
Abstract
Contrast-induced acute kidney injury (CI-AKI) is a form of kidney damage by recent exposure of iodinated contrast media (CM) without another clear cause for AKI. Now serum creatinine-based definition is generally used; however, there are some problems on the definition and differentiation of CI-AKI. CM is known to induce a variety of alterations in the kidney. The new mechanism of direct tubular injury, specifically the role of inflammatory pathway, has recently been characterized to explain CI-AKI in clinical setting. This might lead to new therapeutic strategy. Both patient-related and procedure-related risk factors for CI-AKI have been identified, and volume depletion and chronic kidney disease (CKD) are known to be high risks for CI-AKI.
It is increasingly recognized that old data from cardiac angiography studies may overestimate the risk of CI-AKI for patients undergoing intravenous contrast-enhanced studies. Recent well-designed studies addressed the incidence of CI-AKI after intravenous administration of CM for computed tomography was quite low. At present the only available preventive action to reduce the risk for CI-AKI is to provide intravenous volume expansion before, during, and after CM administration. Reevaluation of definition, the risk factors, the true impact, and preventative measures for CI-AKI are required in order to better understand CI-AKI.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Chronic kidney disease
- Contrast-induced acute kidney injury
- Contrast media
- Dehydration
- Hydration
- Inflammation
- Intravenous administration
1 Introduction
There is significant evidence for renal injury resulting from contrast media (CM) administration in both animal models and humans. Contrast-induced acute kidney injury (CI-AKI) or contrast-associated nephropathy, also commonly referred to as contrast-induced nephropathy, is caused by recent CM administration for diagnostic or therapeutic imaging.
Most people with normal kidney function who receive CM usually do not experience any renal complications. However, patients with volume depletion or chronic kidney disease (CKD) are recognized to be at increased risk for CI-AKI [1,2,3]. CI-AKI occurs within 24–72 h following CM administration and could be associated with poor outcomes including AKI requiring dialysis, worsening of CKD, cardiovascular events, and increased medical expenses [2]. Numerous clinical and epidemiological studies have characterized the risk factors and incidence rates for CI-AKI. Pre-existing renal dysfunction (estimated glomerular filtration rate (eGFR) <60 mL/min/1.73 m2) and diabetes mellitus are the most important risk factors for further deterioration in renal function induced by CM [2, 4]. However, the type of CM administration procedure seems to be an important determinant of CI-AKI. Recent well-designed studies have shown that CI-AKI is much less common than previously believed even in patients with CKD undergoing intravenous (IV) contrast-enhanced computed tomography (CT) [5, 6].
CM is known to induce various alterations in the kidney. Among them, the mechanism of direct tubular injury due to inflammatory pathway has recently been attracted attention for. IV hydration, optimization of hemodynamic status, and minimization of CM volume have been considered for preventive strategies of CI-AKI at present. Any drugs with vasodilative or anti-oxidative effects have not been established to reduce the risk for CI-AKI. The incomplete understanding of CI-AKI has hampered the development of new strategies to prevent or mitigate CI-AKI.
In this review, we aimed to mainly discuss the current understanding and controversy of CI-AKI.
2 Manifestations of CI-AKI
In general, CI-AKI is considered to occur when a patient receives CM either intravenously or intra-arterially for diagnostic or therapeutic imaging and subsequently demonstrates a rise in the serum creatinine concentration. In most CI-AKI, accumulation of serum creatinine typically requires 48–72 h. Serum creatinine peaks at 4–5 days and returns to baseline in 7–14 days. A low urine sodium concentration (urine Na < 10 mEq/L) or a low fractional excretion of sodium (<1%) may be found owing to the vasoconstrictive properties of CM. Acute tubular necrosis occurs as a form of AKI; thus tubular epithelial cells and granular “muddy brown” casts can be seen on urinary microscopy. However, CI-AKI shows broad clinical manifestations, ranging from a mild to moderate increase in serum creatinine to oliguric or non-oliguric AKI requiring temporary or permanent dialysis in small cases.
3 Definition of CI-AKI
There is no specific definition of CI-AKI that is generally agreed worldwide. However, it is widely used that CI-AKI is defined by “a condition in which an impairment in renal function (an increase in serum creatinine by more than 25% or 0.5 mg/dL (44 mmol/L)) from baseline occurs within 48–72 h following the intra-vascular administration of CM in the absence of an alternative etiology” [7]. On the other hand, CI-AKI is evaluated with the Kidney Disease: Improving Global Outcomes (KDIGO) AKI criteria as any of the following: increase in serum creatinine by ≥0.3 mg/dL (≥26.5 μmol/L) within 48 h or increase in serum creatinine to ≥1.5 times baseline that is known or presumed to have occurred within the prior 7 days or urine volume <0.5 mL/kg/h for 6 h [8].
