
Abstract
In the post genomic era, the finding of new therapeutic targets has hugely been accelerated by the use of bioinformatics tools. The availability of genome sequences of pathogenic microbes has led to an increased finding of genes and proteins that could be potential targets for drug or vaccine design. The tools made available by bioinformatics have played a central role in the analysis of the genome and protein sequences for finding immunogenic proteins among the repertoire possessed by the organisms. The methods for prediction of immunogenicity are automated, and the whole proteome can be analyzed to find the top candidates that could have immunity inducing properties. Not only finding of immunogenic proteins has been achieved, but the mapping of the individual epitopes is also being done. The availability of methods for finding T and B cell epitopes can lead to the design of epitope-based vaccines. The description of different bioinformatics tools that are available for determining the immunogenic properties, finding of T and B cell epitopes, and in silico tools that are used in vaccine design is given in here. An account of epitope-based vaccine design employing bioinformatics methods reported in the literature is discussed. There are many shortcomings associated with these methods, which are discussed in the chapter. As is the case with other bioinformatics methods, there exist issues of prediction accuracy. Achievement of higher accuracy in predictions and their translation into in vivo/in vitro conditions still requires improvement. The chapter intends to provide the list of freely accessible software for epitope prediction and vaccine design with their merits/demerits and also throwing light on their applicability in vaccine research.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
11.1 Introduction
Treatment for infectious diseases is one of the most important aspects to improve the quality of human existence on the planet. However, the fight between the host and pathogen is not static but of a highly dynamic nature. The major pathogens threatening mankind are viruses, bacteria, and parasites. The co-evolution of pathogens and the emergence of new diseases have made the battle against pathogens continuous in nature. The issue has been further complicated by pathogens crossing the generic barriers, and pathogens from other animals have been showing presence in humans causing diseases. The most common pathogens that migrate from animals to humans are viruses and events of such transfer are reported. Animal viruses have infected humans, and caused diseases like SARS, MERS, SARS-CoV2 (coronaviruses), H1N1 (swine flu), and H5N1 (avian flu). SARS coronavirus originated most probably in bats though many argue its origin as uncertain (Chen et al. 2013) whereas MERS coronavirus was transmitted to humans from dromedary camels and its probable origin may be bats (Mohd et al. 2016). The most recent outbreak of pandemic novel coronavirus (SARS-CoV2) is still prevailing throughout the world and the reports to date suggest its bat or pangolin origin (Andersen et al. 2020; Zhang et al. 2020). The origins of H1N1 and H5N1 viruses are believed to be pigs and birds, respectively (Mena et al. 2016; Sims et al. 2005). All these viral outbreaks have happened in the first 20 years of this century.
Following the discovery of penicillin, the treatment for bacterial diseases has grown by leaps and bounds. With a battery of antibiotics available for therapy, the mortality and morbidity caused by bacterial diseases have been controlled. However, the drug resistance is increasing in bacteria and strains of multidrug resistant bacteria have been found, particularly in Mycobacterium tuberculosis. The therapeutic measures for parasitic disease treatment are limited by the availability of a few drugs. For example, nifurtimox and benznidazole are the only two drugs available for the treatment of Chagas disease. Furthermore, parasites have also started developing resistance against the current drugs used for treatment. Leishmania strains are showing increasing resistance to antimonates used for treatment. In addition, many of the drugs used for parasitic disease treatment are toxic including those mentioned in preceding sentences. This necessitates the development of better methods for finding a cure for pathogenic diseases and among these prophylactic measures like vaccines find an important place. Vaccines can help in reducing the disease burden by priming the immune system and inducing protective immunity against diseases. The success of vaccines has been well proven in eradicating smallpox and near elimination of polio from the world.
The modern era of vaccines started with the observation of cross-protection of smallpox through cowpox infection. It was Edward Jenner who observed that dairymaids who contracted cowpox subsequently never suffered from smallpox. He opined that cowpox somehow protected against smallpox and validated his theory experimentally by inoculating a boy first with cowpox pustule followed by smallpox pustule and the boy did not contract smallpox. This study published in 1798 was met with mixed reactions at that time (Riedel 2005). Louis Pasteur in 1879 accidentally discovered the “attenuation” while working on chicken cholera. His observations that chicken injected with old cultures of disease causing bacteria developed protection against subsequent injection of virulent cultures laid the foundation for a vaccination with attenuated organisms. Attenuation may thus be defined as the decrease in pathogenicity of a microbe without comprising its immune response generating properties. Later on, he used the same principle to develop protection against anthrax bacteria (Schwartz 2001). These discoveries towards the end of the nineteenth century paved the way for advances in immunology and vaccine development. In the forthcoming years, several new principles of developing vaccines were illustrated. Today vaccines based on various design platforms are being used commercially in the immunization regimens all over the world.
11.1.1 Live Attenuated Vaccine
The vaccines developed on the attenuation principle include the BCG vaccine for tuberculosis, Sabin polio vaccine (oral polio vaccine), measles vaccine, rotavirus vaccine, mumps vaccine, and varicella zoster (chickenpox) vaccine. Attenuation is generally achieved by growing the pathogen in abnormal conditions for long durations. In the case of Pasteur’s chicken cholera vaccine, it was found subsequently that aerobic culture conditions were responsible for attenuation. These vaccines though efficient, yet require considerable time for development. BCG is an attenuated strain of Mycobacterium bovis, which took 13 long years for development. The attenuation was achieved by growing Mycobacterium bovis in increasing concentrations of bile salts by Albert Calmette and Camille Guerin (Luca and Mihaescu 2013). Sabin polio vaccine was developed by culturing poliovirus in monkey kidney epithelial cells. The reversion of attenuated organisms into virulent forms can occur thereby causing disease rather than providing immunity. Sometimes the administration of these vaccines has led to conditions like natural disease in a small percentage of recipient population like in the measles vaccine (Kindt et al. 2007).
11.1.2 Inactivated Vaccine
Inactivated vaccines contain the killed pathogen and hence are also called killed vaccines. This class of vaccines includes hepatitis A vaccine, Salk polio vaccine, rabies vaccine, etc. Inactivation is generally mediated by chemicals like formaldehyde that was used for the inactivation of poliovirus to produce the Salk vaccine. In the case of killed vaccines, the process involves killing or inactivation of pathogens, thus the workers involved in the process are exposed to pathogenic microbes posing a serious health challenge. Further, these individuals, if infected, can serve as a reservoir for other populations and can lead to the spread of disease. Sometimes, there can be a failure in the inactivation or killing of the pathogenic organism, which leads to disease outbreak upon vaccination. This has happened with the first Salk polio vaccine where the virus was not killed by formaldehyde, and a high number of recipients of the vaccine developed paralytic polio (Fitzpatrick 2006).
11.1.3 Subunit Vaccine
The dangers associated with killed and inactivated vaccines have led to the development of vaccines that do not use the whole organism but the parts of the organism, which are sufficient to generate immunity. The subunit vaccines have been developed, which use macromolecules like protein (Hepatitis B vaccine) or carbohydrates (Pneumococcal vaccine) for inducing protective immunity. Hepatitis B virus surface antigen (HBsAg) gene has been cloned into yeast and mammalian cells and this recombinant protein is used as a licensed vaccine. There is no handling of the virus involved during vaccine production (WHO Data n.d.). However, in the case of subunit vaccines comprised of carbohydrate moieties like a pneumococcal vaccine, the bacteria Streptococcus pneumonia is cultured and the polysaccharides are purified for use in vaccine formulations (Morais et al. 2018). Thus, handling is involved during the production process, which can make workers involved in production exposed to the pathogen. The subunit vaccines involving the use of immunogenic proteins are preferable as genes for proteins can easily be cloned in high expression vectors, and the production of such vaccines can be carried out with ease. Toxoid vaccines are produced by inactivating the exotoxin produced by bacteria. Tetanus and diphtheria toxoid vaccines were developed by inactivating the exotoxin with formaldehyde.
11.1.4 Recombinant Vector and DNA Vaccines
The knowledge that the proteins rather than the whole organism can provide immunity has led to the development of recombinant vector and DNA vaccines. The genes for immunogenic proteins can be cloned into attenuated viral or bacterial strains and are expressed for longer duration as the vector used replicates in the host. Adenoviruses, vaccinia virus, attenuated strains of Salmonella, BCG strain of Mycobacterium bovis are some examples of the vectors that can be used. The vaccine for SARS-CoV-2 being developed by Prof. Sarah Gilbert at the University of Oxford contains a gene sequence of spike glycoprotein cloned into the chimpanzee adenovirus vector. This vaccine is undergoing accelerated clinical trials for the remedy of the current prevailing COVID-19 pandemic (https://www.ovg.ox.ac.uk/news/covid-19-vaccine-development). The development of DNA vaccines involves the cloning of a gene for an antigenic protein in a plasmid that can be directly injected into a muscle. The muscle cells take up the DNA and express the protein to induce protective immunity by priming the immune system. Though there are no licensed vaccines based on these approaches yet they are very promising for future vaccine applications (Kindt et al. 2007).
