
Abstract
We reviewed epidemiological studies of gene–environment interactions to detect adverse health effects of environmental chemicals on the next generation in 2008, more than 10 years ago. Since then, researches on gene–environment interactions have continued via small-scale epidemiological studies seeking to elucidate associations between tobacco and environmental chemicals and candidate genes such as those encoding metabolic enzymes. In the last 10 years, extensive innovation in research designs and methods, accompanied by recent rapid advances in analytical technologies, has occurred. Specifically, genome-wide association studies (GWASs) and epigenome-wide association studies (EWASs) have become mainstream in genome cohort studies using advanced genomics and epigenomics. These have made it possible to better understand the genetic basis of diseases. Furthermore, in addition to GWASs and meta-analyses, Mendelian randomization has emerged as a GWAS-based theoretical method for environmental risk assessment that uses genetic factors associated with environmental factors. Although the concept of exposome was initially proposed as an improved tool to quantify total environmental contributions, linking it with genomics is expected to additionally enable the elucidation of the origins of multiple complex diseases. We have reviewed researches on gene–environment interactions and discussed recently developed approaches, such as GWAS, Mendelian randomization, and exposome linked with genomics, for evaluating genetic susceptibility.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Susceptibility
- Genome-wide association study (GWAS)
- Meta-analysis
- Non-communicable disease (NCD)
- Developmental origins of health and disease (DOHaD)
1 Introduction
In 2008, more than 10 years ago, we reviewed epidemiological studies of gene–environment interactions to detect the adverse health effects of environmental chemicals on the next generation [1]. As is well documented, numerous chemical compounds such as polycyclic aromatic hydrocarbons (PHA) in tobacco smoke are activated and detoxified by xenobiotic-metabolizing enzymes [1]. Xenobiotic-metabolizing genes appear to be influenced functionally by maternal smoking during pregnancy, which may be a significant risk factor for low birth weight (LBW) and/or intrauterine growth restriction (IUGR) [1,2,3,4]. The Hokkaido Study, a pioneering work in this field, examined the effects of environmental factors together with a genetic predisposition on the health and development of about 20,000 children across the Hokkaido prefecture, the northern part of Japan, from the prenatal period onward [5, 6].
The last decade has witnessed major innovations in research design and methods, accompanied by recent advances in technologies for statistical and biological analyses and measurement [7,8,9,10]. In particular, genome-wide association studies (GWASs), including epigenome-wide association studies (EWASs), have become mainstream in genome cohort studies using advanced genomics and epigenomics techniques, which has made it possible to better understand the genetic basis of diseases [7]. Furthermore, Mendelian randomization is a fast-growing area that involves the analysis of genetic variants to assess the causal relationships between exposure and outcome [8]. The field of exposure science, termed exposome has emerged, which involves the study of mechanisms by which “non-genetic” exogenous and endogenous exposure influence the risk of disease [9, 10]; linking this field with genomics is expected to enable elucidation of the origins of multiple complex diseases [9]. The methods for evaluating gene–environment interactions have advanced rapidly because of the accumulation of large-scale data, termed big data. Using GWASs, we have obtained data for hundreds of complex traits across a wide range of domains, including common diseases and quantitative traits that are risk factors for diseases, enabling us to better define the relative role of genes and the environment in disease risk [7]. Examination of the impact of early life exposure and maternal physical and mental conditions during pregnancy is a topic of interest for investigation by exposomics, using environmental factors and biological samples from cohorts [9, 10]. Thus, it is recognized that numerous human diseases arise from the complex interplay between environmental exposure and host susceptibilities.
In the present work, we introduce recent progress in evaluating gene–environment interactions in large-scale genome epidemiological studies as well as in small-scale single studies targeting candidate genes.
2 Infant Birth Size, Including Low Birth Weight (LBW), Preterm Birth (PB), Small-for-Gestational-Age (SGA), Gestational Age, and Intrauterine Growth Restriction (IUGR) in Relation to Maternal Smoking
It is known that maternal smoking during pregnancy may lead to a reduction in infant birth size including LBW, PB, SGA, gestational age, and IUGR. In recent years, it has been found that this association is modified by genetic factors. In research studies published up to 2018, this association is reported to be modified by maternal genotypes of genes encoding the xenobiotic receptor (aromatic hydrocarbon receptor [AHR]), enzymes (cytochrome P450 [CYP] 1A1, CYP2A6, CYP2E1, glutathione S-transferase [GST] mu 1 [GSTM1], GST theta 1 [GSTT1], GST theta 2 [GSTT2], epoxide hydrolase 1 [EPHX1], 5,10-methylenetetrahydrofolate reductase [MTHFR], and NAD(P)H dehydrogenase [NQO1]), DNA repair proteins (X-ray repair cross-complementing gene 1 [XRCC1], XRCC3, 8-oxoguanine glycosylase [OGG1]), oncogenes (MDM4), and tumor suppressor genes (TP53) [11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33] (Table 19.1). Moreover, this association has been observed to be modified by fetal genotypes of genes encoding xenobiotic-metabolizing enzymes (GSTT1 and NQO1) and cell-division-related genes (adenosine deaminase 1 [ADA1]) [14, 16, 29]. Most of these studies assessed the smoking status of pregnant women using a questionnaire. A few studies still evaluate the smoking status of pregnant women using objective indicators.
Although studies that have evaluated pregnant women by smoking status using objective indicators are limited, three previous reports have examined the effects of gene–environment interactions of maternal smoking on infant birth size using biomarkers [21, 24, 27]. Only one publication has examined the dose-dependent association of gene–environment interactions and maternal smoking with infant birth size among 3263 Japanese pregnant women enrolled in the prospective birth cohort of the Hokkaido Study on Environment and Children’s Health. Without consideration of genotype, maternal passive smoker levels (plasma cotinine levels: 0.22–11.48 ng/mL) were associated with a mean reduction of 55–57 g in birth weight, and maternal active smoker levels (plasma cotinine levels: ≥11.49 ng/mL) were associated with a mean reduction of 93–171 g in birth weight, compared with that elicited by non-passive smoker levels (plasma cotinine levels: ≤0.21 ng/mL). When maternal AHR (rs2066853) genotypes were considered, active smoker levels were associated with a mean reduction in birth weight of up to 102 g compared with that caused by non-passive smoker levels among maternal AA genotypes. However, active smoker levels were associated with a mean reduction of 182–217 g compared with that associated with non-passive smoker levels among maternal GG genotypes. Differences have also been observed in maternal XRCC1 (rs1799782) genotypes when examining the dose-dependent association between plasma cotinine levels and birth weight reduction (Fig. 19.1) [24]. Further, it has been shown that this association is not modified by maternal or fetal genotypes of genes encoding xenobiotic enzymes (CYP1A2, CYP1B1, cystathionine beta-synthase [CBS], GST theta pseudogene 1 [GSTTP1], methylenetetrahydrofolate dehydrogenase 1 [MTHFD1], 5-methyltetrahydrofolate-homocysteine methyltransferase [MTR], 5-methyltetrahydrofolate-homocysteine methyltransferase reductase [MTRR], N-acetyltransferase 2 [NAT2], and serine hydroxymethyltransferase 1 [SHMT1]), hormone-related factors and receptors (inhibinα [INHA], luteinizing hormone/chorionic gonadotropin receptor [LHCGR], and transforming growth factor-β receptor type 1 [TGFBR1]), or by none of these factors [21, 24, 29, 30, 32].

