
Abstract
Coherent multidimensional spectroscopy is a state-of-the-art technique with applications in a variety of subjects, such as chemistry, molecular physics, biochemistry, biophysics, and materials science. Due to dramatic advances in ultrafast laser technologies, a diverse range of coherent multidimensional spectroscopic methods utilizing combinations of THz, infrared, visible, UV, and X-ray radiation sources have been developed and used to study the real-time dynamics of small molecules in solutions, proteins and nucleic acids in condensed phases and membranes, single and multiple exciton states in functional materials like semiconductors, quantum dots, and solar cells, photo-excited states in light-harvesting complexes, ions in battery electrolytes, electronic and conformational changes in charge or proton transfer systems, and excess electrons and protons in water and biological systems. In this chapter, we introduce the theory behind coherent multidimensional spectroscopy and a summary of recent experiments.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

1.1 Introduction
A spectrometer is a device that measures radiation intensity as a function of electromagnetic field frequency or wavelength, and it has become indispensable in modern-day chemical and biological research. A variety of spectroscopic techniques have been developed to study the interaction between matter and electromagnetic waves, in particular investigating the spectra or distribution of the quantum eigenstates of molecules and materials in the frequency domain and their population evolution and coherent vibrational/electronic oscillatory motion in the time domain.
On a microscopic level, a number of fundamental molecular processes of interest occur over a broad dynamic range of timescales, from femtoseconds (10−15 s) to nanoseconds (10−9 s) and even microseconds [1, 2]. For example, electrons transfer from an electron-donating group to an electron-accepting group in femtoseconds to nanoseconds, depending on the distance between them. In addition, energy transfer and migration processes in light-harvesting protein complexes after photoexcitation have a broad distribution of kinetic rate constants on a picosecond timescale. Similarly, in aqueous solutions, water molecular reorientation takes place on a picosecond timescale, and H-bonding network dynamics are subpicosecond processes that can be slowed down when hydrophobic molecules are nearby. Dihedral rotations about single chemical bonds are another important elementary process involved in a variety of conformational transitions of flexible polyatomic molecules and proteins. The internal rotation of a small molecule about a carbon-carbon bond in solution, which represents a barrier-crossing process, takes tens of picoseconds, which is clearly dependent on the potential energy barrier along the reaction coordinate connecting the two conformer states [3]. Small solvent molecules in solution around electronic chromophore molecules (e.g., dyes) can reorganize themselves on a subpicosecond to several picosecond timescale when the chromophore is electronically excited by an impulsive light pulse [4, 5].
To understand the molecular processes taking place on femtosecond to nanosecond timescales in chemistry, physics, and biology, ultrafast nonlinear spectroscopy is a powerful experimental technique that has been used extensively [1, 2, 6,7,8]. For example, Ti:Sapphire oscillator and amplifier systems are commercially available, producing pulses with a duration in the tens of femtoseconds and a few mJ of energy per pulse. Femtosecond pulses in the frequency range from UV-vis to mid-IR and THz can be readily generated by various optical processes using nonlinear crystals. Therefore, multiple time-separated coherent laser pulses whose amplitudes and relative phases can be precisely controlled have been used to electronically or vibrationally perturb molecular systems under investigation. These field-matter interactions put the systems into superposition states, where each of them can be described as a linear combination of molecular eigenstates. The non-stationary states evolve over time, and the randomly fluctuating solute-solvent interactions in solution allow the systems to relax to a new thermal equilibrium state. These transitions and relaxations can be monitored with a probe pulse using other field-matter interactions.
Naturally, a variety of ultrafast nonlinear spectroscopic techniques have been used to investigate the structure and dynamics of molecular systems. For instance, in the pump-probe spectroscopy of chromophores in condensed phases, a strong pump pulse coherently excites many molecules, and a time-delayed probe pulse is used to monitor the collective relaxation of the non-equilibrium molecular systems as a function of delay time. Because the probe absorption spectrum is recorded at a given waiting time using dispersive optics and an array detector, the measured signals are time and frequency-resolved pump-probe spectra that are plotted against probe frequency.
To extend pump-probe spectroscopy to two-dimensional (2D) measurement methods, it is necessary to include an additional control variable in the time or frequency domain. Over the past two decades, many attempts have been made to develop 2D electronic, vibrational, and electronic-vibrational hybrid spectroscopic techniques that can be used to study the congested dynamic information of molecular systems that cannot be extracted from one-dimensional spectra. This involves disentangling the complex nonlinear response functions and susceptibilities of materials or molecules of interest into two-dimensional (excitation and emission) frequency space. A natural extension of widely used 2D spectroscopy is to add more time-delayed pulses or frequency-scanning schemes with one or more phase-stabilized coherent lasers to develop novel coherent multidimensional spectroscopy (CMS) [7, 8].
Of the many possible N-dimensional spectroscopic techniques, two-dimensional (2D) optical and/or IR spectroscopy, which can be regarded as an ultrafast optical analog of 2D nuclear magnetic resonance (2D NMR), is one of the most widely used. Including the emissive field-matter interaction that produces the signal electric field under detection, four-wave-mixing (4WM) 2D spectroscopy involves four field-matter interactions, which are separated in time by three time intervals, denoted as τ, T, and t, where the incident pulses are assumed to be Dirac delta function-like. Thus, the measured signal can be expressed as S(t, T, τ). The 2D spectrum S(ωt, T, ωτ), which is obtained from the double Fourier transformation of S(t, T, τ) with respect to τ and t, provides information on the electronically/vibrationally resonant transitions of molecules by pump fields at waiting time (T) zero and those by a probe field at a later waiting time. By monitoring the 2D spectra at different waiting times, dynamic information on the system can be extracted. Time-dependent diagonal peak intensities are determined by the survival probabilities of the excited states with regard to population and orientation, whereas the time dependence of off-diagonal peak intensities is associated with the conditional probability of finding the state in resonance with a probe field at time T when the initial state at time zero was in resonance with a pump field. Therefore, 2D electronic/vibrational spectroscopy is capable of time-resolving the molecular dynamics of the relaxation of excited states and state-to-state transitions between molecular quantum states.
