
Abstract
Nutritional immunity refers to the ability of the host to sequester nutrients from pathogens during infection. Metal ions are important for microbial survival and pathogenesis as well as for host defenses. For example, while zinc (Zn2+) is crucial for microbial fitness within the host, immune cells deprive microbes of these metal ions and retain it for their own defense. However, excess Zn2+ may be toxic to both the host and the pathogen. Therefore, Zn2+ regulation is a central component of host-pathogen interactions and antimicrobial immunity. Metallothioneins (MTs) are a family of highly conserved cysteine-rich proteins that are ubiquitously expressed in most organisms. Immune cells express MTs in response to a variety of stimuli including cytokines, chemokines, and infectious agents. They regulate intracellular Zn2+ homeostasis, protect from oxidative stress, and modulate host immunity during infection. Although Zn2+ signals are well known to alter immunological processes, our knowledge of how the MTs-Zn2+ axis affects immune response to infections is relatively scarce. Emerging evidence points to a significant role for MTs in regulating host immunity. Thus, this chapter discusses immunomodulatory roles of MTs with a focus on Zn2+ regulation in response to pathogen attack.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
4.1 Introduction
Nutritional immunity is a mechanism of defense by which the host restricts nutrient access to pathogens to inhibit their growth. Metal ions are micronutrients that are essential for life. They regulate important functions of immune cells as well as that of invading microbes. One form of nutritional immunity is limitation of metal ions by modulating their availability, concentration, and distribution inside the cell. Zn2+ is an important metal ion whose concentration in circulation (plasma and serum) rapidly declines during infection (Besecker et al. 2011; Uttra et al. 2011). This phenomenon is postulated to deprive invading pathogens of Zn2+, control their survival, and prevent dissemination (Hennigar and McClung 2016). Zn2+ deficiency or excess alters the number and activities of immune cells thereby modulating host susceptibility to infection. A decrease in dietary Zn2+ uptake increases the risk of infectious diseases such as tuberculosis, shigellosis, pneumonia, measles, human immunodeficiency virus (HIV), acute cutaneous leishmaniosis, and malaria (Maywald et al. 2017). On the other hand, dietary Zn2+ supplementation aids in improving immune defenses against some of the microbes that cause the aforementioned infections as well as diarrhea, leprosy, chronic hepatitis C, and acute lower respiratory infection (Overbeck et al. 2008). However, there may be a very narrow window within which Zn2+ exerts beneficial effects. Recent data show that excess dietary Zn2+ uptake increases susceptibility to Clostridioides difficile and intensifies disease severity suggesting that exogenous Zn2+ administration may adversely impact the clearance of some pathogens (Zackular et al. 2016).
Effects of Zn2+ on immunity are complex and have been under investigation for many years. Zn2+ deprivation or supplementation in vitro and in vivo affects the expression of several genes in immune cells associated with Zn2+ homeostasis, cytokine response, stress responses, reactive oxygen species and reactive nitrogen species (ROS and RNS) signaling, metabolism, and survival (Beck et al. 2006; Cousins 1998; Cousins et al. 2003; Haase et al. 2007). Zn2+ deficiency inhibits differentiation, proliferation, and survival of monocytes, polymorphonuclear leukocytes (PMN), natural killer cells, and T and B cells (Bonaventura et al. 2015). However, an excess of Zn2+ may lead to toxicity. Therefore, Zn2+ homeostasis is tightly regulated in immune cells by Zn2+ binding proteins such as metallothioneins (MTs), glutathione, the Zn2+-responsive transcription factor metal-response element-binding transcription factor-1 (MTF-1), Zn2+-transporters, Zn2+-permeable ion channels such as transient receptor potential mucolipin 1 (TRPML1), and Zn2+ storage organelles such as zincosomes (Andrews 2001; Crawford et al. 2018; Eide 2004; Inoue et al. 2015; Liu et al. 2012; Palmiter 2004; Palmiter and Huang 2004; Vallee 1995). Taken together, regulation of Zn2+ is necessary for adequate immune function and aberrant homeostasis of these metal ions may have adverse effects on the host’s ability to defend microbial invaders.
Microorganisms require Zn2+ for survival. Thus, restriction of Zn2+ during infection may be an effective strategy that immune cells utilize to inhibit microbial growth. Several recent studies have brought to light the importance of Zn2+ limitation by MTs in immune cells. Zn2+ is bound to MTs through seven binding sites with picomolar binding affinity. This attribute facilitates controlled Zn2+ exchange between proteins and promotes nutritional immunity in host cells, where accessibility of Zn2+ to pathogens must be restricted. Our work revealed that MT1 and MT2 inhibit fungal growth via sequestration of Zn2+ in infected macrophages (Subramanian Vignesh et al. 2013). The MT3 isoform has a very distinct role: it facilitates Zn2+ uptake by intracellular fungi (Subramanian Vignesh et al. 2016). Nonetheless, the finding that MTs are an important component of the antimicrobial defense arsenal raises interesting questions about their mechanisms of action. In sum, MTs are expressed in immune cells, regulate inflammatory responses, and control host-pathogen interactions indicating that they may be at the forefront of immunological fitness in the host. Given the role of Zn2+ in pathogen virulence and the emerging importance of MTs in immune responses, this chapter focuses on the roles of Zn2+ and the MT-Zn2+ landscape in antimicrobial immunity.
4.2 Low Zinc Spells a High Infection Risk
Zn2+ has vital roles in biochemical processes and is crucial for maintaining the structure, stability, and adequate activity of macromolecules, such as proteins and nucleic acids. About 10% of the human proteome consists of Zn2+ binding proteins which require this ion for proper physiological function (Andreini et al. 2006). Zn2+ is the second most abundant (total concentration in the human body 2–3 g) transition metal after Fe2+ in humans (Kehl-Fie and Skaar 2010).
Prasad et al. first discovered Zn2+ deficiency in human male dwarfs in the Middle East in 1963 (Prasad et al. 1963). Zn2+ deficiency is associated with susceptibility to infections, memory impairment, and growth retardation. Zn2+ deficient animals have decreased immunity to viral infections such as Herpes simplex (Feiler et al. 1982) and Semliki forest (Singh et al. 1992); bacterial infections such as Listeria monocytogenes (Carlomagno et al. 1986; Coghlan et al. 1988), Francisella tularensis (Pekarek et al. 1977), Mycobacterium tuberculosis (McMurray et al. 1990), and Salmonella enteritidis (Kidd et al. 1994); parasitic infections such as Trypanosoma cruzi (Fraker et al. 1982), T. musculi (Lee et al. 1983), Toxoplasma gondii (Tasci et al. 1995), and Plasmodium yoelii (Shankar et al. 1995); fungal infections such as Candida albicans (Salvin et al. 1987); and helminthic infections such as Fasciola hepatica (Flagstad et al. 1972), Heligmosomoides polygyrus (Minkus et al. 1992; Shi et al. 1994), Strongyloides ratti (Fenwick et al. 1990b), Schistosoma mansoni (Nawar et al. 1992), and Trichinella spiralis (Fenwick et al. 1990a). Therefore, deficiency of Zn2+ cripples the host’s ability to clear infections and has a considerable impact on human health.
4.3 The Zinc Pill: To Take or Not to Take?
Zn2+ supplementation in general has a beneficial role in antimicrobial immunity. Acrodermatitis enteropathica (caused by mutations in the Zn2+ importer, ZIP4) is a rare and severe genetic autosomal recessive disorder characterized by acral and periorificial dermatitis, alopecia, and diarrhea. Weakened resistance to fungi, bacteria, and viruses is observed in bovine acrodermatitis enteropathica during Zn2+ deficiency. In humans, oral Zn2+ supplementation ameliorates symptoms associated with this disorder (Ciampo et al. 2018; Hambidge et al. 1977). Moreover, Zn2+ supplementation reduces the incidence of acute and chronic persistent diarrhea, dysentery (DD), acute lower respiratory infections (Ruel et al. 1997; Sazawal et al. 1995, 1996, 1998), malaria (Bates et al. 1993; Shankar et al. 2000), recurrent furunculosis (Brody 1977), and infection caused by the parasite S. mansoni (Friis et al. 1997).
