
Abstract
Arsenic became a serious problem in various countries, affecting millions of people as it has major exposure root through drinking water. Arsenic is a non-threshold carcinogen. Many a time, its threat is overlooked only because of little knowledge about its estimation techniques. Various estimation techniques and instruments are available for risk quantification of arsenic. In many undeveloped countries where instrumental facilities are not available, a rapid arsenic testing technique through color development principle is beneficial there. Modern instruments mainly vary in their level of sensitivity. Arsenic estimation of groundwater samples required pre acid treatment before analysis to prevent oxidation of arsenic. Olsen reagent is most widely used for extracting plant available arsenic from soil. Digestion is preferred when we want to know the total amount of arsenic in samples. Among the various instruments available, hydride generation-atomic absorption spectroscopy (HG-AAS) is most widely used for estimating total arsenic and inductively couple plasma-mass spectroscopy (ICP-MS) hyphenated high pressure liquid chromatography (HPLC) is used for arsenic speciation analysis.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
7.1 Introduction
Arsenic is widely dispersed and ubiquitous in environment. Average concentration of As in earth’s crust is approximately 5 mg kg−1. Under oxidizing conditions such as those prevailing in surface waters, the principal species is pentavalent arsenic and under reduce condition trivalent arsenic is dominant. Arsenic, the king of poison, is a non-threshold carcinogen and 20th most abundant element in the earth’s crust. In the field of arsenic research, there are also significant advances in analytical chemistry to open new areas. Around 200 years back, Dr. Marsh in 1830s firstly started the arsenic analysis process. Last few decades, arsenic research grew continuously in accord with the discovery of new arsenic species and the importance of their environmental and biological activities. Recently, the growing interest of arsenic is increased because of its carcinogenic and leukemic effects on human body due to the drinking of groundwater (Mahfuzar 2007). A wide variety of methods to determine arsenic have been used: ultraviolet spectrometry, atomic absorption spectroscopy methods (AAS) coupled with hydride generation (HG-AAS), electrothermal AAS in graphite furnace (ETAAS), atomic fluorescence spectrometry (AFS), atomic emission spectrometry (AES), usually coupled with inductively coupled plasma (ICP-MS), X-ray spectrometry, neutron activation analysis (NAA), capillary electrophoresis, collision induced dissociation (CID), gas chromatography (GC), size exclusion chromatography (SEC), high-performance liquid chromatography (HPLC), Fourier transform ion cyclotron resonance-mass spectroscopy (FTICR-MS), stripping potentiometry, electroanalytical detection on gold plate, and gold film electrode preparation for anodic stripping voltammetric determination of arsenic. Methods involved in these techniques require expensive instrumentation, complicated procedures, and special sample pretreatment. Overall, all these methods are essentially sensitive to total arsenic. Growing interests in the determination of different arsenic species in groundwaters are caused by the fact that toxic effects of arsenic are solely connected with its chemical forms and oxidation states. The toxicity and bioavailability of arsenic can only be determined if all its forms can be identified and quantified. Several techniques including high-performance liquid chromatography separation joined with inductively coupled mass spectrometry, hydride generation atomic spectrometry and electrospray mass spectrometry detection have shown most powerful methods for arsenic speciation in environmental and biological matrices. These methods provide strong reliability on understanding of arsenic metabolism and biological cycling. In this review, we are trying to include recent developments and applications of analytical methods for the detection and speciation of groundwater arsenic.
7.2 Techniques for Arsenic Extraction
Estimation of arsenic level in soil sample comprises two major steps, one is extraction followed by determination. Extraction methodologies varies according to the fraction of arsenic we concern; like plant available, total, oxide bound or organically bound etc., the extraction methodologies varies accordingly.