Two-dimensional electronic spectroscopy (2D ES) is based on the electronic transition of molecular systems of interest induced by their interactions with femtosecond UV or visible pulses [7]. Often, the UV-vis absorption spectra of chromophores in condensed phases are broad and featureless due to ultrafast electronic dephasing and large inhomogeneous line-broadening. Interestingly, photon echo spectroscopy has been shown to be useful for selectively measuring the pure dephasing rate and spectral diffusion. In addition, if chromophores are electronically coupled to produce delocalized exciton states, 2D ES can be used to measure the electronic coupling strength between chromophores, as well as exciton annihilation, migration, and coherence transfer in coupled multi-chromophore systems. One of the most successful uses of 2D ES is in the investigation of photosynthetic light-harvesting complexes and exciton dynamics in semiconductors, where the relaxation, energy transfer, and coherent and incoherent evolution of created single and multiple exciton states have been of great interest.
Two-dimensional infrared (2D IR) spectroscopy utilizing multiple IR pulses has been widely used to analyze the structure and dynamics of molecular systems and to probe the chemical exchange and conformational transition processes of complicated molecular systems in real time [8]. The IR absorption bandwidth of a given normal mode is typically much narrower than that of a broadband mid-IR pulse (>250 cm−1), so the entire band shape and intensity can be monitored with 2D IR spectroscopy. Because vibrational frequency, intensity, and line shape are strongly affected by the local environment and chemical structure, small IR probes that can be easily incorporated into biomolecules and reactive species are excellent reporters for the structure and dynamics of systems under study [9, 10]. For example, femtosecond IR pulses are used to excite various fundamental vibrational modes of molecules in the wavelength range of 2.5–7 μm (mid-IR range), such as −OH, −NH, −CH, −CD, −CN, −SCN, −N3, and −C=O. Because each individual mid-IR photon has an energy that is almost one order of magnitude smaller than that of UV or visible photons, photochemical damage is not a serious issue even though multiple IR excitation-dissipation processes cause an increase in local temperature. To broaden the probe spectral window, a plasma-generated continuum IR pulse has been used to obtain a 2D IR spectrum that covers the entire mid-IR frequency range.
For coupled multi-oscillator systems, vibrational frequency and dynamics are strongly affected by vibrational couplings between local modes through space via intermolecular interactions and/or through bonds via anharmonicities on the multidimensional potential energy surface. However, because the linear vibrational spectrum is mainly determined by harmonic properties such as fundamental transition frequency and the 0–1 transition dipole moment, it is difficult to quantitatively extract weak features like the vibrational coupling constants and potential anharmonic coefficients of coupled oscillators from the linear vibrational spectra. In contrast, coherent multidimensional vibrational spectroscopic methods have been found to be exceptionally useful for estimating vibrational coupling constants by analyzing cross peaks in 2D IR spectra [7, 8]. The changes in the line shapes and intensities of the cross peaks provide crucial information on structural dynamics involving time-dependent changes in the spatial proximity and relative orientation of vibrational chromophores.
Although 2D IR spectroscopy that employs three incident IR laser pulses is one of the most widely used forms of coherent multidimensional vibrational spectroscopy, variations of this method have also been developed for specific purposes. Examples include surface-specific 2D sum-frequency-generation spectroscopy, 2D Raman and terahertz spectroscopy, and 2D IR-IR-visible spectroscopy. (See the other chapters in this book for more details on these.)
In parallel with the advances in experimental techniques, extensive theoretical and computational research on coherent 2D electronic/vibrational spectroscopy has been carried out over the years. Theory and computation can provide valuable insights into the microscopic origin of 2D spectroscopic features, aid in the interpretation of experimental spectra, predict spectra for new systems, and, in some cases, even suggest potentially useful novel nonlinear spectroscopy techniques [11]. Two-dimensional spectroscopic observables are completely determined by the nonlinear response functions of a system. This response function theory provides a framework for the description of the 2D spectroscopy in general, and explicit expressions are available for a number of important model systems [6]. In accordance with the theory, the molecular response to multiple incident light pulses can be decomposed into quantum transition pathways with different time evolution patterns. In 2D spectroscopy, these nonlinear electronic/vibrational transition pathways can be selectively measured, thus microscopic information on the system itself and the influence of the environment can be extracted [7]. For more realistic systems composed of multiple chromophores, the Frenkel exciton model provides an adequate description. The nonlinear response functions derived from this model reflect the delocalization of quantum states due to inter-chromophore couplings, i.e., resonance effects, and the fluctuation in transition frequencies due to chromophore-solvent interactions, i.e., dephasing and rephasing phenomena. The individual components of these nonlinear response functions can be calculated separately using electronic structure calculation methods and molecular dynamics (MD) simulation methods. The former approaches provide information on the transition energies, electronic couplings, and involved transition dipole strengths, whereas the latter approaches are useful for taking into account solute-solvent interaction-induced dephasing and line-broadening effects. An alternative approach is also available for numerically simulating 2D vibrational spectra of complex systems, which is based on the classical limit of the nonlinear vibrational response function and utilizes MD simulations with hybrid quantum mechanical/molecular mechanical (QM/MM) force fields.
In this chapter, a brief historical account of coherent 2D spectroscopy will be presented in Sect. 2. Following this, a general theoretical framework and numerical calculation methods for coherent 2D spectroscopy will be presented and discussed in Sect. 3. Critical experimental techniques that have been developed over the past decade will then be briefly discussed in Sect. 4. Finally, Sect. 5 will summarize the various perspectives on coherent multidimensional spectroscopy experimentation and theory and offer concluding remarks.
1.2 A Brief Account of the Early Developments in Coherent Two-Dimensional Spectroscopy
Coherent 2D spectroscopy has rapidly developed over the past two decades, emerging as one of the most widely used nonlinear spectroscopic techniques. Older reports alluded to the possibility of using multiple laser pulses to realize coherent 2D optical spectroscopy. However, the experimental feasibility of this idea was only demonstrated after lasers that were capable of generating ultrashort pulses had been developed. Today, two of the most popular techniques are 2D electronic spectroscopy and 2D IR spectroscopy. They are an extension of three-pulse stimulated photon echoes, where three time-delayed pulses are used to create third-order material polarization in the sample, which then acts as a radiation source. Weiner, De Silvestri, and Ippen experimentally demonstrated three-pulse scattering spectroscopy [12], which was later found to be of use for analyzing chromophore-solvent dynamics in condensed phases. In addition, a few interesting theoretical studies on the principles underlying photon echo phenomena arising from chromophores with a heterogeneous distribution of transition frequencies have been reported.