4 Differential Diagnosis
An alternative diagnosis of AKI should be suspected when renal injury develops more than 7–10 days following CM administration. In such instances, careful evaluation for alternative causes of AKI, including cholesterol crystal embolization, should be considered especially in patients after cardiac catheterization. However, of note, the American College of Radiology (ACR) Manual on Contrast Media from May 2017 makes a differentiation between CI-AKI and post-contrast acute kidney injury (PC-AKI) [9]. The reason for this differentiation is that we do not recognize at present whether the AKI is actually due to CM administration or if there are concomitant disease processes creating this effect. PC-AKI may occur regardless of whether the CM is the cause of the deterioration of renal function; therefore, CI-AKI is a subgroup of PC-AKI.
In order to evaluate the data across the literature on CI-AKI, it is necessary to establish standardized definition of CI-AKI. Like AKI in general, urinary liver fatty acid binding protein (L-FABP) and serum cystatin C were also reported as an early biomarker for CI-AKI [10, 11]. Recent report indicated serum neutrophil gelatinase-associated lipocalin (NGAL) and serum fibroblast growth factor (FGF)23 might have certain values in early diagnosis of CI-AKI [12]. Onset can be predictable, and the mechanism should be similar among CI-AKI; mechanistic biomarkers that can be applied for CI-AKI diagnosis and prognosis are expected.
5 Incidence
CI-AKI is an important complication that accounts for a significant number of patients of hospital-acquired renal failure. Although all of AKI after CM administration are not caused directly by CM, there are several reports on incidence of CI-AKI in various clinical settings. Incidence of CI-AKI is <1% in patients undergoing non-emergent contrast-enhanced CT [13] and is 4% in CKD patients undergoing non-emergent contrast-enhanced CT [14], whereas incidence of CI-AKI is >10% in patients after IV CM administration in an emergency setting [15]. As for intra-arterial CM administration, incidence of CI-AKI is <3% in patients undergoing percutaneous transluminal catheter angioplasty (PTCA) with normal baseline renal function [16]. On the other hand, the incidence of CI-AKI was reported to be around 40% in CKD patients undergoing PTCA, and the reduction of eGFR among these patients was associated with the increase in incidence of CI-AKI [17, 18].
6 Prognosis
Some studies have demonstrated that both short-term and long-term mortality rates have been found to be significantly higher in patients with CI-AKI compared with patients without CI-AKI [7]. Furthermore, a prognosis of CI-AKI may be associated with development of CKD and progression to end-stage renal disease in long term [19, 20] like other causes of AKI. However, very recent study reported that the administration of both IV and intra-arterial CM was associated with a risk of AKI and multifactorial AKI was associated with worse outcomes, while CI-AKI was associated with better outcomes [21]. The true impact of CI-AKI seems not to be clear.
7 Mechanisms
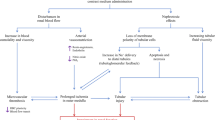
Many complex pathways possibly involved in the development of CI-AKI have been proposed [4, 22]. Under normal conditions, the renal medulla is poorly oxygenated and works in a less oxygenated condition. After CM administration, renal blood flow decreases due to renal vasoconstriction over a prolonged period, leading to ischemic injury to the renal medulla. Vasoactive substances such as prostaglandins, nitric oxide, endothelin, and adenosine may play a major role in this process. Direct cytotoxicity of CM due to free radical formation and osmotic effects of CM on renal tubular cells also play a role. Among these factors, sterile inflammatory damages attracted attention [23]. Very recently Lau et al., using a mouse CI-AKI model with volume depletion, reported that immune activation occurs in distinct compartments and depends on uptake of CM both by resident and infiltrating phagocytes and by tubular epithelial cells, leading to a massive influx of leukocytes and an excessive inflammatory response that ultimately induces AKI (Fig. 7.1) [24]. They also reported levels of the inflammasome-related urinary biomarkers IL-18 and caspase-1 were increased immediately following CM administration in patients undergoing coronary angiography (CAG), consistent with the acute renal effects observed in mice [24]. The discovery of this pathway might provide opportunities to develop specific and effective therapies for CI-AKI. Moreover, Atkinson SJ commented that the report by Lau et al. has an important implication of the clear mechanistic explanation for the contribution of volume depletion to the activation of a sterile inflammatory state and the likelihood of CI-AKI in patients receiving CM [25].

Schematic of CI-AKI. (1) Intravenous or intra-arterial contrast agents enter the renal circulation and peritubular capillaries and are taken up by resident renal phagocytes. (2) Contrast is filtered at the glomerulus and enters the urine and renal tubule. In the hydrated state, contrast is rapidly excreted. (3) Contrast uptake by the resident renal phagocytes activates the Nlrp3 inflammasome to generate IL-1β. (4) IL-1β mediates leukocyte recruitment from the circulation into the kidney. (5) Contrast in the urine is taken up by the tubular cell by the brush border enzyme DPEP-1. The tubular uptake of contrast is enhanced in the volume-depleted state. (6) Recruited leukocytes ingest contrast transported from the urine through a direct interaction with tubular cells that further activates the inflammasome. (7) The activation of resident renal phagocytes, tubular uptake of contrast, and leukocyte recruitment are all necessary, but none alone is sufficient to induce AKI. (From Lau A et al. J Clin Invest. Renal immune surveillance and dipeptidase-1 contribute to contrast-induced acute kidney injury. J Clin Invest. 2018;128:2894–913)
8 Risk Factors
Patients at risk for CI-AKI should be identified before administration of CM. Previously reported factors influencing the incidence of CI-AKI are listed in Table 7.1.
