
Abstract
Ceramic powders are synthesized using chemical solution methods, such as organic acid salt, precipitant generation, alkaline hydrolysis, and alkoxide hydrolysis. The powder synthesis by precipitation and hydrolysis and the synthesis of monodispersed particles are described. The formation of complex alkoxide of lithium niobate (LN) precursor is confirmed by 93Nb nuclear magnetic resonance spectroscopy. Stoichiometric LN films are synthesized on various substrates via chemical solution route. The synthesis of the designed precursor in solution is a key for the low-temperature crystallization of high-quality LN films with preferred orientation. Several functional ceramic thin films of multicomponent oxides are also synthesized using chemical solution process.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
4.1 Fabrication Processes for Ceramic Powders and Films
Ceramic powders are mainly synthesized via three methods: solid phase, liquid phase, and gas phase reaction. In the solid phase method, a raw material oxide is calcined at temperatures of 1000 °C or above, and a highly crystalline product is obtained. In the liquid phase method, raw material components dissolved into a liquid solution are used as starting material, and the synthesized ceramic powder component is homogenous and has an active particulate surface and controllable powder characteristics. In the gas phase method, since the starting compounds are reacted in an inert gas, a high-purity product is more easily obtained. The gas phase also enables synthesis of fine powders of nitrides, carbides, etc. which are difficult to synthesize using liquid phase reactions.
Two major methods are used for ceramic thin film synthesis: physical methods and chemical methods. Physical methods include heating thin film raw materials in a vacuum apparatus or vaporizing the thin film raw material via sputtering, laser ablation, etc., to deposit the vapor particulate onto a substrate to make the film. Chemical methods consist of vapor phase growth methods and chemical solution methods. In vapor phase growth, thin film raw materials are reacted with halides or hydrides in the gas phase, and the thin film compound is vapor-deposited onto a substrate. In the chemical solution method, a film is formed on a substrate using different methods of coating of the raw material solution and crystallized to form an oxide thin film.
This chapter describes the synthesis methods of ceramic powders and thin films with the liquid phase method using chemical reactions in solution.
4.2 Powder Synthesis with the Chemical Solution Method
4.2.1 Characteristics of Synthesis with the Chemical Solution Method
Crystallization of a product is often used to realize desired physical characteristics in ceramic materials. To do so, the starting compounds must undergo heat treatment at high temperatures of 1000 °C or above. In the ordinary solid phase method, a mechanical mixture of raw material oxide powder and other materials is calcined; however, the component elements are not mixed homogeneously. Even after high-temperature processing, segregations and second-phase remnants can be present.
In contrast, by controlling solution reactions in the chemical solution method, the component elements can be mixed in the preheat treatment stage to homogenize them at the molecular level, thereby resolving the above-described problem points. The chemical solution method is characterized as follows: crystal growth is possible at low temperatures, component control is easy, homogeneous additions of trace elements are possible, and there is high homogeneity of product. Another major characteristic of the solution method is that, in addition to powders, porous materials, films, fibers, and a variety of other material configurations, can also be synthesized. The preparation of such films and fibers from solid phase reactions using oxides is difficult.
4.2.2 Powder Synthesis via Precipitation
Figure 4.1 shows the classifications of ceramics precipitated from solution and formed by desolventizing. One ceramic powder synthesis reaction from solution is the precipitation reaction, where coprecipitation, hydrolysis, etc. are used to obtain a multicomponent hydroxide powder. Spray-drying and freeze-drying are means of removing the solvent from a solution containing metal ions. Another liquid phase method is the hydrothermal technique for synthesizing a single crystal such as quartz under pressure; this technique is also used to synthesize nanocrystalline powders. This section describes precipitation reactions under normal pressure.

Classifications of ceramics precipitated from solution and formed by desolventizing
To precipitate a powder from solution, in some cases the precipitant is added from outside, and in others the precipitate is formed within the solution via chemical reactions. For metal-organic acid salt method, the precipitants, such as oxalate and citrate, are added externally. When sodium citrate is added to an aqueous solution with a dissolved mineral salt, a metal citrate salt is precipitated [1]. For example, when aqueous oxalic acid solution is reacted with an aqueous solution containing barium chloride and titanium oxychloride solution such that the Ba/Ti ratio is 1.0, a white BaTiO(C2O4)2 ⋅ 4H2O precipitate is synthesized. Stoichiometric barium titanate (BaTiO3) powder is prepared by heating this precipitate in air and removing its organic constituent. This powder has superior sinterability and is used as a high-purity, dielectric base powder.