Before the development of spectral interferometric detection, 4WM spectroscopy experiments were performed by measuring the generated third-order (in electric fields) signal field intensity |Es(r, t)|2, i.e., homodyne detection. Because the phase information of the signal field is lost in this case, it was not possible to obtain the complex 2D electronic response from optical chromophores in condensed phases. However, the inhomogeneous distribution of the transition frequencies of chromophores in solution and its time-dependent change could be successfully investigated using photon echo peak shift (PEPS) measurements. As shown by Cho and Fleming, the PEPS signal with respect to waiting time T is directly related to the transition frequency-frequency correlation function and spectral diffusion process [5]. The Fleming research group showed that PEPS measurement is an exceptionally useful method for studying ultrafast chromophore-solvent dynamics. As an extension of this optical photon echo technique into the IR frequency domain, vibrational photon echo measurements, which are an IR analog of the optical photon echo, were experimentally demonstrated by the Fayer research group in 1993, who used IR pulses from a free-electron laser [13].
Theoretically, Tanimura and Mukamel in 1993 proposed fifth-order 2D Raman scattering spectroscopy, which allows the analysis of intermolecular vibrational rephasing phenomena from Raman-active molecules whose intermolecular vibrational frequencies are inhomogeneously distributed in liquids [14]. The fifth-order Raman response function, which represents the nonlinear correlation of polarizabilities at different times, depends on two time variables, which is why it was referred to as the 2D time-domain Raman response function. Later, Tominaga, Yoshihara, Fleming, Tokmakoff, Blank, Dwayne-Miller and many others attempted to measure fifth-order Raman signals from neat liquids such as CS2. However, undesired cascading contributions to the detected signal (i.e., two third-order rather than truly fifth-order) are often dominant, as shown by Blank, Fleming, and coworkers [15, 16]. Because the direct fifth-order Raman signal becomes large and dominant as the field frequencies approach electronic transition frequencies, the resonant version of fifth-order Raman scattering spectroscopy theoretically proposed by Cho in 1998 could be of use in investigating 2D Raman responses from chromophores in condensed phases [17].
Independently, Cho and Fleming in 1994 theoretically demonstrated that fifth-order three-pulse scattering spectroscopy probing correlations of electronic transition frequencies at two different times could be useful when investigating the inhomogeneous line-broadening effect on the optical spectrum of chromophores in condensed phases [18]. In contrast to traditional integrated intensity photon echo spectroscopy, which probes one electronic coherence evolution, fifth-order three-pulse scattering spectroscopy can be considered fifth-order 2D electronic spectroscopy that is capable of measuring time-dependent changes in optical transition frequencies. However, due to limitations in controlling the center frequencies of multiple pulses, only degenerate fifth-order three-pulse scattering experiments were performed.
After array detectors in the visible and IR frequency domains became available, heterodyne-detected 2D spectroscopic techniques were developed. Once a generated nonlinear signal electric field is allowed to interfere with an added local oscillator field, a spectral interferogram can be obtained using a monochromator (e.g., a grating) and an array detector. Spectral interferometric detection has been particularly useful in developing coherent multidimensional spectroscopy (CMS) because it enables the simultaneous characterization of the phase and amplitude of a generated 4WM signal electric field. Jonas and coworkers in 1998 showed that the spectral interferometric detection of three-pulse scattering or a photon echo signal field is experimentally feasible [19]. In 2D IR spectroscopy, Hamm, Lim, and Hochstrasser used a frequency-tunable IR pump-probe spectroscopic method to obtain the reconstructed 2D amide I IR spectra of polypeptides in solution, where amide I vibration is mainly carbonyl stretch mode of an amide group or a peptide bond [20]. In their study, a narrowband IR pump pulse was used to selectively excite resonant oscillators, and the transient probe absorption spectrum was then measured. Scanning the pump frequency, they obtained a series of time- and probe frequency-resolved pump-probe spectra, which were used to construct time-resolved 2D IR spectra. Later, 2D IR photon echo experiments were performed by Zanni, Hochstrasser, and coworkers and Tokmakoff and coworkers to determine the solution structure of small oligopeptides.
In parallel with experimental studies using 2D IR spectroscopy, there have been efforts to numerically simulate the 2D vibrational spectra of a variety of molecular systems. Two-dimensional vibrational spectroscopy can be used to achieve both ultrafast time resolution and high spectral resolution. Therefore, this method provides rich information on molecular systems, such as homogeneous (anti-diagonal) and inhomogeneous (diagonal) spectral broadening, vibrational anharmonicity, spectral diffusion, vibrational mode-mode coupling strength, and their time-dependent changes [7, 8].
Although the 2D IR and 2D ES methods, which are based on heterodyne-detected three-pulse scattering geometry, have been found to be useful, the center frequencies of the pulses used were the same, i.e., degenerate. To study a wider range of vibrational dynamics and intramolecular vibrational relaxations, two-color 2D vibrational spectroscopic techniques have since been developed. Theoretically, Park and Cho proposed a new class of non-degenerate 4WM 2D vibrational spectroscopy that requires both IR and visible pulses [21]. Two IR pulses could be used to create two consecutive vibrational coherences or super-position state evolutions. An incident visible pulse, whose frequency is electronically non-resonant, puts the molecular system into a state of electronic coherence. This third-order polarization radiates an IR-IR-vis sum or a difference frequency field that provides information on the 2D vibrational responses of electronically ground-state molecules. The Wright research group experimentally demonstrated that an IR-IR-vis difference frequency generation scheme can be used to measure the cross peak between the C-C stretch and C-N stretch of acetonitrile, which results from both the mechanical and electronic anharmonic couplings between the two modes [22].
In the present section, a brief account of the early developments in coherent 2D spectroscopy in the 1990s was presented. It should be emphasized that this account is far from complete, so readers are strongly recommended to read the representative review articles cited in this chapter and other chapters in this book.
1.3 Theoretical Description and Numerical Simulation Methods
In general, spectroscopic measurement involves both excitation and detection. In 2D spectroscopy, the molecular system of interest interacts with three coherent laser pulses and the generated signal field is then detected and presented with respect to the excitation and detection frequencies [7]. In each of the four field-matter interaction events, a quantum transition takes place between the eigenstates of the system. Depending on both the configuration of the optical laser pulses, including the frequency, direction of propagation (i.e., wave vector), and beam polarization, and the detection method employed, different quantum transition pathways can occasionally be selectively measured.