Zn2+ lozenges reduce the duration of common cold (Mossad et al. 1996) by blocking the binding of HRV14 on the viral surface to the adhesion molecule ICAM-1 on the nasal mucosal surface, ultimately leading to reduced viral uptake (Novick et al. 1996). Low Zn2+ levels commonly detected in the plasma and serum of human HIV patients is associated with disease progression (Bogden et al. 1990; Khalili et al. 2008). Zn2+ supplementation partially reverses these effects (Falutz et al. 1988; Shankar and Prasad 1998), not by curtailing viral load but by dampening the frequency of diarrheal episodes and delaying immunological failure. This evidence suggests that adequate Zn2+ supplementation may be used as an adjunct therapy in HIV-infected adults (Baum et al. 2010). However, poor survival has also been reported in HIV-infected patients with high Zn2+ intake (Tang et al. 1996). In addition, as noted above, excess dietary Zn2+ intake may adversely affect C. difficile clearance. Clearly, determining accurate Zn2+ dosage is an indispensable step in the success of Zn2+ therapy for infection control. While optimal Zn2+ availability promotes resistance to infection, Zn2+ excess may be an Achilles’ heel in maximizing the therapeutic potential of this metal ion.
4.4 Zinc: A Prominent Driver on the Road to Innate Defense
Innate immunity delivers a rapid first line of defense against invading pathogens. Activation of myeloid cells with pathogen-associated molecular patterns (PAMPs) or danger-associated molecular patterns (DAMPs) triggers phagocytosis, cytokine release, antigen presentation to T cells, and bolsters antimicrobial defenses. In the following sections, we discuss the molecular mechanisms of Zn2+ regulation in innate immune cells and counter-defense mechanisms utilized by pathogens.
Polymorphonuclear leucocytes (PMNs) or granulocytes such as neutrophils, eosinophils, and basophils exert robust antimicrobial functions. Neutrophils are the most abundant PMNs. Shortly after phagocytosis of microbes, PMNs migrate into the infected tissue via adhesion and chemotaxis. They generate reactive oxygen species (ROS) through nicotinamide adenine dinucleotide phosphate-oxidase (NADPH oxidase) and kill the invading pathogens. Oxidative burst is an important defense mechanism of activated PMNs that is impaired by Zn2+ chelation. Conversely, Zn2+ is redox-inert, it acts as an antioxidant via catalytic action of the Cu2+ (copper)/Zn2+-superoxide dismutase. Moreover, Zn2+ dampens inflammatory responses that would otherwise augment oxidative stress (Lee 2018).
Zn2+ has an important role in the regulation of number and activities of myeloid cells. Rats fed a Zn2+ deficient diet have increased total white blood cells and granulocytes (neutrophils, eosinophils, and basophils) in blood (Someya et al. 2009). Decreased dietary Zn2+ intake or absorption diminishes eosinophil numbers in blood, liver, and lungs of mice and impairs their ability to clear H. polygyrus or Ascaris suum, a parasitic nematode (large roundworm) that causes ascariasis in pigs (Laubach 1990; Scott and Koski 2000). In contrast, Zn2+ deficiency augments eosinophilic allergic inflammation, and dietary Zn2+ supplementation reduces its intensity (Richter et al. 2003). Eosinophil cationic protein (ECP) is a potent secretory cytotoxic granule that has bactericidal and antiviral activities. Zn2+ inhibits the release of ECP from eosinophils in culture (Winqvist et al. 1985). Likewise, Zn2+ inhibits the release of granular protein histamine from basophils and mast cells in the human lung (Marone et al. 1986).
Zn2+ regulates important processes in immune cells that are crucial to infection control. Physiological Zn2+ concentration (10−3–10−2 mmol/L) in culture medium facilitates serum opsonic activity and ROS-generating capability in human neutrophils whereas excess Zn2+ (10 mmol/L) subdues it (Hasegawa et al. 2000). Reduced Zn2+ levels in the serum and in neutrophils is associated with impaired phagocytosis and diminished T cell-mediated immunity (Karzakova 2005). Voltage-gated proton (Hv1) channels transport H+ across the phagosomal membrane and regulate NADPH oxidase function. Hv1-mediated proton efflux balances the negative charge translocated by NADPH oxidase and provides substrate H+ for the formation of hydrogen peroxide, hypochlorous acid, and ROS crucial to killing pathogens (Decoursey 2012). Zn2+ inhibits Hv1 via two plausible mechanisms: it binds at low concentrations to one site on the channel which prevents the opening of the Hv1 pore, thereby inhibiting proton conduction. At high concentrations, Zn2+ binds to a second site and thwarts the outward movement of Hv1 voltage sensor (Qiu et al. 2016). In sum, Zn2+ exerts antioxidant functions by mitigating superoxide burst (Maret 2006; Prasad 2014), which may explain increased superoxide stress in Zn2+-deficient cells.
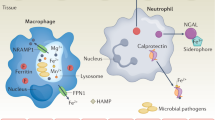
Antimicrobial peptides produced by some myeloid cells can trigger nutritional immunity through Zn2+ restriction. Neutrophils release calprotectin, a heterodimer of S100A8 and S100A9, and S100A12 (calgranulin C) peptides that sequester Zn2+ from pathogens to impair their growth. Calprotectin and calgranulin C restrict Zn2+ access to Staphylococcus aureus, C. albicans, and Helicobacter pylori thus stalling their growth (Besold et al. 2018; Corbin et al. 2008; Kehl-Fie and Skaar 2010). Likewise, S100A7 (psoriasin) released by keratinocytes kills Escherichia coli by withholding Zn2+ (Gläser et al. 2005).
NETosis, defined as the development and secretion of neutrophil extracellular traps (NETs), is a cell death mechanism of neutrophils in response to infections. NETs are composed of DNA, chromatin, and granular proteins which entrap and subsequently kill extracellular bacteria (Brinkmann et al. 2004). Zn2+ acts as a signaling molecule to facilitate NETosis (Hasan et al. 2013). Activated neutrophils elevate intracellular free Zn2+ via a protein-kinase C-ROS-dependent mechanism. While the precise source of this Zn2+ pool in neutrophils is not known, ROS may trigger the release of Zn2+ bound to sulfur on proteins such as MTs or glutathione (Maret 1994, 2000). Perhaps, Zn2+ is released from multiple reservoirs that may include Zn2+-bound proteins, Zn2+ storage organelles, or may be imported from the extracellular milieu. Deciphering the origin of the Zn2+ pool may provide clues to the mechanisms by which this ion prepares the neutrophil defense armor that is rapidly deployed during infection. Zn2+ also regulates neutrophil chemotaxis. These cells migrate into the host infected tissues in response to chemoattractants such as the bacterial product, N-formyl-l-methionyl-l-leucyl-l-phenylalanine (fMLF). In vitro, Zn2+ promotes chemotaxis of neutrophils to fMLF (Hasan et al. 2016; Hujanen et al. 1995; Vruwink et al. 1991). In sum, Zn2+ alters several functions of neutrophils such as chemotaxis, phagocytosis, NETosis, and pathogen killing.
Monocytes and macrophages phagocytose microbes, present antigen, and secrete cytokines to shape the immune response. Rats fed a Zn2+-deficient diet have increased number of total monocytes in blood (Someya et al. 2009). A lack of Zn2+ promotes differentiation and maturation of monocytes into macrophages by augmenting cAMP production by adenylate cyclase in vitro (Dubben et al. 2010). Zn2+ depletion in human monocytes improves the clearance of E. coli, S. aureus, and Streptococcus pneumoniae via phagocytosis and oxidative burst (Mayer et al. 2014).
Studies investigating the role of Zn2+ on cytokine expression and secretion have produced varying results depending on Zn2+ concentration, duration of Zn2+ depletion, experimental conditions, and model system. Supplementation of Zn2+ in serum-free media enhances the expression of interleukin 1 beta (IL-1β) and TNF-α in human peripheral blood mononuclear cells (PBMC) (Wellinghausen et al. 1996). Increasing amounts of Zn2+ dose-dependently inhibit monocyte activation caused by the superantigens, staphylococcal enterotoxins A and E (SEA, SEE), the Mycoplasma arthritidis–derived superantigen (MAS), but not toxic shock syndrome toxin-1 (TSST-1). Zn2+ interferes with the interactions between SEA, SEE, and MAS and their major histocompatibility complex class II (MHC-II)-binding sites. These data demonstrate that Zn2+ levels control the secretion of cytokines and response to superantigen challenge (Driessen et al. 1995).