The precipitant generation method involves making the precipitant within a solution and causing a homogeneous precipitation reaction. One such precipitant is ammonia made by hydrolysis after heating an aqueous urea solution above 70 °C [2]. Precipitant generation can be controlled via selection of urea concentration, temperature, pH, etc.
Other precipitants are diethyl sulfate, thiourea, etc., used for precipitant generation of sulfate and sulfide, respectively.
In the alkaline hydrolysis method, alkali is added to an acidic metal salt solution, which then undergoes hydrolysis. A metal salt such as nitrate has high solubility in water, and a homogenous solution can be prepared and includes the target component elements. Making this solution alkaline enables precipitation of a metal hydroxide or oxide. Anions and H2O coordinate with metal ions in the solution to form complex ions. These complex ions form a polynuclear complex via hydrolysis and condensation reactions. For example, Al3+ forms a hydrated ion in aqueous solution and forms a polynuclear complex such as [A1O4A112(OH)24(H2O)12]7+ [3].
The polynuclear complex further undergoes repeated association and dissociation. During this growth when the complex exceeds a certain size (the critical radius), crystal nuclei are formed and precipitated as particles. Precipitated particles can be separated and heat-treated at high temperatures to obtain the metal-oxide powder.
When precipitating a multicomponent hydroxide from a solution including multiple types of metal ions, careful attention is required for differences in the solubility products of respective metal hydroxides. When there are large differences in the solubilities of the metal ions due to pH, one must be aware that the precipitate composition may not match the solution composition.
The alkoxide hydrolysis method is also called the sol-gel method. A metal alkoxide is a compound made of M—O—R bonds (M, metal element; R, alkyl group) and is expressed by M(OR)n. The M—O bond is a bond between an electropositive metal and a negative oxygen and is readily susceptible to hydrolysis, yielding an oxide as follows [4]:
-
Hydrolysis reaction
$$ \mathrm{M}{\left(\mathrm{OR}\right)}_{\mathrm{n}}+{\mathrm{H}}_2\mathrm{O}\to \mathrm{M}{\left(\mathrm{OR}\right)}_{\mathrm{n}-1}\left(\mathrm{OH}\right)+\mathrm{ROH} $$(4.4)$$ \mathrm{M}{\left(\mathrm{OR}\right)}_{\mathrm{n}-1}\left(\mathrm{OH}\right)+\left(\mathrm{n}-1\right){\mathrm{H}}_2\mathrm{O}\to \mathrm{M}{\left(\mathrm{OH}\right)}_{\mathrm{n}}+\left(\mathrm{n}-1\right)\mathrm{ROH} $$(4.5) -
Polycondensation reaction
$$ \mathrm{mM}{\left(\mathrm{OH}\right)}_{\mathrm{n}}\to {\mathrm{mMO}}_{\mathrm{n}/2}+\mathrm{nm}/2{\mathrm{H}}_2\mathrm{O} $$(4.6)
The formation of a complex alkoxide by the reaction of two or more metal alkoxides is also known. Synthesis of an appropriate complex alkoxide and its use as a starting material enable attainment of a complex oxide as shown in the following formula:
Hydrolysis and polycondensation
The alkoxide hydrolysis product easily forms a network structure, and after passing through a sol phase, it changes to a gel. Using these structural changes, inorganic materials having a variety of shapes can be synthesized. Three-dimensional structures of products can be controlled by adjusting the alkoxide hydrolysis conditions. Hydrolysis of alkoxysilane (silicon alkoxide, Si(OR)4) is known to progress under acidic conditions via electrophilic substitution reactions and under basic conditions by nucleophilic substitution reactions [5]. Under acidic conditions, H3O+ electrophilically attacks the oxygen of the Si—OR bond alkoxy groups, and with the elimination of ROH, Si—OH bonds are formed. Si—OH bonds condense with other Si—OH bonds, and a chain polymer is formed. Under basic conditions, however, OH- ions nucleophilically attack Si, and OR- groups are eliminated. Due to steric hindrance of the substituent groups around the central Si, “bulkier” R groups show a more rapid decrease in reactivity. Generally, under acidic conditions, a network structure develops, and it is easy for a gel to form, while under basic conditions hydrated particles are produced. In this way, the hydrolysis reaction rate and resultant product of alkoxide depend on a variety of factors including acidity, basicity, as well as substituent type, solvent, reaction temperature, water quantity used for hydrolysis, concentrations, etc. Appropriate selection of these conditions enables control of the product’s three-dimensional structure, as well as adjustment of sol viscosity. Spinnable gel fibers can be obtained from a precursor sol whose viscosity has been controlled, enabling preparation of ceramic fibers.