One of the most widely used methods for theoretically describing various nonlinear spectroscopic observables is to use the formalism of the nonlinear response function [6], which naturally emerges from the application of quantum mechanical time-dependent perturbation theory to the molecular system in the presence of perturbative light-matter interactions during the preparation step. In this section, I sketch the theoretical analysis in a stepwise manner and present key results that are particularly relevant to coherent 2D spectroscopy, though the formal theory can easily be generalized to describe other CMS measurement methods.
1.3.1 Third-Order Response Functions
In coherent 2D spectroscopy, the molecular system interacts with the incident electric field and, in the electric dipole approximation, the interaction Hamiltonian can be written as
where \( {\hat{\varvec{\upmu}}} \) is the electric dipole operator and E(r, t) is the superposition of the three X-ray, UV-visible, IR, or THz pulses (depending on the specific experiment), which are generally denoted as E1, E2, and E3. The approximate Hamiltonian of the composite system is the sum of the molecular or material Hamiltonian H0 in the absence of radiation and the field-matter interaction Hamiltonian Hint(r, t). The system evolves over time according to the quantum Liouville equation for the density operator ρ(r, t) of the system, where ρ(r, t) is the state vector in the Liouville space, as follows:
The solution to this equation provides information about any physical observable A(r, t) of the system through the expectation value of \( {\text{Tr}}[\hat{A}\rho ({\mathbf{r}},t)] \). Here, Tr denotes the trace of the matrix and \( \hat{A} \) is the operator associated with the observable A. Diagonal element ρaa of the density matrix represents the probability that the system is in state a, or the population of the system in state a. Off-diagonal element ρab of the density matrix, which is related to the coherence or distinguishability of the two quantum states, gives rise to the temporal oscillation of the aforementioned probability with the frequency \( \omega \approx \omega_{ab} = \, \left( {E_{a} - E_{b} } \right)/\hbar \) determined by the energy difference of the two eigenstates a and b.
Treating Hint(r, t) as a perturbation to the system described by the molecular Hamiltonian H0, (1.2) can be solved by applying time-dependent perturbation theory. The solution is expressed as a power series expansion of ρ(r, t) with respect to the perturbation energy, the zeroth-order term of which is the equilibrium density operator for the unperturbed system ρ(0)(t) = ρeq. The nth-order term ρ(n)(r, t) contains n factors of Hint(r, t) and is given by [6].
where \( G_{0} \left( t \right) \, = \, \text{exp}( - iL_{0} t/\hbar ) \) is the time-evolution operator in the absence of radiation. The Liouville operators are defined as LaA = [Ha, A] for a = 0 or int. According to (1.3), the system initially defined by ρ(t0) evolves freely without perturbation for τ1 − t0 as given by G0(τ1 − t0) and then interacts with the radiation at time τ1 as given by Lint(τ1). This propagation-interaction sequence is repeated n times until the final field-matter interaction at τn, as given by Lint(τn). Finally, the system evolves freely until observation time t for t − τn according to G0(t − τn). The multiple integrals over τ1, …, τn account for all possible interaction times under the time ordering condition t0 ≤ τ1 ≤ … ≤ τn ≤ t.
Each term of the power series expansion of ρ(r, t) in (1.3) gives rise to the corresponding nth-order polarization \( {\mathbf{P}}^{(n)} ({\mathbf{r}},t) = {\text{Tr}}\left[ {{\hat{\varvec{\upmu}}}\rho^{(n)} ({\mathbf{r}},t)} \right] \) in the system as follows:
The nth-order response function is formally given by
where \( {\hat{\varvec{\upmu}}}(t) = \exp ({{iH_{0} t} \mathord{\left/ {\vphantom {{iH_{0} t} \hbar }} \right. \kern-0pt} \hbar }){\hat{\varvec{\upmu}}}\exp ( - {{iH_{0} t} \mathord{\left/ {\vphantom {{iH_{0} t} \hbar }} \right. \kern-0pt} \hbar }) \) is the dipole operator in the interaction picture and the angular bracket in (1.5) denotes the trace of a matrix. The linear response can be obtained by setting n = 1 in (1.5). The 2D spectroscopy signal is determined by the third-order polarization P(3)(r, t) and the third-order response function R(3)(t3, t2, t1) [23]. Here, the latter is a fourth-rank tensor. Note that the time variables t1, …, tn−1 in (1.4) and (1.5) are the time intervals between consecutive field-matter interactions related to τ1, …, τn in (1.3) as tm = τm+1 − τm (1 ≤ m ≤ n − 1), while tn = t − τn is the time elapsed after the last field-matter interaction. Therefore, t1, …, tn are all positive and the response function must vanish if any of its time arguments tm (m = 1…n) are negative in accordance with the causality principle, as imposed by the Heaviside step function s θ(t) in (1.5). In addition, R(n) is a real function because P(n)(r, t) and E(r, t) in (1.4) are both real quantities, though individual terms comprising R(n) are complex in general and represent different quantum transition pathways.
The signal electric field \( {\text{E}}_{S}^{(n)} ({\mathbf{r}},t) \) detected in nth-order nonlinear spectroscopy is obtained by solving Maxwell’s equation where the material nonlinear polarization P(n)(r, t) acts as a radiation source. After making the simplifying assumptions that (i) the generated signal field is only weakly absorbed by the medium, (ii) the temporal envelopes of the polarization and signal fields vary slowly in time compared to the optical period, (iii) the signal field envelope spatially varies slowly compared to its wavelength, and (iv) the frequency dispersion of the medium refractive index is weak, the approximate solution can be obtained as [6, 7].
Here, n(ω) is the refractive index of the medium and \( {\mathbf{P}}_{S}^{(n)} (t) \) is the polarization component propagating with wave vector kS and frequency ωS, which represent one of the combinations \( \pm {\mathbf{k}}_{1} \pm {\mathbf{k}}_{2} \cdots \pm {\mathbf{k}}_{n} \) and \( \pm \omega_{1} \pm \omega_{2} \cdots \pm \omega_{n} \), respectively. These components make up the total nth-order polarization as
By appropriately changing the location of the detector, individual components of the polarization with different wave vectors can be selectively measured.