7.2.1 Extraction of Total Soil Arsenic
For total arsenic estimation from soil or sediment sample, wet digestion technique is adopted. In this technique, various acid mixtures are used (Table 7.1) to achieve complete destruction of all As-bearing phases. The digestion methods can be carried out using a hotplate or microwave-digestion ovens to eliminate loss of volatile arsenic during the extraction.
7.2.2 Extracting Plant Available Arsenic from Soils and Sediments
Extraction of available arsenic is based on solubility product principle. Choice of method will depend on the types of soils and sediments being analyzed. Reduction of such As-bearing amorphous Fe oxides releases As to water systems. Acid ammonium oxalate is used for this purpose (Hudson-Edwards et al. 2004). Hydroxylamine hydrochloride is also used to extract Fe oxide-associated As (Montperrus et al. 2002). Extraction of plant available As from soil is done on a routine basis by Olsen extractant (0.5M NaHCO3, pH 8.5) (Table 7.2).
7.3 Techniques for Arsenic Determination
A variety of analytical techniques have been already applied for arsenic species determination. In the following paragraphs, we will summarize the major methods for the determination of arsenic.
7.3.1 Rapid Arsenic Test
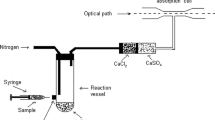
First step reduction: Reducing agent is “nascent” hydrogen which is generated through the reaction of zinc metal and hydrochloric acid. The reduction may be accelerated by adding a small amount of potassium iodide and stannous chloride.
Second step checking interference: Sulfur is most ubiquitous in nature and causes serious interference in arsine gas generation by forming hydrogen sulfide. One way of checking this interference is by passing the gas stream through a filter impregnated with lead acetate which will form insoluble lead sulfide. An alternative way is to use cupric chloride in combination with ferric chloride. Ferric salts (FeCl3) enhance the arsenic evolution and compensate the suppression effect of CuCl2 (Cherukuri and Anjaneyulu 2005).
Third step volatilization: Both the tri and penta valent species of arsenic [As (V) and As (III)] generated arsine gas by the reduction with reducing agent (zinc dust) under acidic conditions (hydrochloric acid). The reactions that could occur are as follows:
Fourth step color development: Color stripe may be of two types, one is mercuric bromide (HgBr2) and another is silver nitrate (AgNO3) (Das et al. 2014). When the arsine (AsH3) gas reacted with mercuric bromide (HgBr2) a yellow-to-brown colored compound is formed (depending upon arsenic concentration) (Fig. 7.1). Reaction on the paper strip is as follows:

Color chart for arsenic in mercuric bromide method. Color chart Kearns and Tyson (2012)
In case of silver nitrate method, the arsine (AsH3) when reacted with silver nitrate (AgNO3) formed a grey-to-black to a silver-black colored complex (depending upon arsenic concentration) (Fig. 7.2). Reaction on the paper strip is as follows:

Color chart for arsenic in silver nitrate method. Authors tested color chart
7.3.2 Spectrophotometric Determination of Arsenic by Silver Diethyldithiocarbamate
In this method, arsenic present in solution is reduced to arsine (AsH3) by reducing agent. Then, the arsine gas bubble absorbed through 0.5% silver diethyldithiocarbamate which produces red color, and the intensity of the red color is measured at 540 nm. If the sample inherently contains sulfur, then it will produce sulfide gas before the formation of arsine and this interference can be removed by passing AsH3 through lead acetate saturated glass wool. Contamination is the chief source of error of this process and this can be checked by rinsing the sample with 4% HNO3. The sensitivity of this method is <0.1 ppm, thus it fail to measure very low level of arsenic; although coprecipitation and adsorption (Talmi and Norvell 1975) and vapor-phase separation (Denyszyn et al. 1978) could somehow manage this problem.
7.3.3 Spectrophotometric Determination of As Using Molybdenum Blue Method
In this method arsenic is treated with molybdenum solution and a heteropolymolybdoarsenate complex is formed which is reduced by hydrazinium sulphate or tin (II) chloride to form blue colour soluble complex known as “molybdenum blue”. Intensity of the blue color is measured at 840 nm. To remove H2S interference, gas is passed through a tube which is loosely packed with cotton wool soaked in lead ethanoate (Dhar et al. 2004) (Table 7.3).
