
Abstract
Autophagy is a major intracellular degradative process that delivers cytoplasmic materials to the lysosome for degradation. An increasing number of studies on the physiological and pathological roles of autophagy in a variety of autophagy knockout models and human diseases were carried out. Among them, the clearance of misfolded proteins is the important function of autophagy. Impairment at different steps of the autophagy system, such as the ubiquitin-proteasome and the autophagy-lysosome pathways, may result in the accumulation of misfolded proteins in insoluble aggregates. Abnormal accumulation of misfolded proteins in cells can lead to a variety of human diseases. Here, we review the major advances in autophagy and the metabolism of misfolding protein in human diseases. Current studies about the promising therapeutic strategy in autophagy-modulating are also summarized.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Intracellular proteins are in a dynamic balance of constant synthesis and degradation, which facilitates the implementation of cell-specific functions and maintenance of homeostasis. However, human cells are often subjected to various stressors (such as oxygen-free radicals, ultraviolet radiation, etc.), and these challenges can lead to various types of protein damage that can damage normal cell function and disrupt cell homeostasis. When a specific protein is abnormal in the intracellular structure and aggregates in a toxic structure, it will collect and inactivate the normal functional protein, cause cell damage and eventually cause cell death, and cause degenerative diseases. For example, some neurodegenerative diseases are characterized by abnormal protein conformation in pathology. The change of protein conformation may be caused by insufficient folding during protein synthesis. Some sites of protein are not folded; Abnormal cell division; Gene insertion, deletion or abnormal modification of amino acid sequence, etc. In the cell, from protein synthesis to release, the intracellular system constantly checks the quality of synthetic proteins in order to timely repair and remove abnormal proteins. Drugs aimed at improving the quality control system of intracellular proteins are being developed, which may bring new hope for the clinical treatment of this disease.
Protein misfolding is a common form of abnormal protein conformation, this chapter mainly introduces the occurrence of protein misfolding and the corresponding degradation pathways, including the ubiquitin proteasome system (ubiquitin-proteasome system, UPS), autophagy-lysosomal pathway (autophagy-lysosome system), also introduces the degradation pathway caused by abnormal nerve vascular lesion and its mechanism of the system, may potentially effective treatment strategies, etc.
2 Protein Misfolding and Degradation Pathways
2.1 Protein Misfolding and Protein Polymer Formation
2.1.1 Basic Principles of Protein Folding and Molecular Chaperones
Protein folding refers to the physical process by which proteins form their intrinsic three-dimensional structure from polypeptide chains. According to the law of genetic center, the transmission of biological genetic information is generated by DNA transcription into RNA, which is then translated into polypeptide chains. Polypeptide chains with the complete primary structure are processed and modified, and finally folded to form proteins with specific spatial conformation. Protein usually has a quaternary structure, among which, the primary structure of a protein refers to the sequence of amino acids in the peptide chain, which is the basic structure of protein molecules and the basis of protein spatial structure and function. Protein spatial structure also includes secondary, tertiary and quaternary structure, which is a three-dimensional conformation formed by folding and coiling on the basis of the primary structure. Newly synthesized polypeptide chains in cells can only perform specific biological functions if they are folded correctly to form specific proteins with three-dimensional structures. So why do newborn peptides spontaneously fold to form proteins with a specific spatial conformation? What is the regulatory mechanism for this folding process? At present, it is believed that protein folding is a spontaneous process, which is determined by hydrophobic bond interaction, hydrogen bond formation, van der Waals force, and other factors in the polypeptide chain. The folding process of the new peptide chain follows the “thermodynamic hypothesis” and is controlled by kinetics.
In the 1960s, Anfinsen et al. found in the denaturation and renaturation experiments of bovine pancreas RNA hydrolases that the denaturated RNA hydrolases could renature only by removing the denaturants and reducing agents without the help of any other substances, and the bioactive RNA hydrolases could be formed again. Based on this, Anfinsen proposed the classical “thermodynamic hypothesis” of protein folding, and believed that natural protein polypeptide chain is the most thermodynamically stable form under certain environmental conditions. Since the conformation of natural protein is the lowest or the most stable thermodynamic free energy under certain environmental conditions (such as solution component, PH, temperature, ionic strength, etc.), denatured protein can spontaneously fold to form its natural conformation with biological activity under appropriate environmental conditions. The “thermodynamic hypothesis” of protein folding has been confirmed by some experiments and widely accepted.
With the development of research, it has been found that many protein polypeptides have low renaturation efficiency in vitro and form some non-natural conformations or nonspecific polymers. And the renaturation rate is much lower than the body level. In the 1990s, Joseph et al. proposed the protein folding energy theory and introduced the principle of minimum frustration. Bakei et al. thought that the natural conformation of some proteins might not be the lowest energy or the most stable energy state, and proposed that a protein polypeptide chain might have two low-energy states: one is a kind of natural conformation, the other is a kind of non-natural conformation, and the two mutual transformations in a lower energy state of a polypeptide chain need to overcome a high energy barrier, thus both mutual transformation is often difficult to complete, so there are two ways in the process of protein folding competing with each other, one way is correct folding form stable natural conformation, another kind is abnormal folding form stable than native conformation. For example, human insulin growth factor type I has two stable conformations, one natural and one non-natural conformation with mismatched disulfide bonds, both of which are in a similarly low-energy state.
At present, the theoretical model of protein folding assumes that the local conformation of a protein depends on the local amino acid sequence. Finally, the secondary structure frames were spliced with each other, and the peptide chain was gradually tightened to form the framework model of the tertiary structure of the protein. Hydrophobic collapse model in which hydrophobic forces play a decisive role in protein folding; A diffusion–collision–adhesion model of complex structures was established by the diffusion, collision and mutual adhesion of unstable secondary structural units generated by several sites of the extensional peptide chain in a nonspecific Brownian motion model. A nucleation–condensation–growth model in which a region of the peptide chain forms a “folded crystal nucleus” and continues to fold to form a natural conformation. Polypeptide chains can be folded along several different pathways, and in the process of folding along each pathway are more and more natural structures, eventually forming a natural conformational puzzle model.
Studies in the 1990s found that intracellular peptide folding was generally helpful, and some cofactors have been isolated that can promote the correct folding of polypeptide chains on the kinetics. For example, molecular chaperones (a particular protein) and folding enzymes (catalyze the covalent bond changes necessary to form a functional conformation directly related to protein folding). These facts strongly show that the correct folding of the polypeptide chain is controlled by dynamics as well as thermodynamics.
In 1978, Laskey et al. called the particular protein that binds to histone and mediates the ordered assembly of nucleosomes as molecular chaperone. In 1993, Ellis extended the concept to “a class of proteins that are sequentially unrelated but have common function: they help other structures containing polypeptides to complete the correct assembly, and separate from these structures after assembly, not forming the functional parts of these proteins” (Finn et al. 2005). Molecular chaperones are required to fold about 10–20% of the new peptide chains into proteins with specific spatial structures. Researchers found, significantly different from the condition of in vitro environment where protein polypeptide chain to fold, the environment inside the cell is very crowded, full of high concentrations of proteins, nucleic acids and other molecules, so the interaction between the molecules is very frequent, and the environmental conditions in the cell aggregation is easy to cause the nascent peptide chain or interaction between different protein peptides that hinder their correct folding of the natural conformation. In order to minimize the risk of protein misfolding, molecular chaperones are involved in a complex regulatory system. On the one hand, they can help ribosome-synthesized polypeptide chains fold as quickly as possible to form natural proteins; on the other hand, they can also make the hydrophobic surface of the aggregation protein recede and promote correct folding. In the cell fluids of prokaryotic and eukaryotic cells, molecular chaperones of different structural types enable the transition of substrate polypeptide chains from the primary synthesis state of ribosomes to the final folding state. Therefore, molecular chaperones play an important role in the interpretation of genetic information and the formation of biologically functional proteins. If the function of the molecular chaperone is inhibited, the misfolded protein will accumulate and deposit in the cell and directly cause toxicity to the cell.
Molecular chaperones are found in a wide variety of organisms, including many types of proteins, such as Chaperonin family (Cpn), heat shock protein 100 (HSP100), heat shock protein 90 (HSP90), heat shock protein 70 (HSP70), heat shock protein 60 (HSP60), small heat shock protein (smHSP), nucleoplasmin and chaperonin containing t-complex polypeptide 1 (CCT) (Finn et al. 2005). Molecular chaperone HSP90 is the most abundant cytoplasm, which consists of two highly similar to 90 kDa subunit HSP90 alpha and HSP90 beta form dimers, HSP90 by combination with ATP hydrolysis and conformational change and plays its role, its activity is a steroid hormone receptor and protein kinase must be mature, HSP90 is able to combine with hundreds of the substrate at the same time, participate in DNA damage repair, immune response process. Mammalian cells contain six HSP70 family members, and HSP70 and its homologous HSC70 are the main members, which are very similar and have similar activity. They can recognize misrecognized proteins and hydrophobic surfaces of some misfolded proteins, and regulate their binding and release to substrate proteins through ATP binding and hydrolytic activity. Increased intracellular HSP70 expression will reduce apoptosis. CCT is a chaperone located in eukaryotic cytoplasm. The substrate protein is folded and trapped in the central lumen of CCT protein, and CCT wraps the substrate protein through its helix protrusions.
Co-chaperone regulates its activity by interacting with molecular chaperones. These include HSP40/DnaJ family protein, bcl2-related athanogene (BAG) family protein, HSP70/HSP90 tissue protein (Hop), HSP110, HSP70 binding protein carboxyl terminal (CHIP), and GimC. HSP40 is located in the endoplasmic reticulum, mitochondria, and nucleus, and is involved in protein transport. DnaJ was first discovered in E. coli, which is called HSP40 in eukaryotes. HSP40/DnaJ family proteins contain J binding region and the ATPase binding region of HSP70 to bind and promote the ATPase activity. These proteins can also bind to the substrate protein and regulate the activity of HSP70. The protein family is divided into three categories: DnaJA, DnaJB, and DnaJC, which play various roles in protein folding, assembly, translocation, and degradation, respectively. All the BAG family proteins have a conservative BAG binding region and bind to the ATPase binding region of HSP70. They affect the exchange of HSP70 nucleotide and its binding/release with the substrate protein. HSP110 is also a nucleotide exchange factor of HSP70 and regulates the activity of HSP70. Both Hop and CHIP are common molecular chaperones that can interact with two different molecular chaperones. Hop is HSC70/HSP90 tissue protein, also known as stress-inducible protein 1 (STI1). Hop binds to HSP90 and HSC70/HSP90 through the tetratricopeptide repeat (TRP) binding region and regulates the interaction between molecular chaperones, so that it is easier to produce accurately folded proteins and form functional protein complexes. These effects include the transport of substrate proteins between them. CHIP, or C terminal HSP70 binding protein, can interact with HSP70 and HSP90 and play an important role in the quality control of ubiquitination of misfolded proteins. For example, Bag2 overexpression will inhibit CHIP activity and stimulate the maturation of other molecular chaperone functions. GimC helps to correctly fold CCT-dependent actin and microtubule proteins by transferring folded protein intermediates to CCT. GimC has six tentacle-like structures like jellyfish that trap unfolded proteins inside.
Under certain environmental conditions, newborn polypeptide chains are folded correctly to form natural proteins with biological activities. In general, the formation of the protein’s natural conformation is determined by the linear amino acid sequence of its primary structure. During the folding process, the protein polypeptide chain follows the “thermodynamic hypothesis” to change from high energy state to low-energy state and is controlled by the dynamics. For some proteins, the correct folding of cellular proteins requires the assistance of molecular chaperones.
2.1.2 Production and Recognition of Misfolded Proteins
The natural conformation of the protein is mainly composed of α-helix and irregular crimp structure, while the conformation of the misfolded protein is mainly composed of β-folding structure which is rich in hydrophobic. For example, neurodegenerative diseases caused by prions are caused by the accumulation in brain tissue of prion proteins (PrP), the pathogenic proteins formed by abnormal folding of normal proteins. There are two forms of PrP: wild-type PrPc and mutant PrPsc. Among them, the sequences of wild-type PrPc were dominated by α-helix and only 11.9% by β-folding, when the α-helix structure in wild-type PrPc is converted to β-folding, it becomes a mutant PrPsc. At this time, β-folding accounts for 43% of the protein structure and aggregates are formed outside the cell. The α-helix/β-folding structural transformation results in the exposure of hydrophobic groups and the embedding of hydrophilic groups in the protein, resulting in the formation of cross-over β-folding structures between protein molecules. β-folding structures are linked together by side chains and hydrogen bonds in the main chain to form a polymer dominated by β-folding, leading to disease.
Polypeptide chains with complete primary structure can only play their biological functions when they are folded correctly to form specific spatial conformation. Once the folding is abnormal, the wrong spatial structure will be formed, leading to the loss of biological functions and the occurrence of a series of diseases. Misfolding of proteins in vivo is caused by mutation induction, increased protein concentration, oxidative stress, aging, and other related reasons. In the cell, there is a protein folding quality control system, which can monitor the folding of proteins in the cytoplasm and timely remove misfolded proteins. This system mainly includes molecular chaperones and protease systems. Their action process is divided into two steps: One is to identify errors; Second, correct mistakes. First, the molecular chaperone recognizes misfolded protein monomers and attaches them to the hydrophobic terminal surface to prevent their polymerization and promote protein refolding and assembly. If the misfolded protein cannot be repaired, it will be delivered to the ubiquitin-proteasome system and the chaperon-mediated autophagy system for degradation under the molecular chaperone mediation. If the misfolded protein monomers accumulate, the proteasome system will lose its function, and the protein aggregates will be degraded and cleared by the macroautophagy pathway. In addition, the microtubule-dependent transport system can transport soluble oligomers/aggregates to inclusion bodies for degradation. The intracellular quality control system is regulated by stress-induced transcription factors, co-chaperone, and other cofactors. If the quality control system obstacle, protein misfolding hydrophobic surface cannot be exposed by molecular partner or protease recognition, or the formation of aggregates is faster than molecular chaperone and protease recognition speed, those who are not protected by molecular chaperone, or who has not been protease degradation of abnormally folded proteins may occur polymerization, leading to the accumulation of abnormal folding proteins inside cells and causing cell damage and even death (Kubota 2009).
So how do these toxic misfolded proteins cause cell death? One of the most important mechanisms is that misfolded proteins induce endoplasmic reticulum stress (ER stress), increasing of protein synthesis, the expression of misfolded proteins, calcium ion imbalance, virus infection and nutrition deprivation, glycosylation changes and cholesterol overload, and then these conditions affect the endoplasmic reticulum folding ability, leading to unfolded protein accumulation in endoplasmic reticulum. In order to maintain the balance of demand and endoplasmic reticulum protein folding ability, endoplasmic reticulum evolved highly specific intracellular signaling pathways—the unfolded protein response (UPR), and the induction of UPR also suggests the activation of the compensatory mechanism, which affects the normal physiological function (Senft and Ronai 2015). In addition, because the degradation of misfolded proteins occurs in the endoplasmic reticulum, it is also known as ER-associated degradation (ERAD). Misfolded proteins inhibit the proteasome function and inhibit the protective effect of ERAD. Some proteins, such as 1-antitrypsin Z mutants, activate ER related caspase 4 and caspase 12, although they do not induce the UPR reaction.