8.1 Patient-Related Risk Factors
In addition to states of reduced renal perfusion, pre-existing CKD (eGFR < 60) and diabetes mellitus are the most important patient-related risk factors for further deterioration in renal function induced by CM [26, 27]. Both of them may have additive effect on CI-AKI [28]; however, it is not clear whether the risk of CI-AKI is significantly increased in patients with diabetes mellitus without renal dysfunction. Older age is also a risk for CI-AKI because it is postulated to predispose patients to renal sodium and water wasting due to reduction in renal mass and function.
Comedication with loop diuretics had no difference of a risk for CI-AKI when compared with discontinuation of loop diuretics [29]. In contrast prophylactic use of loop diuretics increased the incidence of CI-AKI despite adjusting dehydration [30]. The meta-analysis showed the risk of comedication with non-steroidal anti-inflammatory drugs (NSAIDs) for CI-AKI [27] probably due to reduction of intra-renal blood flow. Thus, discontinuation of NSAIDs 24 h before CM administration should be done [31]. Lately, renin-angiotensin system (RAS) inhibitors have been used often for CKD patients. At present, there is no evidence that RAS inhibitors increase the incidence of CI-AKI. Although metformin is not a nephrotoxic agent, it causes life-threatening lactic acidosis in a patient who develops CI-AKI. Most guidelines recommended transient discontinuation of metformin especially in CKD patients who are planned to have CM administration.
8.2 CM- or Procedure-Related Risk Factors
It is well known that high-osmolar CM (HOCM) more frequently causes CI-AKI compared with low-osmolar CM (LOCM) (780–800 mOsm/kg) [32]. Nowadays only iso-osmolar CM (IOCM) and LOCM are used in clinical practice instead of HOCM. In most of studies, there is no difference on the incidence of CI-AKI between IOCM and LOCM. The KDIGO guidelines for AKI stated no recommendation was made about the preference of IOCM or LOCM due to lack of reliable evidence [8].
Lower doses of CM were found to be less nephrotoxic in patients undergoing CAG [33, 34]. Cigarroa advocated maximum allowable CM dose was defined as <5 mL/kg per serum creatinine [33]. A CM dose on the basis of estimated renal function with a planned CM volume restricted to less than preferably twice the calculated creatinine clearance might be valuable in reducing the risk for CI-AKI requiring dialysis in patients undergoing percutaneous coronary intervention (PCI) [35]. However, it is not clear whether low dose of CM is significantly less nephrotoxic in patients undergoing contrast-enhanced CT. It is reported that use of >100 mL of CM was associated with increased risk of CI-AKI among outpatients with mild baseline kidney disease [13]. Though there is not enough evidence, use of lower dose of CM is recommended in patients undergoing contrast-enhanced CT, especially patients with CKD. There are conflicting reports as to whether repeated contrast-enhanced CT scan within 24–72-h interval increases the risk for CI-AKI.
Recently, newer CT modalities have been developed using low tube voltage and low CM dose to reduce radiation exposure and the risk for CI-AKI without sacrificing image quality [36, 37]. However it should be kept in mind that even very low doses of CM may lead to CI-AKI in patients with high-risk factors. The volume of CM should be minimized as much as possible at any time.
9 Route of CM Administration
It is increasingly recognized that CI-AKI has been overestimated in clinical practice. LOCM is proved to be less risk for CI-AKI than HOCM [32]. Older studies on CIN frequently include the patients using HOCM, which has since been replaced by LOCM. In addition, the risk for CI-AKI has been known to be different between routes of CM administration. However, comparing the risks for CI-AKI after IV CM administration and after cardiac catheterization (intra-arterial CM) is difficult because there are so many different conditions, including underlying illness, pre-/comedications, dose of CM, site and number of CM injection, and prophylactic measures. Specifically, CAG differs from IV CM administration as follows: (1) the injection is intra-arterial and supra-renal, (2) CM dose is usually more concentrated, and (3) use of a catheter can induce atheroembolization. Thus, similar patient cohorts for studies on CI-AKI cannot be enrolled adequately. Furthermore, it had been recognized that a high incidence of transient serum creatinine fluctuations exists in hospitalized patients who had not received CM [38] and that the fluctuations are larger in patients with kidney failure than in those with normal kidney function [39]. Although there is no conclusive evidence, some studies have provided circumstantial evidence that the risk for CI-AKI may be higher after intra-arterial than after IV injection [40, 41].