11.1.5 Epitope-Based Vaccines
From the preceding two sections (Sects. 11.1.3 and 11.1.4) it becomes clear that bio-macromolecules particularly proteins alone are capable of generating protective immunity provided they are good antigens. Antigens may be defined as those molecules that can be recognized by B cell receptors (antibodies/immunoglobulins) or T cell receptors. The antigen-antibody binding is direct without the mediation of any other molecule. However, the recognition of the antigen by the T cell receptor requires that the antigen is presented by MHC (Major histocompatibility complex) protein molecule. The antigen loaded in the MHC molecule cleft interacts with the T cell receptor present on T cells. Immune cells, both B and T cells do not interact with the whole antigen molecule but on certain discrete sites present on the antigen called epitopes. Epitopes may be defined as antigenic determinants present in the antigen that directly interact with the antigen-specific receptors present on B and T cells. Epitopes are of immense importance as they can be potentially used in epitope-based vaccine design. Epitopes are regions of immune specificity within a protein and can elicit a protective immune response. Epitope-based vaccines comprise immuno-dominant epitopes of a pathogen. Epitope-based vaccines are considered to be safer than traditional vaccines and focus on the most crucial antigenic elements of the pathogen to generate protective immunity (De Groot et al. 2009). Furthermore, epitope-based vaccines have provided the opportunity to design multi-epitopic immunogens that contain epitopes from different proteins. Such chimeric vaccines generated can have a combined protective effect, which otherwise would have required all the proteins whose epitopes are incorporated in the vaccine, which is a difficult process. This approach derives the benefit of using epitopes derived from multiple proteins rather than focusing on a single protein molecule. The use of bioinformatics has been extensively made in designing such vaccines. There are no commercially available vaccines based on this strategy yet epitope-based vaccines hold a great promise for the future.
11.2 B and T Cell Epitopes
The prerequisite for epitope-based vaccines is the availability of epitopes. The nature of epitopes present in an antigen needs to be understood for such vaccine design. There is a difference between the recognition of epitopes by B and T cells. B cell receptors can bind to epitopes in antigen present either in soluble form or on the surface of pathogen and there is no requirement of mediation by any other molecule for this binding. However, the binding mechanism for T cell epitopes is different, as they require an epitope to be presented by MHC molecules for binding to the T cell receptor. The nature of B and T cells of epitopes and their interactions are detailed in the next sections.
11.2.1 B Cell Epitopes
B cell epitopes are located on the native protein and are both continuous and conformational. The continuous epitopes are also known as linear, or sequential epitopes comprise amino acids present sequentially in the protein. The conformational epitopes also called structural or discontinuous epitopes can comprise amino acids that are located distantly in sequence, but because of protein folding come close together to form a particular protein structure. B cell epitopes are mostly surface accessible, hydrophilic, polar regions of the antigens that can readily bind to the respective antibody molecule (Zobayer et al. 2019). The epitope and the antibody binding site are complementary and the epitope fits into the complementarity determining region (CDR) of the antibody molecule. The interactions between them are stabilized by weak forces like electrostatic interactions, hydrogen bonds, van der Waals forces, and hydrophobic interactions.
11.2.2 T Cell Epitopes and Their Processing
Unlike B cell epitopes that can be recognized directly, T cell epitopes require presentation of epitope with MHC molecules. T cell epitopes are only linear or sequential and the antigens need to undergo processing before being recognized by their receptors. The protein is first degraded into small peptides; these peptides bind to MHC molecule and subsequently form a trimolecular complex with T cell receptors. There are two types of T cells viz Tc cells or cytotoxic T cells that display CD8 protein molecule on their surface and Th cells or helper T cells displaying CD4 surface protein. The epitopes that are presented to Tc cells are displayed by Class I MHC molecules whereas Th cell epitopes are displayed by Class II MHC molecules. The pathways of processing and presenting epitopes to both types of T cells are different.
Tc cells recognize epitopes arising from proteins processed by the cytosolic pathway, which involves processing through proteasome and subsequent binding of the cleaved peptides to class I MHC molecule before presentation and recognition. Concisely, the proteasome (a multimeric protein complex) cleaves the protein into small peptides; these peptides are transported by TAP proteins (transporters associated with antigen processing) into the ER (endoplasmic reticulum) lumen. Class I MHC molecules are undergoing folding in the ER lumen where they bind to these transported peptides with the help of tapasin. The MHC-peptide complex is then transported by the secretory pathway to the cell surface (Hewitt 2003). Class I MHC glycoproteins are expressed by all nucleated cells and present antigen to cytotoxic T (Tc) cells. The peptide binding cleft of Class I MHC molecule is closed at both the ends and can bind peptides with 8–10 amino acids in length with nonamers showing best binding. The structure of Class I MHC with peptide bound in its cleft is shown in Fig. 11.1, whereas its antigen processing pathway is shown in Fig. 11.2.

Class I MHC molecule with peptide bound in the cleft (α chain: green, β2 microglobulin: blue and the peptide: purple color)

Processing of antigen, binding of epitope to Class I MHC, and its display on cell surface
Antigen processing for epitopes binding to Th cells takes place by the endocytic pathway involving phagocytosis and lysosomal cleavage of protein followed by binding to the Class II MHC molecule for presentation and recognition. Briefly, antigens are internalized into the cell by phagocytosis and it proceeds sequentially through early endosomes, late endosomes, and finally to lysosomes. In these increasingly acidic compartments, antigen gets cut into small peptides by the inherent proteases present there. Class II MHC molecules are transported from the Golgi complex to the endocytic pathway by an invariant chain. As the MHC II molecule moves through the endocytic pathway invariant chain gets cleaved leaving CLIP (class II-associated invariant chain peptide) occupying the peptide binding cleft of MHC II. HLA-DM catalyzes the exchange of CLIP with antigenic peptide and finally, Class II MHC molecule moves to the cell surface (Kindt et al. 2007). Class II MHC glycoproteins expressed on the surface of antigen presenting cells (dendritic cells, macrophages, and B cells) present antigen to helper T cells (Th). The peptide binding site of Class II MHC is open at both ends and can bind peptides of 13–18 amino acids length. Figure 11.3 depicts the Class II MHC molecule with a bound peptide in its cleft and its antigen processing pathway is depicted in Fig. 11.4. Thus, the prerequisite for any protein which can be a possible T cell antigen is that it should be comprised of peptides that show binding affinity to MHC molecules. The proteins that upon passing through the antigen processing pathway generate peptides having an affinity for binding to the cleft of MHC molecules can be classified as T cell antigen proteins.

Class II MHC molecule with peptide bound in the cleft (α chain in blue, β chain in orange, and the peptide in yellow color)

Processing of antigen, binding of epitope to Class II MHC, and its display on cell surface
11.3 Bioinformatics in Vaccine Design
With the advance in genomic technologies in the recent past, the genomes of organisms are being sequenced at an unprecedented pace. The amount of the data available is immense and can provide insights into finding unexplored genome regions in search of novel targets for the treatment of diseases. The wealth of available genomic data has to be analyzed for deciphering the encoded proteins, and for vaccinology purposes. The total proteins encoded by the genome can be screened for finding out immunogenic proteins using bioinformatics tools. These antigenic proteins can further be used to find out epitopes located in them. Many of the genome databases have constructed proteomes of the sequenced genomes by automated methods. The repertoire of proteins encoded by the genome can be analyzed by bioinformatics servers to find antigenic proteins. The filtered antigenic proteins can be used to find specific B and T cell epitopes in these proteins by the available epitope prediction methods. A new branch called immunoinformatics has come into existence, which deals with the application of computational tools to immunologic problems (Backert and Kohlbacher 2015).
Locating T and B cell epitopes in the proteins of a pathogen is the major job of immunoinformatics. The tools for finding B and T cell epitopes among the cohort of proteins encoded by the genome of an organism have been available in the public domain for almost more than two decades now. These tools are based on various machine learning methods. The availability of experimental data about T and B cell epitopes has also increased, which has also enhanced the accuracy of prediction methods as most of the methods use this data as a training set for developing tools. The mechanisms of recognition of B and T cell epitopes are different and their properties also vary. T cell epitopes are linear in nature and need to bind with MHC molecules for their presentation to T cell receptors whereas B cell epitopes are linear and conformational, and are recognized in their native position in the protein. The prediction methods, therefore, have to take into account these different properties of the epitopes.
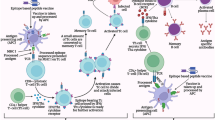
For any T cell epitope, the binding affinity to the MHC molecule is immensely important, as this is the first step that qualifies it to be an epitope. The prediction methods for finding such an affinity of peptides first progressively break antigenic protein into peptides and analyze their affinity for a particular MHC molecule. The diversity of MHC molecules further complicates the situation as the affinity for peptides changes with change in the molecule. The studies on the peptides eluted from MHC molecules reveal that there are differences in the properties of peptides bound in the cleft of different MHC proteins. The alleles for MHC are designated as HLA alleles; for class I these alleles are HLA- A, B, and C and for class II HLA-DP, DQ, and DR. In the human population, the number of HLA class I alleles is 14,800, and that of HLA class II alleles is 5288 (Statistics of HLA alleleshttps://www.ebi.ac.uk/ipd/imgt/hla/stats.html). Further, the distribution of HLA alleles differs among different population groups of the world. Thus, any software tool that is developed for the T cell epitope determination needs to consider these points. The epitope prediction tools used for B and T cell epitopes are discussed in subsequent sections. The B and T cell epitope prediction process is shown in Fig. 11.5.