In order to stabilize their environment, cells degrade abnormally folded proteins through a variety of pathways (as described below), and conformational changes in proteins are key factors for degradation. At present, most proteins that can be degraded and cleared by autophagy are known to be mutated proteins with unstable conformation and easy to form oligomers. There are many reasons for protein conformation instability, not only gene mutation. In mouse models lacking the autophagy regulatory gene atg5 or atg7, it was found that the nerve and liver tissues of the mice had accumulated a large amount of ubiquitinated proteins, and these proteins were not mutated. The accumulation of these proteins suggests that they cannot be cleared by the proteasome pathway, at least this pathway is not very efficient. However, the exact mechanism that causes these proteins conformational instability is unclear. It has been found that disrupting the environment or function of ER can lead to the accumulation of misfolded proteins in cells. In the process of cell development, some special metabolic changes will cause stress of ER, which will lead to the accumulation of misfolded proteins. In addition, oxidative stress, hunger, and other stress factors can also induce the production of misfolded proteins, which can also be ubiquitinated and formed into polymers, similar to polymers formed by mutant conformation changes. Aggresome-like induced structure (ALIS) is the structure of these protein polymers (Szeto et al. 2006). But, how did these proteins turn into misfolded proteins under stress is unclear. Dendritic cell ALIS, a structure called dendritic cell ALIS, is produced when the ribosome products of dendritic cells are insufficient in the process of maturation. Thus, it is possible that ALIS proteins are modified or destroyed under stress, leading to protein misfolding and ubiquitination.
2.2 Degradation of Misfolded Proteins
Cell proteins are in a dynamic balance of continuous synthesis and degradation, which is related to the specific biological functions of cells and the maintenance of cell homeostasis. However, cells are often affected by a variety of environmental factors, such as oxidative stress and ultraviolet radiation. Excessive environmental stress will cause cell protein damage, affect the cell’s normal function and cell homeostasis, and eventually lead to cell death. Therefore, timely clearance of damaged and harmful proteins in cells is crucial, especially for those non-proliferating cells such as neurons.
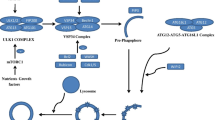
Normally, misfolded proteins in a cell are first identified by a molecular chaperone, and then refolded and assembled with the help of a molecular chaperone. In some cases, however, misfolded proteins mutate so much that the intracellular conditions are insufficient for selective refolding, then molecular chaperones will transport the misfolded protein products to the proteolytic system for degradation. The intracellular proteolytic system consists of two major pathways: the UPS pathway and the autophagy pathway (Martinez-Vicente and Cuervo 2007) (as shown in Fig. 18.1).

Proteolytic systems in mammalian cells (Martinez-Vicente and Cuervo 2007)
Lysosome protein substrates have two sources: from extracellular (swallowed foreign body) and from cells (autophagy). In mammalian cells, there are three types of autophagy, namely macroautophagy, microautophagy and chaperone-mediated autophagy (CMA). In the macroautophagy pathway, intracellular substances can be separated by some double membrane structure and autophagy vesicle formation, and then merge with lysosomes. In microautophagy pathway, lysosome membrane directly by swallowing function within the substrate protein intake. CMA pathway is different from the former two ways of “batch” degradation, CMA-degraded substrate proteins are selective and bind to lysosomal receptors (lamp-2a) before translocation into lysosomes. Ubiquitin-proteasome pathway (Daw et al. 2015) is another important pathway for intracellular protein degradation. The UPS pathway substrate protein is ubiquitinated and then degraded by protease recognition.
2.2.1 UPS Pathway
The UPS pathway, also known as the ubiquitin-proteasome pathway, is an important pathway for protein selective degradation in cells (Martinez-Vicente and Cuervo 2007). Proteasome exists in both the nucleus and the cytoplasm and it is a proteolytic system other than lysosome. The 26S proteasome, the most common form of proteasome, has a molecular weight of about 2.5MDa and contains one 20S core particle and two 19S regulatory particles. The core particle (CP) is a double-sided symmetric hollow cylindrical structure, which surrounds the active site of the spliced protein in the “hole”. By opening the ends of the core particles, the target protein can enter the “hole”. Each end of the core particles is connected with a 19S regulating particles (RP), the RP consists of a central part and a cap-like part, the central part is formed by 6 ATPases, which can hook CP through the C terminal and regulate the switch of degradation channel. The cap-like structure of RP is composed of 9 non-ATPase subunits, and the regulatory particles can recognize the ubiquitinated proteins and deliver them to the core particle degradation chamber. The phosphorylation of CP and RP subunits is related to the activity and stability of proteasome. Proteasomes hydrolyze a variety of ubiquitinated protein substrates from the carboxyl side of basic, acidic, and neutral amino acids. Protein degradation by proteasome is isolated from the intracellular environment. The UPS pathway degrades two types of proteins: proteins that are misfolded and proteins that require quantitative regulation.
The ubiquitin-proteasome system is an important part of the intracellular quality control system, it includes ubiquitin and its promoter system and proteasome system. The ubiquitin promoter enzyme system is responsible for activating ubiquitin and binding it to proteins to be degraded to form target protein polyubiquitin chain, namely ubiquitination. Proteasome systems can recognize and degrade ubiquitinated proteins. Proteasome degradation regulation is an important way to control the proteasome substrate pathway. Ubiquitinated proteins are directly recognized by proteasomes through three ubiquitin receptors: Rpn1, Rpn10, and Rpn13. More and more evidence shows that cells regulate proteasome-mediated protein degradation to meet their own needs by coordinating the expression of proteasome subunits and molecular chaperones, and the core of proteasome-mediated assembly regulation is TOR complex 1 (TORC1), which is a major regulator of cell growth and stress. The ubiquitin-proteasome pathway is involved in a variety of intracellular processes, including apoptosis, MHC I antigen presentation, cell cycle, intracellular signal transduction, etc., and is closely related to some physiological functions and pathological states of cells. For example, some misfolded proteins still maintain their solubility, and these proteins can be selectively degraded by the UPS pathway. The high selectivity of the UPS pathway ensures that the abnormally folded proteins in the cell can be degraded and cleared without affecting the normal cellular components. However, the specific pathway by which an abnormal protein is degraded is not clear, which may be related to the effectiveness of the proteolytic system at a specific time or the characteristics of the substrate protein. For example, certain proteolytic enzymes of the lysosome may be more effective at degrading certain proteins; Only unfolded monomer proteins can be degraded by both the UPS pathway and the autophagy pathway, while proteins that have formed oligomers, fibrils, or fibrous structures can only be processed by batch degradation pathway, namely the autophagy pathway (Martinez-Vicente and Cuervo 2007).
2.2.2 Macroautophagy-Lysosome System (MALS)
Lysosomes, an organelle used for degradation and recycling of intracellular and extracellular substances, were first described and identified more than 60 years ago. The lysosomal system plays an important role in regulating cell surface molecules and plasma membrane receptors as well as resisting the invasion of extracellular substances. The degradation of exogenous substances by lysosomes is called phagocytosis. In contrast, autophagy refers to the degradation of intracellular components by lysosomes (Fig. 18.1). In the past 10 years, some molecular characteristics of autophagy have been discovered by using relatively simple experimental models of gene manipulation such as yeast, worms, and flies and so on, which is of great help for us to better understand and understand autophagy.
In summary, autophagy is a “batch” protein degradation process involving lysosomes. At present, studies have confirmed that almost all neurodegenerative diseases have the accumulation of autophagic vesicles and the aggregation of proteins in the cytoplasm. Autophagy in mammals can be divided into three types according to the route of intracellular material transport to lysosomal degradation: macrophage, microautophagy and molecular chaperon-mediated autophagy (Fig. 18.1) (Martinez-Vicente and Cuervo 2007). All three autophagy pathways have the ability to transport cytoplasmic substrates to lysosomes for degradation. The three types of autophagy have a common endpoint, that is, they all form autophagosomes, but their substrates, regulatory modes, and activation conditions are different.
2.2.2.1 Macroautophagy Pathway
The macroautophagy pathway can degrade a whole region of the cytoplasm in the cell in batches. This region is first surrounded and isolated by a plasma membrane, forming a closed chamber with a bilayer membrane, namely the autophagosome. Autophagosome membrane is formed by coupling microtubule-associated protein 1 light chain 3 with lipid, Atg5, Atg16, Atg12, and other autophagocyte-related proteins. These bilayer membrane vesicles are formed by intracellular lipid phosphorylation of organelles such as the endoplasmic reticulum, mitochondria, and Golgi bodies, triggered by a kinase complex regulated by beclin-1. There are no enzymes in autophagosome, and lysosomes contain all the enzymes needed for content degradation, so the content can only be degraded after the fusion of autophagosome and lysosomes. Autophagy-related protein (Atg) family is involved in the whole process of autophagy degradation, which involves a series of interactions (including protein–protein, protein–lipid, etc.) and several major kinase families in the cell. Autophagy is most easily activated under stress conditions, and its activation has two main functions: one is that autophagy can be used as a source of macromolecules and energy in cells in the state of nutritional deficiency, and the other is to remove abnormal intracellular components. Many tissues have persistent autophagy activity, which is essential to maintain cell stability. Changes in the autophagy pathway have been shown to be strongly associated with tumorigenesis, bacterial and viral infections, severe myopathy, metabolic diseases such as diabetes and neurodegenerative diseases.
Macroautophagy is the main pathway of autophagy, and recent studies on autophagy mainly focus on this pathway (Menzies et al. 2011). Macroautophagy is an important pathway of protein degradation in vivo. According to different protein degradation pathway, the protein can be divided into three types, namely type I, type II, type III, and classification through the ubiquitin-proteasome pathway or Macroautophagy pathway for degradation (Fig. 18.2). These two degradation pathways are related to each other in mechanism, but their ability to degrade different kinds of proteins seems to be different. Macroautophagy degrades almost all forms of misfolded proteins, whereas the ubiquitin-proteasome pathway degrades only those proteins that are soluble. In the state of starvation, the protein in the cell will be degraded in batch without selectivity. Although it is generally believed that the autophagy degradation pathway is nonselective to the substrate protein, the conformational changes of the protein and subsequent ubiquitination and further autophagy degradation suggest that the autophagy pathway has a certain degree of specificity in the degradation of misfolded proteins. At present, it is believed that this specificity is closely related to the recognition of ubiquitinated proteins P62/SQSTM1 and HDAC6 and the promotion of the degradation of these substrate proteins by autophagy pathways. These two molecules can recognize the ubiquitin sites of misfolded proteins and LC3/Atg8 of autophagosomes. The C terminal of P62 contains a ubiquitin-associated ubiquitin-binding region, and the cytoplasm of P62 overexpression can form a large number of p62 positive corpuscles, which are located in the UBA region and are also ubiquitin-positive. This suggests that P62 attracts ubiquitinated proteins to form large complexes. Currently, P62 has been found in the inclusion bodies of various protein aggregation diseases, including Lewy body that is Parkinson’s disease, tau protein that is Alzheimer’s disease, and Huntington’s aggregates. These protein aggregates are composed of misfolded mutant proteins and ubiquitin is often positively altered. Mutant protein aggregates (II b and II c protein) is the target of autophagic degradation, thus P62 also played an important role in it. P62 can also bind to LC3/Atg8 directly through a special sequence near the UBA region (aa.321–342), LC3 and P62 protein co-localizes and is degraded by autophagic lysosome in Huntington. Interestingly, even large p62-positive structures (2 μm) can be degraded by autophagy, suggesting that autophagy can degrade not only aggregates but also some large inclusions and aggregates. In addition, P62 was also involved in autophagy clearance of ALIS. The inhibition of autophagy P62 was significantly up-regulated, suggesting that even at the basic level, P62 was continuously degraded by the autophagy pathway. HDAC6 is another protein that binds to ubiquitinated proteins through the BUZ region, and has been found to co-localize with Lewy bodies and ubiquitin (Pandey et al. 2007). Inhibition of HDAC6 significantly slows down the formation of autophagosome aggregates, indicating that HDAC6 is important for the formation of aggregates. HDAC6 is a microtubule-associated deacetylase that binds to dynein, which has been found that the protein is closely related to the reverse transport of the aggregation protein to the center of the microtubule to form a cluster. HDAC6 can directly act on dynein to lead to the formation of aggregates, and the autophagy degradation of mutant huntington’s protein aggregates also requires the participation of HDAC6. In addition, the activation of autophagy after proteasome inhibition also depends on HDAC6. These findings suggest that HDAC6 is important for the degradation of typeIIb misfolding proteins and type IIa soluble misfolded proteins. Because both of these proteins can be produced after proteasome inhibition, but the degradation of HDAC6 whether involved in IIc aggregated proteins is unclear, and how HDAC6 is involved in autophagic degradation of misfolded proteins remains also unclear. Some evidence suggests that this may be related to HDAC6-mediated microtubule activity, which allows some autophagic components, such as lysosomes, to be reversely transported to the microtubule center for autophagy degradation. P62 and HDAC6 have similar and synergistic effects, and both interact with misfolded proteins and autophagic vesicles, which also show some specificity in autophagy degradation.

Protein classification based on different degradation pathways
In autophagy studies, another autophagy carrier receptor NBR1 (neighbor of BRCA1 gene 1) was also found. NBR1 receptor may be related to the degradation of the ubiquitin-labeled target in autophagosomes. Studies show that when autophagy occurs, NBR1 began to accumulate, it is linked to autophagy-related ubiquitin through its own UBA domain (Ub-associated domain) at the C-terminal, and the proteins to be degraded by ubiquitin are also linked to and aggregated with NBR1 through ubiquitin, and then NBR1 interacts with LC3. The autophagy carrier receptor is formed and the ubiquitin marker is transported to the autophagy for degradation.
In addition, NBR1 has been found to have a variety of biological functions. In the M line of the skeletal muscle segment, it can be observed that NBR1 is directly connected to the giant sarcomeric protein kinase titin and P62. If the mutation of the giant sarcomeric protein kinase breaks the connection with NBR1, it will cause genetic muscle diseases, and NBR1 and P62 will form the “P62 corpuscle” mentioned above. However, it is still uncertain whether autophagosomes play a role in this disease. Knock out of NBR1 in mouse inhibited T cells activation and Th2 cell differentiation, while T cells play an important role in allergic airway inflammation. Therefore, it can be inferred that NBR1 may play a role in allergic airway inflammation by regulating the function of T cells. It can be seen from the above studies that compared with P62, NBR1 undoubtedly has more complex biological functions.
It can be seen from the above that both P62 and NBR1 can serve as the carrier receptors for autophagosomes to degrade the ubiquitinated target, so how do these ubiquitinated degradation targets enter the autophagosome? In general, there are three pathways: P62 body pathway, HDAC6-p62 pathway, and ATGs degradation pathway.
Abnormal folding proteins are generally degraded by the P62 corpuscles, pathway. Studies have shown that P62, ALFY (autophagy-linked FYVE protein), and NBR1 are assembled into P62 corpuscles, which act as autophagy carrier receptors and mediate misfolded proteins into autophagy vesicles. ALFY is a protein molecule with large structure, which plays the role of the skeleton in P62 corpuscles. As cells age, more and more misfolded proteins accumulate, and the stress effect of these misfolded proteins can induce the high expression of P62. In addition, the formation of P62 bodies also requires the assistance of specific molecular chaperones. Recent studies have shown that Hsp70 (Heat Shock Protein70) and its molecular chaperone Bag3 and HspB8 can induce the formation of P62 bodies. Endoplasmic reticulum stress caused by protein misfolding will induce Bag1 into Bag3, and Bag3, Hsp70, HspB8 and E3 ubiquitin kinase CHIP (carboxyl terminus of Hsc70-interacting protein) constitute a complex. The complex can recognize folded abnormal proteins, then the composite experience gathered P62, ALFY and NBR1 form P62 small body, P62 bodies bind to the folded abnormal protein labeled by ubiquitinated protein 1–4 to form autophagic vesicles of different sizes. Finally, P62, as the carrier receptor of the autophagosome, enables autophagic vesicles to enter autophagosome by endocytosis.