9.1 Intra-arterial CM Administration
With a retrospective analysis of the Mayo Clinic PCI Registry, of 7586 patients, 254 (3.3%) experienced AKI. Diabetic patients were at higher risk than non-diabetic patients, whereas all patients with a serum creatinine >2.0 mg/dL are at high risk for AKI. AKI was highly correlated with death [16]. The incidence of CI-AKI after cardiac catheterization was 4.0% in 1157 patients in Japan [42]. Multivariate logistic models revealed that pre-existing renal insufficiency, serum creatinine 1.2 mg/dL or higher, and the use of high-volume (more than 200 mL) CM were independently associated with CI-AKI [42]. A multicenter prospective observational study that enrolled 906 patients with cardiac catheterization showed the incidence of CI-AKI in patients with eGFR <30 mL/min/1.73 m2 was significantly higher than that in patients with eGFR ≥60 mL/min/1.73 m2. CI-AKI was found in patient with normal renal function, but incidence of CI-AKI was increased with the reduction of eGFR. Proteinuria and reduced eGFR were independent risk factors for CI-AKI after cardiac catheterization [43]. These data and others indicated intra-arterial, suprarenal CM administration is a risk for CI-AKI especially among patients with CKD.
9.2 Intravenous CM Administration
Imaging examinations requiring the use of IV CM administration are sometimes avoided for fear of CI-AKI in clinical practice. Previous studies had problems on the risk determination for CI-AKI affected by lacking controls to cases and many confounders among people undergoing IV contrast-enhanced CT scan. Recent well-designed studies overcame those problems.
McDonald et al. reported the results of a large retrospective analysis with propensity score matching controls to cases to compare risk for CI-AKI after IV contrast-enhanced CT and non-contrast-enhanced CT scanning, and no increased risk for CI-AKI could be found from the contrast-enhanced CT scan, even among high-risk groups [44]. In other studies using propensity score matching, most cases of AKI after contrast-enhanced CT were found not to be attributable to CM. The risk for AKI was independent of CM exposure, even in patients with eGFR of less than 30 mL/min/1.73 m2 [5], whereas IV LOCM does not appear to be a nephrotoxic risk factor in patients with a pre-CT eGFR of 45 mL/min/1.73 m2 or greater but a risk factor for CI-AKI in patients with a stable eGFR less than 30 mL/min/1.73 m2 [45].
To create multiple estimates of the risk for CI-AKI, 5.9 million Nationwide Inpatient Sample in the United States in 2009 was stratified according to the presence or absence of 12 common conditions associated with AKI, and the rate of AKI between strata was evaluated. Then, a logistic regression model was created, controlling for comorbidity and acuity of illness, to estimate the risk of AKI associated with CM administration within each stratum [46]. The authors found that the risk for AKI in patients receiving than not receiving CM was nearly identical, 5.5% vs. 5.6%, respectively [46]. IV CM administration was not associated with an excess risk for dialysis or death, even among patients with CKD [6]. Very recent meta-analysis reported that contrast-enhanced CT scan vs. non-contrast CT did not show significant differences in rates of AKI, need for renal replacement therapy, or mortality [47].
These and other recent well-conducted studies have shown that the risk for CI-AKI, especially after IV CM administration, is much lower than has been commonly thought and might barely exist at all; on the other hand, CI-AKI is unpredictable and largely depending on confounders with sometimes bidirectional roles.
10 Prevention and Treatment
In order to prevent CI-AKI, concomitant use of other known nephrotoxic drugs should be withdrawn, or they should not be used if clinically possible. Based on the possible mechanisms of CI-AKI, drugs, which have vasodilative or anti-oxidative effects, they have been considered for preventive strategies of CI-AKI. However, most of them including N-acetylcysteine, human atrial natriuretic peptide, theophylline, endothelin-1, fenoldopam, ascorbic acid, and stain were not reported to significantly decrease the risk for CI-AKI.
IV hydration is recognized as the only effective preventive strategy for CI-AKI at present. Saline before, during, and after exposure to CM can increase tubular fluid volume depending on infusion rate and reduce the concentration of CM in the tubular fluid, which might lead to reduced formation of reactive oxygen species. IV saline hydration was reported to decrease both the incidence and severity of CI-AKI in patients undergoing cardiac catheterization from 12 h before the procedure for 24 h (at a rate of 1 mL/kg per hour) when compared with unrestricted oral fluid intake [48]. There is little evidence about oral fluid loading being inferior to IV saline loading especially in patients with mild to moderate renal dysfunction. Isotonic hydration (0.9% saline) was found to be superior to half-isotonic hydration (0.45% sodium chloride plus 5% glucose) for the prevention of CI-AKI in patients undergoing CAG [49]. However, a recent prospective, randomized controlled study in patients with CKD stage G3 showed that no prophylaxis group was found to be non-inferior to prophylaxis group (0.9% saline) [50].