Schematic process flow of B and T cell epitope prediction for epitope-based vaccine design
11.4 Prediction Tools for Class I and II MHC Binding
A comprehensive list of the freely accessible tools available for determining the binding affinity of peptides in a protein to different MHC molecules is listed in Table 11.1. These tools are based on different machine learning methods like support vector machine (SVM), artificial neural networks (ANN), hidden Markov models (HMM), and position-specific scoring matrices (PSSM). Some tools can carry out the peptide binding predictions for both class I and II MHC molecules, whereas some of the tools are exclusive. Tools like NetMHC, NetMHCPan, ProPred-I, EpiJen, and nHLAPred carry out the binding affinity prediction of peptides to class I MHC molecules, and NetMHCII, NetMHCIIPan, and ProPred are exclusively used for class II MHC binding predictions. Most of the other tools have the capability of carrying out a prediction for both classes of MHC molecules. The number of alleles available for running predictions is different in each tool.
11.4.1 NetMHC
NetMHC utilizes the ANN approach to predict the binding affinity of a peptide for different class I MHC molecules. This predictive model has been trained for 81 different MHC alleles of humans, including HLA-A, HLA-B, HLA-C, and HLA-E (Andreatta and Nielsen 2016).
11.4.2 NetMHCPan
It predicts the binding affinity of peptides to any MHC of the known sequence. This ANN-based method is trained by more than 180,000 binding data, and MHC eluted ligands. The binding affinity data covers 172 MHC molecules from human, mouse (H-2), Cattle (BoLA), primates, and swine (SLA). It provides information about the likelihood of a peptide to be a natural ligand or the binding affinity (Jurtz et al. 2017).
11.4.3 SYFPEITHI
This database contains MHC class I and class II ligands, peptide motifs of humans and other species, natural ligands, and T cell epitopes. It also provides connectivity to resources available at EMBL and PubMed databases (Rammensee et al. 1999).
11.4.4 ProPred-I
ProPred-I is used to identify the MHC class-I binding regions in antigens. It also helps the researcher to identify the promiscuous regions (Singh and Raghava 2003).
11.4.5 RANKPEP
It predicts the peptide binders to MHCI and MHCII from protein sequence information. It also identifies the MHCI ligands, whose C terminal end is likely to be the result of proteasomal cleavage (Reche et al. 2002).
11.4.6 MHCPred
This method assumes that each substituent present in a molecule has an additive and independent contribution to the biological activity. It considers the interaction between individual amino acids and the binding site, the interaction between adjacent and every second amino acids, and their effects on binding (Guan et al. 2003).
11.4.7 EpiJen
This method considers proteasome cleavage and TAP binding and can mimic the MHC binding mechanism in a real way (Doytchinova et al. 2006).
11.4.8 SVMHC
This tool is based on the SVM approach and used to predict both class I and class II MHC binding epitopes. This server is based on (Dönnes and Kohlbacher 2006).
11.4.9 MULTIPRED2
It is used to screen peptide that binds to multiple alleles belonging to HLA class I and class II DR super types. It performs binding predictions on 1077 alleles related to 26 HLA super types (Zhang et al. 2011).
11.4.10 ProPred
ProPred predicts class II MHC binding regions in the antigenic sequence. It assists in locating promiscuous binding regions which are useful in screening vaccine candidates (Singh and Raghava 2001).
11.4.11 MHC2Pred
This tool is used to predict promiscuous class II MHC binding peptides. For algorithm designing, the information of binders and non-binders for different alleles were taken from the MHCBN and JenPep database. The average accuracy of this method is ~80% (Lata et al. 2007).
11.5 CTL Epitope Prediction
Though multiple tools are available for prediction of binding affinity of peptides to different class I MHC molecules, yet only binding to a particular MHC is not sufficient to qualify a peptide to be a Tc cell epitope. In other words, not all class I MHC binders are Tc cell epitopes, whereas all Tc cell epitopes are good MHC binders. Also, the peptide should be amenable to the antigen processing pathway of class I MHC i.e. cytosolic pathway. Proteasomal cleavage and transport of peptides into the rough endoplasmic reticulum (RER) by TAP are other important steps involved in the cytosolic pathway of antigen processing and presentation (Hewitt 2003).
All intracellular proteins after spending a fixed time in the cell are marked for degradation by a small protein called ubiquitin. The marked proteins are then cleaved by proteasome into small peptides within its central hollow. The immune system modifies proteasome by the addition of extra protein molecules called LMP7, LMP2, and LMP10 to generate peptides having a preferential affinity for class I MHC molecules. Thus, for any peptide to act as a Tc cell epitope, it should be processed by the proteasome. The transport of peptides generated by the proteasome to RER is carried out by the transport by TAP. TAP also shows preference to transport peptides of 8–13 amino acid residues in length (Kindt et al. 2007). The peptide should have these properties to get transported from the cytosol to RER. These requirements are not as specific as binding to class I MHC molecule yet play an important role in making a peptide a Tc cell epitope.
There are bioinformatics tools that carry out proteasomal cleavage and TAP transport prediction and are listed in Table 11.2. Some of the class I MHC binding prediction tools have these two functions inbuilt in them. EpiJen server, in addition to MHC binding also uses proteasomal cleavage and TAP transport for predicting Tc cell epitopes (Doytchinova et al. 2006). nHLAPred also uses proteasomal cleavage matrices to refine the results of epitope prediction. ProPred-I uses the proteasomal model and immunoproteasome models for finding the epitopes. RANKPEP predicts class I MHC binding peptides whose C terminal end is likely to be the result of proteasomal cleavage. The description of tools that exclusively serve the purpose of proteasomal cleavage and TAP transport prediction is provided below.
11.5.1 NetCTL
It is used to predict peptide MHC class I binding, proteasomal C terminal cleavage, and efficiency of TAP transport. MHC class I binding and proteasomal cleavage is based on the ANN approach while the efficiency of TAP transport uses a weight matrix (Larsen et al. 2007).
11.5.2 CTLPred
This tool uses a quantitative matrix, SVM, and ANN approach for prediction. It has been developed by training and testing the results from the dataset of T cell epitopes and non-epitopes (Bhasin and Raghava 2004a).
11.5.3 NetChop
NetChop is based on the ANN method to predict the cleavage sites of the human proteasome. Since the method is trained using human data, therefore, it shows better performance in predicting sites of proteasomal cleavage for humans. The method is used by NetCTL for predicting proteasomal cleavage sites (Keşmir et al. 2002; Nielsen et al. 2005).
11.5.4 MAPPP
MAPPP is used to predict antigenic epitopes on the cell surface by class I MHC to CD8 positive T lymphocytes. It also predicts the proteasomal cleavage with peptide anchoring to MHC I molecules (Hakenberg et al. 2003).
11.5.5 Pcleavage
It is an SVM based method used to predict constitutive and immunoproteasome cleavage sites in the antigenic molecule. The method only predicts proteasomal cleavage sites, but no prediction of TAP transport is available (Bhasin and Raghava 2005).
11.6 B Cell Epitope Prediction
B cell epitopes are recognized by the B cell receptors i.e. antibodies without the process of processing and presentation, unlike T cell epitopes. Linear B cell epitopes, in principle, can be predicted by the same methods as used for T cell epitopes. However, the prediction of conformational or structural epitopes is a challenging job. The requirement of structural data of a protein is absolute for finding discontinuous epitopes. There are methods available for prediction of both continuous and discontinuous B cell epitopes (Table 11.3), but the efficiency of these methods is less when compared to T cell epitope prediction methods. The methods use many different approaches like ANN, SVM, HMMs for predictions.
11.6.1 BCPred
In BCPred server, the user can select the method such as amino acids pair scaling (AAP), BCPred, and FBCPred for prediction. AAP has good accuracy in the prediction of antigenicity, hydrophilicity, and flexibility (Chen et al. 2007; EL-Manzalawy et al. 2008).
11.6.2 LBtope
LBtope is based on data of B cell epitopes and non-B cell epitopes from the immune epitope database. Models like SVM and K-nearest neighbor are used in discriminating epitopes and non-epitopes. The features like binary profile, dipeptide composition, AAP (amino acid pair) profile have been used in design of the method, and the accuracy of prediction ranges from 54% to 86% (Singh et al. 2013).
11.6.3 ABCPred
It is an ANN-based approach used to predict continuous B cell epitopes using a fixed length pattern. This tool is developed using the dataset of epitopes from parasites, viruses, bacteria, and fungi from the BciPep database, and it has a prediction accuracy of 65.9% (Saha and Raghava 2006a).
11.6.4 BepiPred 2.0
It is based on the random forest algorithm and developed from a dataset of epitopes annotated from the antibody-antigen structure from PDB. This tool requires a FASTA format of the protein as input (Jespersen et al. 2017).
11.6.5 Bcepred
Bcepred predicts the linear B cell epitopes using physicochemical properties, such as hydrophilicity, accessibility, flexibility, polarity, exposed surface, etc. The accuracy of this server is 58.7% (Saha and Raghava 2004).
11.6.6 DiscoTope
This server is used for the prediction of discontinuous B cell epitopes from protein 3D structures using surface accessibility and a novel epitope propensity score of residues (Kringelum et al. 2012).
11.6.7 ElliPro
ElliPro is used for prediction and analysis of antibody epitopes in a protein structure. Here, PDB ID or a PDB file of a protein is used as input. It has been designed using the information of discontinuous epitopes present in antibody-protein complexes (Ponomarenko et al. 2008).
11.6.8 PEASE
This server predicts antibody-specific epitopes using sequence information of the antibody. The epitopes related information is provided at the residue level and also on the structure of antigen (Sela-Culang et al. 2014a, b).