Macrophages deploy two divergent Zn2+-associated defense mechanisms against intracellular pathogens. On the one hand, these cells may intoxicate M. tuberculosis or E. coli with excess Zn2+ to kill it (Botella et al. 2011). On the other hand, macrophages sequester Zn2+ from Histoplasma capsulatum residing within phagosomes (Subramanian Vignesh et al. 2013). Thus, one may speculate that Zn2+ acts as a double-edged sword: inadequate amounts arrest microbial growth, while an excess intoxicates them. What mechanisms influence the immune system’s decision to “withhold” versus “intoxicate” to overcome microbial pathogenesis remains a conundrum.
T cell–derived IFNγ is important for macrophage activation (Prasad 2014). Insufficient dietary Zn2+ intake compromises IFNγ production by T helper type 1 (Th1) cells resulting in impaired activation of monocytes/macrophages (Agnello et al. 2003; Prasad 2000). Moreover, Zn2+ deficiency reduces the production of IL-12 by monocytes/macrophages, yielding poor Th1 differentiation (Bao et al. 2011; Langrish et al. 2004). Thus, changes in the Zn2+ status not only affect differentiation and activation of monocytes/macrophages but may also compromise adaptive immunity.
4.5 The MT-Zinc Immune-Landscape: An Old Axis with a New Tale
In 1957, Margoshe and Vallee discovered MTs as Cd2+ (cadmium)-binding proteins from the horse renal cortex (Margoshes and Vallee 1957). MTs sense intracellular as well as environmental cues and regulate cellular Zn2+ homeostasis through sequestration, mobilization, and release (Subramanian Vignesh and Deepe Jr 2017). MTs have a higher Zn2+-binding constant (KZn = 3.2 × 1013 M−1 at pH 7.4) than most Zn2+-binding proteins. Despite this property, they facilitate controlled Zn2+ release to proteins with a lower stability constant for Zn2+ (Jacob et al. 1998). This metal is readily released from only one of the sites on MT1 and MT2 through interactions with adenosine triphosphate (ATP), guanosine triphosphate (GTP), or glutathione (Maret 2000).
There are 4 MT isoforms in mice and over 16 in humans (Quaife et al. 1994; Uchida et al. 1991). MTs are present in immune cells including those in the bone marrow (Liu et al. 2004), axillary lymph nodes (Haerslev et al. 1994), spleen (Huang et al. 2019; Mita et al. 2002), and thymus (Savino et al. 1984). In immune cells, MTs are induced by Zn2+, Cu2+, and Cd2+ (Aydemir et al. 2006; Huber and Cousins 1993; Makhijani 1998; Thorvaldsen et al. 1995); cytokines such as GM-CSF, TNFα, IFNγ, IL-1, IL-4, IL-6, and IL-27 (Cousins and Leinart 1988; Schroeder and Cousins 1990; Sciavolino and Vilček 1995; Ullio et al. 2015; Subramanian Vignesh et al. 2013, 2016; Chuan Wu et al. 2013a, b); and microbial ligands such as lipopolysaccharide (LPS) (Arizono et al. 1995; Leibbrandt and Koropatnick 1994), ROS (Dalton et al. 1994; Nourani et al. 2011; Tate et al. 1995), and nitric oxide (NO) (Arizono et al. 1995). In turn, MTs may regulate the activity of some of these immune modulators. For example, MTs scavenge ROS and regulate the function of GM-CSF and IL-4-polarized macrophages (Li et al. 2004; Subramanian Vignesh et al. 2013, 2016). The interrelationship between immune modulators and MTs is schematically represented in Fig. 4.1.

The association between MTs and immune mediators. MTs are induced by metal ions (Zn2+, Cu2+), infection, pathogen associate molecular patterns (PAMPs), cytokines, and superoxide radicals. The protein family in turn controls responses to each of these stimuli, thereby establishing a feedback loop between immune mediators and MTs
MTs influence a variety of immune responses in vivo and in vitro. For example, IL-27 induces MT1 and MT2 that prevent type 1 regulatory T (Tr1) cell development. This effect of MTs is due to negative feedback inhibition of signal transducer and activator of transcription (STAT)1 and STAT3 phosphorylation, resulting in diminished Tr1 differentiation and IL-10 production. Thus, the dynamic balance between STATs and MTs calibrates the development and suppressive function of Tr1 cells. The control of Zn2+ within the intracellular milieu may arm MTs with the ability to control STAT phosphorylation. This is plausible because Zn2+ inhibits the function of phosphatases that downmodulate STAT signaling. By sequestering the ion, MTs may render phosphatases active, leading to increased STAT dephosphorylation (Supasai et al. 2017; Chuan Wu et al. 2013a, b). From an antimicrobial immunity standpoint, this attribute of MTs in Tr1 cells may benefit the host in clearing infection rapidly, before suppressive immunity emerges to subdue inflammation and promote tissue repair.
Mt1−/−Mt2−/− mice exhibit stronger humoral responses through the elevation of nuclear factor-kappaB (NF-κB) transcription factor activity in splenocytes. These knockout mice display higher circulatory immunoglobulin levels, enhanced B cell differentiation upon OVA challenge, and lymphoproliferative responses to mitogenic stimulation (Crowthers et al. 2000). Exogenous administration of MTs into these mice dampens humoral immunity (Lynes et al. 1993). Thus, MTs temper antibody production by B cells. MTs also influence cytokine production by basophils. Stimulation of the Fc epsilon receptor 1 (FcɛRI) induces MT1 and MT2 in mouse basophils to regulate intracellular Zn2+. Lack of MTs increases intracellular free Zn2+ which inhibits calcineurin (CaN) phosphatase activity and thus impacts FcɛRI-induced nuclear factor of activated T-cell (NFAT)-dependent IL-4 production (Ugajin et al. 2015).
4.6 The MT-Zinc Axis in Infection
Several studies have brought to light the complex functions of MTs and the MT-Zn2+ axis in antimicrobial defense. Below, we discuss how the control of signaling pathways, Zn2+ homeostasis, and inflammatory responses by different MTs converge to dictate the outcome of host–pathogen interactions.
4.6.1 Bacterial Infection
NF-κB is essential for adequate innate immunity to infection. Zn2+ is an important negative regulator of NF-κB, while ROS is a positive regulator. MTs subvert the action of Zn2+, possibly by sequestering the intracellular free Zn2+ pool indicating that MT is an important intracellular modulator of NF-κB activation (Kim et al. 2003).
In polymicrobial sepsis in mice, deficiency of Zn2+ promotes systemic infection and NF-κB activation leading to elevated inflammation, lung injury, and mortality. Zn2+ supplementation prior to initiation of sepsis effectively reverses these effects (Bao et al. 2010). NF-κB induces the expression of the Zn2+ importer SLC39A8 (ZIP8) that imports Zn2+ to inhibit IκB kinase (Iκκ) activity. These findings identify a negative feedback loop that directly regulates a master transcription factor via coordination of Zn2+ metabolism (Liu et al. 2013). Salmonella typhimurium is a causative agent for inflamed gut. Macrophages infected with this pathogen exhibit elevated levels of free cytoplasmic Zn2+ that downmodulates NF-κB activity, as a result affecting the expression of reactive species (ROS and RNS) -forming enzymes phos47 (an NADPH oxidase subunit), inducible NO synthase (iNOS), and proinflammatory cytokines. Macrophages counter this change in Zn2+ homeostasis by augmenting MT1 and MT2 that scavenge free Zn2+ and restore ROS and RNS production to kill the pathogen. Thus, the limitation of free Zn2+ by MTs facilitates the control of intestinal colonization by S. typhimurium (Wu et al. 2017). In contrast, M. tuberculosis–infected macrophages rapidly increase free Zn2+ to poison this intracellular pathogen. This phenomenon is associated with an increase in MTs, MTF-1, and ZnT1, an exporter of Zn2+. MTF-1 translocates to the nucleus upon infection to induce MTs and ZnT1, suggesting that the host mounts a direct and quick response to protect itself from Zn2+ intoxication (Botella et al. 2011). Together, these studies suggest that macrophages possess two opposing mechanisms to exert antimicrobial immunity: Zn2+ depletion and Zn2+ poisoning. The effect of MTs on NF-κB activation is paradoxical depending on whether MTs scavenge Zn2+ or ROS. By scavenging ROS, MTs stabilize Iκκ, an inhibitor of NF-κB, ultimately downmodulating activation of this transcription factor. In gastric cells of Mt1−/−Mt2−/− mice with Helicobacter pylori infection, NF-кB activation and downstream production of macrophage inflammatory protein (MIP)-1α and monocyte chemoattractant protein (MCP)-1 is increased. The absence of MT1 and MT2 leads to erosive lesions and elevates infiltration of inflammatory leukocytes in the gastric mucosa. Thus, MTs protect against gastric ulceration during H. pylori infection by negatively regulating NF-кB activation (Mita et al. 2008).