Sodium bicarbonate infusion can produce the same effects of systemic volume expansion as saline, with the additional benefit of an increase in the bicarbonate anion buffer within the renal tubules. This might lead to alkalization of tubular fluid, which may protect the tubular cells against free radical injury. However, the effectiveness of IV sodium bicarbonate to prevent CI-AKI is controversial. Most of recent meta-analysis indicated a preventive effect of the use of sodium bicarbonate on the risk for CI-AKI when compared with saline was borderline statistical significance [51, 52]. It is noteworthy that the use of sodium bicarbonate had no beneficial effect on requiring renal replacement therapy or mortality. Among the large randomized trials, there was no evidence of benefit for hydration with sodium bicarbonate compared with sodium chloride for the prevention of CI-AKI [53].
At present in clinical practice, hydration is recommended to prevent CI-AKI especially for patients with high risk for CI-AKI. Further examinations are necessary as to which patients with risk factors for CI-AKI get benefit by hydration before and after CM administration because achievement of enough hydration is not easy for outpatients.
There was an exacerbation of renal dysfunction when furosemide was used in addition to IV saline solution. However, there is the RenalGuard System (PLC Medical Systems, Milford, Massachusetts) for very high-risk patients to prevent CI-AKI, which delivers IV fluids matched to urine output with a combination of hydration with normal saline at an initial dose bolus plus a low dose of furosemide and continuous monitoring for a urine output flow of >300 mL/h sustained for 6 h. The meta-analysis reported that furosemide with matched hydration by the RenalGuard System may reduce the incidence of CI-AKI in high-risk patients undergoing PCI or transcatheter aortic valve replacement [54].
Although there is no established therapy using single drug to prevent CI-AKI, combination of drugs and hydration may be effective on CI-AKI prevention. Interestingly, randomized, controlled trials of N-acetylcysteine, statins, ascorbic acid, sodium bicarbonate, or saline that used IV or intra-arterial CM and defined CI-AKI indicated that the greatest reduction in risk for CI-AKI has been achieved with low-dose N-acetylcysteine plus IV saline or with statins plus N-acetylcysteine plus IV saline in patients receiving LOCM [55]. Moreover, a comprehensive analysis of currently utilized CI-AKI prevention interventions by the systematic review and network meta-analysis suggested that some options (particularly allopurinol, prostaglandin E1, and oxygen) deserve further evaluation in larger well-designed retrospective control trials [56].
In the meta-analysis including prophylactic hemodialysis and hemofiltration, these therapies were not found to be protective against CI-AKI [57]. Of note, at least hemodialysis was found to increase the risk for CI-AKI. Thus prophylactic renal replacement therapy should not be done.
11 Perspective
CI-AKI is one of the preventable forms of AKI because the timing of renal insult is known in patients with CI-AKI. In order to specify the modifiable risk factors, we still need to revise several factors, which influence on evaluating the CI-AKI incidence, including definition of CI-AKI, underlying risks associated with various conditions, and type of CM administration. Further study is needed to clarify the true impact of CI-AKI, especially after IV CM administration, in light of a growing CKD population and radiological imaging using CM being increased. On the other hand, basic experiments have been revealing the new mechanisms of CI-AKI; thus specific preventive measures and treatments are expected to develop in the future.
References
Thomsen HS, Morcos SK. Risk of contrast-medium-induced nephropathy in high-risk patients undergoing MDCT—a pooled analysis of two randomized trials. Eur Radiol. 2009;19:891–7.
James MT, Samuel SM, Manning MA, Tonelli M, Ghali WA, Faris P, Knudtson ML, Pannu N, Hemmelgarn BR. Contrast-induced acute kidney injury and risk of adverse clinical outcomes after coronary angiography: a systematic review and meta-analysis. Circ Cardiovasc Interv. 2013;6:37–43.
McDonald JS, Leake CB, McDonald RJ, Gulati R, Katzberg RW, Williamson EE, Kallmes DF. Acute kidney injury after intravenous versus intra-arterial contrast material administration in a paired cohort. Investig Radiol. 2016;51:804–9.
Azzalini L, Spagnoli V, Ly HQ. Contrast-induced nephropathy: from pathophysiology to preventive strategies. Can J Cardiol. 2016;32:247–55.
McDonald JS, McDonald RJ, Carter RE, Katzberg RW, Kallmes DF, Williamson EE. Risk of intravenous contrast material-mediated acute kidney injury: a propensity score-matched study stratified by baseline-estimated glomerular filtration rate. Radiology. 2014;271:65–73.
McDonald RJ, McDonald JS, Carter RE, Hartman RP, Katzberg RW, Kallmes DF, Williamson EE. Intravenous contrast material exposure is not an independent risk factor for dialysis or mortality. Radiology. 2014;273:714–25.
Morcos SK, Thomsen HS, Webb JA. Contrast-media-induced nephrotoxicity: a consensus report. Contrast media safety committee, European Society of Urogenital Radiology (ESUR). Eur Radiol. 1999;9:1602–13.