The HDAC6-P62 pathway is mainly used for aggresome degradation. Abnormal protein aggresome refers to the structure formed when proteins are labeled by ubiquitin and transported to microtubule organizing center (MTOC) through microtubule and then encased by intermediate filaments so that they can be effectively degraded by autophagy. HDAC6 is critical for the formation of abnormal protein aggresome by binding to ubiquitinated and dynein proteins, allowing it to transport soluble ubiquitin markers into the abnormal protein aggresome. Another of its roles is to induce the construction of dynein networks to promote the fusion of autophagosomes with abnormal aggresome and lysosomes. When proteins accumulate in large quantities, ubiquitinated proteins are labeled and bound to HDAC6, which is transported to the nucleus of autophagosomes via microtubules to form abnormal protein aggresome, and mediate the fusion of autophagosomes containing abnormal protein aggresome with lysosomes. The degradation process after binding with lysosome is related to P62 and its related factors. After the formation of a complex between ALFY and ATG5, it binds with P62 as a carrier that mediates the fusion of autophagosomes and lysosomes, and after autophagy-lysosome formation, the abnormal protein aggresome degraded in autophagy.
The selective autophagy pathway independent of ubiquitin-protein has been most thoroughly studied in yeast, which can be divided into the following three pathways: Cvt pathway, mitochondrial autophagy pathway, and peroxisomal pathway. Cvt pathway is the most mature selective autophagy pathway. Its main function is to transport cytoplasmic and autophagosome related substrates, such as aminopeptidase (Ape1), to phagophore assembly site (PAS) for its hydrolytic enzyme activity. Ape1 is present in the cytoplasm as an inactive precursor of PrApe1. When PrApe1 forms Ape1 complex with Atg11 and Atg8, Atg11 leads the complex into PAS, and Atg8 leads the complex to bind Atg19 as carrier receptor, which mediates the complex into an autophagosome. The mitochondrial autophagy pathway is easily activated during nutrient deficiency or when cells enter the quiescent phase. This pathway is mainly regulated by a protein on the outer membrane of mitochondria: Atg32. After the damaged mitochondria were combined with Atg11 and Atg8 to form a complex, Atg32 was used as the carrier receptor to mediate the damaged mitochondria into PAS. The peroxisomal pathway refers to the combination of intracellular peroxides with Atg11 to form a complex, and Atg30 as the carrier receptor mediates the peroxisomal entry into PAS.
Activation of autophagy pathway is related to the stress of endoplasmic reticulum (ER). ER controls the degradation of abnormally folded proteins through the proteasome system and macroautophagy-lysosome system (MALS). Both pathways are activated under ER stress induced by misfolded proteins. When the UPS pathway is inhibited, the MALS pathway will be compensatorily activated. However, when the MALS pathway is inhibited, whether the UPS pathway can be compensatorily activated is uncertain, suggesting that the ubiquitin-proteasome pathway may be more limited in the degradation of misfolded proteins. ER stress activates the proteasome pathway through ER-associated degradation (ERAD). On the other hand, ER stress activates the autophagy system through ER-activated autophagy (ERAA). In addition, the release of calcium ions in ER to activate multiple downstream regions including CaMMK/AMPK, calcium protease, and DAPK is also an important mechanism of ERAA. In addition, molecular regulation of G protein signals (e.g., RGS16) may also be involved in ERAA activation. In rapidly growing tumor cells, the rapid metabolism of unfolded or abnormally folded proteins makes endoplasmic reticulum more prone to excessive load. Therefore, compared with normal cells, tumor cells are more prone to endoplasmic reticulum stress. Cells respond to endoplasmic reticulum stress with unfolded protein response (UPR) and enhanced autophagy. The unfolded protein reaction is also an important pathway of ERAD and plays an important role in ERAA. UPR is the main protective and compensatory mechanism in ER stress, it has three main feelings of endoplasmic reticulum stress sensors, endoplasmic reticulum protein kinase sample kinase (pancreatic ER kinase-like ER kinase, PERK), activation of transcription factor 6 (ATF6), and the endoplasmic reticulum inositol-requiring enzyme 1 (IRE1). The accumulation of unfolded or abnormally folded proteins in the endoplasmic reticulum induces the dissociation and activation of these receptors and their molecular chaperones in the endoplasmic reticulum, glucose-regulated protein78 (GRP78). Activated PERK has been found to inhibit the synthesis of unfolded or misfolded proteins by phosphorylating the subunit of eukaryotic initiation factor2 (eIF2). In addition, the activated PERK and its molecular targets eIF2α in the process of autophagy LC3-I to LC3-II played an important role in transformation. The Huntington mutant protein transcriptionally up-regulates CHOP and Atg12 in an eIF2α phosphorylation-dependent manner, whereas Atg12 is a ubiquitin-like protein that modifies Atg5 by covalently binding. These processes are important for autophagosome formation. Activated ATF6 can regulate the expression of endoplasmic reticulum molecular chaperones (e.g., GRP78 and GRP94), X box-binding protein 1 (XBP1) and protein disulfide isomerase (PDI) to reduce the folding, secretion, and degradation of unfolded and abnormal folded proteins in the endoplasmic reticulum. In addition, autophagy can be induced by some drugs, such as A23187, tunicamycin, and toxic carotene, that directly interfere with the environment and function of ER, and this process seems to be dependent on the IRE1 signaling pathway. IRE1 also plays an important role in autophagy induced by proteasome inhibitors. In mammals, autophagy induction requires IRE1 kinase activity. IRE1 can activate the transcription factor XBP-1 to form sXBP1 (spliced XBP1) through the endoribonuclease activity, and sXBP1 can activate the transcription-translation of related proteins in the ER-associated protein degradation pathway (ERAD), promoting the degradation of unfolded or abnormally folded proteins. In addition, IRE1α can also activate ASK-1 and stress kinase JNK by binding to TRAF2. Overexpression of ASK-1 (Apoptosis signal, regulating kinase I) can mediate several cell death, suggesting that ASK-1 is significantly correlated with cell apoptosis induced by ER stress. It has been found that JNK is not only involved in autophagy induced by protease inhibitors but also plays an important role in autophagy induced by ER stress inducers. CHOP (C/EBP homologous protein) and JNK are the main factors in this process. Intense ER stress causes CHOP up-regulation of GADD34 (growth arrest and DNA damage-inducible gene 34) expression. GADD34 proteins can interact with protein phosphatase 1 (PP1) to dephosphorylate eIF2α, resulting in the uninhibited synthesis of folded or abnormally folded proteins. After the phosphorylation of Bcl-2 (B-cell lymphoma 2) by JNK, Beclin1, an autophagy gene, was released and activated from its inhibitory factors to induce autophagy, leading to apoptosis.
Recent studies have found that many factors in the UPR pathway play a role in tumor formation and development. For example, GRP78 has been found to play a role in tumor progression, including the proliferation of tumor cells, escape of apoptosis, tumor angiogenesis, tumor metastasis, and resistance of tumor cells to chemical agents. In addition, GRP78 protects quiescent tumor cells from chemical damage by inhibiting apoptosis. In liver cancer tissues, the GRP78 expression level was higher in low-differentiated liver cancer cells than in high-differentiated liver cancer cells.
The IRE1/XBP1 axis in UPR is another mediator that has been found to play a wide role in tumors. It was found that the growth of fibroblasts in transgenic mice with XBP1 dysfunction was significantly inhibited. On the contrary, the growth of multiple myeloma tumor cells in transgenic mice with sXBP1 overexpression was exuberant after human multiple myeloma cell transplantation. In breast cancer patients, the accumulation of estrogen caused by breast cancer can promote the overexpression of XBP1, and the overexpression of XBP1 can induce the chemical resistance of tumor cells.
The autophagosome pathway is another pathway for the endoplasmic reticulum to degrade unfolded or abnormally folded proteins, which includes the following processes: autophagy guidance, nuclear formation of autophagosome, autophagy vesicle growth, and vesicle formation. ULK1/2 (Unc-51-like kinase 1/2) activates autophagy induction. Beclin1-VPS34 (phosphatidylinositol-3-kinase, PI3KIII)-p150 nuclear complex mediates autophagosome nuclear formation. Ubiquitinated protein is a key factor in the growth stage of autophagic vesicles. It can be mediated into autophagosome by Atg3, Atg5, Atg7, Atg12, LC3 (microtubule-associated protein 1 light chain 3) to promote the growth of autophagosome and eventually form autophagosome. Endoplasmic reticulum stress induced by unfolded or misfolded proteins up-regulated the expression of Atg12 and promoted the transformation of LC3-I into LC3-II. In addition, the endoplasmic reticulum stress can lead to caspase (cysteinyl aspartate specific proteinase) activation of autophagy function can make the Bcl-2 phosphorylation, and activated caspase and phosphorylated Bcl-2 can separate Beclin1 from its inhibitor to form a state of shear activation, and activated Beclin1 is then transported to mitochondria to promote the release of cytochrome C, which itself becomes a free form of Beclin1, free Beclin1 can induce autophagy again and form a positive feedback pathway.
Endoplasmic reticulum stress caused by unfolded or abnormally folded proteins can lead to the occurrence of autophagy. The endocytic degradation of autophagosomes to intracellular abnormal proteins or senescent organelles is an important source of cellular amino acids under endoplasmic reticulum stress. Therefore, in more metabolically active tumor cells, autophagy can increase the resistance of tumor cells to stress such as chemotherapy drugs and radiotherapy; similar studies have shown that inhibition of autophagy can increase the efficiency of chemotherapy drugs. For example, in tumor cells with Ras gene mutations, autophagy is still at a high level even under nutrient-rich conditions. The effect of chemotherapy drugs is not obvious, but if the autophagy pathway is blocked, the inhibition of tumor cell growth is significantly improved. In addition, malignant melanoma has a higher level of expression of LC3, and LC3 levels in advanced malignant melanoma cells are higher than the early stage. The expression of the autophagy gene Beclin-1 can be used as a prognosis for patients with nasopharyngeal carcinoma. The higher the expression level of Beclin-1, the worse the prognosis of patients.
So, what is the significance of ER-activated autophagy in clearing misfolded proteins? Clearly, the separation and removal of misfolded proteins reduce the toxicity of these proteins. The accumulation of misfolded proteins that have been cleared will eventually lead to cell death. The formation of aggregates avoids the distribution of misfolded proteins throughout the cell and is believed to have a protective effect on the cells. Inhibition of HDAC6 interferes with the formation of aggregates and increases cell death. Autophagy separates the misfolded protein and also protects the cells. In some over-expressed misfolded proteins, such as mutant Huntington protein cells, autophagy can be promoted to reduce cell death, while autophagy inhibition can increase cell death (Arrasate et al. 2004). Autophagy has the ability to ameliorate the pathological changes caused by misfolded proteins, which has been confirmed in the animal models of Huntington’s and Kennedy’s diseases. So how does autophagy inhibit cell death? This is related to the upstream signaling pathway of autophagy inhibiting cell death. ER stress-induced apoptosis is largely mediated by the mitochondrial pathway and ultimately depends on the Bcl-2 family proteins, Bax and Bak. Current research suggests that Bax and Bak are not equivalent in response to a death signal stimulus. Bax appears to be more sensitive. When autophagy decreased, the death signal stimulation also acted more on sensitive Bax and less on Bak.
In summary, autophagy and UPS pathways work together to provide another compensatory mechanism for the degradation of misfolded proteins, which attenuates ER stress and reduces cell death. Among them, ER plays an important regulatory center in various degradation pathways. At present, the mechanism of autophagy induction has been continuously discovered. Of course, it is necessary to clarify how various abnormal folding proteins activate autophagy and the recognition mechanism of these proteins.
Proteins can be divided into three types based on the pathway of protein degradation: type I, type II, and type III. Type I proteins are completely degraded by the ubiquitin-proteasome pathway (Daw et al. 2015) and are used to regulate cellular homeostasis; type III proteins are completely degraded by large autophagy pathways for nutrient cycling; type II proteins are nonfunctional, abnormally folded proteins that are capable of being cleared by the UPS pathway, and the macroautophagy pathway. According to different protein solubilities, protein structures, and subcellular localizations, type II proteins can be divided into three subtypes: type IIa, type IIb, and type IIc. Type IIa is a soluble type II protein that is degraded by the UPS pathway; where protein degradation from the endoplasmic reticulum (ER) requires the involvement of the endoplasmic reticulum-associated degradation pathway (ERAD). Macroautophagy can degrade all abnormally folded proteins, but is more important for highly folded proteins (types IIb and IIc). Most misfolded proteins can also be ubiquitinated, and ubiquitinated proteins can be degraded by the protease pathway (pathway 1) and autophagy pathways (pathways 2 and 3). Among them, P62/SQSTM1 and HDAC6 promote the process of path 3. Some type IIa proteins, such as IKK complexes; IIc type proteins, such as a1-antitrypsin mutants, are not clear whether they are ubiquitinated. If they are ubiquitinated, they are degraded via pathways 2 and 3; if there is no ubiquitination, they are degraded via other degradation pathways (pathways 4 and 5).
2.2.2.2 Microautophagy Pathway
More than 50 years ago, Duve and Wattiaux first proposed the concept of microautophagy, in which a small portion of mammalian cytoplasm can directly form vesicles and be engulfed by lysosomes. The microautophagy pathway was first identified in the liver. Microautophagy has a variety of molecular mechanisms. In early studies, it was noted that the cytoplasm was partially extended and encapsulated by the lysosomal membrane. Degradation of vacuolar membrane structure was observed in yeast. Therefore, the definition of microautophagy was redefined in terms of membrane kinetics. Microautophagy was divided into three types: microautophagy with lysosomal membrane protrusion, microautophagy with lysosomal membrane entrapment, and microautophagy with nuclear endosomal membrane entrapment. In yeast, microautophagy involves degradation of a variety of substrates including peroxidase, nuclear fragments, mitochondrial fragments, lipid droplets, and so on. In plants, microautophagy mediates the degradation of anthocyanins. With the endosome membrane invagination, microautophagy depends on the endosome transport complexes (ESCRT) system, resulting in a major or selective cytoplasm protein degradation. Some small molecular mechanisms across different types of autophagy, the formation of vesicle membrane structure is a mammalian cell with lysosome membrane outstanding small autophagy and yeast cells with lysosome membrane invagination of the common features of autophagy, and ESCRT system in yeast cells with small lysosome membrane invagination autophagy and tracing the endosome membrane invagination little autophagy plays a key role. Autophagy-related proteins, such as Hsc70, interact in different types of autophagy pathways, so microautophagy is associated with other types of autophagy. In microautophagy, Hsc70 is recruited to mature endosome through the electrostatic interaction between protein and phosphatidylserine, so that it shows membrane deformation. In the microautophagy pathway, lysosomes deliver substances from different regions of the cell fluid into the lysosomal cavity for rapid degradation through endocytosis or tubulogenesis. Microautophagy is involved in the recycling of intracellular components under normal cell conditions. In addition, autophagy is also responsible for selectively removing excess organelles within the cell. For example, clinical drugs induce the production of peroxisomal enzymes, which maintain a normal number of intracellular organelles through the degradation of the microautophagy pathway. The microautophagy pathway also plays a role in maintaining the stability of the intracellular environment. However, so far, there is still a lack of effective methods to understand the relationship between microautophagy and human diseases.
