cell lung cancer. J Clin Oncol 34(31):3740–3748 Official Journal of the American Society of Clinical Oncology
Reilley MJ, Bailey A, Subbiah V, Janku F, Naing A, Falchook G et al (2017) Phase I clinical trial of combination imatinib and ipilimumab in patients with advanced malignancies. J Immunother Cancer 5:35
Ribas A (2010) Clinical development of the anti-CTLA-4 antibody tremelimumab. Semin Oncol 37(5):450–454
Ribas A, Kefford R, Marshall MA, Punt CJ, Haanen JB, Marmol M et al (2013) Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J Clin Oncol 31(5):616–622 Official Journal of the American Society of Clinical Oncology
Rittmeyer A, Barlesi F, Waterkamp D, Park K, Ciardiello F, von Pawel J et al (2017) Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet 389(10066):255–265
Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al (2015) Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348(6230):124–128
Rizvi NA, Mazieres J, Planchard D, Stinchcombe TE, Dy GK, Antonia SJ et al (2015b) Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): a phase 2, single-arm trial. Lancet Oncol 16(3):257–265
Robert C, Thomas L, Bondarenko I, O’Day S, Weber J, Garbe C et al (2011) Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med 364(26):2517–2526
Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L et al (2015) Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med 372(4):320–330
Roberto A, Di Vito C, Zaghi E, Mazza EMC, Capucetti A, Calvi M et al (2018) The early expansion of anergic NKG2A(pos)/CD56(dim)/CD16(neg) natural killer represents a therapeutic target in haploidentical hematopoietic stem cell transplantation. Haematologica 103(8):1390–1402
Rosenberg JE, Hoffman-Censits J, Powles T, van der Heijden MS, Balar AV, Necchi A et al (2016) Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet 387(10031):1909–1920
Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillere R et al (2018) Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 359(6371):91–97
Rowshanravan B, Halliday N, Sansom DM (2018) CTLA-4: a moving target in immunotherapy. Blood 131(1):58–67
Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US et al (2010) Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother 33(8):828–833
Sakamuri D, Glitza IC, Betancourt Cuellar SL, Subbiah V, Fu S, Tsimberidou AM et al (2018) Phase I dose-escalation study of anti-CTLA-4 antibody ipilimumab and lenalidomide in patients with advanced cancers. Mol Cancer Ther 17(3):671–676
Samstein RM, Lee CH, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY et al (2019) Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet 51(2):202–206
Sanchez-Correa B, Valhondo I, Hassouneh F, Lopez-Sejas N, Pera A, Bergua JM, et al (2019) DNAM-1 and the TIGIT/PVRIG/TACTILE axis: novel immune checkpoints for natural killer cell-based cancer immunotherapy. Cancers 11(6)
Sangro B, Gomez-Martin C, de la Mata M, Inarrairaegui M, Garralda E, Barrera P et al (2013) A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol 59(1):81–88
Sansom DM (2000) CD28, CTLA-4 and their ligands: who does what and to whom? Immunology 101(2):169–177
Schachter J, Ribas A, Long GV, Arance A, Grob JJ, Mortier L et al (2017) Pembrolizumab versus ipilimumab for advanced melanoma: final overall survival results of a multicentre, randomised, open-label phase 3 study (KEYNOTE-006). Lancet 390(10105):1853–1862
Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O et al (2015) Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol 33(17):1889–1894 Official Journal of the American Society of Clinical Oncology
Scherpereel A, Mazieres J, Greillier L, Lantuejoul S, Do P, Bylicki O et al (2019) Nivolumab or nivolumab plus ipilimumab in patients with relapsed malignant pleural mesothelioma (IFCT-1501 MAPS2): a multicentre, open-label, randomised, non-comparative, phase 2 trial. Lancet Oncol 20(2):239–253
Schleypen JS, Von Geldern M, Weiss EH, Kotzias N, Rohrmann K, Schendel DJ et al (2003) Renal cell carcinoma-infiltrating natural killer cells express differential repertoires of activating and inhibitory receptors and are inhibited by specific HLA class I allotypes. Int J Cancer 106(6):905–912
Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H et al (2018) Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med 379(22):2108–2121
Sharma P, Retz M, Siefker-Radtke A, Baron A, Necchi A, Bedke J et al (2017) Nivolumab in metastatic urothelial carcinoma after platinum therapy (CheckMate 275): a multicentre, single-arm, phase 2 trial. Lancet Oncol 18(3):312–322
Sim F, Leidner R, Bell RB (2019) Immunotherapy for head and neck cancer. Oral Maxillofac Surg Clin N Am 31(1):85–100
Slovin SF, Higano CS, Hamid O, Tejwani S, Harzstark A, Alumkal JJ et al (2013) Ipilimumab alone or in combination with radiotherapy in metastatic castration-resistant prostate cancer: results from an open-label, multicenter phase I/II study. Ann Oncol 24(7):1813–1821 Official Journal of the European Society for Medical Oncology
Smith KN, Llosa NJ, Cottrell TR, Siegel N, Fan H, Suri P et al (2019) Persistent mutant oncogene specific T cells in two patients benefitting from anti-PD-1. J Immunother Cancer 7(1):40
Solomon BL, Garrido-Laguna I (2018) TIGIT: a novel immunotherapy target moving from bench to bedside. Cancer Immunol Immunother CII. 67(11):1659–1667
Topalian SL, Drake CG, Pardoll DM (2015) Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 27(4):450–461
Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L et al (2014) PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515(7528):568–571
Vigano S, Alatzoglou D, Irving M, Menetrier-Caux C, Caux C, Romero P et al (2019) Targeting adenosine in cancer immunotherapy to enhance T-cell function. Front Immunol 10:925
Wang L, Rubinstein R, Lines JL, Wasiuk A, Ahonen C, Guo Y et al (2011) VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J Exper Med 208(3):577–592
Wang Z, Zhu J, Gu H, Yuan Y, Zhang B, Zhu D et al (2015) The clinical significance of abnormal Tim-3 expression on NK cells from patients with gastric cancer. Immunol Investig 44(6):578–589
Wang M, Bu J, Zhou M, Sido J, Lin Y, Liu G et al (2018) CD8(+)T cells expressing both PD-1 and TIGIT but not CD226 are dysfunctional in acute myeloid leukemia (AML) patients. Clin Immunol 190:64–73
Wang C, Sandhu J, Fakih M (2019a) Complete response to pembrolizumab in a patient with metastatic colon cancer with microsatellite instability and a history of Guillain-Barre syndrome. J Gastrointest Oncol 10(1):161–165
Wang J, Sanmamed MF, Datar I, Su TT, Ji L, Sun J, et al (2019) Fibrinogen-like protein 1 is a major immune inhibitory ligand of LAG-3. Cell 176(1–2):334–347 e12
Wang J, Wu G, Manick B, Hernandez V, Renelt M, Erickson C et al (2019c) VSIG-3 as a ligand of VISTA inhibits human T-cell function. Immunology 156(1):74–85
Weber JS, Kudchadkar RR, Yu B, Gallenstein D, Horak CE, Inzunza HD et al (2013) Safety, efficacy, and biomarkers of nivolumab with vaccine in ipilimumab-refractory or -naive melanoma. J Clin Oncol 31(34):4311–4318 Official Journal of the American Society of Clinical Oncology
Weber JS, D’Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B et al (2015) Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol 16(4):375–384
Wolchok JD, Neyns B, Linette G, Negrier S, Lutzky J, Thomas L et al (2010) Ipilimumab monotherapy in patients with pretreated advanced melanoma: a randomised, double-blind, multicentre, phase 2, dose-ranging study. Lancet Oncol 11(2):155–164
Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschl CJ et al (2012) Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res 72(4):917–927
Xie C, Duffy AG, Mabry-Hrones D, Wood B, Levy E, Krishnasamy V et al (2019) Tremelimumab in combination with microwave ablation in patients with refractory biliary tract cancer. Hepatology 69(5):2048–2060
Xu B, Yuan L, Gao Q, Yuan P, Zhao P, Yuan H et al (2015) Circulating and tumor-infiltrating Tim-3 in patients with colorectal cancer. Oncotarget 6(24):20592–20603
Yearley JH, Gibson C, Yu N, Moon C, Murphy E, Juco J et al (2017) PD-L2 expression in human tumors: relevance to anti-PD-1 therapy in cancer. Clin Cancer Res 23(12):3158–3167 An Official Journal of the American Association for Cancer Research
Yoo SH, Keam B, Kim M, Kim TM, Kim DW, Heo DS (2018) Generalization and representativeness of phase III immune checkpoint blockade trials in non-small cell lung cancer. Thorac Cancer 9(6):736–744
Yu GT, Mao L, Wu L, Deng WW, Bu LL, Liu JF et al (2018) Inhibition of SRC family kinases facilitates anti-CTLA4 immunotherapy in head and neck squamous cell carcinoma. Cell Mol Life Sci CMLS 75(22):4223–4234
Zandberg DP, Algazi AP, Jimeno A, Good JS, Fayette J, Bouganim N et al (2019) Durvalumab for recurrent or metastatic head and neck squamous cell carcinoma: Results from a single-arm, phase II study in patients with ≥ 25% tumour cell PD-L1 expression who have progressed on platinum-based chemotherapy. Eur J Cancer 107:142–152
Zarek PE, Huang CT, Lutz ER, Kowalski J, Horton MR, Linden J et al (2008) A2A receptor signaling promotes peripheral tolerance by inducing T-cell anergy and the generation of adaptive regulatory T cells. Blood 111(1):251–259
Zhang T, Wang J, Zhou X, Liang R, Bai Q, Yang L et al (2014) Increased expression of TIGIT on CD4 + T cells ameliorates immune-mediated bone marrow failure of aplastic anemia. J Cell Biochem 115(11):1918–1927
Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W et al (2018) Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol 19(7):723–732
Zhou G, Sprengers D, Boor PPC, Doukas M, Schutz H, Mancham S, et al (2017) Antibodies against immune checkpoint molecules restore functions of tumor-infiltrating T cells in hepatocellular carcinomas. Gastroenterology 153(4):1107–1119 e10
Zhu C, Sakuishi K, Xiao S, Sun Z, Zaghouani S, Gu G et al (2015) An IL-27/NFIL3 signalling axis drives Tim-3 and IL-10 expression and T-cell dysfunction. Nat Commun 6:6072
Zimmer L, Eigentler TK, Kiecker F, Simon J, Utikal J, Mohr P et al (2015a) Open-label, multicenter, single-arm phase II DeCOG-study of ipilimumab in pretreated patients with different subtypes of metastatic melanoma. J Transl Med 13:351
Zimmer L, Vaubel J, Mohr P, Hauschild A, Utikal J, Simon J et al (2015b) Phase II DeCOG-study of ipilimumab in pretreated and treatment-naive patients with metastatic uveal melanoma. PLoS ONE 10(3):e0118564
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Wang, J., Yang, T., Xu, J. (2020). Therapeutic Development of Immune Checkpoint Inhibitors. In: Xu, J. (eds) Regulation of Cancer Immune Checkpoints. Advances in Experimental Medicine and Biology, vol 1248. Springer, Singapore. https://doi.org/10.1007/978-981-15-3266-5_23
Download citation
DOI: https://doi.org/10.1007/978-981-15-3266-5_23
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-3265-8
Online ISBN: 978-981-15-3266-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)