Kellum JA, Lameire N, Aspelin P, Barsoum RS, Burdmann EA, Goldstein SL, Herzog CA, Joannidis M, Kribben A, Levey AS. Kidney disease: improving global outcomes (KDIGO) acute kidney injury work group. KDIGO clinical practice guideline for acute kidney injury. Kidney Int Suppl. 2012;2:1–138.
Ellis JH, Davenport MS, Dillman JR, Hartman RP, Herts BR, Jafri SZ, Kolbe AB, Laroia A, Cohan RH, MacDonald RJ, Needleman L, Newhouse JH, Pahade JK, Sirlin CB, Wang CL, Wasserman N, Weinreb JC. ACR manual on contrast media. American College of Radiology. Version 10.3; 2017. p. 35–46. ISBN 978-1-55903-012-0.
Nakamura T, Sugaya T, Node K, Ueda Y, Koide H. Urinary excretion of liver-type fatty acid-binding protein in contrast medium-induced nephropathy. Am J Kidney Dis. 2006;47:439–44.
Briguori C, Visconti G, Rivera NV, Focaccio A, Golia B, Giannone R, Castaldo D, De Micco F, Ricciardelli B, Colombo A. Cystatin C and contrast-induced acute kidney injury. Circulation. 2010;121:2117–22.
Li H, Yu Z, Gan L, Peng L, Zhou Q. Serum NGAL and FGF23 may have certain value in early diagnosis of CIN. Ren Fail. 2018;40:547–53.
Weisbord SD, Mor MK, Resnick AL, Hartwig KC, Palevsky PM, Fine MJ. Incidence and outcomes of contrast-induced AKI following computed tomography. Clin J Am Soc Nephrol. 2008;3:1274–81.
Barrett BJ, Katzberg RW, Thomsen HS, Chen N, Sahani D, Soulez G, Heiken JP, Lepanto L, Ni ZH, Ni ZH, Nelson R. Contrast-induced nephropathy in patients with chronic kidney disease undergoing computed tomography: a double-blind comparison of iodixanol and iopamidol. Invest Radiol. 2006;41:815–21. Erratum in: Invest Radiol. 2007;42:94.
Mitchell AM, Jones AE, Tumlin JA, Kline JA. Incidence of contrast-induced nephropathy after contrast-enhanced computed tomography in the outpatient setting. Clin J Am Soc Nephrol. 2010;5:4–9.
Rihal CS, Textor SC, Grill DE, Berger PB, Ting HH, Best PJ, Singh M, Bell MR, Barsness GW, Mathew V, Garratt KN, Holmes DR Jr. Incidence and prognostic importance of acute renal failure after percutaneous coronary intervention. Circulation. 2002;105:2259–64.
Chong E, Shen L, Poh KK, Tan HC. Risk scoring system for prediction of contrast-induced nephropathy in patients with pre-existing renal impairment undergoing percutaneous coronary intervention. Singap Med J. 2012;53:164–9.
Marenzi G, Lauri G, Assanelli E, Campodonico J, De Metrio M, Marana I, Grazi M, Veglia F, Bartorelli AL. Contrast-induced nephropathy in patients undergoing primary angioplasty for acute myocardial infarction. J Am Coll Cardiol. 2004;44:1780–5.
Maioli M, Toso A, Leoncini M, Gallopin M, Musilli N, Bellandi F. Persistent renal damage after contrast-induced acute kidney injury: incidence, evolution, risk factors, and prognosis. Circulation. 2012;125:3099–107.
Nemoto N, Iwasaki M, Nakanishi M, Araki T, Utsunomiya M, Hori M, Ikeda N, Makino K, Itaya H, Iijima R, Hara H, Takagi T, Joki N, Sugi K, Nakamura M. Impact of continuous deterioration of kidney function 6 to 8 months after percutaneous coronary intervention for acute coronary syndrome. Am J Cardiol. 2014;113:1647–51.
Chaudhury P, Armanyous S, Harb SC, Ferreira Provenzano L, Ashour T, Jolly SE, Arrigain S, Konig V, Schold JD, Navaneethan SD, Nally JV Jr, Nakhoul GN. Intra-arterial versus intravenous contrast and renal injury in chronic kidney disease: a propensity-matched analysis. Nephron. 2019;141:31–40.
Rudnick MR, Kesselheim A, Goldfarb S. Contrast-induced nephropathy: how it develops, how to prevent it. Cleve Clin J Med. 2006;73:75–80, 83–7.
Tan X, Zheng X, Huang Z, Lin J, Xie C, Lin Y. Involvement of S100A8/A9-TLR4-NLRP3 inflammasome pathway in contrast-induced acute kidney injury. Cell Physiol Biochem. 2017;43:209–22.
Lau A, Chung H, Komada T, Platnich JM, Sandall CF, Choudhury SR, Chun J, Naumenko V, Surewaard BG, Nelson MC, Ulke-Lemée A, Beck PL, Benediktsson H, Jevnikar AM, Snelgrove SL, Hickey MJ, Senger DL, James MT, Macdonald JA, Kubes P, Jenne CN, Muruve DA. Renal immune surveillance and dipeptidase-1 contribute to contrast-induced acute kidney injury. J Clin Invest. 2018;128:2894–913.