1.3.2 Nonlinear Response Function Components
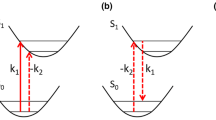
Two-dimensional vibrational spectroscopy usually induces transitions up to the second vibrational excited state. Therefore, a three-level system with eigenstates |g>, |e>, and |f> is a useful model for the nonlinear response function relevant to coherent 2D spectroscopy. Because the third-order response function vanishes for a harmonic oscillator (i.e., a molecular vibration or an electronic Lorentz oscillator), the model system must represent an anharmonic oscillator where the fundamental transition frequency ωeg differs from ωfe.
The evaluation of a realistic response function critically depends on an accurate description of the system-bath interaction, which is essentially responsible for dephasing, relaxation, spectral diffusion, and population and coherence transfers. To highlight the structure of the response function, I consider a simple model where a single three-level chromophore interacts with the environment according to the following Hamiltonian:
where \( \hbar \omega_{m} \) is the energy of the mth state in the absence of a bath. Vm(q) is the chromophore-bath interaction energy of the state, where q represents the bath coordinates. In (1.8), HB(q) is the energy of the bath. The off-diagonal elements of the chromophore-bath interaction are assumed to be negligible for the sake of simplicity. Using this Hamiltonian, the three nested commutators in the response function in (1.5) can be expanded as the sum of eight terms:
where c.c. denotes the complex conjugate and the fourth-rank tensor components \( {\mathbf{R}}_{i} (t_{3} ,t_{2} ,t_{1} ) \) are given by
Here, it is assumed that the system is initially in the ground state g. In (1.10), \( {\varvec{\upmu}}_{ab} \) is the transition dipole moment between states a and b, which is often assumed to be independent of the bath coordinates (i.e., the Condon approximation). In (1.10), the energy gap averaged over bath degrees of freedom is defined as \( \hbar \bar{\omega }_{ab} = \hbar (\omega_{a} - \omega_{b} ) + < V_{a} ({\mathbf{q}}) - V_{b} ({\mathbf{q}}) >_{B} \). \( F_{n}^{gabc} (t_{3} ,t_{2} ,t_{1} ) \) is the line shape function expressed in terms of the time-ordered exponentials of the fluctuations in the system-bath interactions, \( U_{m} ({\mathbf{q}}) = V_{m} ({\mathbf{q}}) - < V_{m} ({\mathbf{q}}) >_{B} \). The total response function is composed of multiple quantum transition pathways represented by individual Ri, each of which is the product of three factors determining the transition strength (i.e., the products of transition moments), the transition frequency (i.e., coherence oscillation), and the line shape function (F1–4). To facilitate the computation of \( F_{n}^{gabc} (t_{3} ,t_{2} ,t_{1} ) \), the time-ordered exponential operators can be approximated by normal exponential functions containing the difference potential energies \( U_{ab} ({\mathbf{q}}) = U_{a} ({\mathbf{q}}) - U_{b} ({\mathbf{q}}) \). Alternatively, the nonlinear line shape function can be approximately described by invoking second-order cumulant expansion technique, which becomes exact when the fluctuation of the transition frequency obeys Gaussian statistics [7, 11]. Because detailed theoretical expressions of line shape functions can be found in other review articles and books [6,7,8, 23], I will not present them here.
The general formulation for multi-level systems has been reported and provides an excellent framework for interpreting experimental results and for understanding the effect of chromophore-solvent interaction dynamics on the diagonal and off-diagonal peak shapes in 2D spectra. For instance, a distinction between homogenous and inhomogeneous line broadening can be achieved by analyzing the extent of the diagonal elongation of a given diagonal peak. Furthermore, time-dependent 2D peak shape analysis of both diagonal and cross peaks provides critical information on the timescale of solvent dynamics affecting transition frequency fluctuations and on the correlation or anticorrelation of the solvent-induced frequency fluctuations of the two associated states, respectively.
1.3.3 Classical Approximation to 2D Vibrational Response Functions
Although the nonlinear response function formalism is exact, fully quantum mechanical simulations of 2D peak shape functions remain difficult and impractical for systems with many degrees of freedom. Thus, applicable and efficient methods based on the classical mechanical description of molecular vibrations have been developed and used to calculate 2D vibrational response functions for coupled oscillator systems [24].
One of these is to use trajectories of equilibrium MD simulations with classical approximations of the vibrational response functions. First, the classical mechanical response functions are derived using the relationship between the quantum mechanical commutator and the Poisson bracket, i.e., \( (i\hbar )^{ - 1} [X,Y] = \{ X,Y\}_{\text{PB}} \), where X and Y are physical variables and the Poisson bracket is expressed as \( \{ X,Y\}_{\text{PB}} = (\partial X/\partial {\mathbf{q}})(\partial Y/\partial {\mathbf{p}}) - (\partial X/\partial {\mathbf{p}})(\partial Y/\partial {\mathbf{q}}) \). For instance, the classical linear response function of a physical quantity A to a perturbation B is given as \( (1/k_{B} T)\langle B^{\prime}(0)A(t)\rangle \), where \( B^{\prime}\) is the time derivative of B. The above relationship between the quantum mechanical commutator and the classical mechanical Poisson bracket can be applied to derive the classical nonlinear response function [25]. For 4WM-based 2D vibrational spectroscopy, the corresponding classical third-order response function that is related to 2D IR spectroscopy is expressed as
Here, the Poisson brackets of the physical variables at two different times, e.g., \( \{ \mu (t),\mu (t^{\prime})\}_{\text{PB}} \), are calculated using equilibrium MD trajectories. This involves the calculation of the stability matrix representing the transformation of the phase space along the trajectory. Using this method, Jeon and Cho investigated 2D IR spectra, employing a quantum mechanical/molecular mechanical (QM/MM) simulation method for an accurate description of the intramolecular vibrations of the solute molecules (deuterated N-methylacetamide [d-NMA] and HOD) [24]. They were able to calculate the 2D IR spectra of the OD stretch of HOD molecules in water and demonstrated that various 2D IR spectroscopic features can be successfully reproduced by this classical approach to calculate the 2D IR spectra.