4.6.2 Fungal Infection
How macrophages utilize the Zn2+ pool to resolve mycobacterial versus fungal infection presents an interesting paradox. Granulocyte macrophage-colony stimulating factor (GM-CSF) augments antimicrobial defenses against the intracellular fungus H. capsulatum. GM-CSF activated, infected macrophages increase MT1 and MT2 expression via activation of STAT3 and STAT5 transcriptional factors. These MTs bind to the macrophage free Zn2+ pool, denying Zn2+ access to the pathogen residing within phagosomes. In fact, Zn2+ is mobilized into the Golgi apparatus in association with an increase in the Zn2+ exporters, ZnT4 and ZnT7 that are expressed on the Golgi membrane. The Zn2+ sequestration “feat” by MTs simultaneously boosts ROS to stall fungal growth. Intriguingly, GM-CSF also elevates Zn2+ import via the importer Zip2, perhaps, to support an increased demand for Zn2+-dependent host processes during pathogen insult (Subramanian Vignesh et al. 2013).
IL-4 and IL-13 are cytokines that shape macrophage polarization to the M(IL-4) and M(IL-13) phenotypes, respectively. Studies on how Zn2+ levels influence macrophage polarization in rodents and human cell lines have produced distinct results. Zn2+ deficiency in rodents diminishes IL-4 production by Th2 cells and the proportion of M(IL-4) polarized macrophages in the spleen (Kido et al. 2019). In contrast, in human THP1 monocyte-derived macrophages, Zn2+ deficiency inhibits M1 polarization by IFNγ and LPS but does not affect M2 polarization by IL-4. Exogenous addition of Zn2+ suppresses the emergence of M(IL-4) macrophages in vitro (Dierichs et al. 2018). The use of different experimental models (rodents versus human cell line and dietary Zn2+ deficiency versus Zn2+ depletion/supplementation in culture media) may explain some of these findings. Nonetheless, these studies suggest that Zn2+ impacts macrophage polarization and can regulate the balance between M1 and M2 polarization states.
M2 macrophages aid in parasite clearance but harbor a permissive milieu for persistence of intracellular pathogens. Recent literature has demonstrated that IL-4 augments intracellular free Zn2+ in bone marrow–derived macrophages, microglia, and human monocyte-derived macrophages (Aratake et al. 2018; Subramanian Vignesh et al. 2016). M(IL-4) macrophages from the bone marrow specifically upregulate the MT3 isoform via STAT6 and interferon regulatory factor (IRF)4 signaling. The relationship between MT1/MT2 and MT3 is dichotomous, in that the latter expands the free Zn2+ pool while the former shrinks it in macrophages (Subramanian Vignesh et al. 2013, 2016). The action of cathepsin proteases enhances Zn2+ release from MT3 (Subramanian Vignesh et al. 2016). This is notable because such an increase in the free Zn2+ reservoir places the intracellular pathogen at an advantage: it assimilates a pool of Zn2+ that was once a part of the host. The finding establishes a link between MT-Zn2+ metabolism and the permissive nature of M(IL-4) macrophages to intracellular microbes. Figure 4.2 schematically outlines the distinct functions of MT1, MT2, and MT3 in macrophage defenses.

MT isoforms have distinct roles in host–pathogen interactions. GM-CSF and IL-6 elevate MT1 and MT2 that promote intracellular Zn2+ sequestration. Limitation of free Zn2+ in the host inhibits intracellular microbial growth via the induction of oxidative burst and restriction of Zn2+ access to the microbes. In contrast, IL-4 and IL-13 augment MT3 expression that increases the intracellular free Zn2+ pool and may dampen superoxide defenses. Moreover, intracellular pathogens may exploit this mechanism for acquisition of the metal ion for survival
4.6.3 Viral and Parasitic Infections
Our knowledge of how the MT-Zn2+ axis controls immune responses to viruses and parasites is limited. Infection with viruses including coxsackievirus B type 3 and influenza A/PR8 upregulates MTs in the liver, lung, kidney, and spleen. The mechanism of induction involves MTF-1, STAT3 signaling, and glucocorticoids (Ghoshal et al. 2001; Ilbäck et al. 2004). In contrast, infection with the hepatitis C virus is associated with a reduction in MT expression in the liver. Increasing MT levels through Zn2+ supplementation decreases viral load pointing at a protective function of the MT-Zn2+ axis in viral clearance (Carrera et al. 2003; Read et al. 2018). Further studies are required to elucidate the precise mechanisms by which MTs influence the immune systems’ ability to curtail viral uptake, replication, and shedding.
The parasite T. cruzi, the causative agent of Chagas disease, leads to cardiomyopathy and gastrointestinal inflammation. Infection with this pathogen reduces the expression of MT1 in the liver, while augmenting NO levels and oxidative stress. Whether the benefits of reducing NO are conferred by restoration of MTs is unknown, but chemically scavenging NO in animals infected with T. cruzi restores MT1 expression and arrests the growth of this parasite (Gonzalez-Mejia et al. 2014).
4.7 Survival Edge: Microbes (Aim to) Get the Upper Hand
Several pathogens have developed counter-defense mechanisms to thrive within the host (Fig. 4.3). For example, to circumvent Zn intoxication by macrophages, M. tuberculosis induces heavy metal efflux P-type ATPases. CtpC, a P-type ATPase, is upregulated rapidly to expel Zn2+ from the microbe. A lack of CtpC causes Zn2+ retention within the mycobacterial cytoplasm, thereby poisoning it. Therefore, P1-type ATPases contribute to the defense armor of M. tuberculosis by dampening the toxic effects of Zn2+ (Botella et al. 2011). Group A Streptococcus growth is restricted by Zn2+ limitation caused by neutrophil-derived calprotectin. Streptococcus pyogenes encodes the Zn2+ importer AdcA and a Zn2+ sensor AdcR to compete with Zn2+ sequestration by calprotectin (Makthal et al. 2017). ZrfC, a plasma membrane Zn2+ transporter of Aspergillus fumigatus, has the ability to scavenge Zn2+ efficiently from lungs enabling it to grow even in the presence of calprotectin (Amich et al. 2014). Neisseria meningitides uses ZnuD, a high-affinity Zn2+ transporter, to circumvent Zn2+ deprivation (Lappann et al. 2013). This pathogen also responds to low Zn2+ by expression of curved DNA binding protein A (CbpA), which is a calprotectin receptor, on its outer membrane. This molecule facilitates the acquisition of Zn2+ bound to calprotectin by N. meningitides. Thus, the microbe defies a vital host defense mechanism and exploits it for its benefit (Stork et al. 2013). Yersinia pestis utilizes a zincophore, yersiniabactin (Ybt) synthetase, and the high-affinity Zn2+ transporter, ZnuABC, to obtain Zn2+. These are crucial in the progression of lethal septicemic plague in mice (Bobrov et al. 2014). S. typhimurium can also express ZnuABC and thrives by subduing the host’s Zn2+ deprivation strategies. These studies indicate that Zn2+ acquisition may be a “virulence determinant” in some pathogens (Liu et al. 2012).

Zn2+-associated defense mechanisms. Immune cells employ various pathways (phagocytosis, oxidative burst, NETosis, Zn2+ depletion, and Zn2+ intoxication) to defend microbial invaders, whereas microbes also utilize counter-defense mechanisms including P1-type ATPases, Zn2+ acquisition transporters/proteins, and biofilm formation
Biofilms contain microbial communities associated with a polymeric matrix structure composed of factors such as extracellular DNA, polysaccharides, and proteins. Micromolar (100–250 μmol l−1) concentrations of Zn2+ block biofilm formation by Actinobacillus pleuropneumoniae, Haemophilus parasuis, S. typhimurium, Escherichia coli, S. aureus, Streptococcus suis, and Klebsiella pneumoniae strains. Mechanistically, Zn2+ may interfere with the stability of extracellular DNA and polymers contained within the biofilm matrix and impede critical microbial processes such as iron homeostasis and energy metabolism of associated microbes (Hancock et al. 2010; Polyudova et al. 2018; Chan Wu et al. 2013a, b). Of note, in the context of biofilms, an excess of Zn2+ or a deficiency of it may exert the same effect: inhibition of biofilm formation. For example, biofilm formation by Staphylococcus epidermidis, S. aureus, and S. pneumoniae is inhibited by Zn2+ chelation or Zn2+ excess (Brown et al. 2017; Conrady et al. 2008; Formosa-Dague et al. 2016). It is plausible that Zn2+ concentrations within a narrow range are necessary to maintain structural integrity of the biofilm, while intoxicating amounts of the metal ion adversely impact the growth of microbes that facilitate biofilm development.