Association of maternal AHR (G>A, Arg554Lys; db SNP ID: rs2066853) and XRCC1 (C>T, Arg194Trp; db SNP ID: rs1799782 and G>A, Arg399Gln; db SNP ID: rs25487) genotype with maternal cotinine levels in relation to infant birth weight (n = 3263) [24]. Ninety-five percent confidence intervals (CI). Maternal plasma cotinine levels: level 1, 0.12–0.21 ng/mL; level 2, 0.22–0.55 ng/mL; level 3, 0.56–11.48 ng/mL; level 4, 11.49–101.66 ng/mL; level 5, 101.67–635.25 ng/mL. Multiple linear regression models are adjusted for maternal age, height, weight before pregnancy, parity, alcohol intake during the first trimester of pregnancy, education level, annual household income, infant gender, and gestational age. β represents the change in infant birth weight (g) in comparison with level 1 as the reference. Dot represents β values (95% CI); ∗P < 0.05; ∗∗P < 0.01; ∗∗∗P < 0.001
The following points of the problems should be recognized when evaluating gene–environment interactions of an association between maternal smoking and infant birth size. Some of these problems include: (1) differences in time of evaluation (e.g., first trimester of pregnancy or the 8th month of pregnancy), (2) differences in the questions used for assessment (e.g., cigarettes/day or a choice between yes and no), (3) differences in evaluation methods (e.g., the use of objective biomarkers or subjective questionnaires), (4) differences in study design (e.g., case–control study or prospective birth cohort study), and (5) differences in the location of gene polymorphisms (e.g., rs4646903 or rs1048943 polymorphism of the CYP1A1 gene). Furthermore, maternal exposure levels such as through active smoking or passive smoking (second-hand smoke; SHS) must be considered.
Finally, it is necessary to focus on the components of tobacco smoke, which contains about 4000 substances. Receptors bind the chemical substances contained in tobacco smoke and initiate intracellular reactions. Based on findings from animal studies and cell experiments, it is important to examine gene–environment interactions focusing on single-nucleotide polymorphisms (SNPs) of genes involved in biological mechanisms, e.g., tobacco smoke induces toxicity. Therefore, studies should aim to elucidate the molecular epidemiology underlying the association between maternal smoking and birth outcome to identify the receptors in either the mother or infant (or both) that modify the association between components of tobacco smoke and adverse health effects on infants and children.
3 Effects of Exposure to Other Chemicals on Infant Birth Size
In studies performed up to 2018, many substances that pregnant women are exposed to have been found to modulate gene–environment interactions in fetal growth. These include environmental pollutants (benzo(a)pyrene), disinfection by-products of drinking water (trihalomethanes, chloroforms, and haloacetic acids), fatty acids (cholesterols, triglyceride, and docosahexaenoic acid [DHA]), vitamins (carotenes, vitamin C, vitamin D, and vitamin E), beverage-derived substances (alcohol and caffeine), types of particle matters (PMs) (PMs of aerodynamic diameter <10 μm [PM10], <2.5 μm [PM2.5], and nitrogen oxides [NOx]), metals (lead, mercury, and iron), short half-life chemicals (alkyl phosphates and phthalates), pesticides including those used in floriculture (organochlorines), and persistent organic pollutants (POPs; perfluoroalkyl substances (PFASs), and dioxins) [34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56] (Table 19.2). Reduced birth weight is caused by the association between PM10 levels and maternal CYP1A1 genotype [34], caffeine levels and maternal CYP1A2 genotype [37], benzo(a)pyrene levels and maternal GSTP1 genotype [36], vitamin D levels and maternal genotypes of the gene encoding group-specific component (vitamin D binding protein) locus (GC) [38], cholesterol levels and maternal apolipoprotein E (APOE) and apolipoprotein C3 (APOC3) genotype [39], DHA levels and maternal fatty acid desaturase (FADS) genotype [40], lead levels and maternal HFE genotype related to human hemochromatosis and transferrin (TF) genotypes [41], mercury levels and maternal GSTM1 and GSTT1 genotypes [42], iron levels and maternal GSTM1 genotypes [43], organochlorine pesticide levels and maternal GSTM1 and CYP17A1 genotypes [44, 45], perfluoroalkyl substance levels and maternal GSTM1 genotypes [46], dioxin levels and maternal AHR and GSTM1 genotypes [47, 57], PM2.5 levels and fetal CYP2D6 and GSTP1 genotypes [35], benzo(a)pyrene levels and fetal GSTP1 genotypes [36], and the association between alcohol consumption and fetal ADH2 genotype [50]. Increased risk of LBW is affected by the association between floriculture chemicals and maternal paraoxonase 1 (PON1) genotype [52], disinfection by-product levels in drinking water and maternal GSTM1 genotype [54], and between phthalate levels and fetal paraoxonase 2 (PON2) genotypes [52]. Increased risk of SGA is affected by the association between disinfection by-product levels in drinking water and maternal CYP2E1 and GSTT1 genotypes [55], and that between disinfection by-product levels in drinking water and fetal CYP17A1 genotypes [58]. No risk of SGA is affected by the association between caffeine levels and maternal and infant CYP1A2 genotypes, and the association between caffeine levels and maternal and infant CYP2E1 genotypes [59]. Reduced gestational age is affected by the association between NOx levels and maternal interleukin (IL)-17A genotype [48], and the association between alkyl phosphate levels and maternal PON1 genotypes [49]. Increased risk of IUGR is affected by the association between alcohol consumption and maternal CYP17 genotypes [55], and that between disinfection by-product levels of drinking water and fetal CYP2E1 genotype [56].
Many maternal and infant genes related to chemical substances have never been examined in relation to those involved in xenobiotic metabolism and hormone biosynthesis. However, at present, epidemiological evidence of disease susceptibility genes is limited with regard to gene–environment interactions for maternal chemical exposure and infant growth.
Additional studies are required to examine not only the genotypes of exposure susceptibility genes encoding metabolizing enzymes and receptors related to extraneous substances, but also genetic polymorphisms of disease susceptibility genes such as growth- and obesity-related genes. The results from epidemiological studies on the effects of gene–environment interactions on infant growth may also be of value for planning environmental policies involving genetically high-risk groups and public health programs, as well as for preventive medicine.
4 Recent Advances in Genome Birth Cohort Studies and Evaluation of Genetic Associations with Birth Weight
In large-scale epidemiological studies such as genome birth cohort studies, evaluation of gene–environment interactions is different from that in small-scale epidemiological studies (described in Sects. 19.2 and 19.3). The main reason for this difference is that, unlike hundreds of thousands of genome and epigenome data, it is difficult to obtain environmental data for each subject, except for their cigarette smoking or alcohol intake status. The development of strategies to address this difference in data availability is one of the most important challenges in large-scale epidemiological studies. Therefore, various methods for analyzing genetic susceptibility, such as GWAS, Mendelian randomization, and exposome linked with genomics, have been implemented.