7.3.4 As (III) Determination by Anode Stripping Voltammetry
Anodic stripping voltammetry is a voltammetric technique used to determine particular ionic species quantitatively (Copeland and Skogerboe 1974). Samples are electroplated on the working electrode during a deposition step and oxidized from the electrode during the stripping step. The current is measured during the stripping step. The oxidized species is registered as a peak of the current signal in their own potential range which is suitable for their oxidation. Stripping steps can be linear, square waves, stairs, or pulse. The peak widths and stripping peak currents on the electrode surface (Hg or alternate) are function of the coverage, size, and distribution of the metal phase. This technique is improved over previous technologies in the areas of better sensitivity (ppt level), reproducibility and provides real-time data of in situ measurement.
7.3.5 Hydride Generation: Atomic Absorption Spectroscopy (HG-AAS)
The most widely accepted method for arsenic analysis in ppb level is based on the principle of hydride gas generation of arsenic. After generation of hydride gas, it is then thermally decomposed to give elemental arsenic for atomic detection in AAS. Combined mixture of sodium borohydrate and sodium hydroxide solution is used as reductant, and hydrochloric acid is used to acidifying the solution. Combination of these two solutions acts as carrier solution and helps to reduce the analyte in hydride form. A cathode lamp is used to produce characteristic resonance frequency of arsenic (193.7 nm) and light absorption is measured by following the Bear–Lambert rule. Little bit speciation analysis could be possible in hydride generation AAS as the process of formation of arsine follows two steps reaction. First step is the reduction of As(V) to As(III) and the second step is the formation of AsH. The levels of the redox reaction involving transferring electrons are rather slow and pH-dependent, so it could be possible to distinguish between the two species if the first phase of the response at elevated pH values is slower than the second. Therefore, species differentiation could be possible using pH-selective arsine generation technique. In this methodology, strongly acidic solutions (pH ≤ 1) are required for the determination of As(V) and mild acidic solution (pH 5) is suitable for hydride formation of As(III) (Howard and Comber 1992). In lieu of total arsenic estimation, pretreatment of the sample is done by applying acid mixture of hydrochloric acid, potassium iodide, and ascorbic acid for at least 45 min. There are some major interferences of this method. Cu (II), Co (II), and Ni (II) form specific chemical species between As and their reduction products decompose NaBH4 and this problem could be solved by applying relatively low concentration of NaBH4. During the hydride atomization, interferences from flame radical absorption of resonance lines could be managed by administrating the hydride in a heated quartz tube.
7.3.6 Chromatographic Methods
In case of chromatographic methods, gas chromatography (GC) and high-performance liquid chromatography (HPLC) deliver more detailed information about arsenic estimation (Niedzielski and Siepak 2003). Gas liquid chromatography runs on the principle of partitioning. In GLC, the components of vaporized samples are fraction due to partition between gaseous and mobile phase (unreacted carrier gas, e.g., N2, He, Ar) and liquid stationary phase (nonvolatile liquid) held in a column. The column separate of compounds according to their different strength of interaction with the stationary phase. Strong affinity between stationary and mobile phases result extended retention of mobile phase, thus more time required to migrate through the column. In high pressure liquid chromatography (HPLC) separation of a sample into its constituent parts on the basis of difference in the relative affinities of different molecules for the mobile phase and stationary phase. Various detectors used in gas liquid chromatographic method are ECD, TCD, FID, PID, UV-Vis and IR these are non selective to compounds of different metals, whereas refractive index detector, ultra-violate detector, and luminescence detectors are used in HPLC. Chromatography is very much useful in speciation analysis of arsenic, and major disadvantage of this method is it does not have sufficiently low detection limits (Table 7.4).