2.2.2.3 Chaperon-Mediated Autophagy Pathway
Molecular chaperone-mediated autophagy pathway is abbreviated as CMA. The most important characteristic of CMA is its selective substrate. Some proteins in the cell have lysosomal target sequences that molecular chaperones can recognize and bind to and transport to the lysosomal surface. Once the substrate protein reaches the lysosomal membrane, the substrate protein will interact with the receptor protein on the lysosomal membrane, and with the assistance of the molecular chaperone in the lysosomal body, the substrate protein passes through the plasma membrane into the lysosomal body for degradation. Like the macroautophagy pathway, CMA activity exists in most tissue cells, but the maximum activity of CMA occurs in the stress state. As mentioned earlier, the initial response to nutrient deficiency is the activation of the autophagy pathway to provide an amino acid source for tissues to maintain normal physiological functions of the body. However, if nutrient deficiency persists, it is difficult to maintain cell metabolism merely by the degradation of intracellular components, and then CMA becomes a new source of amino acids needed for protein synthesis. When certain conditions (oxidative stress, exposure to toxins, etc.) cause protein damage, the selectivity of the CMA pathway can specifically remove the damaged protein without affecting the surrounding normal components.
The CMA action process is sophisticated and complex, which requires the coordination of multiple components. The specific functions and related mechanisms are described as follows (Fig. 18.3):

CMA pathway
(1) HSC70 and common molecular chaperones (including hip, hop, hsp40, hsp90, bag-1, etc.) identify the region containing KFERQ related peptide in the substrate protein and form a complex. (2) the complex binds to the subunits of LAMP-2A on the lysosomal membrane. (3) the substrate protein was folded before translocation through the lysosomal membrane, and then entered the lysosome via the transmembrane segment (transposon) of LAMP-2A. (4) lysosomal HSC70 (ly-HSC70) is necessary for the transmembrane transport of substrate proteins. (5) once the substrate protein enters the lysosomal cavity, it is rapidly degraded by enzymes. (6) HSC70 molecular chaperone complex is released from the lysosome. (7) HSC70 molecular chaperone complex binds to other CMA pathway substrate proteins.
(1) The KFERQ-like motif of substrate protein (KFERQ is a lysosomal targeted signal peptide). Most known CMA substrate proteins contain the KFERQ peptide sequence, and only protein substrates with this sequence can bind and interact with molecular chaperone specificity. Accurate information on this sequence has previously been reported. Specifically, the sequence is centered on glutamine (Tabone et al. 1994), and four amino acid compounds form four flanks, including an alkaline end (K, R), an acidic end (D, E), a large hydrophobic end (F, I, L, V), and a repeating basic or large hydrophobic end (K, R, F, I, L, V). Such sequences are present in 30% of cytoplasmic proteins, some of which may produce kFERQ-like sequences through posttranslational modifications such as deamidation of posttranslational proteins.
(2) Association of molecular chaperone complexes in the cytoplasm with lysosomal membranes. There are a variety of molecular chaperones in the cytoplasm, and HSP70 is taken as an example to illustrate the process of the molecular chaperone. Continuously expressed heat shock protein 70 (HSP70) is a molecular chaperone that plays an important role in substrate proteins entering the lysosome through the plasma membrane. Cytoplasmic HSP70 can recognize the KFERQ sequence of the substrate protein. Co-chaperones interact with HSP70 to form a molecular chaperone complex that binds to the substrate protein recognized by HSP70 and transports it to the lysosomal surface. In addition, multiple mechanisms are involved in the regulation of this process. Heat shock protein 40 (HSP40) can stimulate the ATPase activity of HSP70 and increase the binding and release of HSP70 to a substrate protein. Hip (Huntington-interacting protein) promotes protein and HSP70, HSP40 substrate assembly; Heat shock protein 90 (HSP90) can prevent the aggregation and/or refolding of unfolded proteins. Bcl-2-associated immortal gene-1 (bag-1) is composed of several subtypes that positively or negatively regulate HSP70 activity.
The molecular chaperone complex is also closely related to the substrate protein located in the cytoplasmic surface of the lysosomal membrane and the LAMP-2A on the lysosomal membrane. Substrate proteins need to be folded and unfolded before they can be translocated into the lysosome, a process that requires the assistance of most molecular chaperones. Therefore, HSP70, HSP40, hip, and hop antibodies can prevent substrate proteins from entering the lysosome to varying degrees.
(3) HSP70 in the lysosomal cavity. The HSP70 in the lysosomes is called Ly-HSP70. Ly-HSP70 is not associated with other molecular chaperones, and most of them are acidic HSP70 subtypes. It is possible that all of the HSP70 subtypes and the substrate protein and the molecular chaperone complex can enter the lysosomes through a variety of routes, such as by autophagy, but only the acidic HSP70 subtype can be stably present in the lysosomes. The most reasonable explanation is that all HSP70 subtypes can enter lysosomes, but more basic HSP70 subtypes are transformed into acidic HSP70 subtypes by lysosome modification. An increase in Ly-HSP70 was found in the liver of chronically hungry rats when the CMA pathway was activated, and one of the characteristics of activated lysosomes was the presence of a large amount of Ly-HSP70 in the liver. Just as proteins enter ER and mitochondria require the involvement of Ly-HSP70s, Ly-HSP70 is necessary for CMA-mediated substrate proteins to enter lysosomes.
(4) LAMP-2A. LAMP-2A is a receptor on the lysosome membrane and plays an important role in substrate binding and translocation. In the CMA pathway, the substrate protein binding to the molecular chaperone complex binds to the LAMP-2A on the lysosome membrane, and the binding is saturated. The binding of one substrate protein to LAMP-2A is inhibited by the competitive inhibition of other substrate proteins, but there is no competitive binding site between one substrate protein and other non-substrate proteins. These characteristics of LAMP-2A directly affect the protein degradation efficiency of the CMA pathway. Binding to LAMP-2A is the rate-limiting step of CMA, so LAMP-2A can up-regulate or down-regulate the level of CMA on the lysosome membrane. During oxidative stress or T cell activation, the synthesis of LAMP-2A increased, but the degradation of LAMP-2A decreased under the condition of starvation. LAMP-2 A receptor has three different subtypes, namely, LAMP-2A, LAMP-2B, LAMP-2C. But only LAMP-2A can be used as the receptor of CMA, and the function of the other two subtypes is not clear.
The number of LAMP-2A on the lysosomal membrane was regulated by several different pathways. A complete LAMP-2A can form complex with liposomes in the lysosomal cavity. When the CMA pathway is activated, these molecules can be transposed and reinserted into the lysosomal membrane. By regulating the degradation rate of LAMP-2A, the cells could better facilitate the transport of substrate protein to lysozyme for degradation. The LAMP-2A subregion on the lysosomal membrane is responsible for its dynamic regulation. In a resting state, the LAMP-2A is periodically separated into lipid regions for degradation which is released by the enzyme A on the membrane and rapidly degraded in the lumen. The transport of LAMP-2A may be a regulatory mechanism of CMA. In addition, two different proteases are responsible for the degradation of LAMP-2A, one of which is lysosome cathepsin A. The cleavage of cathepsin A determines the stability of monomer LAMP-2A, and HSP 90 located on the membrane of cytoplasm and lysosome can help stabilize the structure of LAMP2A.
The CMA substrate binds to LAMP-2A with 12 short amino acid chains. Using specific antibodies to block this region, the amino acid residues were exchanged with the corresponding region in LAMP-2B or LAMP-2C to reduce the CMA level. Therefore, the short chain of amino acids in this region exists only in the tail of LAMP-2A. When the substrate binds LAMP-2A, it triggers its multistep conversion from monomer to polymer to form a 700 kDa CMA transposition complex. The transposition complex decomposes immediately after the substrate is transferred to the lysosome.
At present, through the research on a transgenic mouse model, the important role of CMA-related molecules has been established, the kinetic characteristics of CMA substrate degradation have been clarified, and the physiological role of CMA has been verified in vivo. It was found that the regulation of LAMP-2A on the lysosomal membrane made the CMA mechanism better understood. However, the number of known signaling pathways in CMA regulation is still incomplete. In species without LAMP-2A, whether microautophagy is an alternative to CMA or whether there are other CMA equivalent autophagy pathways in these species remains controversial. More and more studies have proved that there is a common autophagy-related protein between CMA and the macroautophagy pathway, and these problems provide new research directions for the autophagy mechanism.
2.2.2.4 Autophagy and the Quality Control System
Autophagy system is an important part of the intracellular quality control system, which is of great significance for maintaining the stability of the intracellular environment (Kubota 2009). The intracellular quality control system also includes another important proteolytic system, ubiquitin-proteasome system, and molecular chaperone.
Changes in intracellular proteins are attributed to intracellular and extracellular influences (e.g., oxidative stress, ultraviolet radiation, exposure to toxic substances, etc.). In addition, the protein translated by the mutant gene is prone to aggregation due to incorrect folding. These abnormal proteins tend to form complex structures such as oligomers, aggregates, and fibrous structures. These form complex structures that can be identified by molecular chaperones. Molecular chaperones can prevent the aggregation of abnormal proteins by facilitating their refolding into proteins with normal structures. In some cases, the intracellular abnormally folded proteins exceed the repair capacity of the molecular chaperone, which will choose to transport these abnormal proteins directly to the proteolytic system (ubiquitin-proteasome system and autophagy system) for degradation and clearance. The imbalance of various clearance systems in the quantity and quality control system of intracellular abnormal proteins will lead to the accumulation and aggregation of intracellular abnormal products. Although the exact mechanism by which abnormal proteins cause cytotoxicity remains controversial, it has been demonstrated that the greatest pattern of cytotoxicity is in complex tissue structures, such as oligomers or fibrous structures. The quality control system is a strong defense against the cytotoxicity of misfolded protein aggregates. If the system fails to function properly, cells are more likely to form protein aggregates rather than oligomers or fibrous structures. Protein aggregates interfere with the normal transport of cell contents and occupy space within the cell and can form an absorption pool. Proteins with normal functions often fall into the absorption pool and affect their normal function.
UPS and autophagy are two major evolutionarily conserved degradation and circulatory systems in eukaryotes. Early studies have shown that their work is not interdependent, but recent studies have shown that there are connections and overlaps between the two systems. Mitochondrial autophagy is an example of two interconnected systems. Functional studies have shown that damage to one of the UPS or autophagy degradation systems leads to compensatory up-regulation in the other system. In order to maintain homeostasis, cellular material accumulated after one degradation system is inhibited needs to be cleared by another system, thus forming a compensatory mechanism. However, this compensation is not always effective and largely depends on the cell type, the intracellular environment, and the load of the target protein. Inhibition of the UPS pathway or gene regulation by different compounds will lead to up-regulation of autophagy. For example, proteasome inhibitors and chemotherapy drugs can lead to increased expression of autophagy-related genes ATG5 and ATG7 and induce autophagy. The up-regulation of autophagy-related gene expression was caused by the activation of ER stress-related pathways and AMPK activation (Kouroku et al. 2007). Similarly, decreased autophagy levels were associated with UPS activation. In colon cancer tumor cells, chemical inhibition of autophagy and ATG knockout will lead to increased levels of proteasome subunits, which in turn activate UPS. Since proteasome is the target of autophagy degradation, the enhancement of proteasome activity after autophagy inhibition may be related to the continuous accumulation of proteasome. In some cases, however, autophagy inhibition is associated with the accumulation of ubiquitinated proteins. For example, accumulation of ubiquitination conjugates was observed in the brains and livers of ATG5 or ATG7 knockout mice. Ubiquitination is considered to be a link between the substrate and the appropriate degradation system or even the UPS system and the autophagy system.
So how do proteasome and autophagy systems cooperate in protein degradation? Firstly, most soluble abnormally folded proteins are preferentially degraded by the proteasome, and only when the proteasome system is overloaded can autophagy pathways be initiated to compensate. For example, after inhibiting the activity of protease, the autophagy pathway is activated and the intracellular ubiquitinated abnormally folded protein is degraded and cleared by the autophagy pathway. Second, whether the autophagy pathway is activated may depend on the stress level of the ER. When the stress level of ER reaches a certain level, the autophagy degradation pathway is activated. Third, the composition of misfolded proteins themselves is also critical. For example, misfolded proteins in the form of soluble monomers can be degraded by proteases, while insoluble misfolded proteins in the form of polymers cannot be degraded by proteases. In addition, misfolded proteins in the form of polymers are harmful to proteases. At present, it has been found that the proteasome function is significantly weakened in the presence of a large number of aggregates. Some misfolded proteins exist in both soluble and insoluble forms, such as α1-antitrypsin Z mutant, whose soluble form can be degraded by proteasome and autophagy system, while the insoluble form can only be degraded by autophagy.
In cells, proteolytic systems are usually rapidly adapted to changes in the concentration of abnormal constituents. However, the accumulation of protein aggregates occurs when a large amount of protein damage occurs in a short time and exceeds the degradation capacity of the proteolytic system. In most cases, cells will eventually overcome this problem. Accumulation of toxic protein products or protein aggregates can have different consequences depending on the extent to which the cell is affected. In cells that are dividing rapidly such as skin fibroblasts, aggregates are distributed to mother and daughter cells to dilute the aggregates. However, in some anaphase tissue cells (those that do not divide) such as brain neurons, the persistence of cytotoxic protein products results in cell death. We all know that neurons don’t regenerate, so the constant loss of neurons eventually leads to disease. This explains why the central nervous system is often affected by a high concentration of proteins with harmful conformational abnormalities in cells. Proteasome system and autophagy system exist at all levels of protein degradation. In addition to ubiquitination, some proteins and signaling pathways are involved in the communication and mutual regulation of these two systems. Discussing the interconnection between the two systems is helpful to understand the biological significance and clinical value of protein quality control from the perspective of basic medicine.
3 Autophagy, Proteins Misfolding, and Diseases
3.1 Factors Affecting the Function of Autophagy and Related Diseases Caused by Its Abnormalities
3.1.1 Dynein Abnormality-Associated Diseases
Signal transmission within and between cells depends on efficient internal transport systems. Neurons accurately convert external stimulus into various reactions between cells through the movement of internal substances. In numbers of motor neurons, long-distance transportation of substances depends on microtubules, dynein, and kinesin. The dynein pushes the substance centripetally to the negative end of the microtubule, and transports the substance to the central part of the nucleus. While the kinesin moves centrifugally to the positive end of the microtubule, transporting the substance into the cytoplasm.
Nocodazole is a kind of antitumor drug and also an antiparasitic. It can depolymerized microtubules and inhibit the fusion of autophagosome and lysosome. Thus, the flow of autophagosome is also associated with microtubules. The disintegration of microtubules results in decreasing ability of autophagy in clearing the substrate. Under these circumstances, the maturity of autophagosome and the fusion of autophagolysosome are affected, so the autophagosomes cannot pass through the microtubules to the microtubule organizing center (MTOC). Similar results are obtained by attenuating the effects of dynein, suggesting that dynein is a key protein that transports intracellular substances along the microtubules to lysosomes. In addition, HDAC6, a histone deacetylase-like protein is associated with microtubules and can regulate autophagy. However, it is not clear that how dynein and HDAC6 participate in the transportation of autophagosomes on microtubules.