4.1 GWAS and Meta-Analyses
A GWAS implements a population-based experimental design to detect associations between genetic variants and diseases or traits in biological samples from various human genome cohorts [7]. Meta-analyses of GWAS have identified numerous genetic variants associated with birth weight [60,61,62,63,64,65,66,67,68,69,70,71,72] (Table 19.3). Two SNPs, rs900400 near CCNL1 and rs9883204 in ADCY5, were identified to be robustly associated with birth weight [60]. Another related SNP in ADCY5 is considered to be implicated in the regulation of glucose levels and susceptibility to type 2 diabetes based on findings of an adult GWAS [73]. This may be the first evidence that associations between lower birth weight and subsequent non-communicable diseases (NCDs) such as type 2 diabetes have a genetic component; this is known as the developmental origins of health and disease (DOHaD) concept. Furthermore, an expanded GWAS meta-analysis and follow-up study involving 69,308 individuals of European descent from 43 studies revealed seven loci (CCNL1, ADCY5, HMGA2, CDKAL1, LCORL, ADRB1, and 5q11.2) associated with birth weight with genome-wide significance [66]. Among them, five loci are known to be associated with other adult phenotypes: ADCY5 and CDKAL1 with type 2 diabetes, ADRB1 with blood pressure, and HMGA2 and LCORL with height. These findings highlight multiple genetic links between birth weight and postnatal growth and metabolism, especially later in life, which are important in accordance with the DOHaD concept from a genetic point of view. Moreover, a multi-ancestry GWAS meta-analysis of birth weight in 153,781 individuals from the EGG Consortium and the UK Biobank identified 60 loci where fetal genotype was associated with birth weight with genome-wide significance [70]. This study revealed strong inverse genetic correlations between birth weight and adult cardiometabolic diseases and traits such as type 2 diabetes and coronary artery disease, blood pressure, cholesterol levels, and triglyceride levels (Fig. 19.2). Thus, a series of GWAS meta-analyses have confirmed genetic involvement in life-course associations between early growth phenotypes and adult cardiometabolic diseases and traits. GWASs of birth weight have thus far focused on fetal genetics, whereas relatively little is known about the role of maternal genetic variation. Recently, similar GWAS meta-analyses in up to 86,577 women of European descent from the same population revealed maternal loci associated with offspring’s birth weight, such as MTNR1B, HMGA2, SH2B3, KCNAB1, L3MBTL3, GCK, EBF1, TCF7L2, ACTL9, and CYP3A7, at GWAS significance [71]. Interestingly, maternal genetic factors associated with glucose metabolism, blood pressure, immune function, cytochrome P450 activity, and gestational duration affect the offspring’s birth weight. In a recent study, expanded GWASs of the same population examining maternal (n = 321,223) and offspring birth weight (n = 230,069 mothers) revealed 190 independent loci associated with both the mother and offspring’s birth weight [72]. In this study, structural equation modeling was used to evaluate the contribution of direct fetal and indirect maternal genetic effects, and Mendelian randomization was applied to reveal causal pathways. Surprisingly, maternal birth weight-lowering genotypes as proxy for an adverse intrauterine environment were found not to affect offspring blood pressure at all [72]. This finding indicates that some exceptions cannot be explained by the DOHaD concept.

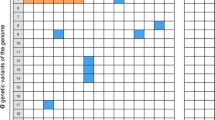
Hierarchical clustering of birth weight (BW) loci based on the similarity of overlap with adult diseases, and metabolic and anthropometric traits [70]. “For the lead SNP at each BW locus (x axis), Z scores (aligned to BW-raising allele) were obtained from publicly available GWAS for various traits (y axis). A positive Z score (red) indicates a positive association between the BW-raising allele and the outcome trait, whereas a negative Z score (blue) indicates an inverse association. BW loci and traits were clustered according to the Euclidean distance among Z scores. Squares are outlined with a solid black line if the BW locus is significantly (P < 5 × 10−8) associated with the trait in publicly available GWAS, or with a dashed line if reported significant elsewhere” [70].
There are few GWAS meta-analyses that have examined the influence of smoking on birth weight; therefore, it is necessary to systematically evaluate gene–environment interactions in relation to smoking, especially genome-wide gene-smoking interactions. Notably, genome-wide gene-smoking interaction studies have become more common in adult GWAS meta-analyses to identify new loci associated with adult traits such as obesity, blood pressure, and serum lipids [74,75,76].
4.2 Mendelian Randomization
An alternative method, Mendelian randomization, entails the use of genetic variants as proxies for the environmental exposure under investigation [77]. Mendelian randomization is a GWAS-based theoretical method for environmental risk assessment that uses genetic factors associated with environmental factors to assess the causal effect on internal biomarkers such as body mass index (BMI), systolic blood pressure, and fasting glucose levels [8, 78]. Birth weight was used as both the outcome of maternal internal biomarkers and the internal marker of fetal intrauterine environment [78,79,80,81,82,83,84,85,86] (Table 19.4). Until recently, genetic risk scores were preferred over SNPs in Mendelian randomization studies [68, 78, 82, 83]. Novel approaches to obtaining genetic risk scores include assessments of the genetic contribution of certain intermediate traits or risk factors to cardiometabolic disease, risk prediction in high-risk populations, studies of gene–environment interactions, and Mendelian randomization [87].
Mendelian randomization is a recent developing field that involves the use of genetic variation to assess the causal relationship between exposure and outcome, where genetic variants within or near coding loci related to protein concentrations enable assessment of their causal role in disease. However, the more complex relationship between genetic variation and exposure makes the findings from Mendelian randomization more difficult to interpret [87]. Recently, the use of maternal birth weight-lowering genotypes to proxy for an adverse intrauterine environment in Mendelian randomization analyses yielded no evidence that such genotype causally raises offspring blood pressure [72]. Mendelian randomization is considered an established method for strengthening causal inference and estimating causal effects; however, the use of genetic instruments, which lack direct links to measurement in most cases, may be one of the limitations of Mendelian randomization.
4.3 Exposome Linked with Genomics
Although GWASs have revealed genetic associations and networks that improve the understanding of diseases, these findings only elucidate a small part of overall disease risk [88]. As disease causation is largely non-genetic, the need for improved tools to quantify environmental contributions seems obvious [89]. The “exposome” was originally defined as representing all kinds of environmental exposure, including those from diet, lifestyle, and endogenous sources, during the entire lifespan from the prenatal period onward, as a quantity of critical interest to disease etiology [90]. Three overlapping domains within the exposome have been described as follows: (1) a general external environment to include factors such as the urban environment, climate factors, social capital, and stress; (2) a specific external environment with specific contaminants, diet, physical activity, tobacco, and infections; and (3) an internal environment to include internal biological factors such as metabolic factors, gut microflora, inflammation, and oxidative stress [91]. Although it is difficult to elucidate the exposome entirely at present, the inherent value of exposomic data in cohort studies is that they can provide a greater understanding of relationships between a wide range of exposure and health conditions, and ultimately lead to more effective and efficient disease prevention and control [92]. For example, the NIH Child Health Environmental Analysis Resource (CHEAR) is a major step toward providing the infrastructure needed to study the exposome in relation to child health [9]. Furthermore, an EU-funded project, EXPOsOMICS, aims to develop a novel approach for assessing exposure to high-priority environmental pollutants, such as air and water contaminants, during critical periods of life, by characterizing the external and internal components of the exposome [93, 94]. Exposome research in the context of developing interventions is targeted at the population level to improve public health, whereas the application of genomics lies in preventive and therapeutic interventions targeted at individuals [94].