11.7 Methods for In Silico Designing of Epitope-Based Vaccines
Vaccines designing using immunoinformatics tools have come a long way, and many strategies have been employed for this purpose. Before the advent of such tools and precedent to the availability of genome, the classical vaccinology approaches were used which required more time and labor. The prime requirement in the case of subunit vaccines is a biomolecule, mostly proteins that have the potential to induce immunity. In the case of epitope-based vaccines, epitopes from more than one protein can be amalgamated in a single construct for enhancing immunity. The general account of approaches used is given below. The process and tools used for epitope analysis and selection of epitopes for vaccine design are displayed in Fig. 11.6.

Graphic representation of steps involved in the analysis of predicted B and T cell epitopes for designing of in silico vaccines
11.7.1 Selection of Proteins
As mentioned earlier, the requirement of immunogenic proteins is prime for epitope-based vaccine designing. The databases like NCBI Protein and UniProt can serve as the source of proteins for analysis. NCBI Protein database is a collection of protein sequences from SwissProt, PIR (Protein Information Resource), PRF (Protein Research Foundation), and PDB (Protein Data Bank) in addition to translated sequences obtained from annotated coding regions of GenBank sequences. UniProt contains protein sequences obtained from SwissProt and translated EMBL (trEMBL) database. The finding of immunogenic proteins from the genome can be achieved by using various criteria. The total proteins encoded by the genome of a pathogen i.e. its proteome can be analyzed for immunogenic proteins by using servers like VaxiJen (Doytchinova and Flower 2007). This server can take as input multiple protein sequences from bacteria, viruses, fungi, parasites, and the threshold value can be controlled by the user. The total proteome of an organism can be provided as input and depending upon the threshold, antigenic proteins can be selected. Proteomics approaches to find the stage-specific expression of proteins can also aid in vaccine development (Soria-Guerra et al. 2015). The other approach for fishing proteins from the proteome is to find the surface proteins. The surface proteins are easily accessible to immune effector molecules particularly to antibodies and can suffice the purpose. The servers like CELLO (Yu et al. 2006), Cell-PLoc (Chou and Shen 2008) that predict the subcellular localization of proteins can help in finding the surface proteins from the proteome. The combined approach in which first surface proteins are predicted from the proteome and these proteins are then subjected to immunogenicity prediction by VaxiJen has also been employed (Pritam et al. 2019). There is another server by the name of Vaxign which provides two modes of usage; one in which pre-computed results of more than 350 genomes are available and can be used for finding immunogenic proteins, and the second involves the protein input to be provided by the user and results are computed by the server (He et al. 2010). This server can also assist in protein selection. Literature studies are also a good source for finding immunogenic proteins. The previously reported proteins capable of generating immunity can also be used for epitope prediction and a vaccine can be designed from the epitopes derived from multiple proteins.
In some cases, several variant sequences exist for a single immunogenic protein. This could be due to the protein sequences arising from different strains of a pathogen or the variability induced by the pathogen itself in its surface proteins for evading the immune response. This variability provides an advantage to the pathogen and poses a major hindrance in vaccine development. The conserved regions in such a protein are deciphered by multiple sequence alignment of the different variant sequences. Tools like Clustal Omega, TCOFFEE, etc. can be employed to carry out multiple sequence alignment. These conserved regions can then serve as the source for the prediction of epitopes.
11.7.2 Epitope Prediction and Analysis
Once the protein/proteins have been selected the subsequent step involves the prediction of B and T cell epitopes. The prediction of epitopes can be carried out using the tools mentioned in Sects. 11.4, 11.5, and 11.6. Tc cell epitope prediction involves predicting the affinity of peptides (by tools in Sect. 11.4) for the respective HLA allele (Class I MHC) followed by the proteasomal cleavage and TAP transport prediction (tools in Sect. 11.5). The epitopes qualifying these criteria are generally selected for the vaccine designing process. For helper T cells the epitope prediction involves ascertaining the binding affinity of peptides for HLA alleles (Class II MHC) and there are no methods available for determining the antigen processing and presentation prediction by endocytic pathway. The tools presented in Sect. 11.6 can predict continuous and discontinuous B cell epitopes. Thus, a pool of B and T cell epitopes can be generated which can be further analyzed.
Many a time the epitopes particularly, T cell epitopes predicted for different HLA alleles can share considerable sequence similarity. Such epitopes can be clustered together and a single representative of this cluster can be used in the final design. The process of clustering removes the unwanted repetitiveness of epitopes and prevents the vaccine construct from unnecessary elongation. Epitope cluster analysis tool (Dhanda et al. 2018) can be used to carry out this step as the epitopes are clustered together based on the sequence identity threshold set by the user. Another important aspect is to check the similarity of the epitopes with the host proteins and this can be achieved by using BLAST (Basic Local Alignment Search Tool) available at NCBI (National Center for Biotechnology Information). After BLAST analysis the epitopes sharing similarity with host proteins need to be omitted, as they may not generate any response. Population coverage tool helps in finding the predicted immune response to T cell epitopes in a population group based on HLA allele distribution (Bui et al. 2006). The Allele Frequency Net Database is the source of HLA allele distribution frequencies in different populations of the world used in the tool. The epitopes should be able to provide a higher percentage of population coverage to be used in vaccine design as this ensures immune response generation in most of the individuals in a population group. To check the conservancy of epitopes across the variants of a protein, Epitope conservancy analysis tool can be used which calculates the degree of the conservancy of a particular epitope in the cohort of protein sequences (Bui et al. 2007). This tool becomes an important asset when different variants of a protein exist as mentioned in Sect. 11.7.1.
The epitopes can also be checked for the presence of any allergenic and toxic peptides among them. The tools for allergenicity prediction like AlgPred (Saha and Raghava 2006b), AllergenFP (Dimitrov et al. 2014a), AllerCatPro (Maurer-Stroh et al. 2019), and AllerTop (Dimitrov et al. 2014b) are freely available and can be used to remove the epitopes possessing allergenic nature from the group to be used in vaccine design. The toxic peptides can be predicted by the ToxinPred server (Gupta et al. 2013) and any epitopes that are toxic in nature have to be omitted from the final construct. The epitopes after filtration by the above methods can be used further.
11.7.3 Molecular Docking and Molecular Dynamics Simulation
The use of molecular docking and simulation to find interactions between epitopes and immune effector molecules is an important aspect of in silico vaccine designing. T cell epitopes should bind to MHC molecules for presentation to T cell receptors. This binding can be studied by molecular docking, for which the structure of epitope needs to be determined. The servers like PEPFOLD (Lamiable et al. 2016), QUARK (Xu and Zhang 2012), etc. are freely available for modeling of small peptides and can provide models of T cell epitopes. The structures of MHC molecules (Class I and II) can either be obtained from Protein Databank (PDB) if available or models of these proteins be generated by homology modeling servers like SwissModel (Waterhouse et al. 2018) and many others. The docking of the epitopes with respective MHC molecule/HLA allele can be carried out by protein-protein docking servers like ZDOCK (Pierce et al. 2014), ClusPro (Kozakov et al. 2017), etc. The docking results can validate whether the epitope binds in the cleft of the MHC molecule. The docked complexes can further be analyzed by molecular dynamics simulation to explore the interaction between the epitope and MHC molecule in conformational space. These results, if positive can further strengthen the possibility of epitopes being presented by MHC molecules to T cells. Software suites like GROMACS (van der Spoel et al. 2005) are freely available that can be used for simulation studies. T cell epitopes that dock into the peptide binding cleft of MHC molecules are selected for vaccine designing. The docking and simulation studies for B cell epitopes are not required as they bind directly to the antibody molecules and are not presented through MHC molecules. The complementarity determining regions (CDRs) of antibody molecules are highly diverse and thus binding studies cannot be carried out.
11.7.4 Construction of Vaccine
Many studies culminate at the finding of epitopes that qualify the processes mentioned in Sects. 11.7.1–11.7.3 and the resultant cohort of epitopes is left for the designing of vaccines in future studies followed by experimental validations. In some studies, the epitopes are used for cell culture based studies, and their ability to initiate an immune response is validated by the cytokine response generated by them in peripheral blood mononuclear cells (PBMCs). However, in silico vaccine designing based on the predicted epitopes is also widely carried out. The epitopes are joined in tandem with the insertion of specific linkers for efficient processing of epitopes such as AAY linker is generally used between two CTL epitopes. AAY linker possesses the cleavage site of proteasomes, which leads to the generation of natural epitopes and it can also reduce the unwanted joining of two neighboring epitopes in the vaccine construct. Similarly, the GPGPG linker is used for the separation of T helper cell epitopes as this linker is reported to facilitate immune processing and prevent the joining of two epitopes. In many of the vaccine constructs protein adjuvants like cytokines have also been fused with the epitopes and in these constructs linker like EAAAK has been widely used. The linker EAAAK causes the separation of fusion proteins (Arai et al. 2001) and can prevent the interaction between the vaccine and adjuvant domains of the vaccine protein construct. Thus, the final fusion protein obtained as vaccine consists of epitopes, linkers, and may be adjuvants in certain cases. The structure of this fusion protein can be deduced using web-based protein modeling severs like I-TASSER (Roy et al. 2010). In some instances, homology modeling servers can successfully model the vaccine protein structure, whereas for some structures ab initio modeling approaches have to be used. The protein sequence can be reverse translated into DNA and thus gene constructs of vaccine can be made. JCat tool can be used for reverse translation as well as for codon optimization for efficient translation in the host cells (Grote et al. 2005). The protein sequence of the vaccine construct can be reverse translated using codon bias for expression in eukaryotic or prokaryotic cells for heterologous production of the vaccine. Alternatively, the gene for human expression can be codon optimized for direct use as a DNA vaccine in humans. Thus, this section summarizes the methods that can be used for designing of in silico vaccines. Some examples of the development of vaccines using these approaches are depicted in Sect. 11.8.