4.2.3 Synthesis of Monodispersed Spherical Particles
Important particle properties are shape, particle size, and particle size distribution. These properties impact the electrical and optical properties of the particles themselves. A well-known example is that the coercivity of a magnetic particle depends on its particle size (particle diameter). When the particle size decreases and its size changes from a multi-magnetic domain structure to a single domain structure, a large coercive force is observed [6]. Another example of size effect is that the optical absorption edge of a semiconductor particle is dependent on particle size, and at nanosizes, the short wavelength shift also depends on the particle size [7]. In the preparation of a ceramic sintered body, to ensure that it has the desired properties, it is necessary to control the particle size and size distribution of the raw material particles. When particles of submicron size have a narrow size distribution, the sintering temperature can be lowered, and a dense sintered body with controlled microstructure can be prepared. Thus, particles with a controlled particle shape, size, and size distribution are required to meet a desired goal. In a solution reaction, monodispersed spherical particles with a sharp size distribution can be prepared by controlling nucleation and growth.
Figure 4.2 shows the relationship between time and solute concentration when synthesizing monodispersed spherical particles [8]. Here, “solute” means the precursor materials such as polynuclear complexes and polycondensates dissolved in the solution prior to precipitation. As the solute concentration changes, crystal nuclei form and particle growth occurs. In the initial growth process, t n is the time from when the solute concentration rises and exceeds the degree of saturation C s, until it reaches the supersaturated concentration C ss. From this t n to the t g stage, self-nucleation occurs, and during the period until the concentration again declines to C s, the dispersion of chemical species to crystal nuclei occurs leading to crystal growth. Nuclei generated in the first period continue to exist, and their particle diameters (sizes) become bigger than the nuclei generated later. Thus, in the brief time period from t n to t g, if large numbers of nuclei are explosively generated to promote uniform growth, monodispersed spherical particles can be produced. In the growth period, small-sized (small-diameter) particles are redissolved and disappear, and the solute component precipitates as large-sized (large-diameter) particles. After passing through these processes, the maximum of the particle size distribution curve changes from small particle diameters to large particle diameters, a process known as “Ostwald ripening.”

Relationship between time and solute concentration for the synthesis of monodispersed spherical particle. (Reprinted with permission from Ref. [8]. Copyright 1950 American Chemical Society)
To speed up the nucleation rate so as to complete nucleation at an early time point, the solute concentration must be raised to speed up particle formation reactions. To slow down the rate of particle growth, one must select conditions such as the supersaturation degree and temperature. The generated nuclei are nanoparticles that agglomerate easily. When particle agglomeration occurs in the early stage and secondary particles are formed, these become polydispersed particles. Since it is extremely difficult to dissociate such agglomerated secondary particles for redispersion, a dispersant is added to the solution beforehand, and this effectively prevents agglomeration. Polyelectrolytes, such as sodium polymethacrylate and ammonium polyacrylate, are used as dispersants. A dispersant is adsorbed at the particle surface, the surface charge is controlled, and—due to steric repulsion of polymer chains and other effects—the dispersion state of nanocrystalline particles is stabilized.
Stöber et al. [9] investigated the hydrolysis of silicon alkoxide and found that controlling the growth of Si(OH)4 produced in the intermediate stage enabled the synthesis of monodispersed spherical silica particles of diameter 0.05 μm–2.0 μm. Small-sized particles were obtained from methanol, and large-sized particles were obtained from butanol. In a comparison of silicon substituents, when Si(OCH3)4 was used, the reaction time was within 1 min, and silica particles with a diameter of 0.2 μm or less were obtained. With Si(OC2H5)4 and a reaction time of 24 h, silica particles of approximately 2 μm were obtained. Monodispersed spherical particles of a variety of functional ceramics including silica, TiO2, ZrO2, etc. can be synthesized [10].
4.3 Thin Film Synthesis by the Chemical Solution Method
4.3.1 Thin Film Synthesis
Utilizing the characteristics of the solution method, numerous kinds of functional ceramic thin films are currently synthesized. Examples of electronic functional thin film types currently synthesized are ferroelectrics (BaTiO3, Pb(Ti,Zr)O3) [11], transparent conductors (Sn-doped In2O3) [12], superionic conductors (Na2O•11Al2O3) [13], superconductors (YBa2CuO7) [14], and others. Optical functional films synthesized by the solution method include light absorption and colored films (FeO, NiO [15], CeO2-TiO2 [16]), reflection coatings (PbO-TiO2 [17], TiO2-SiO2 [18]), second-order nonlinear optical films (KTiOPO4 [19], β-BaB2O4 [20], Ba2NaNb5O15 [21]), etc. As a representative example, the synthesis of a stoichiometric epitaxial LiNbO3 film is described below.