4.4 Perspectives
Analysis of genetic factors and environmental exposure offers evidence-based explanations for the associations between LBW, early growth, and increased propensity to develop NCDs in later life [60, 66, 70,71,72, 95]. Among epidemiological studies, GWASs have revealed the relative role of genes and the environment in disease risk, assisting in risk prediction such as in preemptive and precision medicine [96, 97]. Although Mendelian randomization is a powerful tool that utilizes genetic information to explain the likely causal relevance of an exposure to an outcome and increasingly complex gene-to-exposure and exposure-to-outcome relationships; thus, it is more difficult to perform reliable conduct and interpretation [8]. Tremendous strides are being achieved toward developing an exposomics approach together with infrastructural progress toward the identification of new methods and consortia that can address big-picture questions of how environment impacts health and development [9, 93]. Future studies should aim to develop new methods and analytical approaches for exposure assessment and data harmonization [9].
In Japan, the concept of preemptive medicine, which is a novel medical paradigm that advocates for pre-symptomatic diagnosis or prevention intervention at an early stage to prevent disease onset, has been proposed, and policy strategic proposals based on this approach have been made [97, 98]. Recently, collaborations of birth cohort studies are being supported by the project for babies and infants in research of health and development to adolescent and young adult (BIRTHDAY) of the Japan Agency for Medical Research and Development (AMED) on the basis of the current health policy “Overcoming health issues according to life stage” [99]. The rapid progress of large-scale epidemiological studies for elucidating gene–environment interactions is expected to occur in Japan in the near future.
5 Conclusions
Analysis of gene–environment interactions between tobacco and environmental chemicals and candidate genes, such as those encoding metabolic enzymes including CYPs and GSTs, has been performed via small-scale epidemiological studies. In the last 10 years, major innovations in research designs and methods, accompanied by recent rapid advances of analytical and measurement technologies, have occurred. In particular, GWASs and EWASs have become mainstream in genome cohort studies using advanced genomics and epigenomics, which has made it possible to better understand the genetic basis of diseases. As disease causation is largely non-genetic, the concept of the exposome was proposed as an improved tool to quantify total environmental contributions. Linking the exposome with genomics, in combination with preemptive and precision medicine, is expected to reveal novel insights into the origins of multiple complex diseases.
Abbreviations
- ADA1:
-
Adenosine deaminase 1
- AHR:
-
Aromatic hydrocarbon receptor
- CBS:
-
Cystathionine beta-synthase
- CHEAR:
-
Child Health Environmental Analysis Resource
- CYP:
-
Cytochrome P450
- DOHaD:
-
Developmental origins of health and disease
- EGG:
-
Early growth genetics
- GST:
-
Glutathione S-transferase
- GSTTP1:
-
GST theta pseudogene 1
- EPHX1:
-
Epoxide hydrolase 1
- EWAS Epigenome-wide association study GWAS:
-
Genome-wide association study
- LHCGR:
-
Luteinizing hormone/chorionic gonadotropin receptor
- INHA:
-
Inhibinα
- IUGR:
-
Intrauterine growth restriction
- LBW:
-
Low birth weight
- MTHFD1:
-
Methylenetetrahydrofolate dehydrogenase 1
- MTHFR:
-
5,10-Methylenetetrahydrofolate reductase
- MTR:
-
5-Methyltetrahydrofolate-homocysteine methyltransferase
- MTRR:
-
5-Methyltetrahydrofolate-homocysteine methyltransferase reductase
- NAT2:
-
N-acetyltransferase 2
- NCD:
-
Non-communicable disease
- NQO1:
-
NAD(P)H dehydrogenase
- OGG1:
-
8-Oxoguanine glycosylase
- PB:
-
Preterm birth
- SGA:
-
Small-for-gestational-age
- SHMT1:
-
Serine hydroxymethyltransferase 1
- SNP:
-
Single-nucleotide polymorphism
- TGFBR1:
-
Transforming growth factor-β receptor type 1
- XRCC1:
-
X-ray repair cross-complementing gene 1
- XRCC3:
-
X-ray repair cross-complementing gene 3
References
Kishi R, Sata F, Yoshioka E, Ban S, Sasaki S, Konishi K, et al. Exploiting gene-environment interaction to detect adverse health effects of environmental chemicals on the next generation. Basic Clin Pharmacol Toxicol. 2008;102(2):191–203. https://doi.org/10.1111/j.1742-7843.2007.00201.x.
Kelada SN, Eaton DL, Wang SS, Rothman NR, Khoury MJ. The role of genetic polymorphisms in environmental health. Environ Health Perspect. 2003;111(8):1055–64. https://doi.org/10.1289/ehp.6065.
Edwards TM, Myers JP. Environmental exposures and gene regulation in disease etiology. Environ Health Perspect. 2007;115(9):1264–70. https://doi.org/10.1289/ehp.9951.
Kraft P, Hunter D. Integrating epidemiology and genetic association: the challenge of gene-environment interaction. Philos Trans R Soc Lond B Biol Sci. 2005;360(1460):1609–16. https://doi.org/10.1098/rstb.2005.1692.
Kishi R, Sasaki S, Yoshioka E, Yuasa M, Sata F, Saijo Y, et al. Cohort profile: The Hokkaido Study on Environment and Children’s Health in Japan. Int J Epidemiol. 2011;40(3):611–8. https://doi.org/10.1093/ije/dyq071.
Kishi R, Kobayashi S, Ikeno T, Araki A, Miyashita C, Itoh S, et al. Ten years of progress in The Hokkaido Birth Cohort Study on Environment and Children’s Health: cohort profile--updated 2013. Environ Health Prev Med. 2013;18(6):429–50. https://doi.org/10.1007/s12199-013-0357-3.
Visscher PM, Wray NR, Zhang Q, Sklar P, McCarthy MI, Brown MA, et al. 10 years of GWAS discovery: biology, function, and translation. Am J Hum Genet. 2017;101(1):5–22. https://doi.org/10.1016/j.ajhg.2017.06.005.
Holmes MV, Ala-Korpela M, Smith GD. Mendelian randomization in cardiometabolic disease: challenges in evaluating causality. Nat Rev Cardiol. 2017;14(10):577–90. https://doi.org/10.1038/nrcardio.2017.78.