11.8 Case Studies of Vaccine Designing
There have been various studies on vaccine designing using bioinformatics tools. Immunoinformatics has been widely used for in silico designing of vaccines for various pathogens like viruses, bacteria, and parasites. The details of these vaccine designing studies are given in the forthcoming sections.
11.8.1 Vaccine Designing for Viral Pathogens
Viruses are nucleoprotein particles, which have imposed a heavy disease burden throughout human history. Since, viruses use the host cell machinery for their replication and other functions, it limits the availability of drug targets in them. Vaccines have been the prime means for the treatment of viral diseases. Recently, vaccines for viruses have been designed using in silico methods. The vaccines have been designed based on epitopes derived from a single viral protein. The criteria used for the selection of protein are either immunogenicity or surface accessibility. Ebola virus vaccine was designed using predicted B and T cell epitopes present in the glycoprotein of the virus. VaxiJen server was used to find immunogenic protein followed by epitope predictions, which were further validated by molecular docking and molecular dynamics simulation approach (Dash et al. 2017). T and B cell epitopes (linear and discontinuous) were predicted in the Spike protein of MERS-COV using bioinformatics tools, which could be used in vaccine design (Ul Qamar et al. 2019). In some cases, epitopes have been predicted from more than one viral protein for use in vaccine design. The proteins E, prM, NS1, NS3, and NS5 of Japanese Encephalitis Virus (JEV) were used in a recent study for the prediction of T and B cell epitopes. Based on different parameters assessed four T cell and one B cell epitope were found to have potential in inducing immunity and could be used in vaccines against the virus (Chakraborty et al. 2020). B and T cell epitopes from five structural polyproteins (capsid, E2, 6K, E3, and E1) of the Mayaro virus were predicted using immunoinformatics tools. Multi-epitope vaccine was designed, molecular docking with TLR-3 was done, and finally in silico expression was carried out in E. coli (Khan et al. 2019). The phenomenon of cross reactivity found among related viruses has earlier led to the development of immunity as in the case of smallpox (details in Sect. 11.1). With this background, attempts have been made to find common epitopes present in two or more related viruses for designing vaccines that could generate immunity across these viruses.
A study on four antigenically important proteins (HA, NA, NP, and M2) of H1N1, H2N2, H3N2, and H5N1 viruses revealed the presence of 18 conserved epitopes across these viruses which have the potential for future vaccines (Muñoz-Medina et al. 2015). Hendra virus and Nipah virus proteins (F, G, and M), when subjected to B and T cell epitope prediction, showed common epitopes which could be used for vaccine design against both the viruses (Saha et al. 2017). In the envelope protein of the Japanese Encephalitis virus and West Nile virus, a common conserved epitope was detected which contained both B and T cell epitopes that could find use in designing epitope-based vaccines (Slathia and Sharma 2019).
11.8.2 Vaccine Designing for Bacteria
Since the discovery of penicillin, the therapeutic interventions for bacterial diseases have increased by leaps and bounds, and antibiotics remain the most important treatment for bacterial infections. Prophylactic vaccines like DTP (Diphtheria, Tetanus, Pertussis), Hib (Haemophilus influenzae type B), pneumococcal are included in immunization schedules throughout the world and have been helpful in reducing the disease burden considerably. Bacteria have a larger genome and proteome as compared to viruses, therefore finding immunogenic proteins is a little laborious job. The full proteome of bacteria has been studied to find immunogenic proteins, which can be used for vaccine design. The total proteome of M. tuberculosis H37Rv, when used for finding the best vaccine candidates by in silico methods, revealed six novel vaccine candidates, EsxL, PE26, PPE65, PE_PGRS49, PBP1, and Erp, which could be used to design new TB vaccines (Monterrubio-López and Ribas-Aparicio 2015). In another study proteomes of three serotypes of Shigella: S. dysenteriae type1 (sd197), S. flexneri 2a (str. 301 and str. 2457T), and S. sonnei (ss046) were investigated to determine the common proteins of these three bacteria. The epitope prediction for these common proteins was done and five peptides were used for in vivo animal and human serum studies. The peptides elicited antibody and cytokine (Th1 and Th2) response confirming that these cross protective and conserved peptides have the potential to be used in future vaccines (Pahil et al. 2017). Studies have also been focused on a group of proteins or even a single protein for epitope prediction and vaccine design. Essential hypothetical proteins of five Salmonella strains were studied to find out drug and vaccine targets. Out of 106 proteins, 4 proteins were found to be immunogenic for which conserved B and T cell epitopes were predicted which can be used for future vaccine design (Sah et al. 2020). Nine epitopes were predicted from 11 multidrug resistance (MDR) proteins of Salmonella typhi that had the potential to generate B and T cell response and can find use in vaccine design (Jebastin and Narayanan 2019). A DNA vaccine based on cytotoxic T cell epitopes predicted from a single protein Listeriolysin-O of Listeria monocytogenes was constructed using in silico methods. T cell epitopes were fused in tandem, human and mouse gene constructs were made in addition to determining posttranslational modifications like phosphorylation and glycosylation (Jahangiri et al. 2011). An outer membrane protein of Vibrio cholera was used for epitope prediction by different tools and one surface exposed peptide was found containing both B and T cell epitopes, which could have future vaccine design applications (Rauta et al. 2016).
11.8.3 Vaccine Designing for Other Parasites
Parasitic diseases caused by helminths and protozoans are difficult to treat, as these organisms are eukaryotic in nature and drug targets that are non-homologous with host tend to be less in number. The therapeutic measures for their treatment are limited and there are no licensed vaccines for use in humans. A vaccine against Plasmodium falciparum “RTS, S” has been introduced under the aegis of WHO in Ghana, Kenya, and Malawi and is undergoing pilot scale trials since 2019. The efforts, therefore, are required to develop vaccines against parasitic diseases. The use of bioinformatics tools has been made to design vaccines for these diseases. A multi-epitope peptide vaccine derived from epitopes obtained from six proteins of Onchocerca volvulus was designed using in silico methods. The epitopes used in the peptide vaccine showed varying degrees of conservation in related species Onchocerca ochengi, Loa loa, Onchocerca flexuosa, Brugia malayi, and Wuchereria bancrofi indicating its cross protective capability. The peptide vaccine was reverse translated, codon optimized, and conceptually cloned in the pET vector after carrying out other analysis like docking, immune simulation (Shey et al. 2019). The proteome of Taenia solium was used to find surface accessible immunogenic proteins for which B and T cell epitopes were predicted. A peptide construct based on the epitopes was made and the structure was determined by modeling and after that, it was docked with immune receptors and finally, a gene was constructed to express the peptide vaccine (Kaur et al. 2020). B and T cell epitopes were predicted from the enolase protein of Echinococcus granulosus and a multi-epitope vaccine was designed after analyzing its immune response generating properties (Pourseif et al. 2019). Triosephosphate isomerase from the same organism has also been used to predict epitopes for use in vaccines (Wang and Ye 2016).
From the total proteome of Plasmodium falciparum, five surface accessible antigenic proteins were selected for the prediction of T cell epitopes. These epitopes upon docking and population coverage revealed their efficiency to be used in epitope-based vaccines (Pritam et al. 2019). B and T cell epitopes from AMR1, a surface exposed protein of Plasmodium falciparum have been predicted in a study that has the potential for use in future subunit vaccines (Sanasam and Kumar 2019). An approach for developing epitope-based vaccine Trypanosoma cruzi involved epitope prediction from the proteome of the pathogen. mRNA construct and the structure of the peptide vaccine comprising epitopes were made (Michel-Todó et al. 2019). Conserved T cell epitopes were predicted from variants of an amastin protein of Trypanosoma cruzi for future vaccine designing (Slathia and Sharma 2018). In silico prediction of T cell epitopes from promastigote surface antigen (PSA), LmlRAB (L. major large RAB GTPase), and histone (H2B) proteins of Leishmania was done followed by testing of these epitopes for inducing different cytokines in peripheral blood mononuclear cells (PBMCs) isolated from cured and healthy individuals. The epitopes were able to induce specific cytokine producing helper and cytotoxic T cells and could be used in future vaccine design (Hamrouni et al. 2020).
11.9 Limitations and Challenges
The major step involved in epitope-based vaccine designing using bioinformatics tools is the prediction of epitopes. Therefore, the accuracy of epitope prediction methods is of prime importance, as this will govern the success of vaccines in the real world. More is the accuracy of the epitope prediction methods greater are the chances of success of inducing protective immunity by the vaccine. The methods available for epitope prediction have been benchmarked using the experimental steps in many studies. The limitations and their prediction efficiency have been studied. Many new methods have been redesigned as new data becomes available. Generally, it has been observed that modern machine learning methods like SVM and ANN perform better than linear methods like PSSM. The prediction efficiency achieved in class I MHC epitope prediction is better as compared to predictions for class II MHC and B cell epitope prediction. The benchmarking of automated servers for class I MHC prediction is carried out weekly, and the results are available on the immune epitope database (IEDB). These benchmarking results show that among the participating servers, NetMHCPan is the best performing server.