Atkinson SJ. Kidney surveillance in the spotlight: contrast-induced acute kidney injury illuminated. J Clin Invest. 2018;128:2754–6.
Kooiman J, Pasha SM, Zondag W, Sijpkens YW, van der Molen AJ, Huisman MV, Dekkers OM. Meta-analysis: serum creatinine changes following contrast enhanced CT imaging. Eur J Radiol. 2012;81:2554–61.
Moos SI, van Vemde DN, Stoker J, Bipat S. Contrast induced nephropathy in patients undergoing intravenous (IV) contrast enhanced computed tomography (CECT) and the relationship with risk factors: a meta-analysis. Eur J Radiol. 2013;82:e387–99.
McCullough PA. Contrast-induced acute kidney injury. J Am Coll Cardiol. 2008;51:1419–28. Erratum in: J Am Coll Cardiol. 2008;51: 2197.
Shemirani H, Pourrmoghaddas M. A randomized trial of saline hydration to prevent contrast-induced nephropathy in patients on regular captopril or furosemide therapy undergoing percutaneous coronary intervention. Saudi J Kidney Dis Transpl. 2012;23:280–5.
Majumdar SR, Kjellstrand CM, Tymchak WJ, Hervas-Malo M, Taylor DA, Teo KK. Forced euvolemic diuresis with mannitol and furosemide for prevention of contrast-induced nephropathy in patients with CKD undergoing coronary angiography: a randomized controlled trial. Am J Kidney Dis. 2009;54:602–9.
Barrett BJ, Parfrey PS. Clinical practice. Preventing nephropathy induced by contrast medium. N Engl J Med. 2006;354:379–86.
Lautin EM, Freeman NJ, Schoenfeld AH, Bakal CW, Haramati N, Friedman AC, Lautin JL, Braha S, Kadish EG. Radiocontrast associated renal dysfunction: a comparison of lower-osmolality and conventional high-osmolality contrast media. AJR Am J Roentgenol. 1991;157:59–65.
Cigarroa RG, Lange RA, Williams RH, Hillis LD. Dosing of contrast material to prevent contrast nephropathy in patients with renal disease. Am J Med. 1989;86:649–52.
Brown JR, Robb JF, Block CA, Schoolwerth AC, Kaplan AV, O'Connor GT, Solomon RJ, Malenka DJ. Does safe dosing of iodinated contrast prevent contrast-induced acute kidney injury? Circ Cardiovasc Interv. 2010;3:346–50.
Gurm HS, Dixon SR, Smith DE, Share D, Lalonde T, Greenbaum A, Moscucci M, BMC2 (Blue Cross Blue Shield of Michigan Cardiovascular Consortium) Registry. Renal function-based contrast dosing to define safe limits of radiographic contrast media in patients undergoing percutaneous coronary interventions. J Am Coll Cardiol. 2011;58:907–14.
Chen CM, Chu SY, Hsu MY, Liao YL, Tsai HY. Low-tube-voltage (80 kVp) CT aortography using 320-row volume CT with adaptive iterative reconstruction: lower contrast medium and radiation dose. Eur Radiol. 2014;24:460–8.
Taguchi N, Oda S, Utsunomiya D, Funama Y, Nakaura T, Imuta M, Yamamura S, Yuki H, Kidoh M, Hirata K, Namimoto T, Hatemura M, Kai N, Yamashita Y. Using 80 kVp on a 320-row scanner for hepatic multiphasic CT reduces the contrast dose by 50% in patients at risk for contrast-induced nephropathy. Eur Radiol. 2017;27:812–20.
Bruce RJ, Djamali A, Shinki K, Michel SJ, Fine JP, Pozniak MA. Background fluctuation of kidney function versus contrast-induced nephrotoxicity. Am J Roentgenol. 2009;192:711–8.
Ricós C, Iglesias N, García-Lario JV, Simón M, Cava F, Hernández A, Perich C, Minchinela J, Alvarez V, Doménech MV, Jiménez CV, Biosca C, Tena R. Within-subject biological variation in disease: collated data and clinical consequences. Ann Clin Biochem. 2007;44:343–52.
Campbell DR, Flemming BK, Mason WF, Jackson SA, Hirsch DJ, MacDonald KJ. A comparative study of the nephrotoxicity of iohexol, iopamidol and ioxaglate in peripheral angiography. Can Assoc Radiol J. 1990;41:133–7.
Moore RD, Steinberg EP, Powe NR, Brinker JA, Fishman EK, Graziano S, Gopalan R. Nephrotoxicity of high-osmolality versus low-osmolality contrast media: randomized clinical trial. Radiology. 1992;182:649–55.
Abe M, Kimura T, Morimoto T, Furukawa Y, Kita T. Incidence of and risk factors for contrast-induced nephropathy after cardiac catheterization in Japanese patients. Circ J. 2009;73:1518–22.