Wright RO. Environment, susceptibility windows, development, and child health. Curr Opin Pediatr. 2017;29(2):211–7. https://doi.org/10.1097/MOP.0000000000000465.
Smith MT, de la Rosa R, Daniels SI. Using exposomics to assess cumulative risks and promote health. Environ Mol Mutagen. 2015;56(9):715–23. https://doi.org/10.1002/em.21985.
Wang X, Zuckerman B, Pearson C, Kaufman G, Chen C, Wang G, et al. Maternal cigarette smoking, metabolic gene polymorphism, and infant birth weight. JAMA. 2002;287(2):195–202. https://doi.org/10.1001/jama.287.2.195.
Sasaki S, Kondo T, Sata F, Saijo Y, Katoh S, Nakajima S, et al. Maternal smoking during pregnancy and genetic polymorphisms in the Ah receptor, CYP1A1 and GSTM1 affect infant birth size in Japanese subjects. Mol Hum Reprod. 2006;12(2):77–83. https://doi.org/10.1093/molehr/gal013.
Sasaki S, Sata F, Katoh S, Saijo Y, Nakajima S, Washino N, et al. Adverse birth outcomes associated with maternal smoking and polymorphisms in the N-Nitrosamine-metabolizing enzyme genes NQO1 and CYP2E1. Am J Epidemiol. 2008;167(6):719–26. https://doi.org/10.1093/aje/kwm360.
Gloria-Bottini F, Magrini A, Cozzoli E, Bergamaschi A, Bottini E. ADA genetic polymorphism and the effect of smoking on neonatal bilirubinemia and developmental parameters. Early Hum Dev. 2008;84(11):739–43. https://doi.org/10.1016/j.earlhumdev.2008.05.001.
Grazuleviciene R, Nieuwenhuijsen MJ, Danileviciute A, Nadisauskiene R, Buinauskiene J. Gene-environment interaction: maternal smoking and contribution of GSTT1 and GSTM1 polymorphisms to infant birth-weight reduction in a Kaunas Cohort Study. J Epidemiol Community Health. 2010;64(7):648. https://doi.org/10.1136/jech.2009.100859.
Aagaard-Tillery K, Spong CY, Thom E, Sibai B, Wendel G Jr, Wenstrom K, et al. Pharmacogenomics of maternal tobacco use: metabolic gene polymorphisms and risk of adverse pregnancy outcomes. Obstet Gynecol. 2010;115(3):568–77. https://doi.org/10.1097/AOG.0b013e3181d06faf.
Karahalil B, Emerce E, Kocabaş NA, Akkaş E. Associations between GSTM1 and OGG1 Ser326Cys polymorphisms and smoking on chromosomal damage and birth growth in mothers. Mol Biol Rep. 2011;38(5):2911–8. https://doi.org/10.1007/s11033-010-9953-0.
Danileviciute A, Grazuleviciene R, Paulauskas A, Nadisauskiene R, Nieuwenhuijsen MJ. Low level maternal smoking and infant birthweight reduction: genetic contributions of GSTT1 and GSTM1 polymorphisms. BMC Pregnancy Childbirth. 2012;12:161. https://doi.org/10.1186/1471-2393-12-161.
Zheng X, Feingold E, Ryckman KK, Shaffer JR, Boyd HA, Feenstra B, et al. Association of maternal CNVs in GSTT1/GSTT2 with smoking, preterm delivery, and low birth weight. Front Genet. 2013;4:196. https://doi.org/10.3389/fgene.2013.00196.
Yila TA, Sasaki S, Miyashita C, Braimoh TS, Kashino I, Kobayashi S, et al. Effects of maternal 5,10-methylenetetrahydrofolate reductase C677T and A1298C Polymorphisms and tobacco smoking on infant birth weight in a Japanese population. J Epidemiol. 2012;22(2):91–102. https://doi.org/10.2188/jea.JE20110039.
Kobayashi S, Sata F, Sasaki S, Braimoh TS, Araki A, Miyashita C, et al. Combined effects of AHR, CYP1A1, and XRCC1 genotypes and prenatal maternal smoking on infant birth size: biomarker assessment in the Hokkaido Study. Reprod Toxicol. 2016;65:295–306. https://doi.org/10.1016/j.reprotox.2016.08.020.
Hong YC, Lee KH, Son BK, Ha EH, Moon HS, Ha M. Effects of the GSTM1 and GSTT1 polymorphisms on the relationship between maternal exposure to environmental tobacco smoke and neonatal birth weight. J Occup Environ Med. 2003;45(5):492–8. https://doi.org/10.1097/01.jom.0000063627.37065.a1.
Wu T, Hu Y, Chen C, Yang F, Li Z, Fang Z, et al. Passive smoking, metabolic gene polymorphisms, and infant birth weight in a prospective cohort study of Chinese women. Am J Epidemiol. 2007;166(3):313–22. https://doi.org/10.1093/aje/kwm090.
Kobayashi S, Sata F, Sasaki S, Braimoh TS, Araki A, Miyashita C, Goudarzi H, et al. Modification of adverse health effects of maternal active and passive smoking by genetic susceptibility: dose-dependent association of plasma cotinine with infant birth size among Japanese women-The Hokkaido Study. Reprod Toxicol. 2017;74:94–103. https://doi.org/10.1016/j.reprotox.2017.09.002.
Infante-Rivard C, Weinberg CR, Guiguet M. Xenobiotic-metabolizing genes and small-for-gestational-age births: interaction with maternal smoking. Epidemiology. 2006;17(1):38–46. https://doi.org/10.1097/01.ede.0000187669.34003.b1.
Xie C, Wen X, Niu Z, Ding P, Liu T, He Y, et al. Combinations of CYP2A6∗4 and glutathione S-transferases gene polymorphisms modify the association between maternal secondhand smoke exposure during pregnancy and small-for-gestational-age. Nicotine Tob Res. 2015;17(12):1421–7. https://doi.org/10.1093/ntr/ntv072.
Huang KH, Chou AK, Jeng SF, Ng S, Hsieh CJ, Chen MH, Chen PC, Hsieh WS. The impacts of cord blood cotinine and glutathione-S-transferase gene polymorphisms on birth outcome. Pediatr Neonatol. 2017;58(4):362–9. https://doi.org/10.1016/j.pedneo.2016.08.006.
Delpisheh A, Brabin L, Topping J, Reyad M, Tang AW, Brabin BJ. A case-control study of CYP1A1, GSTT1 and GSTM1 gene polymorphisms, pregnancy smoking and fetal growth restriction. Eur J Obstet Gynecol Reprod Biol. 2009;143(1):38–42. https://doi.org/10.1016/j.ejogrb.2008.11.006.
Price TS, Grosser T, Plomin R, Jaffee SR. Fetal genotype for the xenobiotic metabolizing enzyme NQO1 influences intrauterine growth among infants whose mothers smoked during pregnancy. Child Dev. 2010;81(1):101–14. https://doi.org/10.1111/j.1467-8624.2009.01383.x.
Nukui T, Day RD, Sims CS, Ness RB, Romkes M. Maternal/newborn GSTT1 null genotype contributes to risk of preterm, low birthweight infants. Pharmacogenetics. 2004;14(9):569–76.