4.8 Concluding Remarks
The value of dietary Zn2+ intake to maintain immunological robustness has long been appreciated. The highly conserved nature of MTs and their ability to bind Zn2+ across prokaryotes and eukaryotes has prompted scientists to query their importance in cellular functions. Immune cells are no exception. As it turns out, this class of proteins fiercely guards the Zn2+ reservoir in immune cells and can dictate the ions’ spatiotemporal presence both intracellularly and in the extracellular milieu. This attribute of MTs is notable, because the versatility of Zn2+ ions in biochemical processes demands that adequate amounts of Zn2+ be available for immune cells when and where they need it. Recent years have illuminated our knowledge of how the host taps into the MT-Zn2+ landscape to challenge microbial intrusion and overcome inflammatory damage caused by pathogen insult. These findings have opened newer apertures to explore the extent to which MTs orchestrate immunological responses. An important possibility to consider is that MT may function independent of Zn2+, perhaps in its apo-form or by binding to another metal ion. Of note, the protein also interacts with Cu2+ ions to regulate Cu2+ homeostasis. Whether an MT-Cu2+ axis impacts immunological performance or the triad (MT- Zn2+-Cu2+ axis) prevails over the two is unanswered. Nonetheless, MTs have surfaced prominently in the host-pathogen realm and will pave the path to a galvanizing story that continues to be told.
References
Agnello D, Lankford CS, Bream J, Morinobu A, Gadina M, O’shea JJ, Frucht DM (2003) Cytokines and transcription factors that regulate T helper cell differentiation: new players and new insights. J Clin Immunol 23(3):147–161
Amich J, Vicentefranqueira R, Mellado E, Ruiz-Carmuega A, Leal F, Calera JA (2014) The ZrfC alkaline zinc transporter is required for A spergillus fumigatus virulence and its growth in the presence of the Zn/Mn-chelating protein calprotectin. Cell Microbiol 16(4):548–564
Andreini C, Banci L, Bertini I, Rosato A (2006) Counting the zinc-proteins encoded in the human genome. J Proteome Res 5(1):196–201
Andrews GK (2001) Cellular zinc sensors: MTF-1 regulation of gene expression. In: Zinc Biochemistry, Physiology, and Homeostasis. Springer, Tokyo, pp 37–51
Aratake T, Higashi Y, Ueba Y, Hamada T, Shimizu T, Shimizu S et al (2018) The inhibitory role of intracellular free zinc in the regulation of Arg-1 expression in interleukin-4-induced activation of M2 microglia. Metallomics 10(10):1501–1509
Arizono K, Kagawa S-I, Hamada H, Ariyoshi T (1995) Nitric oxide mediated metallothionein induction by lipopolysaccharide. Res Commun Mol Pathol Pharmacol 90(1):49–58
Aydemir TB, Blanchard RK, Cousins RJ (2006) Zinc supplementation of young men alters metallothionein, zinc transporter, and cytokine gene expression in leukocyte populations. Proc Natl Acad Sci 103(6):1699–1704
Bao S, Liu M-J, Lee B, Besecker BY, Lai J-P, Guttridge DC, Knoell DL (2010) Zinc modulates the innate immune response in vivo to polymicrobial sepsis through regulation of NF-κB. Am J Phys Lung Cell Mol Phys 298(6):L744–L754
Bao B, Prasad AS, Beck FW, Bao GW, Singh T, Ali S, Sarkar FH (2011) Intracellular free zinc up-regulates IFN-γ and T-bet essential for Th1 differentiation in Con-A stimulated HUT-78 cells. Biochem Biophys Res Commun 407(4):703–707
Bates C, Bates P, Dardenne M, Prentice A, Lunn P, Northrop-Clewes C et al (1993) A trial of zinc supplementation in young rural Gambian children. Br J Nutr 69(1):243–255
Baum MK, Lai S, Sales S, Page JB, Campa A (2010) Randomized, controlled clinical trial of zinc supplementation to prevent immunological failure in HIV-infected adults. Clin Infect Dis 50(12):1653–1660
Beck FW, Li Y, Bao B, Prasad AS, Sarkar FH (2006) Evidence for reprogramming global gene expression during zinc deficiency in the HUT-78 cell line. Nutrition 22(10):1045–1056
Besecker BY, Exline MC, Hollyfield J, Phillips G, Disilvestro RA, Wewers MD, Knoell DL (2011) A comparison of zinc metabolism, inflammation, and disease severity in critically ill infected and noninfected adults early after intensive care unit admission. Am J Clin Nutr 93(6):1356–1364. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/21525204. https://doi.org/10.3945/ajcn.110.008417
Besold AN, Gilston BA, Radin JN, Ramsoomair C, Culbertson EM, Li CX et al (2018) Role of calprotectin in withholding zinc and copper from Candida albicans. Infect Immun 86(2):e00779–e00717
Bobrov AG, Kirillina O, Fetherston JD, Miller MC, Burlison JA, Perry RD (2014) The Y ersinia pestis siderophore, yersiniabactin, and the ZnuABC system both contribute to zinc acquisition and the development of lethal septicaemic plague in mice. Mol Microbiol 93(4):759–775
Bogden JD, Baker H, Frank O, Perez G, Kemp F, Bruening K, Louria D (1990) Micronutrient status and human immunodeficiency virus (HIV) infection. Ann N Y Acad Sci 587(1):189–195
Bonaventura P, Benedetti G, Albarède F, Miossec P (2015) Zinc and its role in immunity and inflammation. Autoimmun Rev 14(4):277–285
Botella H, Peyron P, Levillain F, Poincloux R, Poquet Y, Brandli I et al (2011) Mycobacterial P1-type ATPases mediate resistance to zinc poisoning in human macrophages. Cell Host Microbe 10(3):248–259
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS et al (2004) Neutrophil extracellular traps kill bacteria. Science 303(5663):1532–1535
Brody I (1977) Treatment of recurrent furunculosis with oral zinc. Lancet 310(8052):1358
Brown LR, Caulkins RC, Schartel TE, Rosch JW, Honsa ES, Schultz-Cherry S et al (2017) Increased zinc availability enhances initial aggregation and biofilm formation of streptococcus pneumoniae. Front Cell Infect Microbiol 7:233
Carlomagno M, Coghlan LG, McMurray D (1986) Chronic zinc deficiency and listeriosis in rats: acquired cellular resistance and response to vaccination. Med Microbiol Immunol 175(5):271–280
Carrera G, Paternain JL, Carrere N, Folch J, Courtade-Saïdi M, Orfila C et al (2003) Hepatic metallothionein in patients with chronic hepatitis C: relationship with severity of liver disease and response to treatment. Am J Gastroenterol 98(5):1142
Ciampo IRLD, Sawamura R, Ciampo LAD, Fernandes MIM (2018) Acrodermatitis enteropathica: clinical manifestations and pediatric diagnosis. Revista Paulista de Pediatria 36(2):238–241
Coghlan LG, Carlomagno MA, McMurray DN (1988) Effect of protein and zinc deficiencies on vaccine efficacy in guinea pigs following pulmonary infection with Listeria. Med Microbiol Immunol 177(5):255–263
Conrady DG, Brescia CC, Horii K, Weiss AA, Hassett DJ, Herr AB (2008) A zinc-dependent adhesion module is responsible for intercellular adhesion in staphylococcal biofilms. Proc Natl Acad Sci 105(49):19456–19461
Corbin BD, Seeley EH, Raab A, Feldmann J, Miller MR, Torres VJ et al (2008) Metal chelation and inhibition of bacterial growth in tissue abscesses. Science 319(5865):962–965
Cousins RJ (1998) A role of zinc in the regulation of gene expression. Proc Nutr Soc 57(2):307–311
Cousins RJ, Leinart AS (1988) Tissue-specific regulation of zinc metabolism and metallothionein genes by interleukin 1. FASEB J 2(13):2884–2890
Cousins RJ, Blanchard RK, Moore JB, Cui L, Green CL, Liuzzi JP et al (2003) Regulation of zinc metabolism and genomic outcomes. J Nutr 133(5):1521S–1526S