The next best performing methods are SMM and ANN. The ranking scores are indicative of the performance of methods among each other and do not indicate the absolute predictive performance. The ranks are concerning each other and not in the context of their prediction efficiency (Trolle et al. 2015). Many of the binding peptides are not immunogenic, and even if they are amenable to processing and presentation, they do not act as epitopes. There are still loopholes in the methods, and the binding stability of peptide and HLA molecule has also to be taken into account. The only tool available for this is NetMHCstab (Jørgensen et al. 2014) which is an ANN-based tool and has only been trained on 13 HLA alleles. With the increase in data about HLA alleles and their binding peptides, these tools are bound to increase their efficiency in the future.
The prediction methods for class II MHC are yet to achieve the efficiency of class I MHC predictors. In most of the methods, the prediction is limited to HLA-DR alleles, and few servers like NetMHCII 2.3, RANKPEP, NetMHCIIPan carry out predictions for HLA-DP and HLA-DQ as well. The nature of peptides binding to class II MHC is different from that showing binding to class I. Peptides binding to class II MHC molecules have a binding core rather than anchor residues seen in class I MHC binding peptides. Besides, the peptides binding to class II MHC are longer, and the position of the binding core is not fixed (Kindt et al. 2007). The peptide binding mode of class II is less specific than class I, and the genotype structure of class II allotypes is more complicated. This makes designing of class II MHC binding prediction methods more challenging. The tools need to address these issues, and the lack of data available makes these prediction tools less efficient.
The benchmarking of class II predictors is also done weekly, and among the different prediction tools, the NN-align method which is the basis of NetMHC2.0 (Nielsen and Lund 2009) outperforms the other methods. NetMHCIIPan is the next best performing method followed by Comblib matrices, (Sidney et al. 2008) a method available at IEDB analysis resource and SMM-align. Next-generation sequencing (NGS) has increased the inflow of genomic data in an unprecedented manner, and the data for HLA alleles is now being generated at a high pace. This data along with other high throughput experimental data about the class II MHC-peptide binding is required to increase the efficiency of prediction tools.
Prediction of continuous B cell epitopes follows the same principles, albeit the length of epitopes is not fixed. For discontinuous epitopes, the prediction requires different approaches as the classic machine learning methods need continuous sequence data (Backert and Kohlbacher 2015). There are fewer benchmarking studies for B cell epitope predictors, and most of them conclude that the efficiency of these methods is yet far from meeting the requirements in the biological context. Since there are no universal properties that are present in antigenic epitopes but absent in other protein surfaces, therefore, designing methods for prediction is a challenging job. The methods for linear epitope prediction are based on the hypothesis that certain amino acids occur more frequently in the epitopic regions. A benchmarking study for linear B cell epitope prediction concluded that these methods require improvement, and new approaches need to be taken into account for devising more efficient methods (Blythe and Flower 2005).
In a study on discontinuous epitope prediction tools, it was found out that DiscoTope and PEPITO have the highest predictive performance (Kringelum et al. 2012). The prediction efficiency of different discontinuous epitope predictors was done by Yao et al. (Yao et al. 2013), wherein they found out that the highest prediction accuracy obtained was only 25.6% by the EPMeta server. In the case of lowering the threshold for prediction, the prediction accuracy rose to 31.6%. There is a huge scope of improvement in the B cell epitope prediction methods to reach the accuracy levels of T cell epitope prediction methods. An important consideration for designing epitope-based vaccines is the prevalence of HLA alleles in the target populations. HLA alleles have a varied affinity towards the binding peptides and their distribution also varies in different population groups. The selection of epitopes for vaccine designing without taking this into account may fail vaccine to provide immunity (Oyarzun and Kobe 2015).
The challenge in vaccine design using only epitopes is that the peptides mostly fail to generate the immune response required for producing long lasting immunity. Because of their small size, the peptides are often weakly immunogenic and this thwarts the basic function of designing vaccines. Epitope-based peptide vaccines are mostly known to initiate antibodies (humoral response) and fail to induce T cell-mediated immunity. The generation of humoral immunity is not enough to protect against disease (Li et al. 2014). Since the molecular size is an important feature for immune response development such as small-sized peptides harboring epitopes need to be conjugated with carriers/adjuvants. The “RTS, S” vaccine for Plasmodium falciparum developed recently is based on truncated (C terminal end) of circumsporozoite protein (CSP) containing B and T cell epitopes. However, this 188 amino acid part of CSP has been fused with HBsAg protein to generate an immunogenic construct (Oyarzún and Kobe 2016). The CSP alone is weakly immunogenic predominantly generating antibody response but its fusion with HBsAg enhances its immunogenicity (Collins et al. 2017). Therefore, suitable carriers are required for vaccines based on epitopes as most of the times they are not enough immunogenic to induce both cell-mediated and humoral immunity.
The use of carriers/adjuvants becomes critical in designing epitope-based vaccines and many studies involving in silico designing of vaccines have taken this into account by the addition of adjuvant in the final vaccine construct (Shey et al. 2019; Khatoon et al. 2017). The usage of adjuvants like toxoids, Freund’s incomplete adjuvant, and the most recent TLR (Toll-like receptor) agonists enhances the immunogenicity of vaccines. These adjuvants are an essential requirement for the success of epitope-based vaccines; however, in silico studies can only design a construct using protein-based adjuvants and for other adjuvants, lab studies need to be undertaken.
11.10 Conclusion
In silico methods can provide a huge impetus to vaccine design and development. The B and T cell epitope prediction methods form the core of in silico epitope-based vaccine designing. The prediction of epitopes reduces the huge cost and labor involved in experimentally finding out the epitopes. These methods ease out the efforts involved in deducing T and B cell epitopes. The methods for T cell epitope prediction are more advanced in terms of prediction accuracy when compared to B cell epitope prediction tools. These methods need to be improved so that prediction accuracy can be increased, and we may be able to design more efficient vaccines in the future. The tools and methods for the analysis of the predicted epitopes though appear to be subsidiary yet their importance cannot be ignored. The checking of epitope clusters to avoid undue repetition of epitopes, checking their conservancy, finding toxic and allergic epitopes are essentiality that cannot be done away with. Analyzing the population coverage that can be achieved by the epitopes has far reaching consequences for the success or failure of vaccines in different population groups. Molecular docking and dynamics simulation strengthen the chances of epitope binding to MHC molecules. Finally, the construction of vaccines using these rational approaches can strengthen the possibility of it being successful, which of course needs to be validated by laboratory studies.
References
Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF (2020) The proximal origin of SARS-CoV-2. Nat Med 26(4):450–452
Andreatta M, Nielsen M (2016) Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics 32(4):511–517
Ansari HR, Raghava GPS (2010) Identification of conformational B-cell Epitopes in an antigen from its primary sequence. Immunome Res 6(1):6
Arai R, Ueda H, Kitayama A, Kamiya N, Nagamune T (2001) Design of the linkers which effectively separate domains of a bifunctional fusion protein. Protein Eng 14(8):529–532
Backert L, Kohlbacher O (2015) Immunoinformatics and epitope prediction in the age of genomic medicine. Genome Med 7(1):119
Bhasin M, Raghava GPS (2004a) Prediction of CTL epitopes using QM, SVM and ANN techniques. Vaccine 22:3195–3204
Bhasin M, Raghava GPS (2004b) Analysis and prediction of affinity of TAP binding peptides using cascade SVM. Protein Sci 13(3):596–607
Bhasin M, Raghava GPS (2005) Pcleavage: an SVM based method for prediction of constitutive proteasome and immunoproteasome cleavage sites in antigenic sequences. Nucleic Acids Res 33(2):W202–W207