Tsai HJ, Liu X, Mestan K, Yu Y, Zhang S, Fang Y, et al. Maternal cigarette smoking, metabolic gene polymorphisms, and preterm delivery: new insights on GxE interactions and pathogenic pathways. Hum Genet. 2008;123(4):359–69. https://doi.org/10.1007/s00439-008-0485-9.
Huang H, Clancy KB, Burhance C, Zhu Y, Madrigal L. Women who deliver twins are more likely to smoke and have high frequencies of specific SNPs: results from a sample of African-American women who delivered preterm, low birth weight babies. Am J Hum Biol. 2015;27(5):605–12. https://doi.org/10.1002/ajhb.22723.
Grazuleviciene R, Danileviciute A, Nadisauskiene R, Vencloviene J. Maternal smoking, GSTM1 and GSTT1 polymorphism and susceptibility to adverse pregnancy outcomes. Int J Environ Res Public Health. 2009;6(3):1282–97. https://doi.org/10.3390/ijerph6031282.
Suh YJ, Kim BM, Park BH, Park H, Kim YJ, Kim H, et al. Cytochrome P450IA1 polymorphisms along with PM(10) exposure contribute to the risk of birth weight reduction. Reprod Toxicol. 2007;24(3-4):281–8. https://doi.org/10.1016/j.reprotox.2007.07.001.
Slama R, Gräbsch C, Lepeule J, Siroux V, Cyrys J, Sausenthaler S, et al. Maternal fine particulate matter exposure, polymorphism in xenobiotic-metabolizing genes and offspring birth weight. Reprod Toxicol. 2010;30(4):600–12. https://doi.org/10.1016/j.reprotox.2010.07.001.
Duarte-Salles T, Mendez MA, Morales E, Bustamante M, Rodríguez-Vicente A, Kogevinas M, et al. Dietary benzo(a)pyrene and fetal growth: effect modification by vitamin C intake and glutathione S-transferase P1 polymorphism. Environ Int. 2012;45:1–8. https://doi.org/10.1016/j.envint.2012.04.002.
Sasaki S, Limpar M, Sata F, Kobayashi S, Kishi R. Interaction between maternal caffeine intake during pregnancy and CYP1A2 C164A polymorphism affects infant birth size in the Hokkaido Study. Pediatr Res. 2017;82(1):19–28. https://doi.org/10.1038/pr.2017.70.
Chun SK, Shin S, Kim MY, Joung H, Chung J. Effects of maternal genetic polymorphisms in vitamin D-binding protein and serum 25-hydroxyvitamin D concentration on infant birth weight. Nutrition. 2017;35:36–42. https://doi.org/10.1016/j.nut.2016.10.006.
Ruiz JR, Labayen I, Ortega FB, Moreno LA, González-Lamuño D, Martí A, et al. Birth weight and blood lipid levels in Spanish adolescents: influence of selected APOE, APOC3 and PPARgamma2 gene polymorphisms. The AVENA Study. BMC Med Genet. 2008;9:98. https://doi.org/10.1186/1471-2350-9-98.
Moltó-Puigmartí C, van Dongen MC, Dagnelie PC, Plat J, Mensink RP, Tan FE, et al. Maternal but not fetal FADS gene variants modify the association between maternal long-chain PUFA intake in pregnancy and birth weight. J Nutr. 2014;144(9):1430–7. https://doi.org/10.3945/jn.114.194035.
Cantonwine D, Hu H, Téllez-Rojo MM, Sánchez BN, Lamadrid-Figueroa H, Ettinger AS, et al. HFE gene variants modify the association between maternal lead burden and infant birthweight: a prospective birth cohort study in Mexico City, Mexico. Environ Health. 2010;9:43. https://doi.org/10.1186/1476-069X-9-43.
Lee BE, Hong YC, Park H, Ha M, Koo BS, Chang N, et al. Interaction between GSTM1/GSTT1 polymorphism and blood mercury on birth weight. Environ Health Perspect. 2010;118(3):437–43. https://doi.org/10.1289/ehp.0900731.
Hur J, Kim H, Ha EH, Park H, Ha M, Kim Y, et al. Birth weight of Korean infants is affected by the interaction of maternal iron intake and GSTM1 polymorphism. J Nutr. 2013;143(1):67–73. https://doi.org/10.3945/jn.112.161638.
Sharma E, Mustafa M, Pathak R, Guleria K, Ahmed RS, Vaid NB, et al. A case control study of gene environmental interaction in fetal growth restriction with special reference to organochlorine pesticides. Eur J Obstet Gynecol Reprod Biol. 2012;161(2):163–9. https://doi.org/10.1016/j.ejogrb.2012.01.008.
Chand S, Mustafa MD, Banerjee BD, Guleria K. CYP17A1 gene polymorphisms and environmental exposure to organochlorine pesticides contribute to the risk of small for gestational age. Eur J Obstet Gynecol Reprod Biol. 2014;180:100–5. https://doi.org/10.1016/j.ejogrb.2014.06.016.
Kwon EJ, Shin JS, Kim BM, Shah-Kulkarni S, Park H, Kho YL, et al. Prenatal exposure to perfluorinated compounds affects birth weight through GSTM1 polymorphism. J Occup Environ Med. 2016;58(6):e198–205. https://doi.org/10.1097/JOM.0000000000000739.
Kobayashi S, Sata F, Miyashita C, Sasaki S, Ban S, Araki A, et al. Dioxin-metabolizing genes in relation to effects of prenatal dioxin levels and reduced birth size: The Hokkaido Study. Reprod Toxicol. 2017;67:111–6. https://doi.org/10.1016/j.reprotox.2016.12.002.
Nansook P, Naidoo RN, Muttoo S, Asharam K, Ramkaran P, Phulukdaree A, et al. IL-17A[G197G]-association between NOx and gestational age in a South African Birth Cohort. Int J Immunogenet. 2018;45(2):54–62. https://doi.org/10.1111/iji.12358.
Harley KG, Huen K, Aguilar Schall R, Holland NT, Bradman A, Barr DB, et al. Association of organophosphate pesticide exposure and paraoxonase with birth outcome in Mexican-American women. PLoS One. 2011;6(8):e23923. https://doi.org/10.1371/journal.pone.0023923.
Arfsten DP, Silbergeld EK, Loffredo CA. Fetal ADH2∗3, maternal alcohol consumption, and fetal growth. Int J Toxicol. 2004;23(1):47–54. https://doi.org/10.1080/10915810490265450.
Kogevinas M, Bustamante M, Gracia-Lavedán E, Ballester F, Cordier S, Costet N, et al. Drinking water disinfection by-products, genetic polymorphisms, and birth outcomes in a European Mother-Child Cohort Study. Epidemiology. 2016;27(6):903–11.
Moreno-Banda G, Blanco-Muñoz J, Lacasaña M, Rothenberg SJ, Aguilar-Garduño C, Gamboa R, et al. Maternal exposure to floricultural work during pregnancy, PON1 Q192R polymorphisms and the risk of low birth weight. Sci Total Environ. 2009;407(21):5478–85. https://doi.org/10.1016/j.scitotenv.2009.06.033.