Crawford AC, Lehtovirta-Morley LE, Alamir O, Niemiec MJ, Alawfi B, Alsarraf M et al (2018) Biphasic zinc compartmentalisation in a human fungal pathogen. PLoS Pathog 14(5):e1007013
Crowthers KC, Kline V, Giardina C, Lynes MA (2000) Augmented humoral immune function in metallothionein-null mice. Toxicol Appl Pharmacol 166(3):161–172
Dalton T, Palmiter RD, Andrews GK (1994) Transcriptional induction of the mouse metallothionein-I gene in hydrogen peroxide-treated Hepa cells involves a composite major late transcription factor/antioxidant response element and metal response promoter elements. Nucleic Acids Res 22(23):5016–5023
Decoursey TE (2012) Voltage-gated proton channels. Compr Physiol 2(2):1355–1385. Retrieved from https://www.ncbi.nlm.nih.gov/pubmed/23798303. https://doi.org/10.1002/cphy.c100071
Dierichs L, Kloubert V, Rink L (2018) Cellular zinc homeostasis modulates polarization of THP-1-derived macrophages. Eur J Nutr 57(6):2161–2169
Driessen C, Hirv K, Kirchner H, Rink L (1995) Divergent effects of zinc on different bacterial pathogenic agents. J Infect Dis 171(2):486–489
Dubben S, Hönscheid A, Winkler K, Rink L, Haase H (2010) Cellular zinc homeostasis is a regulator in monocyte differentiation of HL-60 cells by 1α, 25-dihydroxyvitamin D3. J Leukoc Biol 87(5):833–844
Eide DJ (2004) The SLC39 family of metal ion transporters. Pflugers Arch 447(5):796–800
Falutz J, Tsoukas C, Gold P (1988) Zinc as a cofactor in human immunodeficiency virus—induced immunosuppression. JAMA 259(19):2850–2851
Feiler L, Smolin G, Okumoto M, Condon D (1982) Herpetic keratitis in zinc-deficient rabbits. Invest Ophthalmol Vis Sci 22(6):788–795
Fenwick P, Aggett P, Macdonald D, Huber C, Wakelin D (1990a) Zinc deficiency and zinc repletion: effect on the response of rats to infection with Trichinella spiralis. Am J Clin Nutr 52(1):166–172
Fenwick P, Aggett P, Macdonald DC, Huber C, Wakelin D (1990b) Zinc deprivation and zinc repletion: effect on the response of rats to infection with Strongyloides ratti. Am J Clin Nutr 52(1):173–177
Flagstad T, Andersen S, Nielsen K (1972) The course of experimental Fasciola hepatica infection in calves with a deficient cellular immunity. Res Vet Sci 13(5):468–477
Formosa-Dague C, Speziale P, Foster TJ, Geoghegan JA, Dufrêne YF (2016) Zinc-dependent mechanical properties of Staphylococcus aureus biofilm-forming surface protein SasG. Proc Natl Acad Sci 113(2):410–415
Fraker PJ, Caruso R, Kierszenbaum F (1982) Alteration of the immune and nutritional status of mice by synergy between zinc deficiency and infection with Trypanosoma cruzi. J Nutr 112(6):1224–1229
Friis H, Ndhlovu P, Mduluza T, Kaondera K, Sandström B, Michaelsen KF et al (1997) The impact of zinc supplementation on Schistosoma mansoni reinfection rate and intensities: a randomized, controlled trial among rural Zimbabwean schoolchildren. Eur J Clin Nutr 51(1):33
Ghoshal K, Majumder S, Zhu Q, Hunzeker J, Datta J, Shah M et al (2001) Influenza virus infection induces metallothionein gene expression in the mouse liver and lung by overlapping but distinct molecular mechanisms. Mol Cell Biol 21(24):8301–8317
Gläser R, Harder J, Lange H, Bartels J, Christophers E, Schröder J-M (2005) Antimicrobial psoriasin (S100A7) protects human skin from Escherichia coli infection. Nat Immunol 6(1):57
Gonzalez-Mejia ME, Torres-Rasgado E, Porchia LM, Salgado HR, Totolhua J-L, Ortega A et al (2014) Metallothionein-1 and nitric oxide expression are inversely correlated in a murine model of Chagas disease. Mem Inst Oswaldo Cruz 109(2):174–181
Haase H, Mazzatti DJ, White A, Ibs KH, Engelhardt G, Hebel S, … Rink L (2007) Differential gene expression after zinc supplementation and deprivation in human leukocyte subsets. Paper presented at the molecular medicine
Haerslev T, Jacobsen K, Nedergaard L, Zedeler K (1994) Immunohistochemical detection of metallothionein in primary breast carcinomas and their axillary lymph node metastases. Pathol Res Pract 190(7):675–681
Hambidge K, Walravens P, Neldner K (1977) The role of zinc in the pathogenesis and treatment of acrodermatitis enteropathica. Prog Clin Biol Res 14:329–342
Hancock V, Dahl M, Klemm P (2010) Abolition of biofilm formation in urinary tract Escherichia coli and Klebsiella isolates by metal interference through competition for fur. Appl Environ Microbiol 76(12):3836–3841
Hasan R, Rink L, Haase H (2013) Zinc signals in neutrophil granulocytes are required for the formation of neutrophil extracellular traps. Innate Immun 19(3):253–264
Hasan R, Rink L, Haase H (2016) Chelation of free Zn2+ impairs chemotaxis, phagocytosis, oxidative burst, degranulation, and cytokine production by neutrophil granulocytes. Biol Trace Elem Res 171(1):79–88
Hasegawa H, Suzuki K, Suzuki K, Nakaji S, Sugawara K (2000) Effects of zinc on the reactive oxygen species generating capacity of human neutrophils and on the serum opsonic activity in vitro. Lumin J Biol Chem Lumin 15(5):321–327
Hennigar SR, McClung JP (2016) Nutritional immunity: starving pathogens of trace minerals. Am J Lifestyle Med 10(3):170–173
Huang X, Feng Y, Fan W, Duan J, Duan Y, Xiong G et al (2019) Potential ability for metallothionein and vitamin E protection against cadmium immunotoxicity in head kidney and spleen of grass carp (Ctenopharyngodon idellus). Ecotoxicol Environ Saf 170:246–252
Huber KL, Cousins RJ (1993) Zinc metabolism and metallothionein expression in bone marrow during erythropoiesis. Am J Physiol Endocrinol Metab 264(5):E770–E775
Hujanen ES, Seppä ST, Virtanen K (1995) Polymorphonuclear leukocyte chemotaxis induced by zinc, copper and nickel in vitro. Biochimica et Biophysica Acta (BBA)-Gen Subjects 1245(2):145–152
Ilbäck N-G, Glynn AW, Wikberg L, Netzel E, Lindh U (2004) Metallothionein is induced and trace element balance changed in target organs of a common viral infection. Toxicology 199(2–3):241–250
Inoue K, O’Bryant Z, Xiong Z-G (2015) Zinc-permeable ion channels: effects on intracellular zinc dynamics and potential physiological/pathophysiological significance. Curr Med Chem 22(10):1248–1257
Jacob C, Maret W, Vallee BL (1998) Control of zinc transfer between thionein, metallothionein, and zinc proteins. Proc Natl Acad Sci 95(7):3489–3494
Karzakova L (2005) Laboratory manifestations of asymptomatic zinc deficiency. Klin Lab Diagn 12:39–41
Kehl-Fie TE, Skaar EP (2010) Nutritional immunity beyond iron: a role for manganese and zinc. Curr Opin Chem Biol 14(2):218–224
Khalili H, Soudbakhsh A, Hajiabdolbaghi M, Dashti-Khavidaki S, Poorzare A, Saeedi A, Sharififar R (2008) Nutritional status and serum zinc and selenium levels in Iranian HIV infected individuals. BMC Infect Dis 8(1):165
Kidd M, Qureshi M, Ferket P, Thomas LN (1994) Dietary zinc-methionine enhances mononuclear-phagocytic function in young turkeys. Biol Trace Elem Res 42(3):217–229
Kido T, Ishiwata K, Suka M, Yanagisawa H (2019) Inflammatory response under zinc deficiency is exacerbated by dysfunction of the T helper type 2 lymphocyte–M2 macrophage pathway. Immunology 156(4):356–372
Kim CH, Kim JH, Lee J, Ahn YS (2003) Zinc-induced NF-κB inhibition can be modulated by changes in the intracellular metallothionein level. Toxicol Appl Pharmacol 190(2):189–196