7.3.7 Inductively Coupled Plasma-Mass Spectroscopy
Mass spectroscopy is an analytical technique that ionized the chemical species and sorts ions based on mass to charge ratio. Actually, a mass spectrum measures the mass within the sample. In this spectroscopic technique, a sample is ionized by bombarding it with the electron. It may cause some of the sample molecules to break into charged fragments. These ions are then separated according to mass to charge ratio by accelerating and subjecting them to an electric or magnetic field. Ions having the same mass to charge ratio undergo same amount of deflection, and these ions are detected by an electron multiplier. Results are displayed as spectra or signal of the relative abundance of detected ions as a function of mass to charge ratio. The atoms or molecules in the samples can be identified by correlating known mass to identified mass. ICP-MS undeniably belongs to the most often used hyphenated techniques because of its detection limits equal to or better than AAS, ability to handle both simple and complex matrices, minimum matrix interferences, superior detection capability to ICP-AES, and its ability to obtain isotopic information. Disadvantages and weaknesses of the ICP-MS detection are due to polyatomic interferences (75As+ with 40Ar35Cl+).
7.3.8 Other Methods Available for As Determination
-
Radiochemical methods:
-
Nuclear magnetic resonance (NMR) (Faucher et al. 2014).
-
Hyperspectral remote sensing (Shi et al. 2016).
-
Ion selective electrodes (Kang 1974).
-
Micro XRF for in situ element mapping (Voegelin et al. 2007).
But these methods are lacking in the sensitivity required for ultra-low level detection of arsenic.
7.4 Conclusion
Finally, it can be concluded that the analytical techniques existing for the estimation and speciation of arsenic are diverse in nature. Each method has its own advantages and disadvantages that must be considered with respect to the type of research conducted and laboratory facilities available. When environmental observation is used to evaluate the toxic compound exposure, it is important that instrument can able to differentiate between toxic species and non-toxic species. Suitable methods for speciation analysis are HPLC-HG-AAS, HPLC-ICP, and HPLC-ICP-MS having high sensitivity and selectivity. These instruments are expensive and not available in many laboratories. In such cases, the development of rapid arsenic testing kits with color charts is expected to play a crucial role in the estimation of this dreadful heavy metal.
References
Afkhami A, Madrakian T, Ass AA (2001) Kinetic-spectrophotometric determination of trace amounts of As (III) based on its inhibitory effect on the redox reaction between bromate and hydrochloric acid. Talanta 55(1):55–60
Arbab-Zavar MH, Hashemi M (2000) Evaluation of electrochemical hydride generation for spectrophotometric determination of As (III) by silver diethyldithiocarbamate. Talanta 52(6):1007–1014
Cherukuri J, Anjaneyulu Y (2005) Design and development of low cost, simple, rapid and safe, modified field kits for the visual detection and determination of arsenic in drinking water samples. Int J Environ Res Public Health 2(2):322–327
Copeland TR, Skogerboe RK (1974) Anodic stripping voltammetry. Anal Chem 46(14):1257–1268
Das J, Sarkar P, Panda J, Pal P (2014) Low-cost field test kits for arsenic detection in water. J Environ Sci Health A Tox Hazard Subst Environ Eng 49(1):108–115
Denyszyn RB, Grohse PM, Wagoner DE (1978) Sampling and atomic absorption spectrometric determination of arsine at the 2 mu g/m3 level. Anal Chem 50(8):1094–1096