However, because this approach requires stability matrix calculations, it is computationally expensive and suffers from numerical instability, which makes it difficult to calculate nonlinear response functions accurately. Another approach that has been developed to overcome this problem utilizes the simulation of non-equilibrium MD trajectories. In this case, nonlinear vibrational response functions are evaluated by considering external field-matter interactions directly, in a similar way to real experiments performed with pulsed electromagnetic fields. In other words, instead of using equilibrium MD trajectories, the nonlinear vibrational responses can be calculated by directly taking into consideration radiation-molecule interaction by means of carrying out a number of independent non-equilibrium MD simulations. For instance, the linear response function can be approximately calculated using [26].
where ε is the perturbation parameter. Here, the first term is the expectation value of A(t) on the perturbed trajectory determined by the Hamiltonian H0 − εBδ(t), where B is, for example, \( {\mathbf{\mu (r)}} \cdot {\mathbf{E}}({\mathbf{r}},t) \). The second term \( \langle A\rangle \) is the expectation value of A on the trajectory in the absence of perturbation. In this non-equilibrium finite-field method, the third-order response function for 2D vibrational spectroscopy is expressed as
Jansen and coworkers have further developed an efficient method using positive and negative electric fields [27].
Although non-equilibrium MD simulation methods do not require the direct calculation of a cumbersome stability matrix, it is still expensive computationally because a number of non-equilibrium trajectories need to be obtained to accurately calculate the nonlinear vibrational response functions. To save computational time, the Tanimura research group developed an efficient method that combines the equilibrium MD simulation and non-equilibrium finite perturbation methods [28].
The third-order vibrational response functions obtained from equilibrium and/or non-equilibrium MD simulations in principle allow any third-order vibrational spectra to be predicted. However, despite these recent developments in computational spectroscopy, it is still computationally demanding to calculate the third-order response functions of complicated systems in solution. Furthermore, the fundamental validity of the classical approximations used previously has been re-investigated by several groups and it has been found that classical nonlinear response functions are not stable for integrable systems and systems without dissipation. Sakurai and Tanimura examined the quantum effects on the IR and 2D IR spectra of a Morse oscillator interacting with a collection of harmonic bath oscillators [29]. They showed that the classical 2D IR spectra represent a good approximation of the quantum 2D IR spectra when the system is largely modulated by the bath via a strong system-bath coupling or when the bath modulation is fast even in a weak system-bath coupling regime. Recently, this issue was investigated again by Reppert and Brumer, who showed that the classical 2D IR spectra of a Morse oscillator mimicking amide I mode can reproduce most of the qualitative features of the quantum 2D IR spectra very well [30]. From these studies, it is clear that the validity of the classical approximation when calculating linear and nonlinear IR spectra depends on system-bath coupling.
Although classical nonlinear spectral simulations have been shown to reproduce spectra derived via quantum mechanical calculations reasonably well when system vibrations are largely modulated by the bath, there remain intrinsic differences with regards to the quantum mechanical description of vibrational transitions. In fact, real vibrational spectra are described as transitions from one vibrational level to another in quantum mechanics, whereas they are obtained from the fluctuation of dipole moments in classical mechanics. Furthermore, the anharmonic frequency shift observed in 2D IR spectra is determined by information about the entire potential energy function. In contrast, no discrete vibrational quantum states are considered in any approaches based on classical mechanics. Trajectories in a classical mechanical regime are determined by local information on coordinates and momenta. Consequently, the anharmonic shift corresponding to the frequency difference between the positive and negative peaks in classical 2D IR spectra arises from the difference between the curvatures of the trajectories perturbed by one and multiple electric fields. Therefore, the anharmonic shifts found in the 2D IR spectra obtained with classical approaches are small compared to those in the experimental or quantum mechanically calculated 2D IR spectra [25].
The other important quantity determining each nonlinear vibrational response function component is the transition dipole moment. In quantum mechanics, spectral intensity is related to the transition dipole moments between vibrational levels, whereas classical mechanically calculated spectra are determined by the dipole moments induced by vibrational and conformational changes. Therefore, it is absolutely crucial to model highly accurate potential function and transition (dipole, polarizability, etc.) moments to correctly simulate nonlinear vibrational response functions and corresponding spectra. Of course, fully quantum mechanical MD simulation methods exist, but the calculation of quantum mechanical nonlinear response functions that include all of the effects of the surrounding thermal bath remain challenging and impractical, despite the dramatic advances in computer technology and algorithms. Therefore, other semiclassical approach es like semiclassical initial value representation, centroid MD, and ring-polymer MD, which have been used to calculate the linear spectroscopic properties of molecules in condensed phases, should be employed in the calculation of various coherent 2D vibrational spectra in the near future.
1.3.4 Numerical Integration of the Vibrational Schrödinger Equation
Instead of considering the fluctuation in system-bath interaction-induced frequency and the change in transition dipoles over time using MD simulations, the fluctuating vibrational frequency and the transition dipole of the oscillator of interest in condensed phases can be described using theoretical models. The task remaining is then to solve the time-dependent vibrational Schrödinger equation. This approach has been referred to as the Numerical Integration of the Schrödinger Equation (NISE) theory [31]. In this case, each oscillator is treated as a weakly anharmonic oscillator with three vibrational levels that are coupled to bath degrees of freedom. The latter is taken into account through their time-dependent modulation of the parameters of the quantum oscillator, such as the harmonic frequency and transition dipole. For coupled multi-oscillator systems interacting with external electric fields, the corresponding time-dependent Hamiltonian can be written as
Here, \( a_{n}^{\dag } \) and \( a_{n}^{{}} \) are the creation and annihilation operators of the nth oscillator considered quantum mechanically. The individual local modes are characterized by their frequency ωn(t), transition dipole μn(t), transition polarizability αn(t), and anharmonicity Δn(t). Any pair of local modes can be mixed by their mutual couplings Jnm(t). In this approach, the time dependence of these parameters strictly arises from the coupling of each individual oscillator with bath degrees of freedom. The last two terms in (1.14) account for the interaction of the oscillating dipoles and molecular polarizabilities with the applied electric field(s) E(t), respectively, depending on the specific experimental configuration.
Determining the fluctuating frequencies, transition moments, and coupling constants in the above time-dependent Hamiltonian depends on the system under consideration. Once there exist quantitatively reliable models for these parameters, approximate time-evolution operator approaches can be used to calculate the response functions. The key step is to divide the propagation time into sufficiently short time intervals so that the Hamiltonian during these intervals can be considered time-independent. The solution for the time-dependent Schrödinger equation for each short time interval can then be easily obtained. Successive applications of the finite-difference time-evolution operator s for neighboring time-intervals enable the time-dependent vibrational wavefunction of the coupled multi-oscillator systems to be calculated.