Langrish CL, McKenzie BS, Wilson NJ, de Waal Malefyt R, Kastelein RA, Cua DJ (2004) IL-12 and IL-23: master regulators of innate and adaptive immunity. Immunol Rev 202(1):96–105
Lappann M, Danhof S, Guenther F, Olivares-Florez S, Mordhorst IL, Vogel U (2013) In vitro resistance mechanisms of N eisseria meningitidis against neutrophil extracellular traps. Mol Microbiol 89(3):433–449
Laubach HE (1990) Effect of dietary zinc on larval burdens, tissue eosinophil numbers, and lysophospholipase activity of Ascaris suum infected mice. Acta Trop 47(4):205–211
Lee SR (2018) Critical role of zinc as either an antioxidant or a Prooxidant in cellular systems. Oxidative Med Cell Longev 2018: 9156285, 11. https://doi.org/10.1155/2018/9156285
Lee CM, Humphrey PA, Aboko-Cole GF (1983) Interaction of nutrition and infection: effect of zinc deficiency on resistance to Trypanosoma musculi. Int J Biochem 15(6):841–847
Leibbrandt ME, Koropatnick J (1994) Activation of human monocytes with lipopolysaccharide induces metallothionein expression and is diminished by zinc. Toxicol Appl Pharmacol 124(1):72–81
Li X, Chen H, Epstein PN (2004) Metallothionein protects islets from hypoxia and extends islet graft survival by scavenging most kinds of reactive oxygen species. J Biol Chem 279(1):765–771
Liu AL, Zhang ZM, Zhu BF, Liao ZH, Liu Z (2004) Metallothionein protects bone marrow stromal cells against hydrogen peroxide-induced inhibition of osteoblastic differentiation. Cell Biol Int 28(12):905–911
Liu JZ, Jellbauer S, Poe AJ, Ton V, Pesciaroli M, Kehl-Fie TE et al (2012) Zinc sequestration by the neutrophil protein calprotectin enhances Salmonella growth in the inflamed gut. Cell Host Microbe 11(3):227–239
Liu M-J, Bao S, Gálvez-Peralta M, Pyle CJ, Rudawsky AC, Pavlovicz RE et al (2013) ZIP8 regulates host defense through zinc-mediated inhibition of NF-κB. Cell Rep 3(2):386–400
Lynes MA, Borghesi LA, Youn J, Olson EA (1993) Immunomodulatory activities of extracellular metallothionein I. Metallothionein effects on antibody production. Toxicology 85(2–3):161–177
Makhijani EJYPR (1998) Flow cytometric determination of metallothionein levels in human peripheral blood lymphocytes: utility in environmental exposure assessment. J Toxicol Environ Health A 54(6):445–457
Makthal N, Nguyen K, Do H, Gavagan M, Chandrangsu P, Helmann JD et al (2017) A critical role of zinc importer AdcABC in group A streptococcus-host interactions during infection and its implications for vaccine development. EBioMedicine 21:131–141
Maret W (1994) Oxidative metal release from metallothionein via zinc-thiol/disulfide interchange. Proc Natl Acad Sci 91(1):237–241
Maret W (2000) The function of zinc metallothionein: a link between cellular zinc and redox state. J Nutr 130(5):1455S–1458S
Maret W (2006) Zinc coordination environments in proteins as redox sensors and signal transducers. Antioxid Redox Signal 8(9–10):1419–1441
Margoshes M, Vallee BL (1957) A cadmium protein from equine kidney cortex. J Am Chem Soc 79(17):4813–4814
Marone G, Columbo M, De Paulis A, Cirillo R, Giugliano R, Condorelli M (1986) Physiological concentrations of zinc inhibit the release of histamine from human basophils and lung mast cells. Agents Actions 18(1–2):103–106
Mayer LS, Uciechowski P, Meyer S, Schwerdtle T, Rink L, Haase H (2014) Differential impact of zinc deficiency on phagocytosis, oxidative burst, and production of pro-inflammatory cytokines by human monocytes. Metallomics 6(7):1288–1295
Maywald M, Wessels I, Rink L (2017) Zinc signals and immunity. Int J Mol Sci 18(10):2222
McMurray DN, Bartow RA, Mintzer CL, Hernandez-Frontera E (1990) Micronutrient Status and Immune Function in Tuberculosisa. Ann N Y Acad Sci 587(1):59–69
Minkus TM, Koski KG, Scott ME (1992) Marginal zinc deficiency has no effect on primary or challenge infections in mice with Heligmosomoides polygyrus (Nematoda). J Nutr 122(3):570–579
Mita M, Imura N, Kumazawa Y, Himeno S (2002) Suppressed proliferative response of spleen T cells from metallothionein null mice. Microbiol Immunol 46(2):101–107
Mita M, Satoh M, Shimada A, Okajima M, Azuma S, Suzuki JS et al (2008) Metallothionein is a crucial protective factor against Helicobacter pylori-induced gastric erosive lesions in a mouse model. Am J Physiol Gastrointest Liver Physiol 294(4):G877–G884
Mossad SB, Macknin ML, Mendendorp SV, Mason P (1996) Zinc gluconate lozenges for treating the common cold: a randomized, double-blind, placebo-controlled study. Ann Intern Med 125(2):81–88
Nawar O, Akridge RE, Hassan E, El Gazar R, Doughty BL, Kemp W (1992) The effect of zinc deficiency on granuloma formation, liver fibrosis, and antibody responses in experimental schistosomiasis. Am J Trop Med Hyg 47(3):383–389
Nourani MR, Ebrahimi M, Roudkenar MH, Vahedi E, Ghanei M, Fooladi AAI (2011) Sulfur mustard induces expression of metallothionein-1A in human airway epithelial cells. Int J Gen Med 4:413
Novick S, Godfrey J, Godfrey N, Wilder H (1996) How does zinc modify the common cold? Clinical observations and implications regarding mechanisms of action. Med Hypotheses 46(3):295–302
Overbeck S, Rink L, Haase H (2008) Modulating the immune response by oral zinc supplementation: a single approach for multiple diseases. Arch Immunol Ther Exp 56(1):15–30
Palmiter RD (2004) Protection against zinc toxicity by metallothionein and zinc transporter 1. Proc Natl Acad Sci 101(14):4918–4923
Palmiter RD, Huang L (2004) Efflux and compartmentalization of zinc by members of the SLC30 family of solute carriers. Pflugers Arch 447(5):744–751
Pekarek R, Hoagland A, Powanda M (1977) Humoral and cellular immune responses in zinc deficient rats. Nutr Rep Int 16:267–276
Polyudova TV, Eroshenko DV, Korobov VP (2018) Plasma, serum, albumin, and divalent metal ions inhibit the adhesion and the biofilm formation of Cutibacterium (Propionibacterium) acnes. Aims Microbiol 4(1):165–172
Prasad AS (2000) Effects of zinc deficiency on Th1 and Th2 cytokine shifts. J Infect Dis 182(Supplement_1):S62–S68
Prasad AS (2014) Zinc is an antioxidant and anti-inflammatory agent: its role in human health. Front Nutr 1:14
Prasad AS, Schulert AR, Miale JR, Farid Z, Sandstead HH (1963) Zinc and iron deficiencies in male subjects with dwarfism and hypogonadism but without ancylostomiasis, schistosomiasis or severe anemia. Am J Clin Nutr 12(6):437–444
Qiu F, Chamberlin A, Watkins BM, Ionescu A, Perez ME, Barro-Soria R et al (2016) Molecular mechanism of Zn2+ inhibition of a voltage-gated proton channel. Proc Natl Acad Sci 113(40):E5962–E5971
Quaife CJ, Findley SD, Erickson JC, Froelick GJ, Kelly EJ, Zambrowicz BP, Palmiter RD (1994) Induction of a new metallothionein isoform (MT-IV) occurs during differentiation of stratified squamous epithelia. Biochemistry 33(23):7250–7259
Read SA, Parnell G, Booth D, Douglas MW, George J, Ahlenstiel G (2018) The antiviral role of zinc and metallothioneins in hepatitis C infection. J Viral Hepat 25(5):491–501
Richter M, Bonneau R, Girard M-A, Beaulieu C, Larivée P (2003) Zinc status modulates bronchopulmonary eosinophil infiltration in a murine model of allergic inflammation. Chest 123(3):446S
Ruel MT, Rivera JA, Santizo M-C, Lönnerdal B, Brown KH (1997) Impact of zinc supplementation on morbidity from diarrhea and respiratory infections among rural Guatemalan children. Pediatrics 99(6):808–813