At present, it was found that mutations of certain genes affecting microtubule transportation can cause motor neuron disease (MND) in the mouse model and humans (Cipolat Mis et al. 2016). MND refers to a group of sporadic, familial diseases characterized by motor neuron degeneration. In transgenic mice, material transportation through the axon is affected by overexpression or deletion of dynein, leading to progressive degeneration of motor neuron. Pathological changes and symptom progression observed in MND mouse model were similar to those of MND patients. In MND model, protein aggregates accumulate in neurons, suggesting that autophagic dysfunction is one of the pathogenesis of these diseases. Dynamin-dependent microtubule transportation dysfunction is one of the causes of this kind of diseases. The partial reason for protein aggregates accumulating in motor neurons is the disruption of the links such as microtubule-dependent autophagosome dyskinesia, autophagosome maturation, and autophagosome-lysosomal fusion dysfunction. The importance of autophagy decline in the development and progression of MND remains to be elucidated.
3.1.2 The Role of ESCRT and Related Diseases (Endosomal Sorting Complexes Required for Transport, ESCRT)
Most of the recycling of intramembrane proteins of cell membranes is done by various functions of endocytic pathways, including some receptor proteins, such as the epidermal growth factor receptor (EGFR) protein. Some proteins, called endosomes recycles proteins, perform a simple classification of these membrane proteins and return them to the membrane. More complex degradation systems rely on the multivesicular body (MVB), which is a luminal vesicle formed by the inclusion of body membrane and invaginated to produce a specific porous structure. After MVB is fused with lysosomes, vesicles are released into the acidic lysosomal cavity, where the hydrolytic enzymes degrade the vesicles and the substances they carry with. The classification of membrane proteins and their entry into MVB depend on the ubiquitination of proteins. This specific process can interact specific proteins with lumen like vesicles of MVB. Four kinds of ESCRTs ensure the high fidelity of protein classification to the endosome and fusion with the lysosome. The specific interaction of the ESCRT complexes is necessary for the formation of MVB and the normal fusion of endosome with lysosomal. Each complex performs its specific function through multiple interactions with proteins, cell membranes, endosomes, and other complexes. ESCRT plays an important role in the formation of autophagosomes and the fusion with lysosomes, which may be related to the fact that ESCRT can polymerize SNAREs and Rab7. Soluble N-ethylmaleimide-sensitive fusion attachment protein receptors(SNAREs) in autophagy body play an important role in the process of formation and fusion. In mammalian cells, vesicle-associated membrane protein7 (VAMP7), syntaxin-7 (syntaxin-7), syntaxin-8 (syntaxin-8), VTI1B (vesicle transport through interaction with t-snares 1B) promote the mutual fusion of Atg16L protein on vesicle precursor membrane. And they form a tubular network, and eventually, these networks fuse to form an autophagosome. Vamp3-mediated fusion of endosomes in mature autophagosomes to form Amphisomes has also been observed in some special cells. Inhibition of these SNAREs can lead to a reduction of the volume of autophagy precursors and delayed maturation of autophagosome. In yeast, the functions of Sec9p and Sso2p in SNAREs are similar to those of VAMP7 in mammals, which can promote the fusion of Atg9 proteins in autophagy precursor membrane to form tubular reticular structure and then fuse with each other to form autophagosome. In Yeast, the fusion of autophagosomes and lysosomes requires the Vam3 (vacuolar syntaxin homolog), Vam7, GTP binding protein transport 7 in Rab family, and in mammalian cells, it needs the mediation of VAMP7, syntxin-7, and VTI1B. Studies have shown that when a large amount of cholesterol accumulates in the body, it leads to the immobilization of VAMP7 by abnormal organelles hinders the fusion of autophagic lysosomes, and prevents the entry of substances to the lysosomes by endocytosis. As mentioned above, the acidification of autophagosomes and the activation of hydrolases in lysosomes are important preconditions for autophagosomes to perform their functions and also signs of their maturation. Recent studies have found that syntaxin-5 in SNAREs is involved in this process.
Rab7 is an important GTPase in the Ras family, together with SNAREs playing a role in the process of endosome maturation and lysosomal fusion in autophagosomes. In the process of maturation, an endosome is transported from the cell edge to lysosome by microtubule system, which is called forward transportation. This direction is determined by dynein-dynactin, which is constantly consumed during transportation. ORP1L (oxysterol-binding protein-related protein 1L) and Rab7 together form the complex that positioning on the membrane of the endosome, when it moves forward in the microtube it will activate ORP-Rab7 complexes to connect with RILP (Rab7-interacting lysosomal protein). Then ORP interacts with the cholesterol contributed at the endosome membrane to constitute a relatively stable conformation that can make the Rab7-RILP complex concentration dynein, complementing the consumption. When endosome gets near to the late endocytic compartments forming near the edge of the cell membrane, ORP1L will induce the Rab7-RILP complex to become β-III contractile protein, which is necessary for the extracellular transportation of the endosome through microtubules. In the process of autophagosome-lysosomal fusion, Rab7 first binds to GTP under the induction of HOPS (homotypic fusion and protein sorting complex) and becomes into an activated form. Cis-SNAREs complex which is located at the membrane of autophagosome and lysosome that is about to be fuse with each other dissociated into monomer. Subsequently, under the mediation of rab7-GTP protein complex, monomers of SNAREs on the fusing membrane get close to each other to form a compact trans-SNAREs complex and start to fuse with each other. After Rab7 aggregation was inhibited by toxic carotene, it was found that autophagosome and lysosome could not fuse with each other, which proved the important role of Rab7 in this process.
The classification of the early substances transportation is mainly finished by the ESCRT-0, -I, -II interacting with ubiquitin substances. ESCRT complex decomposition and the involvement of DE-ubiquitinating enzymes (DUBs) make the ubiquitin dissociate from the substrate before the substrate degradation. The process requires ESCRT-III. Although specific ESCRT mutations are associated with neurodegenerative diseases, the mechanisms of the disease are still not well understood. Now it has been found that the ESCRT-III who has the changed gene-splicing sites, its CHMP2B subunits point mutations is associated with frontotemporal dementia linked to chromosome 3(FTD3) at its familial autosomal dominant type.
The expression of deletion mutations and splicing mutations of CHMP2B aggravates the reduction of neurons and dendrites through non-apoptotic pathways, so some current studies aim at observing the effect of CHMP2B deletion mutations on autophagy. In cells and fly model, the expression of the deletion mutation increased raising of LC3-II level caused by autophagy accumulation, and reduce the formation of MVB. The experiment compared CHMP2B wild-type and mutant SKD1, the latter prevents the ESCRT-III complexes separate from the endosome. The experiment shows that proper dissociation of ESCRT-III complex is the key to autophagy body maturation and autophagy-lysosome. The deficiency of msnf7-2 which is the key component of ESCRT-III, or the mutation of CHMP2B protein may lead to the functional deficiency of ESCRT-III which will cause accumulation of ubiquitinating marker protein, and finally leads to FTD3 and amyotrophic lateral sclerosis (ALS). Studies on mature cortical neurons have found that the functional deficiency of ESCRT-III will lead to the accumulation of autophagosomes and block the fusion of autophagosomes and lysosomes leading to the clearance of aging proteins and organelles.
It was found that the autophagy function decreased to different degrees through the construction of the gene mutation expression of ESCRT complex or the reduction of its expression. Data from a variety of cell types showed that the functional deletion of various ESCRT genes would result in immature autophagosomes or failure in fusing with lysosomes. The accumulation of autophagosomes without the ability to degrade the substances leads to neurodegeneration. In addition, polyubiquitinated endosomes increased in these tissues. It is not clear whether the decline in autophagy function is caused by the destruction of autophagosome by the ESCRT complex or by the indirect destruction of the endosome and lysosome. In conclusion, current studies suggest that neural degeneration caused by decreased autophagy function with decreased ESCRT function is one of the important pathogenesis of FTD3 and ALS.
3.1.3 Lysosomal Storage Disorder (LSD)
Lysosomes are the ubiquitous organelles in cells surrounded by monolayer membrane, which contain a variety of acid hydrolytic enzymes which have the functions of dissolution or digestion. Normal functional lysosomes are involved in the circulation of cellular components, cholesterol homeostasis, down-regulation of surface receptors, inactivation of pathogenic microorganisms, repair of the cytoplasmic membrane, and cell bone remodeling. Lysosomes can be divided into two types: primary lysosomes are vesicular structures formed by bulking at the edges of the Golgi capsule, actually they are secretory vesicles containing a variety of hydrolytic enzymes. These enzymes are synthesized on the ribosome of the rough endoplasmic reticulum and transferred to the Golgi capsule. The enzymes in the primary lysozyme do not digest. Secondary lysosomes are the fusion products of phagocytic vesicles and primary lysosomes. In the secondary lysosomes, the remaining substances in phagocytic vesicles are excreted after digestion.
The fusion of autophagosomes and lysosomes is necessary for the degradation of substances in autophagy, so it plays an important role in the process of autophagy. Without this fusion, autophagy accumulates and carries a large amount of undegraded material, which is toxic to cells. Currently, LSDs is considered to be a group of nearly 60 different genetic diseases, each with a single-gene defect that causes the lysosomal system malfunction and fails to degrade specific substances within the cell. As a result, many tissue and organ systems are affected, including the brain, internal organs, bones, and cartilage, with early-onset central nervous system (CNS) dysfunction predominating. Although the clinical characteristics of these diseases vary from each other, most of the survival keep within 20 years from onset. Progressive phenotypic development is one of the hallmarks of LSDs.
Generally speaking, LSD is a single-gene disease caused by complete or partial dysfunction of lysosomal proteins (mainly lysosomal hydrolases) due to gene mutation. It is currently estimated that lysosomes contain 50–60 hydrolases that are active in acid environments in lysosomes. Although LSD varies greatly in pathology, all of these diseases result in the accumulation of undegraded substances in and out of the lysosomal cavity at the molecular level. This accumulation affects the normal functioning of a large number of intracellular signaling pathways. Recent biochemical and cell biological studies on LSDs have shown that LSDs have abnormalities in a variety of cell functions. These defects include signal pathway defects, calcium homeostasis imbalances, lipid biosynthesis and degradation defects, and intracellular transportation disorders. Because lysosomes and autophagosomes fuse and digest their contents, they play a fundamental role in the autophagy pathway, which is called the high integrity of lysosomes and autophagosomes. The researchers believe that the lysosomal accumulation in LSDs will have an impact on autophagy.
Mucopolysaccharidoses (MPSs) is a kind of disorder for glycosaminoglycans (GAGs) degradation caused by the inactivation of hydrolytic enzyme mutation in a set of lysosomes. As long as one of the mucopolysaccharides hydrolytic enzyme is inactivated, mucopolysaccharides will gradually accumulate leading to the occurrence of tissue and organ dysfunction. Deficiency of 11 enzymes is known to be the cause of 7 different MPSs.
Multiple sulfatase deficiency (MSD) is a typical disease of LSD, and the patient has a complex multisystem phenotype due to the impairment of all sulfate esterase activities. It is caused by a gene mutation which code the FGE (formylglycine-generating enzyme) of SUMF1 (sulfatase modifying factor 1). The posttranslational modification of sulfate esterase requires the participation of FGE. Without this modification, the sulfatase activity is reduced and the lysosomal substrate cannot be degraded. Autophagy injury is believed to play an important role in the pathogenesis of this disease.
MSD mouse chondrocytes have severe lysosomal storage defect, which presents as abnormal autophagy activity, and eventually leads to energetic metabolism imbalance and cell death. The number of positive cells in the co-localized staining of LAMP1 and LC3 of the autophagosome marker in MSD mice decreased, suggesting that the fusion of lysosomal and autophagosome may be impaired. In MSD mouse body, we also found that not only the substrate of lysosome and degradable lysosome increased, autophagosome also appears in a large number of accumulation. It can be proved by western blot and immunofluorescence technology which found LC3-II level increasing obviously. The MSD mouse SUMF1 mutation inhibited the degradation of lysosomal substrates and the fusion of lysosomal and autophagosome. Accumulation of ubiquitination aggregates was also found in brain tissues. A large amount of lysosomes in the cell will lead to the inhibition of autophagy, thus affecting two important pathways: (1) protein recycling is inhibited, and toxic protein polymers accumulate in the cell in large quantities, inducing inflammatory reactions, damaging mitochondria, and forming a vicious cycle. (2) the regeneration and circulation function of mitochondria decreases and damaged mitochondria accumulate continuously, which will induce apoptosis.
Two kinds of LSD mouse models which are related to severe nerve degeneration are often used in the study: MSD and mucopolysaccharidosis type IIIA (MPS IIIA) model. The generality of these two models includes: (1) autophagy body increase in number, (2) the clearance of autophagy substrate of endogenous and exogenous decreases, (3) organelles circulatory disturbance.
The mechanism of weakened autophagosome-lysosomal fusion in LSD remains to be elucidated. The possible mechanisms are (1) microtubule-based transportation system dysfunction. (2) lipid composition of the lysosomal membrane was changed. Some lipid components (mainly cholesterol and glycosphingolipids) have been found to accumulate in various LSD diseases. The lipid components are an important part of the cell membrane where is rich in lipid, called lipid rafts. Lipid rafts played an important role in the physiological function of the cell membrane by determining the cell membrane elasticity. The accumulation of lipid raft in LSDs may affect the dynamics of the lysosomal membrane and its fusion with autophagosomes. It has been suggested that the abnormal distribution of cholesterol in the lysosomal membrane and the rupture of lipid rafts in the plasma membrane may be the cause of dysfunction of lysosomal and autophagosome in MSD.
Sphingolipidosis is a heterogeneous inherited sphingolipid metabolic disorders. Children are the main population of s sphingolipidosis. The accumulation of sphingomyelin, glycolipids, glycosides, gangliosides, unesterified cholesterol, sulfides, and other compounds may be caused by abnormal hydrolytic enzyme function or secondary to the accumulation of other lipids. The increase in sphingomyelin will change the level of autophagy. Studies have shown that sphingomyelin addition to cell culture medium can induce autophagy and reduce the clearance efficiency of autophagosomes. It can be observed in multiple models. Therefore, the accumulation of sphingomyelin can change the function of the autophagy pathway, leading to the occurrence of such diseases.
Niemann-pick type C disease (NPC) is an autosomal recessive inherited fatal neurovisceral lipid deposition disease, which is mainly manifested as liver damage, progressive neurological dysfunction, and mental symptoms. Most of them are caused by the mutation of the NPC1 gene on chromosome 18, while a few are caused by the mutation of the NPC2 gene on chromosome 14. The protein products of NPC1 and NPC2 genes are associated with the outflow of cholesterol in the late lysozyme. Researchers found that the fibroblasts of patients with NPC and the brain of NPC mouse can find a large accumulation of autophagosomes. It was regarded that the combination of class III phosphatidylinositol kinase and BECN1 induce the occurrence of autophagy and maturity of the autophagosome. They put forward the deficiency of NPC1 caused the autophagy induction and autophagy pathway is unusual, autophagy substrate degradation dysfunction, neurons, and glial cells eventually develop a large amount of lipid, cause-related symptoms.
Gaucher disease (GD) is a chromosomal recessive inherited disease of glucose and lipid metabolic abnormality. The deficiency of glucocerebrosidase causes the accumulation of glucocerebrosidase in mononuclear macrophages of the liver, spleen, bone and central nervous system, causing related symptoms. Like NPCs, autophagy induction, autophagosome formation, and autophagosome substrate accumulation can be seen in neurons and glial cells in GD model, while lysosomal degradation ability in gaucher’s cells is insufficient.