4.3.2 Synthesis of Stoichiometric Epitaxial Lithium Niobate Thin Film
4.3.2.1 Lithium Niobate
The structure of lithium niobate (LiNbO3) has hexagonal closest packing of oxygen ions, and a lithium ion and a niobium ion occupy two-thirds of the octahedral sites. This crystal structure is called an “ilmenite-type structure” and belongs to the trigonal system. Therefore, at room temperature, the lithium and niobium ions are at locations slightly askew of the center of the tetrahedral position, and spontaneous polarization occurs; thus, at room temperature, LiNbO3 is a ferroelectric. Due to its superior piezoelectric and optical properties, LiNbO3 is a functional inorganic material that has been researched for many years for its optical material applications, for example, as a surface acoustic wave (SAW) element, etc.
Figure 4.3 shows an equilibrium phase diagram of LiNbO3, comprising lithium oxide (Li2O) and niobium oxide (Nb2O5), with magnification of the vicinity of 50 mol% Li2O [22]. While 50 mol% Li2O is equivalent to LiNbO3, there exists a solid solution region of width 2%–3% in the vicinity of this stoichiometric composition. The apex of the dome seen at 1250 °C in this phase diagram is called the “congruent melting composition.” Detailed research has shown that this congruent melting composition is 48.45 mol% Li2O. In other words, at 1253 °C, LiNbO3 does not decompose—rather, it is dissolved as is and becomes LiNbO3 melt; that composition, however, is not the 50.0 mol% stoichiometric composition.

Equilibrium phase diagram of LiNbO3 consisting of lithium oxide (Li2O) and niobium oxide (Nb2O5). (Reprinted with permission from Ref. [22]. Copyright 1971 American Institute of Physics)
The LiNbO3 single crystal is grown using the Czochralski method, by raising a LiNbO3 seed crystal from LiNbO3 melt maintained at high temperatures while rotating it. However, there is a lithium shortage in the melt at 1250 °C. Therefore, a single crystal obtained using the Czochralski method does not have a Li 50% and Nb 50% stoichiometric composition but a nonstoichiometric composition with Li shortage. As shown by the dashed line in Fig. 4.3, the Curie point (the ferroelectric-paraelectric phase-transition temperature) of LiNbO3 is sensitive to the Li2O constituent and changes with composition from 1020 to 1180 °C. The important optical property, the refractive index, changes depending on composition. Further, since this is a nonstoichiometric composition, defects occur within the LiNbO3 crystal, and LiNbO3 crystals can easily undergo optical damage from laser light, etc., used in optical applications. Thus, LiNbO3 with a good stoichiometric composition is desired.
While it is difficult to obtain a stoichiometric composition LiNbO3 using the commonly used Czochralski method, stoichiometric LiNbO3 can be synthesized using the chemical solution method.
The chemical solution method can be used to synthesize an epitaxial thin film, which is also considered to be a single-crystalline film or preferentially oriented film, where the crystal growth surface is oriented in a specific direction.
4.3.2.2 Synthesis and Structure of LiNbO3 (LN) Precursor
For thin film synthesis, a precursor solution containing the component element compounds is coated onto a substrate to prepare the precursor thin film. The following coating methods exist among others: the dip-coating method, in which the precursor film is created by immersing the substrate in the solution and raising it at a fixed speed; the spin-coating method, in which solution droplets are coated onto the substrate via rotation at high speeds; and the spray-coating method, in which the solution is spray-coated onto the substrate. A variety of materials are used for the substrate depending on its purpose and the application in which it will be used; substrates include a metal (platinum, etc.), semiconductor (silicon), silica glass, an insulator (oxide single crystal, etc.), and so on.
When synthesizing an epitaxial thin film, an appropriate single-crystal substrate must be selected with consideration given to lattice matching with the target crystal. Frequently used single-crystal substrates are magnesia and sapphire (α-alumina).
Figure 4.4 shows the synthesis procedure for a LN precursor complex alkoxide, as well as LN powder and thin film synthesis processes [23]. Equimolar lithium ethoxide (LiOC2H5 or LiOEt) and niobium ethoxide (Nb(OC2H5)5 or Nb(OEt)5) are heated to reflux in anhydrous ethanol and made to react for a specified amount of time. Water (1.0 equivalent to LN precursor) to the precursor complex alkoxide diluted with ethanol is added dropwise at room temperature. This is again heated to reflux for 24 h, and a homogeneous solution is prepared. The synthesized precursor solution is concentrated to a concentration of 0.1 mol L−1. The solution is hydrolyzed, and after solvent elimination and drying, heat treatment is performed at a specified temperature resulting in the preparation of LN powder.