Bhasin M, Raghava GPS (2007) A hybrid approach for predicting promiscuous MHC class I restricted T cell epitopes. J Biosci 32(1):31–42
Blythe MJ, Flower DR (2005) Benchmarking B cell epitope prediction: underperformance of existing methods. Protein Sci 14(1):246–248
Bui HH, Sidney J, Dinh K, Southwood S, Newman MJ, Sette A (2006) Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinf 7:153
Bui HH, Sidney J, Li W, Fusseder N, Sette A (2007) Development of an epitope conservancy analysis tool to facilitate the design of epitope-based diagnostics and vaccines. BMC Bioinf 8:361
Chakraborty S, Barman A, Deb B (2020) Japanese encephalitis virus: a multi-epitope loaded peptide vaccine formulation using reverse vaccinology approach. Infect Genet Evol 78:104106
Chen J, Liu H, Yang J, Chou KC (2007) Prediction of linear B-cell epitopes using amino acid pair antigenicity scale. Amino Acids 33(3):423–428
Chen F, Cao S, Xin J, Luo X (2013) Ten years after SARS: where was the virus from? J Thorac Dis 5(2):S163–S167
Chou KC, Shen HB (2008) Cell-PLoc: a package of Web servers for predicting subcellular localization of proteins in various organisms. Nat Protoc 3(2):153–162
Collins KA, Snaith R, Cottingham MG, Gilbert SC, Hill AV (2017) Enhancing protective immunity to malaria with a highly immunogenic virus-like particle vaccine. Sci Rep 7:46621
Dash R, Das R, Junaid M, Akash MF, Islam A, Hosen SZ (2017) In silico-based vaccine design against Ebola virus glycoprotein. Adv Appl Bioinforma Chem 10:11–28
De Groot AS, Moise L, McMurry JA, Martin W (2009) Epitope-based Immunome-derived vaccines: a strategy for improved design and safety. In: Clinical applications of immunomics. Springer, New York, pp 39–69
Dhanda SK, Vaughan K, Schulten V, Grifoni A, Weiskopf D, Sidney J, Peters B, Sette A (2018) Development of a novel clustering tool for linear peptide sequences. Immunology 155(3):331–345
Dimitrov I, Naneva L, Doytchinova I, Bangov I (2014a) AllergenFP: allergenicity prediction by descriptor fingerprints. Bioinformatics 30(6):846–851
Dimitrov I, Bangov I, Flower DR, Doytchinova I (2014b) AllerTOP v.2—a server for in silico prediction of allergens. J Mol Model 20(6):2278
Dönnes P, Kohlbacher O (2006) SVMHC: a server for prediction of MHC-binding peptides. Nucleic Acids Res 34(2):W194–W197
Doytchinova IA, Flower DR (2007) VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinf 8(1):4
Doytchinova IA, Guan P, Flower DR (2006) EpiJen: a server for multistep T cell epitope prediction. BMC Bioinf 7(1):131
EL-Manzalawy Y, Dobbs D, Honavar V (2008) Predicting linear B cell epitopes using string kernels. J Mol Recognit 21(4):243–255
Fitzpatrick M (2006) The cutter incident: how America’s first polio vaccine led to a growing vaccine crisis. J R Soc Med 99(3):156
Grote A, Hiller K, Scheer M, Münch R, Nörtemann B, Hempel DC, Jahn D (2005) JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res 33(2):W526–W531
Guan P, Doytchinova IA, Zygouri C, Flower DR (2003) MHCPred: bringing a quantitative dimension to the online prediction of MHC binding. Appl Bioinforma 2:63–66
Gupta S, Kapoor P, Chaudhary K, Gautam A, Kumar R, Raghava GP, Open Source Drug Discovery Consortium (2013) In silico approach for predicting toxicity of peptides and proteins. PLoS One 8(9):e73957
Hakenberg J, Nussbaum AK, Schild H, Rammensee HG, Kuttler C, Holzhütter HG, Kloetzel PM, Kaufmann SH, Mollenkopf HJ (2003) MAPPP: MHC class I antigenic peptide processing prediction. Appl Bioinforma 2(3):155–158
Hamrouni S, Bras-Gonçalves R, Kidar A, Aoun K, Chamakh-Ayari R, Petitdidier E, Messaoudi Y, Pagniez J, Lemesre JL, Meddeb-Garnaoui A (2020) Design of multi-epitope peptides containing HLA class-I and class-II-restricted epitopes derived from immunogenic Leishmania proteins, and evaluation of CD4+ and CD8+ T cell responses induced in cured cutaneous leishmaniasis subjects. PLoS Negl Trop Dis 14(3):e0008093
He Y, Xiang Z, Mobley HL (2010) Vaxign: the first web-based vaccine design program for reverse vaccinology and applications for vaccine development. J Biomed Biotechnol 2010:297505
Hewitt EW (2003) The MHC class I antigen presentation pathway: strategies for viral immune evasion. Immunology 110(2):163–169
Jahangiri A, Rasooli I, Gargari SL, Owlia P, Rahbar MR, Amani J, Khalili S (2011) An in silico DNA vaccine against Listeria monocytogenes. Vaccine 29(40):6948–6958
Jebastin T, Narayanan S (2019) In silico epitope identification of unique multidrug resistance proteins from Salmonella Typhi for vaccine development. Comput Biol Chem 78:74–80
Jensen KK, Andreatta M, Marcatili P, Buus S, Greenbaum JA, Yan Z, Sette A, Peters B, Nielsen M (2018) Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology 154(3):394–406
Jespersen MC, Peters B, Nielsen M, Marcatili P (2017) BepiPred-2.0: improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res 45(W1):W24–W29
Jørgensen KW, Rasmussen M, Buus S, Nielsen M (2014) Net MHC stab–predicting stability of peptide–MHCI complexes; impacts for cytotoxic T lymphocyte epitope discovery. Immunology 141(1):18–26
Jurtz V, Paul S, Andreatta M, Marcatili P, Peters B, Nielsen M (2017) NetMHCpan-4.0: improved peptide–MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J Immunol 199(9):3360–3368
Kaur R, Arora N, Jamakhani MA, Malik S, Kumar P, Anjum F, Tripathi S, Mishra A, Prasad A (2020) Development of multi-epitope chimeric vaccine against Taenia solium by exploring its proteome: an in silico approach. Expert Rev Vaccines 19(1):105–114
Keşmir C, Nussbaum AK, Schild H, Detours V, Brunak S (2002) Prediction of proteasome cleavage motifs by neural networks. Protein Eng 15(4):287–296
Khan S, Khan A, Rehman AU, Ahmad I, Ullah S, Khan AA, Ali SS, Afridi SG, Wei DQ (2019) Immunoinformatics and structural vaccinology driven prediction of multi-epitope vaccine against Mayaro virus and validation through in-silico expression. Infect Genet Evol 73:390–400
Khatoon N, Pandey RK, Prajapati VK (2017) Exploring Leishmania secretory proteins to design B and T cell multi-epitope subunit vaccine using immunoinformatics approach. Sci Rep 7(1):1–2
Kindt TJ, Goldsby RA, Osborne BA, Kuby J (2007) Kuby immunology. W. H. Freeman, New York
Kozakov D, Hall DR, Xia B, Porter KA, Padhorny D, Yueh C, Beglov D, Vajda S (2017) The ClusPro web server for protein–protein docking. Nat Protoc 12(2):255–278
Krawczyk K, Liu X, Baker T, Shi J, Deane CM (2014) Improving B-cell epitope prediction and its application to global antibody-antigen docking. Bioinformatics 30(16):2288–2294
Kringelum JV, Lundegaard C, Lund O, Nielsen M (2012) Reliable B cell epitope predictions: impacts of method development and improved benchmarking. PLoS Comput Biol 8(12):e1002829
Lamiable A, Thévenet P, Rey J, Vavrusa M, Derreumaux P, Tufféry P (2016) PEP-FOLD3: faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res 44(W1):W449–W454
Larsen MV, Lundegaard C, Lamberth K, Buus S, Lund O, Nielsen M (2007) Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinf 8(1):424
Lata S, Bhasin M, Raghava GPS (2007) Application of machine learning techniques in predicting MHC binders. Methods Mol Biol 409:201–215
Li W, Joshi MD, Singhania S, Ramsey KH, Murthy AK (2014) Peptide vaccine: progress and challenges. Vaccine 2(3):515–536
Liang S, Zheng D, Standley DM, Yao B, Zacharias M, Zhang C (2010) EPSVR and EPMeta: prediction of antigenic epitopes using support vector regression and multiple server results. BMC Bioinf 11(1):381
Liu W, Wan J, Meng X, Flower DR, Li T (2007) In silico prediction of peptide-MHC binding affinity using SVRMHC. Methods Mol Biol 409:283–291
Luca S, Mihaescu T (2013) History of BCG vaccine. Maedica 8(1):53–58
Maurer-Stroh S, Krutz NL, Kern PS, Gunalan V, Nguyen MN, Limviphuvadh V, Eisenhaber F, Gerberick GF (2019) AllerCatPro—prediction of protein allergenicity potential from the protein sequence. Bioinformatics 35(17):3020–3027
Mena I, Nelson MI, Quezada-Monroy F, Dutta J, Cortes-Fernández R, Lara-Puente JH, Castro-Peralta F, Cunha LF, Trovão NS, Lozano-Dubernard B, Rambaut A (2016) Origins of the 2009 H1N1 influenza pandemic in swine in Mexico. elife 5:e16777
Michel-Todó L, Reche PA, Bigey P, Pinazo MJ, Gascón J, Alonso-Padilla J (2019) In silico design of an epitope-based vaccine ensemble for Chagas disease. Front Immunol 10:2698
Mohd HA, Al-Tawfiq JA, Memish ZA (2016) Middle East respiratory syndrome coronavirus (MERS-CoV) origin and animal reservoir. Virol J 13(1):87
Monterrubio-López GP, Ribas-Aparicio RM (2015) Identification of novel potential vaccine candidates against tuberculosis based on reverse vaccinology. Biomed Res Int 2015:483150
Morais V, Dee V, Suárez N (2018) Purification of capsular polysaccharides of Streptococcus pneumoniae: traditional and new methods. Front Bioeng Biotechnol 6:145
Muñoz-Medina JE, Sánchez-Vallejo CJ, Méndez-Tenorio A, Monroy-Muñoz IE, Angeles-Martínez J, Santos Coy-Arechavaleta A, Santacruz-Tinoco CE, González-Ibarra J, Anguiano-Hernández YM, González-Bonilla CR, Ramón-Gallegos E (2015) In silico identification of highly conserved epitopes of influenza A H1N1, H2N2, H3N2, and H5N1 with diagnostic and vaccination potential. Biomed Res Int 2015:813047