Neuronal ceroid lipofuscinoses (NCLs) is the most common neurodegenerative disease in children due to the deficiency of fatty acid peroxidase. Severe pigmentation of the retina, waxy and lipofuscin deposits in the skin, internal organs, and nerve cells, mainly manifesting as blindness, epilepsy, progressive cognitive impairment, and loss of motor function. NCLs are genetically and phenotypically heterogeneous. Autophagosomes were significantly increased in both NCLs animal models and patient cells. Mutations in the CLN3 gene cause juvenile-onset NCL (JNCL). Mitochondrial dysfunction, down-regulation of the MTOR pathway and formation of autophagosome can be seen in JNCL mouse model neurons.
Autophagy-related molecules play different roles in the pathogenesis of different types of LSDs. In most cases, the autophagy pathway is blocked, leading to the accumulation of autophagy substrate and the dysfunction of mitochondria, resulting in an increase of autophagosomes. At the same time, synthesis of related molecules inducing autophagy increases to compensate for the blocked autophagy pathway. Therefore, LSDs is mainly manifested as autophagy disorder.
3.2 Neurodegenerative Diseases Caused by Autophagy and Misfolded Proteins
In the research of special neurodegenerative diseases, it has been found that autophagy dysfunction is closely related to neurodegenerative changes (Choi et al. 2013). Although the pathogenesis of each neurodegenerative disease is complex, they all share some common characteristics, which are related to the passway that neurons process abnormal intracellular proteins. Recent studies have shown some common pathogenesis of this disease. Abnormal proteins that still maintain their own solubility can undergo normal, targeted degradation via the ubiquitin-proteasome system (Daw et al. 2015) or the CMA pathway. These degradation pathways are relatively selective in that they remove abnormal proteins without affecting the surrounding intracellular components. However, as mentioned before, the specific pathway by which an abnormal protein is degraded is not clear, which may be related to the effectiveness of the proteolytic system at a specific time or the characteristics of the substrate protein. For example, certain proteolytic enzymes of the lysosome may be more effective at degrading certain proteins; Only unfolded monomer proteins can be degraded through both the UPS pathway and the autophagy pathway, while proteins that have formed oligomers, fibrils or fibrous structures can only be processed by batch degradation pathways, such as microautophagy or macroautophagy. Now it is thought that macroautophagy can remove the aggregation protein. Although the mechanism that determines macroautophagy activity under these conditions is unclear, it seems that abnormal proteins that cannot be degraded by other proteolytic systems can only be degraded by macroautophagy. In cultured cells, oligomers and specific protein fibers block the activity of the ubiquitin-proteasome system and CMA, and the blocking of these two pathways increases the activity of the macroautophagy pathway (Martinez-Vicente and Cuervo 2007).
In normal individuals or in the early stages of the disease, most soluble abnormal proteins are typically degraded in a targeted manner by either the ubiquitin-proteasome system or the CMA. However, some abnormal proteins sometimes have toxic effects on these two proteolytic pathways and partially inhibit their activity (red arrow). In addition, once these toxic proteins form complexes (oligomers or cellulose), they cannot be degraded through the UPS or CMA pathway. At the compensatory phase, the macroautophagy pathway is activated (green arrow) to compensatorily scavenge toxic proteins. In the late stage, the UPS and CMA pathways are further blocked, the macrophage activity decreases and the toxic proteins accumulate in the cells and the aggregates leak out from the autophagy vesicles. Then the cells begin to show dysfunction or even death (the red circle represents the non-degradable autophagy vesicles). Although the exact cause of proteolytic system failure is still not clear, however, factors such as oxidative stress and senescence promote cells to enter the late stages (Martinez-Vicente and Cuervo 2007).
Studies have shown that macroautophagy consistently clears intracellular abnormal proteins. Thus, the absence of macroautophagy in neurons will lead to accumulation of agrin and neurodegeneration (Metcalf et al. 2012). At the compensatory phase, the appropriate activity of the macroautophagy pathway is essential to maintain cell survival. In fact, if the activity of the macroautophagy pathway is enhanced by drugs at this stage, the accumulation of intracellular aggregates and the occurrence of symptoms will be significantly delayed. Unfortunately, the normal course of these diseases inevitably leads to depletion after the compensatory phase. The ubiquitin-proteasome system and CMA activity are further reduced and the macroautophagy pathway begins to lose activity. With the inactivation of these systems, abnormal proteins and normal intracellular components begin to accumulate, leading to progressive cell damage and death.
For these reasons, studies of clinical treatment have focused on how to prevent primary blocking of the UPS and CMA pathways or remove abnormal proteins in cells by enhancing autophagy activity, thereby prolonging the asymptomatic compensatory period. Following we outline several major neurodegenerative diseases (Parkinson’s disease, Alzheimer’s disease, polyglutamine disease, etc.) to help people understand the role of autophagy in neurodegenerative changes and the characteristics of these diseases (Rubinsztein et al. 2005).
3.2.1 Parkinson Disease
Parkinson’s disease is characterized by progressive diffused loss of the substantia nigra and striatum dopaminergic neurons. A pathological hallmark of the disease is the inclusion bodies and Lewy bodies in the cells, mainly containing aggregated α-synuclein proteins. Oxidative stress (caused by mitochondrial dysfunction) and damage to the proteolytic system may be responsible for the accumulation of these abnormal proteins and other aggregation-prone proteins, but the reason of the loss of selectivity of dopaminergic neurons and the accumulation of α-synuclein protein still not clear (Sheehan and Yue 2019). There is also evidence that neurons with more inclusion bodies can survive. In contrast, large pyramidal neurons and Purkinje cells appear to be vulnerable to be attacked by abnormal proteins and die before large inclusion bodies formed. Therefore, the formation of inclusion bodies in neurons is harmful or beneficial to cells still has much controversy.
The α-synuclein protein is known to be a major component of Lewy bodies, but studies have found that less than 2% of patients (familial Parkinson’s disease) develop mutations in this protein. The increase in the concentration of intracellular non-mutated α-synuclein protein (such as the triploid of α-synuclein) also causes Parkinson’s disease, which confirms that α-synuclein protein plays a key role in the pathogenesis of Parkinson’s disease. Posttranslational modification of the non-mutated α-synuclein protein also promotes the formation of oligomers and fiber intermediates, which usually develop into insoluble α-synuclein fibers, which are the major constituents of Lewy bodies. In the neuron, dopamine reacts with α-synuclein to induce posttranslational modification of α-synuclein, which inhibits fibrosis of α-synuclein protein, resulting in accumulation of α-synuclein protein in cells in a soluble toxic form, thus ultimately causes damage and death of neurons. This may explain why dopaminergic neurons are sensitive to neurodegenerative changes in Parkinson’s disease.
Some symptoms of Parkinson’s disease may result from loss of function due to protein aggregation, and other symptoms may be the result of a direct action of toxic proteins. Although the physiological function of α-synuclein is still unclear, it is generally localized at the presynaptic end of the neuron and has a certain correlation with the vesicle structure in the cell. This is consistent with the previously proposed theory that α-synuclein plays a certain role in the dopamine synaptic sac recycling. The mechanism of action of fibrin structure to produce cytotoxicity is still unclear, but they can bind to and promote the permeabilization of secretory vesicles (increased permeability), which may play an important role in the transmission of neurotransmitters and the maintenance of cell homeostasis.
Whether the α-synuclein protein is degraded by the ubiquitin-proteasome system or the autophagy pathway depends on its conformation and the conditions which the cells are located. Only soluble forms of the protein can be degraded by the proteasome pathway; proteins in the fibrotic form are usually trapped inside certain proteasomes and block the activity of the enzyme. These soluble proteins can also be degraded via the CMA pathway in the lysosome. Pathogenic α-synuclein variants bind tightly to lysosomal membranes, but they are difficult to degrade via the CMA pathway due to their inability to translocate into lysosomes. More importantly, due to its high affinity with the CMA receptor, α-synuclein hinders the uptake and degradation of other substrate proteins by lysosomes, causing blockade of the CMA pathway. Inappropriate posttranslational modifications also affect the degradation of the α-synuclein protein through the CMA pathway. On the one hand, α-synuclein, which cannot be degraded, causes an increase of the concentration of the protein in the cytoplasm, which aggravates the oligomerization or aggregation reaction of the protein; on the other hand, the normal α-synuclein protein accumulates in the cell due to the inability to degrade through this pathway. The ubiquitin-proteasome system and CMA pathway inhibitors promote up-regulation of autophagy, thereby maintaining the function of cell-degrading and removing cytotoxicity, thereby controlling α-synuclein at normal levels. Of course, this process is costly for cells from highly selective protein degradation pathways (the ubiquitin-proteasome pathway and the CMA pathway) to the initiation of bulk nonselective protein degradation pathways. This makes the cells more sensitive to stressors and less resistant to stress states such as oxidative stress. We can imagine that as the disease progresses, autophagy will eventually fail to maintain the stability of the intracellular environment, and neurotoxicity becomes more apparent. Accumulation of abnormal proteins and oxidative stress (typically present in damaged neurons) can directly impair large autophagy, and as the substrate proteins from other degradation pathways increase, the large autophagy pathway may be overloaded and eventually being exhausted. In addition, as the age increases, the activity of the human protein hydrolyzing system will progressively decline, which inevitably leads to cell decline.
In general, Parkinson’s disease mainly manifests as Parkinson’s syndrome or tremor paralysis. All conditions are characterized by the absence of dopaminergic neurons; some of them also have characteristic Lewy bodies. In addition, some abnormal proteins other than α-synuclein were found in patients with Parkinson’s disease, which is helpful for people to further understand the pathogenesis of the disease. Recent studies on Parkinson’s disease (PD) have focused on two genes involved in the occult formation of autophagosomes, PINK1 (PTEN-induced putative kinase 1) and Parkin (Jiang and Mizushima 2014). Among them, PIK1 is associated with mitochondrial clearance, and Parkin is associated with autophagy. Parkin can re-aggregate and clear damaged mitochondria by autophagy, which requires the stable expression of PINK1 in mitochondria. Blockade of the above association can be seen in familial Parkinson’s disease (FPD). Recent studies have also reported that the direct association of PINK1 and Beclin-1 can promote the formation of autophagosomes. Other studies have shown that impaired mitochondrial function is associated with mutations in the PARK7(Parkinson autosomal recessive kinase 7). The loss of expression of this gene increases the oxidative stress of neurons and the sensitivity of cell death, while the expression of PINK1 and Parkin can avoid this phenomenon.
The most common genetic risk factor for PD is the glucocerebrosidase (GBA) mutation. Deletion of GBA results in the accumulation of its substrate glycosylceramide in the lysosome, resulting in lysosomal dysfunction which may lead to autophagic damage. In PD patients without GBA mutations, enzyme levels and activities were reduced in brain regions where a-synuclein protein levels were elevated at an early stage of the disease.
Early-onset Parkinson’s disease (EOPD) is a Parkinson’s disease with an onset age of less than 50 years due to a loss of P-type ATPase ATP13A2, a defective function of PARK9 mutation. ATP13A2 mutant cells have impaired lysosomal degradation ability, causing a-synuclein accumulation, which may lead to toxicity of ATP13A2 mutation. Mutation of ATP13A2 resulted in a decrease in the expression level of another PD-related gene, synaptotagmin 11 (SYT11), which further impaired lysosomal function and interrupt autophagosome degradation. LRRK2 is the most common mutein in late-type familial PD. Mutations in LRRK2 result in hyperactivation of proteases and are involved in the regulation of autophagy activity.
Synaptic vesicles (SVs) are sites where presynaptic membrane nerve endings store and release transmitters. In addition to the autophagy-lysosomal pathway, PD gene studies suggest that synaptic vesicle dysfunction is another neuro membrane transport pathway, and that two pathways may share some regulatory proteins, i.e., synaptic vesicle dysfunction is PD. Potential pathogenesis. Early studies suggest that autophagy and SV transport are two independent pathways, but recent studies suggest that if the SV cycle is abnormal, synapses may lose function over time, causing abnormal neuronal signaling, ultimately leading to neurodegenerative. Many PD-related proteins, including LRRK2, EndophilinA (EndoA), synaptojanin1 (synj1), dynamin, and auxilin, have a clear role in SV endocytosis, and these proteins also play a role in autophagy pathways, some PD-related genes. It also plays a role in regulating SV transport and autophagy, thus suggesting a wide relationship between SV transport and synaptic autophagy, and the mechanism of synaptic autophagy in PD patients may be impaired. Due to the complexity of autophagy regulation at the central nervous system. Further research is needed to address the specific mechanisms of SV transport and autophagy regulation, and to understand the interaction mechanism between SV transport and autophagy, which may help to reveal new therapeutic targets for PD.
3.2.2 Alzheimer’s Disease
The gradual loss of neurons in Alzheimer’s patients and progressive dementia are closely related to the tangles of fibers in neurons and the appearance of extra-neuronal senescence. The tangles are mainly formed by the aggregation of highly phosphorylated taurine (a microtubule-associated protein); the amyloid beta-peptide (Aβ) is a transmembrane protein hydrolysate that is a major component of senile plaques (Choi et al. 2013). Over 80% of Alzheimer’s disease is sporadic, and the rare genetic defects that have been found are mainly caused by mutations in some enzymes that produce Aβ. Among them, soluble amyloid has a high toxic effect on neurons, and the content of insoluble senile plaques is not significantly related to the severity of clinical symptoms.
The association between abnormalities in the ubiquitin-proteasome system and the onset of Alzheimer’s disease is currently uncertain. Although in vitro experimental evidence indicates that amyloid protein has a negative effect on the activity of the proteasome, the intracellular proteasome activity of this type of patient has not changed. In contrast, changes in lysosomal systems and their association with disease pathogenesis have been confirmed and widely accepted. Up-regulation of intracellular lysosomal system activity is an early cellular change in Alzheimer’s disease, which is already obvious before amyloid precipitation. The up-regulation of autophagy in the early stage of the disease is related to the proliferation of lysosomes and the growth of lysosomal enzymes. Up-regulation of autophagy can increase the successful clearance of aggregates and toxic products. As the disease progresses, the removal efficiency of the lysosomal system decreases progressively, resulting in insufficient clearance of harmful components in the cell. Among them, most of the components to be removed are still isolated in autophagic vesicles, and their structures are not significantly affected. Many autophagic vesicles cannot fuse with lysosomes, so they are unable to obtain the enzymes required for protein degradation; although some vesicles can fuse with lysosomes, their contents cannot be degraded by enzymes. The detailed reason is not clear. The result is that a large number of autophagic vesicles accumulate in the neurons. The presence of these autophagic vesicles may interfere with the normal intracellular transportation of the cells, affecting the normal function of the neurons, leading to damage or even death of the neurons. In addition, autophagic vesicles may begin to leak after a period of time in the cells, and the free proteases and undegraded toxic substances are toxic to cells. A new perspective has recently been proposed: in Alzheimer’s disease, persistent autophagic vesicles eventually transform into a source of Aβ because they contain transmembrane proteins and the enzymes which are needed to produce Aβ leading to amyloid accumulation. At the same time, the association between autophagy and apoptosis has been confirmed by more and more studies.