Synthesis procedure for LN precursor, LN powder, and LN thin film
For precursor film formation, dip coating is performed. The substrate is immersed in LN precursor solution, and after raising the substrate at a fixed speed, it is dried to form the precursor film on the substrate. It is then crystallized under controlled conditions to obtain the LN thin film.
Figure 4.5 shows the 93Nb nuclear magnetic resonance spectrum of the starting material niobium ethoxide and the LN precursor measured in ethanol. Within the solution, niobium ethoxide has become an equilibrium mixture of the monomer and dimer (Fig. 4.6a) [24] and shows two resonance lines at −1067 ppm and −1305 ppm (Fig. 4.5a). Meanwhile, the LN precursor—synthesized by reacting niobium ethoxide and lithium ethoxide in ethanol at 80 °C for 24 h—shows only a single resonance at −1156 ppm; its chemical shift also differs from that of niobium ethoxide (Fig. 4.5b). From these facts, it is known that the LN precursor has structures including the Nb(OEt)6 octahedral structure, as shown in Fig. 4.6b. Further, from the 13C NMR results, the LN precursor contains only one type of ethoxy group, which is attributed to Nb-OC2H5. This LN precursor takes a structure in the ethanol solution such as that shown by Li[Nb(OEt)6]. Measurement of these NMR spectra makes it easy to know whether or not the LN precursor has been generated. When composition deviation exists and the precursor has not been sufficiently generated, different resonance lines are observed in the 93Nb NMR spectrum. From the precursor solution that has not taken the desired configuration within the process of crystallization to LiNbO3, a second phase is generated as described below.

93Nb nuclear magnetic resonance spectrum of the starting material niobium ethoxide (Nb(OC2H5)5) and the LN precursor measured in ethanol (a) niobium ethoxide, (b) LN precursor

Molecular structures of niobium ethoxide and LN precursor in ethanol (a) niobium ethoxide, (b) LN precursor. (Reprinted with permission from Ref. [24]. Copyright 1995 John Wiley & Sons)
4.3.2.3 Low Temperature Synthesis of LiNbO3 (LN) Powder
Powder obtained from the hydrolysis of product formed from the LN precursor was heat-treated in air for 2 h at 350 °C, and the obtained product was analyzed using X-ray diffraction; results are shown in Fig. 4.7. In the precursor immediately after mixing, there is still insufficient reaction of the lithium ethoxide and the niobium ethoxide, and the powder after heat treatment is a mixture of LiNbO3, LiNb3O8, and Li3NbO4. Even after 4 h of reaction, although LiNbO3 is the main phase, it is still a mixture. From the precursor synthesized for 22 h by heating to reflux, the powder after heat treatment at 350 °C crystallized to single-phase LiNbO3, as shown in Fig. 4.7c.

X-ray diffraction patterns of the heat-treated products at 350 °C for the 0 h-, 4 h-, and 22 h-reacted precursors in ethanol, (a) 0 h, (b) 4 h, (c) 22 h (Courtesy of the American Ceramic Society from Ref. [23]. Copyright 1987 American Ceramic Society)
When a LN precursor that has undergone hydrolysis with a controlled quantity of water is used, the LN crystallization temperature can be further reduced. During the hydrolysis stage of Fig. 4.4, 1.0 equivalent water is added. When this has been heated to reflux at 80 °C for 24 h, a homogenous precursor solution is obtained. The powder obtained from this precursor is amorphous, as shown in Fig. 4.8b. When this powder is crystallized in a water vapor/oxygen mixture flow, a crystalline LN powder is obtained at 250 °C, as shown in Fig. 4.8a. By using an appropriately configured precursor, and with decomposition of the organic matter in a controlled environment, crystalline LN can be synthesized at temperatures lower than when using conventional processes.