Deuel LE, Swoboda AR (1972) Arsenic solubility in a reduced environment 1. Soil Sci Am J 36(2):276–278
Dhar RK, Zheng Y, Rubenstone J, van Geen A (2004) A rapid colorimetric method for measuring arsenic concentrations in groundwater. Anal Chim Acta 526(2):203–209
Faucher A, Terskikh VV, Wasylishen RE (2014) Feasibility of arsenic and antimony NMR spectroscopy in solids: an investigation of some group 15 compounds. Solid State Nucl Mag 61:54–61
Guerin T, Astruc A, Astruc M, Batel A, Borsier M (1997) Chromatographic ion-exchange simultaneous separation of arsenic and selenium species with inductively coupled plasma-mass spectrometry on line detection. J Chrom Sci 35:213–220
Gupta PK, Gupta PK (1986) Microdetermination of arsenic in water, spectrophotometrically, by arsine-silver diethyldithiocarbamate-morpholine-chloroform system. Microchem J 33(2):243–251
Howard AG, Comber SDW (1992) Hydride-trapping techniques for the speciation of arsenic. Microchim Acta 109(1–4):27–33
Hudson-Edwards KA, Houghton SL, Osborn A (2004) Extraction and analysis of arsenic in soils and sediments. Trends Analyt Chem 23(10–11):745–752
Jacobs LW, Syers JK, Keeney DR (1970) Arsenic sorption by soils. Soil Sci Am J 34(5):750–754
Johnson LR, Hiltbol AE (1969) Arsenic content of soil and crops following use of methanearsonate herbicides. Soil Sci Am J 33(2):279–282
Johnston SE, Barnard WM (1979) Comparative effectiveness of fourteen solutions for extracting arsenic from four Western New York soils. Soil Sci Am J 43(2):304–308
Kang SG (1974) The development of an arsenic (V) selective electrode. Dissertations and Theses, Portland State University
Kearns J, Tyson J (2012) Improving the accuracy and precision of an arsenic field test kit: increased reaction time and digital image analysis. Anal Methods 4(6):1693–1698
Koellensperger G, Nurmi J, Hann S, Stingeder G, Fitz WJ, Wenzel WW (2002) CE–ICP-SFMS and HPIC–ICP-SFMS for arsenic speciation in soil solution and soil water extracts. J Anal Atomic Spectr 17:1042–1047
Kolesnikova AM, Lazarev AI (1991) Spectrophotometric determination of arsenic in copper- and nickel-base alloys by using iodonitrotetrazolium. Zh Anal Khim 46:169–174
Kopp JF (1973) 1-ephedrine in chloroform as a solvent for silver diethyldithiocarbamate in the determination of arsenic. Anal Chem 45(9):1786–1787
Krachler M, Mohl C, Emons H, Shotyk W (2002) Analytical procedures for the determination of selected trace elements in peat and plant samples by inductively coupled plasma mass spectrometry. Spectro Chim Acta 57(8):1277–1289
Kundu S, Ghosh SK, Mandal M, Pal T (2002) Silver and gold nanocluster catalyzed reduction of methylene blue by arsine in micellar medium. Bull Mater Sci 25(6):577–579
Liang QQ, Zhao DY, Qian TW, Freeland K, Feng YC (2012) Effects of stabilizers and water chemistry on arsenate sorption by polysaccharide-stabilized magnetite nanoparticles. Ind Eng Chem Res 51:2407–2418
Mahfuzar R (2007) Arsenic and contamination of drinking-water in Bangladesh: a public-health perspective. J Health Popul Nutr 20:193–197
Montperrus M, Bohari Y, Bueno M, Astruc A, Astruc M (2002) Comparison of extraction procedures for arsenic speciation in environmental solid reference materials by high performance liquid chromatography–hydride generation atomic fluorescence spectroscopy. Appl Organomet Chem 16(7):347–354
Morita K, Kaneko E (2006) Spectrophotometric determination of arsenic in water samples based on micro particle formation of ethyl violet-molybdoarsenate. Anal Sci 22(8):1085–1089