Aβ balance in the brain plays a key role in the pathogenesis of AD. Abnormal brain tissue Aβ deposition can lead to neuronal axoplasmic transportation disorders and trigger neuronal cell death. Different clearance mechanisms of Aβ play different roles in the progression of AD. Aβ is produced by two sequential cleavages of amyloid precursor protein (APP). Aβ and APP are degraded by autophagy, and up-regulation of autophagy levels will reduce Aβ production.
Studies have shown that mitochondrial function is impaired in brain tissue of AD patients, which promotes the production and deposition of Aβ. Inhibition of impaired mitochondrial clearance, while an increase in oxidative stress levels, leads to dysfunction of AD neurons. In order to remove damaged mitochondria, autophagosomes containing mitochondria must fuse with lysosomes to form autolysosomes, which in turn degrade mitochondria. Experiments have found that normal cell mitochondria damage has a phenomenon similar to PD neurons. Autophagy is one of the most characteristic downstream pathways regulated by mTOR and plays an important role in AD neurodegeneration. The accumulation of protein aggregation may be due to excessive activation of the PI3K/Akt/mTOR axis. Induction of autophagy can reduce Aβ accumulation and reduce cognitive decline in transgenic AD mice. As one of the autophagy markers, Beclin1 is an important marker of autophagy and the initiation of autophagosome formation. Decreased expression levels of Beclin1 can lead to Aβ deposition and neurodegeneration in the AD mouse model. In addition, the expression of Beclin1 in the olfactory cortex and hippocampus is also significantly reduced, which can accelerate the progression of AD. Recent studies have found that the expression of Beclin1 is significantly decreased in neurons of AD patients, and the decrease in Beclin1 expression impairs microglial phagocytosis and increases Aβ deposition and neurodegeneration. Inhibitors of mTOR pathway induce neuronal autophagy. For example, rapamycin, a selective inhibitor of TORC1, can reduce Aβ deposition and inhibit Tau protein phosphorylation in an AD mouse model.
On the other hand, in vitro experiments showed that the production of Aβ was reduced after the addition of autophagy inhibitors, Aβ transport and plaque formation after autophagy-related gene ATG5 knockdown were significantly reduced, and Aβ secretion returned to normal after induction of autophagy. It is suggested that Aβ is produced by the autophagy process.
Therefore, the role of autophagy in Aβ deposition is still controversial. Autophagy inducers may provide a new effective therapeutic strategy by degrading AD early Aβ deposition. However, activation of autophagy may increase the deposition of Aβ in AD and aggravate the condition.
3.2.3 Polyglutamine Diseases
Polyglutamine diseases are a general term for a group of diseases characterized by intracellular toxicity of agglomerated abnormal proteins containing abnormally extended glutamine ends. Huntington’s disease is the most characteristic of polyglutamine disease. It also contains a group of diseases such as spinal cord medullary muscle atrophy, spinocerebellar ataxia, and dentate globus pallidus atrophy (Martin et al. 2015).
Huntington’s disease is an autosomal dominant genetic disease caused by the abnormal amplification of the IT-15 gene in the 4p16.3 region of chromosome 4, the CAG trinucleotide repeat in the huntingtin gene. The CAG repeat in the Huntington gene is translated into the Huntington’s protein (HTT) N-terminal polyglutamine (polyQ) sequence extension. Amplified mutations in polyQ are generally considered to be functional manifestations of HD cytotoxicity.
The main features of the disease are neuronal loss and progressive damage, abnormal motor function, and abnormal Huntington’s inclusion body structure in neurons, which are manifested as dance-like symptoms, cognitive and mental disorders. At present, people have insufficient understanding of the physiological functions of Huntington’s protein, so it is not possible to correctly distinguish whether the disease symptoms are caused by the toxic effects of the mutant protein or the malfunction of Huntingtin itself. Huntingtin interacts with other intracellular proteins and participates in different physiological processes of the cell such as gene transcription, signal transduction, intracellular transport. The interaction of most proteins with Huntingtin occurs in the glutamine prolongation region of the n-terminal region; interacting proteins are often dragged into inclusion bodies, which exacerbates the loss of physiological function of the protein.
In Huntington’s disease, the amount of glutamine repeated is directly related to its toxicity. It was initially thought that aggregates would directly lead to cell death; however, there have been recent observations that protein aggregates may have protective functions on neurons. In fact, in a Huntington’s mouse model, Huntington’s inclusions are widely distributed in neurons, and the model does not exhibit any characteristic neurodegenerative changes, while in other mouse models, there is no Huntington inclusion. The neurons also die.
By detecting different proteasomal subunits in Huntington’s aggregates, it has been found that there is a correlation between Huntington’s disease and the abnormal ubiquitin-proteasome system. In cultured cells, filaments formed by the mutated polyQ fragment (formed before the appearance of inclusion bodies) inhibit the ubiquitin-proteasome system. Abnormalities in macroautophagy are also associated with Huntington’s disease. Autophagy is currently considered to play a dual role in toxicity and protection in HD. On the one hand, this pathway can degrade Huntington’s protein aggregates. The expression of autophagy-related genes was altered in HD patients, with increased expression of LAMP2, ULK2, and LC3A, and decreased expression of PINK1, FKBP1A, and EEF1A2. In HD, the recognition function of autophagosomes and the transport efficiency of substrates are decreased due to the failure of autophagy substrate-associated protein p62 to interact with mutant HTT. Mutant HTT can also interact with autophagy regulatory gene Rhes. And inactivate it, so that autophagy activity is inhibited. Mutant HTT clearance is impaired, causing HTT accumulation and neurotoxicity. In fact, blocking large autophagy by drugs or the like in an animal model of Huntington’s disease exacerbates the pathological changes of the disease. Some researchers believe that the activation of large autophagy may be the result of proteasome inhibitors, and some scholars have suggested that aggregates may isolate endogenous autophagy inhibitors (such as mTOR) to activate autophagy pathways, activation of large autophagy. Conducive to the clearance of intracellular toxic Huntington’s protein, thereby improving the symptoms of the Huntington’s mouse model. Because of this, it is envisaged that macroautophagy activators may be used for the treatment of these diseases in the future. Other scholars believe that mTOR function is inhibited in polyQ, causing autophagy to activate, leading to cell death and accelerating neurodegeneration.
As with other neurodegenerative diseases, the specific cause of the disease’s autophagy failure is unclear. It has now been found that isolation of autophagy-associated proteins in aggregates may result in a decrease in autophagy activity.
3.2.4 Amyotrophic Lateral Sclerosis (ALS)
Amyotrophic lateral sclerosis, also known as atrophic lateral sclerosis, is a progressive and fatal neurodegenerative disease. ALS is a kind of chronic progressive degenerative disease involving upper motor neurons (brain, brain stem, spinal cord neurons), which affects lower motor neuron (cranial nucleus, spinal anterior horn cells) and their dominating trunk, limbs with head muscles. Clinically, the combination of upper and lower motor neurons combined with impaired paralysis. Venkatachalam et al. found that abnormal accumulation of macromolecular substances in motor neurons is a direct result of autophagic dysfunction, which leads to the degradation of motor neurons. They found that autophagic dysfunction in motor neurons of ALS rats was accompanied by accumulation of large autophagic vesicles, which often caused significant impairment of motor function (Evans and Holzbaur 2019; Ramesh and Pandey 2017). Superoxide dismutase 1 (SOD1) is a key enzyme that inhibits intracellular toxic superoxide radicals. More than 100 different mutations in SOD1 are associated with the onset of ALS. Alterations in autophagy activity in the mouse ALS model of mutant SOD1 G93A can affect the degradation of motor neurons. When the experimenter gave lithium (lithiium) or rapamycin, some autophagic vacuoles were observed in the cells, and all autophagy markers were up-regulated, indicating that autophagy was enhanced, while damaged cells were observed significantly recovery occurred in pathological morphology. Excessive macroautophagy vesicles in diseased neurons were removed by enhanced autophagy, and large autophagic vesicles were replaced by newly formed small autophagic vesicles. In addition, in the G93A mice (control group) administered with saline, a large amount of damaged mitochondria appeared in the motor neurons; after the ALS mice were treated with the lithium salt, the damaged mitochondria in the motor neurons were cleared. In contrast, when autophagy blocker 3-MA was used to treat G93A mice and wild-type mouse primary motor neurons (which were derived from primary cultured cells of the embryonic abdominal spinal cord), we found that after the autophagy pathway was blocked, only primary motor neurons of G93A mice died. But under basal conditions, there was no significant difference in spontaneous cell death rates between cultured cells of G93A mice and wild-type mice. From the above studies, it can be inferred that autophagic damage leads to a significant increase in motor neuron death of the SOD1 G93A mutation. SOD1 is an antioxidant enzyme that protects neurons from oxygen-free radicals. After the mutation, the accumulated abnormal proteins cause apoptosis due to the continuous attack of oxygen-free radicals. The mutated protein transcribed from the mutated SOD1 is misfolded and aggregated, and the binding of the autophagy receptor p62 to the mutated SOD1 protein enhances the action of the SOD1 protein and LC3. The false aggregation of SOD1 protein will affect multiple early autophagy processes, such as inhibiting mTOR function and inducing autophagy. During the process of binding with lysosomes after autophagosome formation, SOD1 protein impairs the retrograde transport of axons and causes the failure in fusion of autophagosomes and lysosomes, which plays a toxic role.
In the ALS-G93A mutant, the beneficial effects of autophagy were further confirmed by Kabuta et al. They found that autophagy cleared the cells’ toxic substances, including the removal of the mutated SOD1. Similar results were obtained in in vivo experiments by Fornai et al.: Mutant SOD1 aggregates in spinal anterior horn neurons were cleared by induced autophagy. Recent experiments have found that although most familial ALS and sporadic ALS do not have SOD1 mutations, abnormal SOD1 occurs in cells, and administration of SOD1 antibodies can inhibit disease progression. Activated autophagy can accelerate the clearance of intracellular SOD1, so enhancing autophagy function by autophagy inducer can be used to treat familial ALS and sporadic ALS. α-synuclein mutations can accumulate in the spinal cord of ALS and cause motor neuron death, while lithium salts promote the clearance of α-synuclein. Similarly, in the SOD1 mouse model and other forms of ALS, autophagy also clears ubiquitinated aggregates. All of these proteins (SOD1, α-synuclein, ubiquitin, etc.) are characteristically present in the neurons of most familial ALS and sporadic ALS patients, and they are substrates for the autophagy pathway. Therefore, these proteins accumulate when autophagy is depleted, which is very common in familial ALS and sporadic ALS. Only riluzole is currently available for the treatment of this disease, but it only extends the average life expectancy by about 2–3 months. Recent studies have found that due to the more complex relationship between SOD1 and autophagy, SOD1-related autophagy seems to be different at the different stage of ALS progression. In a recent study, inhibition of autophagy activity resulted in the early onset of ALS symptoms in SOD1G93A mice after knockout of the autophagy gene ATG7, suggesting that autophagy has neuroprotective effects in the early stages of the disease. However, subsequent inhibition of autophagy prolonged the lifespan of mice, delayed the progression of the disease and showed that autophagy played a detrimental role in the late stages of the disease. This phase-dependent effect of autophagy was also confirmed in another study. Starvation-induced autophagy significantly reduced the accumulation of neurotoxic mSOD1 in the early pathological stage, but mSOD1 was significantly increased in SOD1G93A mice in the late stage of ALS.
TDP-43 is a regulatory factor of cellular RNA. Recent studies have found that TDP-43 gene mutation in ALS patients impairs its binding and stabilizes the function of autophagy-associated protein ATG7 mRNA, suggesting that the accumulation of TDP-43 in the cytoplasm will lead to autophagy damage, and then leads to the ubiquitinated protein and p62 accumulation. TDP-43 regulates autophagy by affecting the localization of TFEB and regulates DCTN1 levels to help autophagosome-autolysosomal fusion. In mice and drosophila, the deposition of TDP-43 is neurotoxic, and when autophagy is chemically inhibited, TDP-43 deposition is significantly reduced and neuronal survival is improved.
The exact role of autophagy regulation in the pathogenesis of ALS needs further study, but there is a unique common clue between ALS genes. Therefore, it is particularly important to study the interaction between ALS mutant genes or proteins and autophagy. These interactions will be able to bring greater benefits to patients.
3.3 Autophagy and Heart Disease Caused by Abnormal Folding Protein
Heart failure (HF), is often an end-stage change in a variety of heart diseases, including coronary heart disease, hypertension, and idiopathic cardiomyopathy. Mutations in the genes of cardiac muscle fibrin, cytoskeletal proteins, and other related proteins cause a variety of myocardial diseases. These mutations promote or cause abnormal folding of proteins, which in turn affect the normal function, interaction, localization, stability of other proteins. Among them, some gene mutations in cardiomyopathy also involve some molecular chaperone. For example, mutations in the desmin or its chaperone (a small heat shock-like protein αβ-crystalline) can cause desmin-related myopathy (DRM). DRMS is characterized by the accumulation of intracellular insoluble molecules and other interacting proteins, and cause muscle weakness and dilated cardiomyopathy. Protein misfolding can lead to dysplasia of the myocardium and amyloidosis.
The accumulation of incorrect conformational proteins in the intracellular and extracellular space is a common feature of neurodegenerative amyloidosis diseases, including Alzheimer’s disease, Huntington’s disease, and Parkinson’s disease. Recent studies suggest that these insoluble protein aggregates may not be the direct cause of disease, but the toxicity of pre-amyloid oligomer (PAO), an intermediate product of fibril formation, maybe the main cause of disease. Regardless of the gene sequence, these soluble PAOs share a common conformation-dependent protein structure and this structure can be detected by conformation-specific antibodies. Using staining PAOs in DRM and HF model mouse, it was found that there was PAO accumulation in cardiomyocytes of various cardiomyopathy, but not in normal cardiomyocytes. The presence of intracellular PAO in patients with HF suggests that there may be a common pathogenic mechanism between neurodegenerative amyloidosis and some cardiomyopathy that can cause advanced HF. Regrettably, the relationship between intracellular PAO production, protein accumulation, as well as cardiac pathological changes is still unclear.
So, can the high expression of PAO substances in cardiomyocytes cause heart failure? In order to confirm the relationship between the expression and the accumulation of PAO in cardiomyocytes and the development of cardiomyopathy, people use PQ83 (an ectopic peptide containing 83 glutamine repeating structure, similar to PAO) transgenic mice to study this relationship, taking PQ19 (a polypeptide containing 19 repeating inactivated starch polypeptide-like structure) transgenic mice as control to eliminate the effect of ectopic protein expression. If the expression and accumulation of PAO in cells produce toxic effects, the expression of PQ83 will lead to pathological changes in cardiomyocytes, potential cell loss, and successive HF. The results showed that low levels of expression of the peptides PQ83 can cause the deposition of PAOs and PQ83 aggregates, resulting in loss of cardiac cells and heart dilation in mice, and the mice eventually died of HF within 5–7 months. Further study of cardiomyocytes of dead mice revealed increased autophagosome activity and increased lysosomes number. In addition, cell necrosis was evident, but the initiation of apoptosis and the markers of cytoplasmic endoplasmic reticulum stress were not found. Cellular ultrastructure study showed an increase in the number of labeled lysosomes and autophagic vesicles filled with polyglutamate aggregates. However, in the development of PQ83-related cardiomyopathy, the good or bad effect of the increase of intracellular autophagosomes and lysosomal contents on the survival of cardiomyocytes are still unclear. Further research will explore the relationship between autophagy and PAO-induced cardiomyocyte cytotoxicity and death, as well as the specific stage of the disease.