X-ray diffraction patterns of the as-prepared powder from the LN precursor and the heat-treated product at 250 °C in a water gas/oxygen mixture flow (a) heat-treated product at 250 °C, (b) as-prepared powder (Courtesy of the American Ceramic Society from Ref. [23]. Copyright 1987 American Ceramic Society)
4.3.2.4 Formation of Stoichiometric Epitaxial LiNbO3 (LN) Thin Film
LN crystal is trigonal with hexagonal closest packing of the oxide ions. Thus, when sapphire (α-Al2O3) single crystal, also having hexagonal closest packing of oxide ions, is used as the substrate, oriented thin films and epitaxial thin films can be synthesized. There are three types of sapphire substrate: (012) plane, (110) plane, and (001) plane; oriented thin films can be produced on any of these substrates. Figure 4.9 shows X-ray diffraction patterns of LN film crystallized at 400 °C with silicon (100) and each sapphire plane as the substrates. As shown in Fig. 4.9a, a LN thin film on a silicon substrate shows the same diffraction pattern as a polycrystalline powder. Meanwhile, only the strong diffraction lines of LN 012, 011, and 006 are observed for the sapphire (012) plane, (110) plane, and (001) plane, respectively. We thus know that LN oriented in these crystal planes has been produced on these sapphire substrates. When LN thin films crystallized on sapphire (001) are further analyzed using the X-ray pole figure method, three-fold symmetry spots are displayed, showing that this is an epitaxial film. That is, while the LN crystal lattice has three-dimensional regularity vis-à-vis the sapphire (001) plane of the substrate, the LN is also a single-crystalline film.

X-ray diffraction patterns of LN film crystallized at 400 °C with silicon (100) and each sapphire plane as the substrates, (a) Si (100), (b) sapphire (012), (c) sapphire (110), (d) sapphire (001)
Figure 4.10 shows the relationships between ion sites on the sapphire (001) plane and the LN (001) plane. Since LN has a larger lattice constant, the open circles located on the large hexagonal shape show oxide ions. The lattice mismatch between the sapphire and LN is 8.20% in the a-axis direction and 6.67% in the c-axis direction. The refractive indices of the epitaxial LN thin film synthesized on the respective sapphire planes were as follows: 2.308 on the (012) plane, 2.314 on the (110) plane, and 2.386 on the (001) plane; these are appropriate values that show the stoichiometric composition of the LN.

Relationships between ion sites on the sapphire (001) plane and the LN (001) plane
The LN film on the sapphire (001) plane showed the high refractive index of 2.386. This is thought to be because the LN thin film undergoes compressive stress from within the (001) plane of the sapphire substrate due to differences in their coefficients of thermal expansion. Cases of thin film properties being impacted by stress from the substrate are well known. An example is the Curie point shift and increase of coercive filed of BaTiO3 films because of high compressive stresses (≧ 400 MPa) [25]. Of deep interest is the use of stress to control thin film properties.
4.3.3 Synthesis of Metal-Oxide Thin Films Having Complex Crystal Structure
Generally, in the crystallization process using the chemical solution method, it is not in the desired crystal phase but in a low-temperature phase (metastable phase) that is not reported for ordinary synthesis at high temperatures when prior crystallization can occur; the more complex the crystal structure, the more often this trend occurs.
Once this low-temperature phase is produced, heat treatment at high temperatures must be performed again to make the transition to the target crystal phase. At this time, due to reactions of the thin film and substrate, a second phase is produced, and abnormal grain growth occurs. The result is a decline in the translucency and surface smoothness of the thin film; nonuniform particle size distribution occurs, and one can no longer obtain a good-quality epitaxial film having the desired optical and electrical properties. This is the case for Ba2NaNb5O15 (BNN) thin films having a tungsten bronze structure. BNN is a ferroelectric and a crystal that displays electro-optical effects. As the precursor film crystallizes, a crystal hexagonal structure is generated at low temperatures. BNN with this structure has inferior dielectric properties and shows no electro-optical effects. In this case, one uses a thin underlayer film crystallized beforehand with the tungsten bronze structure; on this, a precursor film of ordinary thickness is formed, and crystallization is performed by rapid heating. In this way, an epitaxial BNN thin film displaying dielectric hysteresis and second harmonic generation can be obtained [21, 26].
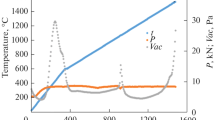
Recently, there has been interest in bismuth-layered perovskite structure oxide (Bi4Ti3O12) as a nonvolatile ferroelectric memory material. Compared with the Pb(Zr,Ti)O3 thin film, oxygen defects do not readily occur even when fabricated on a platinum electrode substrate, and its fatigue properties are superior when there are repeated polarization reversals. As shown in Fig. 4.11, this oxide has a complex crystal structure in which a perovskite lattice is sandwiched between bismuth oxide layers. For application in devices, it will be necessary to form the thin film material on a substrate (like semiconductor silicon) onto which the platinum electrode has been deposited. In this case, crystallization must be performed at temperatures of 650 °C or below to prevent reactions between the thin film and the substrate. Layered compound thin films having this complex composition and structure can also be synthesized at 650 °C or below using the chemical solution method; the favorable hysteresis characteristics of a (Bi,Nd)Ti3O12 thin film are shown in Fig. 4.12 [27]. The residual polarization value (P r) of this film is 21.6 μC cm−2, and its coercive electric field (Ec) is 70 kV cm−1. These +P r and −P r correspond to “1” and “0,” respectively. Also, uniform Ge trace doping utilizing the characteristics of the solution method is applied to crystallize a Ge-doped (Bi,Nd)Ti3O12 thin film with good properties even at 600 °C [28].