The success of this NISE approach relies on the accuracy of the computed parameters needed to construct the time-dependent Schrödinger equation. The vibrational frequency and transition moment of a given oscillator depends on the local environment and is determined by the intermolecular interaction potential and the vibrational anharmonicity of the multidimensional intramolecular vibrational potential. For instance, an early attempt to calculate the solute-solvent interaction-induced shift of vibrational frequency was based on the assumption that the solute-solvent interaction is dictated by electrostatic interactions. The vibrational frequency shift of an oscillator was assumed to be dependent on the solvent electric potential, electric field, or sometimes the electric field gradient on specific sites of the solute molecule. These vibrational frequency mappings have allowed the frequency trajectories of the coupled oscillators to be obtained from equilibrium MD trajectories. However, recently it has been shown that the vibrational solvatochromic frequency shift is determined by not just electrostatic interactions but also dispersive interaction, short-range Pauli repulsion, polarization, and even multipole-multipole interactions [32].
The anharmonicity of a given molecular vibration also depends on its interaction with the solvent molecules. For multi-oscillator systems, the vibrational coupling constant between any pair of local modes should be accurately calculated to describe the delocalized nature of the vibrational modes. One of the most popular models is the transition-dipole coupling model, which is based on the assumption that the two oscillators interact with each other through electric dipole-dipole interactions. So far, this form of semiempirical mapping has been found to be exceptionally useful, achieving a chemical accuracy within a few wavenumbers, something which cannot be achieved using current classical or even ab initio MD simulation methods.
The quantum-classical methods discussed here have a number of crucial advantages. One of the commonly used methods that incorporates second-order cumulant approximation or another method that requires an assumption that the coupled bath degrees of freedom are harmonic oscillators cannot account for intermolecular interaction-induced effects properly. On the other hand, the quantum-classical methods take them correctly. Nevertheless, the quantum-classical methods still have clear limitations. The time-dependent Hamiltonian for NISE does not allow for the relaxations between the different excitation manifolds. Furthermore, while these quantum-classical methods are able to account for the effect that the bath exerts on the system, the feedback of the system to the bath when in an excited state is unable to be considered. As a consequence, the method cannot reproduce the correct thermalization in quantum systems, which results in artifacts at low temperatures. Another inherent difficulty of NISE is that the quantum mechanical oscillators need to be well defined and localized. If the nature of an oscillator changes over time (e.g., H-bond vibrations and delocalized intermolecular modes), it is not possible to treat them quantum mechanically.
As mentioned in this section, despite the prolonged efforts to develop approximate theory and computational methods, clear limitations in the accurate calculation of the coherent multidimensional spectra of molecules in condensed phases exist. The next section will now switch focus to experimental techniques and their underlying principles.
1.4 Experimental Methods
1.4.1 Femtosecond Laser Light Sources
Owing to the recent developments in high-power Ti:Sapphire laser s, stable femtosecond pulses in the 750–900 nm wavelength range have been used in a variety of ultrafast spectroscopic studies. Ti:Sapphire laser systems that produce pulses centered at 800 nm of ~50 fs in duration and a pulse energy of a few mJ are commercially available. The 800-nm pulses can then be used to generate femtosecond pulses in the visible (400–700 nm), near IR (1.2–2.4 μm), mid-IR (2.5–7.0 μm), and even THz (10–30 μm) frequency ranges by employing various nonlinear optical techniques with appropriate nonlinear crystals, such as optical parametric amplification (OPA), second and higher harmonic generation, sum frequency generation, and difference frequency generation. Thus, the generated femtosecond pulses have been used to carry out various forms of time-resolved spectroscopic research. More recently, 100-kHz repetition rate lasers and optical frequency comb lasers with repetition frequencies in the hundreds of MHz have been added to the list of radiation sources for coherent 2D spectroscopy experiments. In fact, two or more phase-stabilized optical frequency comb lasers, with each producing a train of phase-stabilized pulses with well-defined and constant repetition and carrier-envelop-offset frequencies, can be used to construct double- and multiple-comb spectrometers. A review article written by Kim et al. will be of interest for understanding state-of-the-art theories and experiments regarding dual-comb-based nonlinear spectroscopy (see the chapter 16 written by Kim and Cho in this book too) [33]. They present an extensive discussion of the advantages and limitations of this technique compared to conventional time-resolved spectroscopic methods using a single mode-locked laser with beam splitters and translational stages.
1.4.2 Interferometry
In spectroscopy and microscopy, various interferometric approaches to measuring the phase and amplitude of an unknown field by having it interfere with a reference field have been adopted. One of the most popular techniques used in nonlinear spectroscopy is Mach-Zehnder (MZ) interferometry (Fig. 1.1a). A single pulse from a coherent radiation source (e.g., a laser) is split into two daughter pulses that propagate along two different paths. The two beams are then combined by a beam combiner placed before a photon detector. Now, suppose one of the two path lengths is changed intentionally. As long as the two beams remain coherent with each other, the recorded intensity at the detector will exhibit an interference fringe with respect to the difference in the path lengths. Let us denote the waves in the upper and lower paths as \( \psi_{u} \) and \( \psi_{l} \), respectively, and the incident wave as \( \psi_{0} \). For the sake of simplicity, it is assumed that the incident beam is split into two by a 50:50 beam splitter that is fabricated from a lossless material. The wave arriving at the detector is given by

Mach-Zehnder interferometry for spectroscopy. a The incoming wave is divided in two by a beam splitter. The two waves pass through the upper and lower paths and then interfere to produce a fringe of varying path length differences. b The wave on the lower path is modified by the complex susceptibility of the optical sample, which changes the interference pattern
where \( \phi_{u} \,(\phi_{l} ) \) is the phase acquired by the upper (lower) beam. The detected intensity of the total wave is
The phase difference \( \phi_{u} - \phi_{l} \) is primarily determined by the difference in path length and it determines the fringe spacing. The sinusoidal term in (1.16) represents the coherence or distinguishability of the two waves. From the complementarity relation D2 + V2 = 1 for a single particle, distinguishability (D) is related to the visibility (V) of the fringe, where the former is a particle characteristic and the latter a wave characteristic. Thus, any finite visibility in the interference pattern indicates that the particle path is not completely distinguishable, i.e., |D| < 1. Now, suppose that there exist media on one or both of the two paths that can induce random phase fluctuations, \( \varphi_{m} (t) \), in the two waves, i.e., \( \psi_{m} = (1/\sqrt 2 )\psi_{0} \text{e}^{{i\phi_{m} + i\varphi_{m} (t)}} \) for m = l and u. The interference term will then vanish when it is averaged over the broad and randomly fluctuating phases. This is known as the dephasing process, which is measurable with MZ interferometry.