Saito Y, Watanabe M, Aonuma K, Hirayama A, Tamaki N, Tsutsui H, Murohara T, Ogawa H, Akasaka T, Yoshimura M, Sato A, Takayama T, Sakakibara M, Suzuki S, Ishigami K, Onoue K, CINC-J Study Investigators. Proteinuria and reduced estimated glomerular filtration rate are independent risk factors for contrast-induced nephropathy after cardiac catheterization. Circ J. 2015;79:1624–30.
McDonald RJ, McDonald JS, Bida JP, Carter RE, Fleming CJ, Misra S, Williamson EE, Kallmes DF. Intravenous contrast material-induced nephropathy: causal or coincident phenomenon? Radiology. 2013;267:106–18.
Davenport MS, Khalatbari S, Cohan RH, Dillman JR, Myles JD, Ellis JH. Contrast material-induced nephrotoxicity and intravenous low-osmolality iodinated contrast material: risk stratification by using estimated glomerular filtration rate. Radiology. 2013;268:719–28.
Wilhelm-Leen E, Montez-Rath ME, Chertow G. Estimating the risk of Radiocontrast-associated nephropathy. J Am Soc Nephrol. 2017;28:653–9.
Aycock RD, Westafer LM, Boxen JL, Majlesi N, Schoenfeld EM, Bannuru RR. Acute kidney injury after computed tomography: a meta-analysis. Ann Emerg Med. 2018;71:44–53.e4.
Trivedi HS, Moore H, Nasr S, Aggarwal K, Agrawal A, Goel P, Hewett J. A randomized prospective trial to assess the role of saline hydration on the development of contrast nephrotoxicity. Nephron Clin Pract. 2003;93:C29–34.
Mueller C, Buerkle G, Buettner HJ, Petersen J, Perruchoud AP, Eriksson U, Marsch S, Roskamm H. Prevention of contrast media-associated nephropathy: randomized comparison of 2 hydration regimens in 1620 patients undergoing coronary angioplasty. Arch Intern Med. 2002;162:329–36.
Nijssen EC, Rennenberg RJ, Nelemans PJ, Essers BA, Janssen MM, Vermeeren MA, Ommen VV, Wildberger JE. Prophylactic hydration to protect renal function from intravascular iodinated contrast material in patients at high risk of contrast-induced nephropathy (AMACING): a prospective, randomised, phase 3, controlled, open-label, non-inferiority trial. Lancet. 2017;389:1312–22.
Hoste EA, De Waele JJ, Gevaert SA, Uchino S, Kellum JA. Sodium bicarbonate for prevention of contrast-induced acute kidney injury: a systematic review and meta-analysis. Nephrol Dial Transplant. 2010;25:747–58.
Trivedi H, Nadella R, Szabo A. Hydration with sodium bicarbonate for the prevention of contrast-induced nephropathy: a meta-analysis of randomized controlled trials. Clin Nephrol. 2010;74:288–96.
Brar SS, Hiremath S, Dangas G, Mehran R, Brar SK, Leon MB. Sodium bicarbonate for the prevention of contrast induced-acute kidney injury: a systematic review and meta-analysis. Clin J Am Soc Nephrol. 2009;4:1584–92.
Putzu A, Boscolo Berto M, Belletti A, Pasotti E, Cassina T, Moccetti T, Pedrazzini G. Prevention of contrast-induced acute kidney injury by furosemide with matched hydration in patients undergoing interventional procedures: a systematic review and meta-analysis of randomized trials. JACC Cardiovasc Interv. 2017;10:355–63.
Subramaniam RM, Suarez-Cuervo C, Wilson RF, Turban S, Zhang A, Sherrod C, Aboagye J, Eng J, Choi MJ, Hutfless S, Bass EB. Effectiveness of prevention strategies for contrast-induced nephropathy: a systematic review and meta-analysis. Ann Intern Med. 2016;164:406–16.
Ahmed K, McVeigh T, Cerneviciute R, Mohamed S, Tubassam M, Karim M, Walsh S. Effectiveness of contrast-associated acute kidney injury prevention methods; a systematic review and network meta-analysis. BMC Nephrol. 2018;19:323.
Cruz DN, Goh CY, Marenzi G, Corradi V, Ronco C, Perazella MA. Renal replacement therapies for prevention of radiocontrast-induced nephropathy: a systematic review. Am J Med. 2012;125:66–78.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Fujigaki, Y. (2020). Contrast-Induced Acute Kidney Injury. In: Terada, Y., Wada, T., Doi, K. (eds) Acute Kidney Injury and Regenerative Medicine . Springer, Singapore. https://doi.org/10.1007/978-981-15-1108-0_7
Download citation
DOI: https://doi.org/10.1007/978-981-15-1108-0_7
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-1107-3
Online ISBN: 978-981-15-1108-0
eBook Packages: MedicineMedicine (R0)