Structure of layered perovskite, Bi4Ti3O12

Polarization versus electric field hysteresis curve for Bi4Ti3O12 thin film
4.3.4 Synthesis and Applications of Multiferroic Thin Film
Substances with combined properties of ferroelectricity, ferromagnetism, ferroelasticity, etc. are called “multiferroic” and are attracting attention as new functional materials. Bismuth ferrite (BiFeO3, BF) is a perovskite-type oxide and is a multiferroic material that shows superior ferroelectricity and antiferromagnetism in thin films at room temperature. Since the magnetization direction in an electric field and direction of polarization in a magnetic field of BF are controllable, abundant related research is being conducted for BF as a next-generation electronic material [29]. With BF films, however, due to Bi volatilization, Fe ions easily attain a mixed valence state with Fe2+ and Fe3+. Reduction of Fe3+ to Fe2+ results in oxide ion vacancies, and electronic conductance occurs, increasing the leakage current. This problem must be resolved before BF films can be used in ferroelectric device applications. An attempt to control the occurrence of oxide ion vacancies involves a slight doping with aliovalent ions. Addition of even trace levels of such elements is effective in the chemical solution method. For example, when Fe ions located at site A are substituted with Mn and Cr ions at just a few mol%, BF film can be synthesized using the chemical solution method. It has been reported that such a BF film has reduced leakage current and shows good ferroelectric hysteresis at room temperature [30, 31].
Solid solution of other perovskite-type oxides in BF enables the synthesis of perovskite single-phase BF-ABO ferroelectric thin films with improved insulation properties. It has been reported that 0.7BF-0.3PbTiO3 (PT) thin film synthesized by the chemical solution method has a reduced leakage current, with improved ferroelectric properties. Also, 0.7BF-0.3 PT film doped with 5 mol% Mn showed a residual polarization of 40.0 μC cm−2 and a coercive electric field of 100 kV cm-1 at room temperature [32]. It was found that the decreased coercive electric field also enabled easy control of polarization reversal.
Recently, there has been increased interest in energy-harvesting materials, which generate electricity using commonly existing phenomena such as vibration, light, and temperature differences. BF has a comparatively narrow band gap, a shortcoming in a ferroelectric. Because it absorbs visible light, a photocurrent is generated due to the photovoltaic effect, and this photocurrent can be converted into electrical energy. A BF film (500 nm thickness) synthesized on a substrate using the chemical solution method was irradiated with 450 nm wavelength light [33]. The results are shown in Fig. 4.13a. A photocurrent with extremely good responsiveness to on-off light irradiation was detected. The same experiment was performed with a laminated film made with 1 mol% Al-doped ZnO film (35 nm thickness) on BF film (300 nm thickness). As shown in Fig. 4.13b, there is major improvement in the photocurrent value. This is thought to be due to a combination of the photocurrent generated at the pn junction interface between the p-type BF film and the n-type Al-doped ZnO film and the self-bias current within the BF film. This generated photoinduced current showed high stability, continuing for 24 h.

Zero-bias photoelectric current properties (measured at room temperature) of unpoled (a) BiFeO3 single-layer and (b) 1 mol% Al-doped ZnO/BiFeO3-layered thin films on Pt/TiOx/SiO2/Si substrates as a function of time under dark (OFF) and blue light (λ = 450 nm, 1.0 mW/cm2) irradiation (ON) conditions (Copyright 2015 The Japan Society of Applied Physics)
BF is also being investigated as a vibration power generation element for thin film and microelectromechanical system (MEMS) applications. Novel properties are still being discovered, and it is a deeply interesting material.
4.4 Current Trend of Ceramic Synthesis
In the synthesis of functional inorganic material powders and thin films, it is necessary to control the crystal structure and chemical composition. Yet, the control of microstructure is the key to property manifestation. In powders, these properties are structure, particle shape, particle size (diameter), and particle size distribution. In thin films, these properties include post-crystallization particle size (diameter), particle size distribution, surface smoothness, etc. The same holds for nanoparticles and nanomaterials that are of recent particular interest, and the chemical solution method is one important technique for the synthesis of these new materials.