What role does autophagy play in heart failure? Whether the up-regulation of autophagy is a protection or damage to the heart is a controversial topic. Autophagy has been observed in HF myocardium caused by dilated cardiomyopathy, heart valvular disease, and ischemic heart disease. In animal models, autophagy was also observed in death and sudden death of cardiomyocytes. However, it does not mean that autophagy is a sign of failure of myocardial cell repair or a way to remove damaged cardiomyocytes. We already know that autophagy is a key pathway to eliminate abnormal folding proteins and protein aggregates in cells. In some cases, autophagy is a form of cell death but it plays a vital role in cell survival in starvation. Autophagy is very important for the maintenance of the basic functions of cardiomyocytes, and abnormalities in the regulation of autophagy can cause lesions in many types of cells including cardiomyocytes. It has also been shown that the loss of autophagy or lysosomal function is detrimental to the heart in some cases. The production of β-adrenergic antibodies that cause cardiomyocyte death is associated with a decrease in autophagy, and the rate of death of cardiomyocytes due to toxic effects is decreased after autophagy is up-regulated. Activation of autophagy during acute stress, such as ischemia and myocardial perfusion, can play a role in acute cardio-protection. Cardiac hypertrophy is a precursor to HF and is also a hallmark of myocardial remodeling in most cases. Angiotensin II is an important mediator of cardiac remodeling and is involved in hypertrophic responses to stress and acute injury, and is involved in the regulation of cardiomyocytes through the interaction of angiotensin II type 1 (AT1) and type 2 (AT2) receptors. Of course, a significant up-regulation of autophagy or lysosomal function is also detrimental. In cardiomyocytes, direct evidence of autophagy overactivation associated with autologous cell death is unclear. Thus, for the maintenance of the normal function of cardiomyocytes, autophagy may require precise regulation to maintain cell homeostasis.
Some evidence suggests that blocking the formation of amyloidogenic proteins can eliminate their cytotoxic effects. These problems have not been fully confirmed in human diseases. Whether the aggregation of PAO observed in human HF specimens is due to abnormal folding of proteins, loss of function of degradation system, or damage to the transport pathway of protein degradation pathways, has not yet been clearly answered. Abnormal folding of proteins and intracellular accumulation of PAO are the causes of HF progression? In many tissues, cellular stress and aging can lead to aggregation of insoluble proteins and massive formation of abnormally folded proteins. Hundreds of proteins can undergo abnormal folding, amyloidosis, and then produce cytotoxicity. Cardiomyocytes are in a postmitotic state, which reduces the ability of cardiomyocytes to clear these aggregates. This feature is reflected in many cell types in neurodegenerative diseases. The effect of autophagy/lysosomal regulation on cardiomyocyte survival, the relationship between autophagy/lysosomal interaction and myocardial necrosis, the pathway by which PAO aggregation induces cell death, as well as the generation of PAO Commonality, etc., all of these issues need to be further clarified in future research.
At present, through the study of the proteasome pathway, it has been found that the proteasome plays an important role in the pathophysiological processes of various heart diseases. Cardiomyocyte proteases have at least 34 different subunits, and variations in these subunits alter the specificity and selectivity of proteases, so they play an important role in the regulation of degradation pathways in cardiac proteins. Currently, most studies focus on the effects of decreased proteasome function on the myocardial disease, and some studies have shown increased proteasome activity in certain heart diseases. Evidence for a decrease in proteasome function is mainly due to an increase in the concentration of ubiquitinated proteins leading to intracellular accumulation, and the other is the activity of the proteasome in vitro. Proteasome dysfunction exists to varying degrees in myocardial ischemia, heart failure, atherosclerosis, and other heart diseases.
Based on the ischemia-reperfusion injury rat heart model, researchers found the deletion of activity of the proteasome 20S and/or 26S in accordance with the increase of oxidation of the protein ubiquitin-proteasome after myocardial ischemia and reperfusion injury in myocardial ischemia. The mechanism of ischemia-induced proteasome inhibition is still unclear. Studies have found that some proteasome subunits are significantly inhibited or inactivated after oxidative stress. We know that the UPS pathway can degrade a large number of proteins including pre-apoptotic proteins and regulate multiple signaling pathways, so the decline in proteasome function in myocardial ischemia has an important effect on cardiac function. Similarly, a large amount of ubiquitinated protein was found in cardiomyocytes of heart failure, suggesting a decrease in UPS activity in heart failure cells. In the mouse model of pressure-overloaded heart, the level of ubiquitinated protein was increased and the activity of protease was decreased, indicating that the ability of proteasome to clear abnormal proteins in cardiomyocytes of HF was insufficient. In human dilated cardiomyopathy, the expression of the apoptosis regulator P53 was also found to be associated with dysregulation of the UPS system. Abnormal proteasome function in cardiac hypertrophic cells may cause accumulation of pre-apoptotic proteins leading to heart failure. In addition, it has recently been found that the UPS system also plays an important role in atherosclerosis. Atherosclerosis is thought to be induced by oxidative stress. In the high cholesterol-fed pig model, the proteasome inhibitor MLN-273 caused an increase in coronary oxidative stress and led to early atherosclerosis. UPS can also regulate insulin signaling pathways by altering the internalization of insulin receptors, controlling insulin receptor substrate levels, and insulin degradation. Therefore, changes in the UPS system can lead to insulin resistance and diabetic complications. In cardiac pathology, autophagy is a potentially promising therapeutic target. However, since there is no noninvasive imaging method for directly monitoring autophagy, there are difficulties in the experimental intervention targeting autophagy in the treatment of heart disease.
4 Aging and Autophagy
We know that some abnormal proteins exist since birth; However, for some patients with neurodegenerative disorders, pathological changes only occur in the later stages of life. For example, many symptoms of neurodegeneration appear only after 60 years old. Progressive deterioration of intracellular quality control systems with age is considered to be the main cause of these neurodegenerative pathological changes. The homeostasis function associated with the autophagy and the longevity protein metabolism, the removal of damaged organelles and cell debris is considered an anti-aging process.
The ubiquitination system and the decline in autophagy activity are common features of all older animals. The activity decline of the proteasome-degrading protein, the dysfunction of the assembling and disassembling proteasome, and the changes of enzyme activity that catalyzes the proteasome will lead to the whole system activity decline as the age increase. For the autophagy pathway, both large autophagy and CMA activity also decreased with age. For example, an important abnormality in the signaling mechanism of insulin that controls autophagy leads to diminished activity of the entire pathway during the stress. In addition, as aging progresses, the body’s ability to clear autophagosome also decreases. The accumulation of undigested products in the lysosome results in a decrease in the ability of the substance to degrade in the autophagy. With the aging of the body, whether the function change of important autophagy-related proteins, resulting in autophagy failure, is a focus of current research.
An important reason for the decrease in the activity of the CMA pathway with age is that the levels of CMA receptors in lysosomes are significantly reduced in older animal cells (Choi et al. 2013). The substrate proteins that are recognized by molecular chaperones in the cytosol cannot be combined with the lysosomal membrane in the absence of receptors, accumulating in the cytoplasm for a long time, eventually causing irreversible damage to the cells.
5 Treatment Strategy for Misfolded Protein Disease
Many of the neurodegenerative diseases mentioned above are associated with abnormal folding or aggregation of intracellular proteins. At present, there is no effective treatment for slowing or preventing neurodegenerative and muscular atrophy diseases. Due to the narrow scope of action of the proteasome system and the high specificity of the degradation process, most proteins with aggregation tendency (such as Huntingtin) are more dependent on the autophagy pathway for degradation. Certain chemical regulators significantly reduced the aggregation-prone proteins (e.g., mutant huntingtin or α-synuclein protein) removal efficiency after autophagy inhibition, which results in a large accumulation of the protein in the cell.
The induction of autophagy by rapamycin significantly enhances the clearance of proteins with aggregation tendency and also reduces the formation of protein aggregates. In addition, in the Drosophila HD model and the HD transgenic mouse model, it has been found that the symptoms of neurodegenerative diseases are improved after the induction of enhanced autophagy.
The same situation occurs in other abnormally folded protein disease models. For example, rapamycin increases the clearance of aggregation-prone proteins to reduce cytotoxicity, such as tau protein. Autophagy can eliminate aggregation-prone proteins suggesting that the development of drugs that act on autophagy pathways may improve the symptoms of such diseases and prevent disease. Importantly, rapamycin also plays a better role in the fly model of these diseases, which are autophagy-dependent, and rapamycin has little effect.
Unfortunately, long-term use of rapamycin causes complications such as poor wound healing and decreased immune function. mTOR can regulate a series of pathways (such as translation of some proteins, cell division, etc.), and the main side effects of this drug are caused by non-autophagy-dependent pathways. In addition, it is still unclear how mTOR regulates autophagy in mammals. An in-depth study of this problem will help provide a safer, more specific, long-lasting drug target for the clinic.
At present, there are few effective treatments for neurodegenerative proteinopathy. Disaccharide trehalose can induce autophagy with mTOR-dependent manner thereby to reduce the accumulation of intracellular abnormal proteins, but the specific mechanism of action remains unclear. Recently, it was discovered that another non-mTOR-dependent pathway induces autophagy: inhibition of inositol monophosphatase (IMPase) reducing the free inositol and myoinositol-1,4,5-triphosphate, (IP3) levels, and up-regulated autophagy activity. Other drugs are to treat a range of neuropsychiatric disorders by this route induce autophagy, such as lithium, valproate. Like Rapamycin, these drugs in insects HD model increase the clearance of the aggregation-prone proteins (such as mutant huntingtin) and protect the cells.
Recently, it has also been found that lithium also has a good performance in the treatment of ALS diseases. Compared to a single standard drugs riluzole, the combination of Lithium carbonate and riluzole can significantly delay the onset of disability and death in patients with ALS. Lithium carbonate treatment of G93A mutant ALS mice can significantly delay the progression of disease and death in mice. The autophagy activity of these mice is increased, and the accumulation of ubiquitin and α-synuclein is generally reduced.
Thus, induction of autophagy can protect a series of neurodegenerative lesions caused by aggregation-prone proteins (Scrivo et al. 2018). In addition, the combination of rapamycin with another non-mTOR-dependent drug may exert a more effective effect on neurodegenerative diseases through stronger autophagy induction and reduce some of the side effects of rapamycin used alone.
6 Summary and the Prospects
The defect of the cell quality control system is based on the pathogenesis of a series of diseases represented by common neurodegenerative diseases, and this view is strongly confirmed. Among them, some defects are caused by the direct toxic effects of abnormal proteins on the clearance system. In addition, factors such as aging and oxidative stress can aggravate damage to the quality control system and accelerate system failure. Future research should focus on the study of the mechanisms of action of abnormal protein damage clearance systems in order to develop therapeutic drugs that block their toxic effects. Activation of large autophagy is beneficial for the clearance of abnormal proteins in the cell, and this view has been confirmed by recent research, increasing the possibility of treating diseases caused by abnormal proteins such as neurodegenerative diseases, heart diseases, liver diseases, etc. It can also be said that the activation of autophagy can be regarded as a compensatory mechanism induced by the failure of other proteolytic systems in the cell. The biggest challenge now is how to find new ways to activate the autophagy pathway. In summary, a better understanding of the autophagy mechanism may help to discover the new targets for the diagnosis and treatment. Drug screening of autophagy agonists or antagonists, including upstream regulators of autophagy and downstream targets, may provide a more useful treatment for human disease.
References
Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S (2004) Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431:805–810
Choi AM, Ryter SW, Levine B (2013) Autophagy in human health and disease. N Engl J Med 368:651–662
Cipolat Mis MS, Brajkovic S, Frattini E, Di Fonzo A, Corti S (2016) Autophagy in motor neuron disease: key pathogenetic mechanisms and therapeutic targets. Mol Cell Neurosci 72:84–90
Daw NC, Chou AJ, Jaffe N, Rao BN, Billups CA, Rodriguez-Galindo C, Meyers PA, Huh WW (2015) Recurrent osteosarcoma with a single pulmonary metastasis: a multi-institutional review. Br J Cancer 112:278–282
Evans CS, Holzbaur ELF (2019) Autophagy and mitophagy in ALS. Neurobiol Dis 122:35–40
Finn PF, Mesires NT, Vine M, Dice JF (2005) Effects of small molecules on chaperone-mediated autophagy. Autophagy 1:141–145
Jiang P, Mizushima N (2014) Autophagy and human diseases. Cell Res 24:69–79
Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, Ogawa S, Kaufman RJ, Kominami E, Momoi T (2007) ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ 14:230–239
Kubota H (2009) Quality control against misfolded proteins in the cytosol: a network for cell survival. J Biochem 146:609–616
Martin DD, Ladha S, Ehrnhoefer DE, Hayden MR (2015) Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci 38:26–35
Martinez-Vicente M, Cuervo AM (2007) Autophagy and neurodegeneration: when the cleaning crew goes on strike. Lancet Neurol 6:352–361
Menzies FM, Moreau K, Rubinsztein DC (2011) Protein misfolding disorders and macroautophagy. Curr Opin Cell Biol 23:190–197
Metcalf DJ, Garcia-Arencibia M, Hochfeld WE, Rubinsztein DC (2012) Autophagy and misfolded proteins in neurodegeneration. Exp Neurol 238:22–28
Pandey UB, Nie Z, Batlevi Y, McCray BA, Ritson GP, Nedelsky NB, Schwartz SL, Diprospero NA, Knight MA, Schuldiner O, Padmanabhan R, Hild M, Berry DL, Garza D, Hubbert CC, Yao TP, Baehrecke EH, Taylor JP (2007) HDAC6 rescues neurodegeneration and provides an essential link between autophagy and the UPS. Nature 447:859–863
Ramesh N, Pandey UB (2017) Autophagy dysregulation in ALS: when protein aggregates get out of hand. Front Mol Neurosci 10:263
Rubinsztein DC, Difiglia M, Heintz N, Nixon RA, Qin ZH, Ravikumar B, Stefanis L, Tolkovsky A (2005) Autophagy and its possible roles in nervous system diseases, damage and repair. Autophagy 1:11–22
Scrivo A, Bourdenx M, Pampliega O, Cuervo AM (2018) Selective autophagy as a potential therapeutic target for neurodegenerative disorders. Lancet Neurol 17:802–815
Senft D, Ronai ZA (2015) UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem Sci 40:141–148
Sheehan P, Yue Z (2019) Deregulation of autophagy and vesicle trafficking in Parkinson’s disease. Neurosci Lett 697:59–65
Szeto J, Kaniuk NA, Canadien V, Nisman R, Mizushima N, Yoshimori T, Bazett-Jones DP, Brumell JH (2006) ALIS are stress-induced protein storage compartments for substrates of the proteasome and autophagy. Autophagy 2:189–199
Tabone MD, Kalifa C, Rodary C, Raquin M, Valteau-Couanet D, Lemerle J (1994) Osteosarcoma recurrences in pediatric patients previously treated with intensive chemotherapy. J Clin Oncol 12:2614–2620
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Science Press and Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Cheng, C., Liu, ZG. (2019). Autophagy and the Metabolism of Misfolding Protein. In: Qin, ZH. (eds) Autophagy: Biology and Diseases. Advances in Experimental Medicine and Biology, vol 1206. Springer, Singapore. https://doi.org/10.1007/978-981-15-0602-4_18
Download citation
DOI: https://doi.org/10.1007/978-981-15-0602-4_18
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-0601-7
Online ISBN: 978-981-15-0602-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)