Nielsen M, Lund O (2009) NN-align. An artificial neural network-based alignment algorithm for MHC class II peptide binding prediction. BMC Bioinf 10(1):296
Nielsen M, Lundegaard C, Lund O, Keşmir C (2005) The role of the proteasome in generating cytotoxic T-cell epitopes: insights obtained from improved predictions of proteasomal cleavage. Immunogenetics 57(1–2):33–41
Nussbaum AK, Kuttler C, Hadeler KP, Rammensee HG, Schild H (2001) PAProC: a prediction algorithm for proteasomal cleavages available on the www. Immunogenetics 53(2):87–94
Oyarzun P, Kobe B (2015) Computer-aided design of T-cell epitope-based vaccines: addressing population coverage. Int J Immunogenet 42(5):313–321
Oyarzún P, Kobe B (2016) Recombinant and epitope-based vaccines on the road to the market and implications for vaccine design and production. Hum Vaccin Immunother 12(3):763–767
Pahil S, Taneja N, Ansari HR, Raghava GPS (2017) In silico analysis to identify vaccine candidates common to multiple serotypes of Shigella and evaluation of their immunogenicity. PLoS One 12:8
Pierce BG, Wiehe K, Hwang H, Kim BH, Vreven T, Weng Z (2014) ZDOCK server: interactive docking prediction of protein–protein complexes and symmetric multimers. Bioinformatics 30(12):1771–1773
Ponomarenko J, Bui HH, Li W, Fusseder N, Bourne PE, Sette A, Peters B (2008) ElliPro: a new structure-based tool for the prediction of antibody epitopes. BMC Bioinf 9(1):514
Pourseif MM, Yousefpour M, Aminianfar M, Moghaddam G, Nematollahi A (2019) A multi-method and structure-based in silico vaccine designing against Echinococcus granulosus through investigating enolase protein. Bioimpacts 9(3):131–144
Pritam M, Singh G, Swaroop S, Singh AK, Singh SP (2019) Exploitation of reverse vaccinology and immunoinformatics as promising platform for genome-wide screening of new effective vaccine candidates against Plasmodium falciparum. BMC Bioinf 19(13):468
Rammensee HG, Bachmann J, Emmerich NPN, Bachor OA, Stevanović S (1999) SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics 50(3–4):213–219
Rauta PR, Ashe S, Nayak D, Nayak B (2016) In silico identification of outer membrane protein (Omp) and subunit vaccine design against pathogenic Vibrio cholerae. Comput Biol Chem 65:61–68
Reche PA, Glutting JP, Reinherz EL (2002) Prediction of MHC class I binding peptides using profile motifs. Hum Immunol 63(9):701–709
Riedel S (2005) Edward Jenner and the history of smallpox and vaccination. Proc Baylor Univ Med Cent 18(1):21–25
Roy A, Kucukural A, Zhang Y (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5(4):725–738
Rubinstein ND, Mayrose I, Martz E, Pupko T (2009) Epitopia: a web-server for predicting B-cell epitopes. BMC Bioinf 10(1):287
Sah PP, Bhattacharya S, Banerjee A, Ray S (2020) Identification of novel therapeutic target and epitopes through proteome mining from essential hypothetical proteins in Salmonella strains: an in silico approach towards antivirulence therapy and vaccine development. Infect Genet Evol 83:104315
Saha S, Raghava GPS (2004) BcePred: prediction of continuous B-cell epitopes in antigenic sequences using physico-chemical properties. In: International conference on artificial immune systems. Springer, Berlin, pp 197–204
Saha S, Raghava GPS (2006a) Prediction of continuous B cell epitopes in an antigen using recurrent neural network. Proteins 65(1):40–48
Saha S, Raghava GPS (2006b) AlgPred: prediction of allergenic proteins and mapping of IgE epitopes. Nucleic Acids Res 34:W202–W209
Saha CK, Hasan MM, Hossain MS, Jahan MA, Azad AK (2017) In silico identification and characterization of common epitope-based peptide vaccine for Nipah and Hendra viruses. Asian Pac J Trop Med 10(6):529–538
Sanasam BD, Kumar S (2019) In-silico structural modeling and epitope prediction of highly conserved Plasmodium falciparum protein AMR1. Mol Immunol 116:131–139
Schwartz M (2001) The life and works of Louis Pasteur. J Appl Microbiol 91(4):597–601
Sela-Culang I, Ashkenazi S, Peters B, Ofran Y (2014a) PEASE: predicting B-cell epitopes utilizing antibody sequence. Bioinformatics 31(8):1313–1315
Sela-Culang I, Benhnia MRI, Matho MH, Kaever T, Maybeno M, Schlossman A, Nimrod G, Li S, Xiang Y, Zajonc D (2014b) Using a combined computational-experimental approach to predict antibody-specific B cell epitopes. Structure 22(4):646–657
Shey RA, Ghogomu SM, Esoh KK, Nebangwa ND, Shintouo CM, Nongley NF, Asa BF, Ngale FN, Vanhamme L, Souopgui J (2019) In-silico design of a multi-epitope vaccine candidate against onchocerciasis and related filarial diseases. Sci Rep 9(1):4409
Sidney J, Assarsson E, Moore C, Ngo C, Pinilla C, Sette A, Peters B (2008) Quantitative peptide binding motifs for 19 human and mouse MHC class I molecules derived using positional scanning combinatorial peptide libraries. Immunome Res 4(1):2
Sims LD, Domenech J, Benigno C, Kahn S, Kamata A, Lubroth J, Martin V, Roeder P (2005) Origin and evolution of highly pathogenic H5N1 avian influenza in Asia. Vet Rec 157(6):159–164
Singh H, Raghava GPS (2001) ProPred: prediction of HLA-DR binding sites. Bioinformatics 17(12):1236–1237
Singh H, Raghava GPS (2003) ProPred1: prediction of promiscuous MHC Class-I binding sites. Bioinformatics 19(8):1009–1014
Singh H, Ansari HR, Raghava GPS (2013) Improved method for linear B-cell epitope prediction using antigen’s primary sequence. PLoS One 8(5):e62216
Slathia PS, Sharma P (2018) Conserved epitopes in variants of amastin protein of Trypanosoma cruzi for vaccine design: a bioinformatics approach. Microb Pathog 125:423–430
Slathia PS, Sharma P (2019) A common conserved peptide harboring predicted T and B cell epitopes in domain III of envelope protein of Japanese Encephalitis Virus and West Nile Virus for potential use in epitope based vaccines. Comp Immunol Microbiol Infect Dis 65:238–245
Soria-Guerra RE, Nieto-Gomez R, Govea-Alonso DO, Rosales-Mendoza S (2015) An overview of bioinformatics tools for epitope prediction: implications on vaccine development. J Biomed Inform 53:405–414
Sweredoski MJ, Baldi P (2008) PEPITO: improved discontinuous B-cell epitope prediction using multiple distance thresholds and half sphere exposure. Bioinformatics 24(12):1459–1460
Trolle T, Metushi IG, Greenbaum JA, Kim Y, Sidney J, Lund O, Sette A, Peters B, Nielsen M (2015) Automated benchmarking of peptide-MHC class I binding predictions. Bioinformatics 31(13):2174–2181
Ul Qamar MT, Saleem S, Ashfaq UA, Bari A, Anwar F, Alqahtani S (2019) Epitope-based peptide vaccine design and target site depiction against Middle East Respiratory Syndrome Coronavirus: an immune-informatics study. J Transl Med 17:362
Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ (2005) GROMACS: fast, flexible, and free. J Comput Chem 26(16):1701–1718
Wang F, Ye B (2016) In silico cloning and B/T cell epitope prediction of triosephosphate isomerase from Echinococcus granulosus. Parasitol Res 115(10):3991–3998
Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TA, Rempfer C, Bordoli L, Lepore R (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46(W1):W296–W303
WHO Data (n.d.) Hepatitis B. https://www.who.int/biologicals/vaccines/Hepatitis_B/en/
Xu D, Zhang Y (2012) Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins 80(7):1715–1735
Yao B, Zhang L, Liang S, Zhang C (2012) SVMTriP: a method to predict antigenic epitopes using support vector machine to integrate tri-peptide similarity and propensity. PLoS One 7(9):e45152
Yao B, Zheng D, Liang S, Zhang C (2013) Conformational B-cell epitope prediction on antigen protein structures: a review of current algorithms and comparison with common binding site prediction methods. PLoS One 8(4):e62249
Yu CS, Chen YC, Lu CH, Hwang JK (2006) Prediction of protein subcellular localization. Proteins 64(3):643–651
Zhang Q, Wang P, Kim Y, Haste-Andersen P, Beaver J, Bourne PE, Bui HH, Buus S, Frankild S, Greenbaum J, Lund O (2008) Immune epitope database analysis resource (IEDB-AR). Nucleic Acids Res 36(2):W513–W518
Zhang GL, DeLuca DS, Keskin DB, Chitkushev L, Zlateva T, Lund O, Reinherz EL, Brusic V (2011) MULTIPRED2: a computational system for large-scale identification of peptides predicted to bind to HLA supertypes and alleles. J Immunol Methods 374(1–2):53–61
Zhang T, Wu Q, Zhang Z (2020) Probable pangolin origin of SARS-CoV-2 associated with the COVID-19 outbreak. Curr Biol 30(7):1346–1351
Zobayer N, Hossain AA, Rahman MA (2019) A combined view of B-cell epitope features in antigens. Bioinformation 15(7):530–534
Competing Interest
The authors declare that there are no competing interests.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 The Editor(s) (if applicable) and The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Slathia, P.S., Sharma, P. (2020). In Silico Designing of Vaccines: Methods, Tools, and Their Limitations. In: Singh, D.B. (eds) Computer-Aided Drug Design. Springer, Singapore. https://doi.org/10.1007/978-981-15-6815-2_11
Download citation
DOI: https://doi.org/10.1007/978-981-15-6815-2_11
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-6814-5
Online ISBN: 978-981-15-6815-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)