Xie C, Jin R, Zhao Y, Lin L, Li L, Chen J, et al. Paraoxonase 2 gene polymorphisms and prenatal phthalates’ exposure in Chinese newborns. Environ Res. 2015;140:354–9. https://doi.org/10.1016/j.envres.2015.03.028.
Danileviciute A, Grazuleviciene R, Vencloviene J, Paulauskas A, Nieuwenhuijsen MJ. Exposure to drinking water trihalomethanes and their association with low birth weight and small for gestational age in genetically susceptible women. Int J Environ Res Public Health. 2012;9(12):4470–85. https://doi.org/10.3390/ijerph9124470.
Delpisheh A, Topping J, Reyad M, Tang A, Brabin BJ. Prenatal alcohol exposure, CYP17 gene polymorphisms and fetal growth restriction. Eur J Obstet Gynecol Reprod Biol. 2008;138(1):49–53. https://doi.org/10.1016/j.ejogrb.2007.08.006.
Infante-Rivard C. Drinking water contaminants, gene polymorphisms, and fetal growth. Environ Health Perspect. 2004;112(11):1213–6. https://doi.org/10.1289/ehp.7003.
Ames J, Warner M, Mocarelli P, Brambilla P, Signorini S, Siracusa C, et al. AHR gene-dioxin interactions and birthweight in the Seveso Second Generation Health Study. Int J Epidemiol. 2018;47(6):1992–2004. https://doi.org/10.1093/ije/dyy165.
Bonou SG, Levallois P, Giguère Y, Rodriguez M, Bureau A. Prenatal exposure to drinking-water chlorination by-products, cytochrome P450 gene polymorphisms and small-for-gestational-age neonates. Reprod Toxicol. 2017;73:75–86. https://doi.org/10.1016/j.reprotox.2017.07.019.
Infante-Rivard C. Caffeine intake and small-for-gestational-age birth: modifying effects of xenobiotic-metabolising genes and smoking. Paediatr Perinat Epidemiol. 2007;21(4):300–9. https://doi.org/10.1111/j.1365-3016.2007.00825.x.
Freathy RM, Mook-Kanamori DO, Sovio U, Prokopenko I, Timpson NJ, Berry DJ, et al. Variants in ADCY5 and near CCNL1 are associated with fetal growth and birth weight. Nat Genet. 2010;42:430–5. https://doi.org/10.1038/ng.567.
Andersson EA, Pilgaard K, Pisinger C, Harder MN, Grarup N, Faerch K, et al. Type 2 diabetes risk alleles near ADCY5, CDKAL1 and HHEX-IDE are associated with reduced birthweight. Diabetologia. 2010;53(9):1908–16. https://doi.org/10.1007/s00125-010-1790-0.
Andersson EA, Harder MN, Pilgaard K, Pisinger C, Stančáková A, Kuusisto J, et al. The birth weight lowering C-allele of rs900400 near LEKR1 and CCNL1 associates with elevated insulin release following an oral glucose challenge. PLoS One. 2011;6(11):e27096. https://doi.org/10.1371/journal.pone.0027096.
Kilpeläinen TO, den Hoed M, Ong KK, Grøntved A, Brage S, Early Growth Genetics Consortium, et al. Obesity-susceptibility loci have a limited influence on birth weight: a meta-analysis of up to 28,219 individuals. Am J Clin Nutr. 2011;93(4):851–60. https://doi.org/10.3945/ajcn.110.000828.
Ryckman KK, Feenstra B, Shaffer JR, Bream EN, Geller F, Feingold E, et al. Replication of a genome-wide association study of birth weight in preterm neonates. J Pediatr. 2012;160(1):19–24. https://doi.org/10.1016/j.jpeds.2011.07.038.
Urbanek M, Hayes MG, Armstrong LL, Morrison J, Lowe LP, Badon SE, et al. The chromosome 3q25 genomic region is associated with measures of adiposity in newborns in a multi-ethnic genome-wide association study. Hum Mol Genet. 2013;22(17):3583–96. https://doi.org/10.1093/hmg/ddt168.
Horikoshi M, Yaghootkar H, Mook-Kanamori DO, Sovio U, Taal HR, Hennig BJ, et al. New loci associated with birth weight identify genetic links between intrauterine growth and adult height and metabolism. Nat Genet. 2013;45:76–82. https://doi.org/10.1038/ng.2477.
Metrustry SJ, Edwards MH, Medland SE, Holloway JW, Montgomery GW, Martin NG, et al. Variants close to NTRK2 gene are associated with birth weight in female twins. Twin Res Hum Genet. 2014;17(4):254–61. https://doi.org/10.1017/thg.2014.34.
Elks CE, Heude B, de Zegher F, Barton SJ, Clément K, Inskip HM, et al. Associations between genetic obesity susceptibility and early postnatal fat and lean mass: an individual participant meta-analysis. JAMA Pediatr. 2014;168(12):1122–30. https://doi.org/10.1001/jamapediatrics.2014.1619.
Wang T, Huang T, Li Y, Zheng Y, Manson JE, Hu FB, et al. Low birthweight and risk of type 2 diabetes: A Mendelian Randomisation Study. Diabetologia. 2016;59(9):1920–7. https://doi.org/10.1007/s00125-016-4019-z.
Horikoshi M, Beaumont RN, Day FR, Warrington NM, Kooijman MN, Fernandez-Tajes J, et al. Genome-wide associations for birth weight and correlations with adult disease. Nature. 2016;538:248–52.
Beaumont RN, Warrington NM, Cavadino A, Tyrrell J, Nodzenski M, Horikoshi M, et al. Genome-wide association study of offspring birth weight in 86,577 women identifies five novel loci and highlights maternal genetic effects that are independent of fetal genetics. Hum Mol Genet. 2018;27(4):742–56. https://doi.org/10.1093/hmg/ddx429.
Warrington NM, Beaumont RN, Horikoshi M, Day FR, Helgeland Ø, Laurin C, et al. Maternal and fetal genetic effects on birth weight and their relevance to cardio-metabolic risk factors. Nat Genet. 2019;51(5):804–14. https://doi.org/10.1038/s41588-019-0403-1.
Dupuis J, Langenberg C, Prokopenko I, Saxena R, Soranzo N, Jackson AU, et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet. 2010;42(2):105–16. https://doi.org/10.1038/ng.520.
Justice AE, Winkler TW, Feitosa MF, Graff M, Fisher VA, Young K, et al. Genome-wide meta-analysis of 241,258 adults accounting for smoking behaviour identifies novel loci for obesity traits. Nat Commun. 2017;8:14977. https://doi.org/10.1038/ncomms14977.
Sung YJ, Winkler TW, de Las Fuentes L, Bentley AR, Brown MR, Kraja AT, et al. A large-scale multi-ancestry genome-wide study accounting for smoking behavior identifies multiple significant loci for blood pressure. Am J Hum Genet. 2018;102(3):375–400. https://doi.org/10.1016/j.ajhg.2018.01.015.