References
B.J. Mulder, Am. Ceram. Soc. Bull. 49, 990 (1970)
G.J.D.A. Soler-Illia, M. Jobbagy, R.J. Candal, A.E. Regazzoni, M.A. Blesa, J. Dispersion Sci. Technol. 19, 207 (1988)
G. Johansson, Acta. Chem. Scand. 14, 771 (1960)
D.C. Bradley, R.C. Mehrotra, D.P. Gauer, Metal Alkoxide (Academic Press, New York, 1978), p. 149
C.J. Brinker, J. Non-Cryst. Solid 100, 31 (1988)
D.L. Leslie-Pelecky, R.D. Rieke, Chem. Mater. 8, 1770 (1996)
H. Weller, Angew. Chem. Int. Ed. 32, 41 (1993)
V.K. LaMer, R.M. Dinger, J. Am. Chem. Soc. 72, 4847 (1950)
W. Stöber, A. Fink, E. Bohn, J. Colloid Interface Sci. 26, 62 (1968)
E.A. Barringer, H.K. Bowen, J. Am. Ceram. Soc. 65, C199 (1982)
R.W. Schwartz, T. Schneller, R. Waser, Comp. Rend. Chim. 7, 433 (2004)
R.M. Pasquarelli, D.S. Ginley, R. O’Hayre, Chem. Soc. Rev. 40, 5406 (2011)
B.E. Yoldas, D.P. Oatlow, Am. Ceram. Soc. Bull. 59, 640 (1980)
T. Kumagai, H. Yokota, K. Kawaguchi, W. Kondo, S. Mizuta, Chem. Lett. 1987, 1645 (1987)
F. Geotti-Bianchini, M. Guglielmi, P. Polato, G.D. Soraru, J. Non-Cryst. Solid 63, 251 (1984)
A. Makishima, H. Kubo, K. Wada, Y. Kitami, T. Shimohira, J. Am. Ceram. Soc. 69, C127 (1986)
E.R. La Serra, Y. Charbouillot, P. Baudry, M.A. Aegerter, J. Non-Cryst. Solids 121, 323 (1990)
H. Dislich, E. Hussmann, Thin Solid Films 77, 129 (1981)
S. Hirano, T. Yogo, K. Kikuta, K. Noda, M. Ichida, A. Nakamura, J. Am. Ceram. Soc. 72, 2956 (1995)
T. Yogo, K. Niwa, K. Kikuta, M. Ichida, A. Nakamura, S. Hirano, J. Mater. Chem. 7, 929 (1997)
T. Yogo, W. Sakamoto, T. Isaji, M. Ichida, A. Nakamura, S. Hirano, J. Am, Ceram. Soc. 82, 2672 (1999)
J.R. Carruthers, G.E. Peterson, M. Grasso, P.M. Bridenbaugh, J. Appl. Phys. 42, 1846 (1971)
S. Hirano, K. Kato, Adv. Ceram. Mater. 2, 142 (1987)
T. Yogo, K. Kikuta, Y. Ito, S. Hirano, J. Amer. Ceram. Soc. 78, 2175 (1995)
S.B. Desu, Phys. Stat. Solid A 141, 119 (1994)
T. Yogo, W. Sakamoto, T. Isaji, K. Kikuta, S. Hirano, J. Am. Ceram. Soc. 80, 1767 (1997)
W. Sakamoto, Y. Mizutani, N. Iizawa, T. Yogo, T. Hayashi, S. Hirano, Jpn. J. Appl. Phys. 43, 6599 (2004)
W. Sakamoto, Y. Mizutani, N. Iizawa, T. Yogo, T. Hayashi, S. Hirano, Jpn. J. Appl. Phys. 42, L1384 (2003)
G. Catalan, J.F. Scott, Adv. Mater. 21, 2463 (2009)
J.Z. Huang, Y. Wang, Y. Lin, M. Li, C.W. Nan, J. Appl. Phys. 106, 063911 (2009)
J.K. Kim, S.S. Kim, W.J. Kim, A.S. Bhalla, R. Guo, Appl. Phys. Lett. 88, 132901 (2006)
W. Sakamoto, A. Iwata, T. Yogo, J. Appl. Phys. 104, 104106 (2008)
T. Katayama, W. Sakamoto, I. Yuitoo, T. Takeuchi, K. Hayashi, T. Yogo, Jpn. J. Appl. Phys. 54, 10NA05 (2015)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Yogo, T. (2019). Powder and Thin Film Synthesis. In: Hojo, J. (eds) Materials Chemistry of Ceramics. Springer, Singapore. https://doi.org/10.1007/978-981-13-9935-0_4
Download citation
DOI: https://doi.org/10.1007/978-981-13-9935-0_4
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-9934-3
Online ISBN: 978-981-13-9935-0
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)