Niedzielski P, Siepak M (2003) Analytical methods for determining arsenic, antimony and selenium in environmental samples. Pol J Environ Stud 12(6):653–667
Orero Iserte L, Roig-Navarro AF, Hernández F (2004) Simultaneous determination of arsenic and selenium species in phosphoric acid extracts of sediment samples by HPLC-ICP-MS. Anal Chim Acta 527:97–104
Pereira SFP, Ferreira SLC, Oliveira GR, Palheta DC, Barros BC (2008) Spectrophotometric determination of arsenic in soil samples using 2-(5-bromo-2-pyridylazo)-5-di-ethylaminophenol (Br-PADAP). Eclet Quím 33(3):23–28
Reed JF, Sturgis MB (1936) Toxicity from arsenic compounds to rice on flooded soils. Agron J 28(6):432–436
Rosenfels RS, Crafts AS (1939) Arsenic fixation in relation to the sterilization of soils with sodium arsenite. Hilgardia 12(3):201–229
Sanz E, Olivas MR, Cmara C, Sengupta KM, Ahamed S (2007) Arsenic speciation in rice, straw, soil, hair and nails samples from the arsenic-affected areas of middle and lower Ganga plain. J Environ Sci Health A Tox Hazard Subst Environ Eng 42:1–11
Shi T, Liu H, Chen Y, Wang J, Wu G (2016) Estimation of arsenic in agricultural soils using hyperspectral vegetation indices of rice. J Hazard Mater 308:243–252
Talmi Y, Norvell VE (1975) Determination of arsenic and antimony in environmental samples using gas chromatography with a microwave emission spectrometric system. Anal Chem 47(9):1510–1516
Terada K, Okuda K, Maeda K, Kiba T (1978) A neutron activation analysis of selenium, arsenic and antimony in rocks and sediments by simultaneous evolution with bromide-strong phosphoric acid reagent. J Radioanal Nucl Chem 46(2):217–228
Vandecaveye SC, Horner GM, Keaton CM (1936) Unproductiveness of certain orchard soils as related to lead arsenate spray accumulations. Soil Sci 42(3):203–216
Voegelin A, Weber FA, Kretzschmar R (2007) Distribution and speciation of arsenic around roots in a contaminated riparian floodplain soil: micro-XRF element mapping and EXAFS spectroscopy. Geochim Cosmochim Acta 71(23):5804–5820
Woolson EA (1973) Arsenic phytotoxicity and uptake in six vegetable crops. Weed Sci 21(6):524–527
Woolson EA, Axley JH, Kearney PC (1971) Correlation between available soil arsenic, estimated by six methods, and response of corn (Zea mays L.). Soil Sci Soc Am J 35(1):101–105
Woolson EA, Axley JH, Kearney PC (1973) The chemistry and phytotoxicity of arsenic in soils: II. Effects of time and phosphorus. Soil Sci Soc Am J 37(2):254–259
Yamamoto M (1975) Determination of arsenic, methane arsenate and dimethyl arsenite in water and sediment extracts. Soil Sci Soc Am Proc 39:859–861
Yuan CG, He B, Gao EL, Lü JX, Jiang GB (2007) Evaluation of extraction methods for arsenic speciation in polluted soil and rotten ore by HPLC-HG-AFS analysis. Microchim Acta 159(1–2):175–182
Zheng Y, Stute M, Van Geen A, Gavrieli I, Dhar R, Simpson HJ, Ahmed KM (2004) Redox control of arsenic mobilization in Bangladesh groundwater. Appl Geochem 19(2):201–214
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Chattopadhyay, A., Rakshit, A., Singh, A.P., Sarkar, D., Singh, N.K., Chourasia, A. (2020). Analytical Strategies for Arsenic Estimation. In: Rakshit, A., Ghosh, S., Chakraborty, S., Philip, V., Datta, A. (eds) Soil Analysis: Recent Trends and Applications. Springer, Singapore. https://doi.org/10.1007/978-981-15-2039-6_7
Download citation
DOI: https://doi.org/10.1007/978-981-15-2039-6_7
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-2038-9
Online ISBN: 978-981-15-2039-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)