Bentley AR, Sung YJ, Brown MR, Winkler TW, Kraja AT, Ntalla I, et al. Multi-ancestry genome-wide gene-smoking interaction study of 387,272 individuals identifies new loci associated with serum lipids. Nat Genet. 2019;51(4):636–48. https://doi.org/10.1038/s41588-019-0378-y.
Sleiman PM, Grant SF. Mendelian randomization in the era of genomewide association studies. Clin Chem. 2010;56(5):723–8. https://doi.org/10.1373/clinchem.2009.141564.
Tyrrell J, Richmond RC, Palmer TM, Feenstra B, Rangarajan J, Metrustry S, et al. Genetic evidence for causal relationships between maternal obesity-related traits and birth weight. JAMA. 2016;315(11):1129–40. https://doi.org/10.1001/jama.2016.1975.
Lee HA, Park EA, Cho SJ, Kim HS, Kim YJ, Lee H, et al. Mendelian randomization analysis of the effect of maternal homocysteine during pregnancy, as represented by maternal MTHFR C677T genotype, on birth weight. J Epidemiol. 2013;23(5):371–5.
Yajnik CS, Chandak GR, Joglekar C, Katre P, Bhat DS, Singh SN, et al. Maternal homocysteine in pregnancy and offspring birthweight: epidemiological associations and Mendelian randomization analysis. Int J Epidemiol. 2014;43(5):1487–97. https://doi.org/10.1093/ije/dyu132.
Bouthoorn SH, van Lenthe FJ, Kiefte-de Jong JC, Taal HR, Wijtzes AI, Hofman A, et al. Genetic taste blindness to bitter and body composition in childhood: a Mendelian randomization design. Int J Obes. 2014;38(7):1005–10. https://doi.org/10.1038/ijo.2013.141.
Zhang G, Bacelis J, Lengyel C, Teramo K, Hallman M, Helgeland Ø, et al. Assessing the causal relationship of maternal height on birth size and gestational age at birth: a Mendelian randomization analysis. PLoS Med. 2015;12(8):e1001865. https://doi.org/10.1371/journal.pmed.1001865.
Wang T, Huang T, Li Y, Zheng Y, Manson JE, Hu FB, Qi L. Low birthweight and risk of type 2 diabetes: a Mendelian randomisation study. Diabetologia. 2016;59(9):1920–7. https://doi.org/10.1007/s00125-016-4019-z.
Au Yeung SL, Lin SL, Li AM, Schooling CM. Birth weight and risk of ischemic heart disease: a Mendelian randomization study. Sci Rep. 2016;6:38420. https://doi.org/10.1038/srep38420.
Bernard JY, Pan H, Aris IM, Moreno-Betancur M, Soh SE, Yap F, et al. Long-chain polyunsaturated fatty acids, gestation duration, and birth size: a Mendelian randomization study using fatty acid desaturase variants. Am J Clin Nutr. 2018;108(1):92–100. https://doi.org/10.1093/ajcn/nqy079.
Geng TT, Huang T. Maternal central obesity and birth size: a Mendelian randomization analysis. Lipids Health Dis. 2018;17(1):181. https://doi.org/10.1186/s12944-018-0831-4.
Smith JA, Ware EB, Middha P, Beacher L, Kardia SL. Current applications of genetic risk scores to cardiovascular outcomes and subclinical phenotypes. Curr Epidemiol Rep. 2015;2(3):180–90.
Claussnitzer M, Dankel SN, Klocke B, Grallert H, Glunk V, Berulava T, et al. Leveraging cross-species transcription factor binding site patterns: from diabetes risk loci to disease mechanisms. Cell. 2014;156(1-2):343–58. https://doi.org/10.1016/j.cell.2013.10.058.
Miller GW, Jones DP. The nature of nurture: refining the definition of the exposome. Toxicol Sci. 2014;137(1):1–2. https://doi.org/10.1093/toxsci/kft251.
Wild CP. Complementing the genome with an “exposome”: the outstanding challenge of environmental exposure measurement in molecular epidemiology. Cancer Epidemiol Biomarkers Prev. 2005;14(8):1847–50. https://doi.org/10.1158/1055-9965.EPI-05-0456.
Wild CP. The exposome: from concept to utility. Int J Epidemiol. 2012;41(1):24–32. https://doi.org/10.1093/ije/dyr236.
DeBord DG, Carreón T, Lentz TJ, Middendorf PJ, Hoover MD, Schulte PA. Use of the “exposome” in the practice of epidemiology: a primer on -omic technologies. Am J Epidemiol. 2016;184(4):302–14. https://doi.org/10.1093/aje/kwv325.
Vineis P, Chadeau-Hyam M, Gmuender H, Gulliver J, Herceg Z, Kleinjans J, et al. The exposome in practice: design of the EXPOsOMICS project. Int J Hyg Environ Health. 2017;220(2 Pt A):142–51. https://doi.org/10.1016/j.ijheh.2016.08.001.
Turner MC, Vineis P, Seleiro E, Dijmarescu M, Balshaw D, Bertollini R, et al. EXPOsOMICS: final policy workshop and stakeholder consultation. BMC Public Health. 2018;18(1):260. https://doi.org/10.1186/s12889-018-5160-z.
Hanson MA, Gluckman PD. Early developmental conditioning of later health and disease: physiology or pathophysiology? Physiol Rev. 2014;94(4):1027–76. https://doi.org/10.1152/physrev.00029.2013.
Chatterjee N, Shi J, García-Closas M. Developing and evaluating polygenic risk prediction models for stratified disease prevention. Nat Rev Genet. 2016;17(7):392–406. https://doi.org/10.1038/nrg.2016.27.
Imura H. Life course health care and preemptive approach to non-communicable diseases. Proc Jpn Acad Ser B Phys Biol Sci. 2013;89(10):462–73. https://doi.org/10.2183/pjab.89.462.
Sata F. Developmental origins of health and disease (DOHaD) cohorts and interventions: status and perspective. In: Sata F, Fukuoka H, Hanson M, editors. Pre-emptive medicine: public health aspects of developmental origins of health and disease. Singapore: Springer; 2019. p. 53–70.
https://www.amed.go.jp/en/program/list/04/02/002.html. Accessed 23 May 2019.
Acknowledgements
Our work was supported in part by Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science; the Japan Ministry of Health, Labour, and Welfare; and the Japan Agency for Medical Research and Development (AMED) under Grant Number JP18gk0110032.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Sata, F., Kobayashi, S., Kishi, R. (2020). Gene–Environment Interactions to Detect Adverse Health Effects on the Next Generation. In: Kishi, R., Grandjean, P. (eds) Health Impacts of Developmental Exposure to Environmental Chemicals. Current Topics in Environmental Health and Preventive Medicine. Springer, Singapore. https://doi.org/10.1007/978-981-15-0520-1_19
Download citation
DOI: https://doi.org/10.1007/978-981-15-0520-1_19
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-0519-5
Online ISBN: 978-981-15-0520-1
eBook Packages: MedicineMedicine (R0)