Salvin S, Horecker B, Pan L-X, Rabin B (1987) The effect of dietary zinc and prothymosin α on cellular immune responses of RFJ mice. Clin Immunol Immunopathol 43(3):281–288
Savino W, Huang P, Corrigan A, Berrih S, Dardenne M (1984) Thymic hormone-containing cells. V. Immunohistological detection of metallothionein within the cells bearing thymulin (a zinc-containing hormone) in human and mouse thymuses. J Histochem Cytochem 32(9):942–946
Sazawal S, Black RE, Bhan MK, Bhandari N, Sinha A, Jalla S (1995) Zinc supplementation in young children with acute diarrhea in India. N Engl J Med 333(13):839–844
Sazawal S, Black RE, Bhan MK, Jalla S, Bhandari N, Sinha A, Majumdar S (1996) Zinc supplementation reduces the incidence of persistent diarrhea and dysentery among low socioeconomic children in India. J Nutr 126(2):443–450
Sazawal S, Black RE, Jalla S, Mazumdar S, Sinha A, Bhan MK (1998) Zinc supplementation reduces the incidence of acute lower respiratory infections in infants and preschool children: a double-blind, controlled trial. Pediatrics 102(1):1–5
Schroeder JJ, Cousins RJ (1990) Interleukin 6 regulates metallothionein gene expression and zinc metabolism in hepatocyte monolayer cultures. Proc Natl Acad Sci 87(8):3137–3141
Sciavolino PJ, Vilček J (1995) Regulation of metallothionein gene expression by TNF-α and IFN-β in human fibroblasts. Cytokine 7(3):242–250
Scott ME, Koski KG (2000) Zinc deficiency impairs immune responses against parasitic nematode infections at intestinal and systemic sites. J Nutr 130(5):1412S–1420S
Shankar AH, Prasad AS (1998) Zinc and immune function: the biological basis of altered resistance to infection. Am J Clin Nutr 68(2):447S–463S
Shankar A, Kumar N, Scott A (1995) Zinc-deficiency exacerbates experimental malaria infection in mice. Paper presented at the FASEB Journal
Shankar AH, Genton B, Baisor M, Paino J, Tamja S, Adiguma T et al (2000) The influence of zinc supplementation on morbidity due to Plasmodium falciparum: a randomized trial in preschool children in Papua New Guinea. Am J Trop Med Hygiene 62(6):663–669
Shi HN, Scott E, Stevenson M, Koski G (1994) Zinc deficiency impairs T cell function in mice with primary infection of Heligmosomoides polygyrus (Nematoda). Parasite Immunol 16(7):339–350
Singh K, Zaidi S, Raisuddin S, Saxena A, Murthy R, Ray P (1992) Effect of zinc on immune functions and host resistance against infection and tumor challenge. Immunopharmacol Immunotoxicol 14(4):813–840
Someya Y, Tanihata J, Sato S, Kawano F, Shirato K, Sugiyama M et al (2009) Zinc-deficiency induced changes in the distribution of rat white blood cells. J Nutr Sci Vitaminol 55(2):162–169
Stork M, Grijpstra J, Bos MP, Torres CM, Devos N, Poolman JT et al (2013) Zinc piracy as a mechanism of Neisseria meningitidis for evasion of nutritional immunity. PLoS Pathog 9(10):e1003733
Subramanian Vignesh K, Figueroa JAL, Porollo A, Caruso JA, Deepe GS Jr (2013) Granulocyte macrophage-colony stimulating factor induced Zn sequestration enhances macrophage superoxide and limits intracellular pathogen survival. Immunity 39(4):697–710
Subramanian Vignesh K, Figueroa JAL, Porollo A, Divanovic S, Caruso JA, Deepe GS Jr (2016) IL-4 induces metallothionein 3-and SLC30A4-dependent increase in intracellular Zn2+ that promotes pathogen persistence in macrophages. Cell Rep 16(12):3232–3246
Subramanian Vignesh K, Deepe G Jr (2017) Metallothioneins: emerging modulators in immunity and infection. Int J Mol Sci 18(10):2197
Supasai S, Aimo L, Adamo A, Mackenzie G, Oteiza P (2017) Zinc deficiency affects the STAT1/3 signaling pathways in part through redox-mediated mechanisms. Redox Biol 11:469–481
Tang AM, Graham NM, Saah AJ (1996) Effects of micronutrient intake on survival in human immunodeficiency virus type 1 infection. Am J Epidemiol 143(12):1244–1256
Tasci S, Sengil AZ, Altindis M, Arisoy K (1995) The effect of zinc supplementation in experimentally induced Toxoplasma gondii infection. J Egypt Soc Parasitol 25(3):745–751
Tate D, Miceli MV, Newsome DA (1995) Phagocytosis and H2O2 induce catalase and metallothionein gene expression in human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci 36(7):1271–1279
Thorvaldsen JL, Mehra RK, Yu W, Sewell AK, Winge DR (1995) Analysis of copper-induced metallothionein expression using autonomously replicating plasmids in Candida glabrata. Yeast 11(15):1501–1511
Uchida Y, Takio K, Titani K, Ihara Y, Tomonaga M (1991) The growth inhibitory factor that is deficient in the Alzheimer’s disease brain is a 68 amino acid metallothionein-like protein. Neuron 7(2):337–347
Ugajin T, Nishida K, Yamasaki S, Suzuki J, Mita M, Kubo M et al (2015) Zinc-binding metallothioneins are key modulators of IL-4 production by basophils. Mol Immunol 66(2):180–188
Ullio C, Brunk UT, Urani C, Melchioretto P, Bonelli G, Baccino FM, Autelli R (2015) Autophagy of metallothioneins prevents TNF-induced oxidative stress and toxicity in hepatoma cells. Autophagy 11(12):2184–2198
Uttra CKM, Devrajani BR, Shaikh HM, Shah SZA, Devrajani T, Shaikh N, Shah SFA (2011) Serum Zinc level in patients with pulmonary tuberculosis. Adv Biol Res 5(3):174–178
Vallee BL (1995) The function of metallothionein. Neurochem Int 27(1):23–33
Vruwink KG, Fletcher MP, Keen CL, Golub MS, Hendrickx AG, Gershwin ME (1991) Moderate zinc deficiency in rhesus monkeys. An intrinsic defect of neutrophil chemotaxis corrected by zinc repletion. J Immunol 146(1):244–249
Wellinghausen N, Driessen C, Rink L (1996) Stimulation of human peripheral blood mononuclear cells by zinc and related cations. Cytokine 8(10):767–771
Winqvist I, Olofsson T, Persson E (1985) Effect of zinc and other cations on the release of the eosinophil cationic protein. Scand J Clin Lab Invest 45(8):671–677
Wu C, Labrie J, Tremblay Y, Haine D, Mourez M, Jacques M (2013a) Zinc as an agent for the prevention of biofilm formation by pathogenic bacteria. J Appl Microbiol 115(1):30–40
Wu C, Pot C, Apetoh L, Thalhamer T, Zhu B, Murugaiyan G et al (2013b) Metallothioneins negatively regulate IL-27–induced type 1 regulatory T-cell differentiation. Proc Natl Acad Sci 110(19):7802–7807
Wu A, Tymoszuk P, Haschka D, Heeke S, Dichtl S, Petzer V et al (2017) Salmonella utilizes zinc to subvert antimicrobial host defense of macrophages via modulation of NF-κB signaling. Infect Immun 85(12):e00418–e00417
Zackular JP, Moore JL, Jordan AT, Juttukonda LJ, Noto MJ, Nicholson MR et al (2016) Dietary zinc alters the microbiota and decreases resistance to Clostridium difficile infection. Nat Med 22(11):1330
Acknowledgments
This work was supported by 19CDA34770022 American Heart Association grant to KSV and NIH grant AI106269 to GSD. The authors declare no conflicts of interest.
Author Contributions
DC and KSV wrote the manuscript. GD reviewed it and KSV supervised the work.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Chowdhury, D., Deepe, G.S., Subramanian Vignesh, K. (2019). The Metallothionein-Zinc Landscape: How It Shapes Antimicrobial Immunity. In: Fukada, T., Kambe, T. (eds) Zinc Signaling. Springer, Singapore. https://doi.org/10.1007/978-981-15-0557-7_4
Download citation
DOI: https://doi.org/10.1007/978-981-15-0557-7_4
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-0556-0
Online ISBN: 978-981-15-0557-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)