Now, let us consider the case where a real optical sample that resonantly interacts with the incident beam is placed on the lower beam path (Fig. 1.1b). Due to the absorptive and dispersive properties of the sample, the wave passing through the lower path is modified and can be written as \( \psi_{l} = (1/\sqrt 2 )\psi_{0} \text{e}^{{ - \kappa_{l} + i\eta_{l} + i\phi_{l} }} \), where \( \kappa_{l} \) is the attenuation factor (\( \alpha \) extinction coefficient) due to the resonant absorption of the radiation by chromophores in the sample and \( \eta_{l} \) is the phase shift due to the dispersion (\( \alpha \) refractive index) of the sample. These two factors, \( \kappa_{l} \) and \( \eta_{l} \), are related to the imaginary and real parts of the linear susceptibility of the sample, respectively. The measured signal in this case is
In principle, by comparing this interference pattern with that produced without an optical sample (or, more specifically, chromophores in the solution), quantitative information about the real and imaginary parts of the linear susceptibility of chromophores can be extracted.
Heterodyne-detected coherent 2D spectroscopic measurement [19, 34] is also based on MZ interferometry (Fig. 1.2). Instead of placing a sample cell containing molecules of interest on the lower beam path, the optical setup shown in the lower box in Fig. 1.2 is employed there. The pulse incident into the 4WM setup is split into three pulses, the relative delay times of which are controlled by two mechanical delay devices. The three pulses interact with the optical sample of interest, which then generates a third-order signal field propagating along the phase-matching direction. This signal field interferes with the reference beam traveling along the upper path, which closes the MZ interferometry circuit. In this form of coherent 2D spectroscopy, the output wave from the lower path can be generalized as

Modified MZ interferometry. The experimental setup for 4WM scattering is placed on the lower beam path. The generated third-order signal electric field, which results from the desired phase-matching condition, is combined with the local oscillator (i.e., reference) field passing through the upper path. The interference pattern is recorded using a monochromator and array detector pair or a single detector depending on the experimental configuration
where \( \kappa_{l} (t,T,\tau ) \) and \( \eta_{l} (t,T,\tau ) \) represent the attenuation (extinction) factor and the phase shift factor, respectively. Note that they depend on the two pulse-to-pulse delay times τ and T and are related to the imaginary and real parts of the third-order response susceptibility. The wave passing along the lower path interferes with the wave from the upper path, and the interference term is selectively measured by the detector (D). Depending on the specific experimental configuration, the spectral interferogram in the frequency domain or the temporal interferogram in the time domain can be measured. In conventional 2D spectroscopy with laser pulses (Fig. 1.2), the time delay between the local oscillator field and the signal field is usually fixed. Using a monochromator and array detector, the spectral interferogram is measured experimentally, which provides information on the phase and amplitude of the third-order 2D spectroscopic signal field.
Recently, dual frequency comb spectroscopy has been shown to be useful in measuring the nonlinear electronic response functions of atomic (Rb) vapor or chromophores in condensed phases [35, 36]. Because the down conversion factor, which is determined by the ratio of the mean repetition frequency and the difference in repetition frequency between the two comb lasers is in the order of 106–108, it is possible to use a single detector to monitor ultrafast molecular response and relaxation processes with microsecond timescale detectors [37, 38]. Thus, for multiple frequency comb spectroscopy, the time-domain interferogram is recorded with a single detector and its Fourier transformation provides direct information about the spectrum of the sample with respect to the probe frequency. Although the underlying interferometry in dual and even multiple frequency comb spectroscopic techniques is more similar to Michelson interferometry than to MZ interferometry, the underlying principles are the same in the sense that the linear or nonlinear optical signal field is characterized by analyzing the interference patterns produced by the interference between the signal field and the local oscillator (i.e., reference) field.
1.4.3 2D Electronic and Vibrational Spectroscopy
Two-dimensional electronic and vibrational spectroscopy that utilizes multiple laser pulses whose frequencies cover a broad range from THz, UV, to X-ray can be considered a time-domain 4WM process. The first three femtosecond pulses propagate along a non-collinear beam geometry. The pulse-to-pulse time intervals are controlled by changing the relative optical path lengths with motorized translational stages or dispersive materials such as a pair of wedged glasses. In coherent 2D spectroscopy, the molecular system evolves on different density matrix elements. The coherence evolution time τ is almost identical to the delay time between the first and second pulses when they are assumed to be ultrashort compared to the molecular relaxation processes. The waiting time T is the time interval between the second and third pulses, and the detection time t is the time between the third pulse and the emitted signal. Initially, the chromophores in condensed phases are in the thermal equilibrium state. The first E1 field-matter interaction creates a coherence state between the ground and excited states, i.e., the superposition of the ground and excited states. The second interaction with the E2 pulse puts the molecular system back to a population in either an excited or ground state. When there are multiple vibrational states or electronically delocalized exciton states that can be excited by spectrally broadband pulses, the state vector during T will demonstrate an oscillating pattern, i.e., quantum beats. The third pulse E3 shifts the molecular system to another coherence and it evolves for t. If the initial excitation frequency is different from the emission frequency due to changes in the local environment or the chemical structure of the chromophores during the waiting time, quantitative information about these processes can be extracted by analyzing the changes in both the diagonal and cross-peak shapes and intensities.
Various nonlinear response function components can usually be conveniently classified into non-rephasing \( {\mathbf{R}}_{\text{NR}}^{(3)} (\tau ,T,t) \) and rephasing \( {\mathbf{R}}_{\text{R}}^{(3)} (\tau ,T,t) \) transition pathways. In experiments, the non-rephasing and rephasing signals can be selectively measured by changing the time sequence of the first three pulses.
The emitted signal electric field is thus a function of the two delay times τ and T, i.e., Es(t, T, τ). To measure the phase and amplitude of this signal electric field, a successful strategy is to have it interfere with an additional reference field, i.e., local oscillator field ELO(kLO), and to measure and analyze the spectral interferogram (Fig. 1.3). This detection technique is known as heterodyned detection because the spectral distribution of the local oscillator can differ from that of the third-order signal electric field. Because the spectral components of the total electric field are measured using an array detector combined with a monochromator, the measured spectrum is given by

Coherent 2D spectroscopy based on the measurement of a spectral interferogram. Delay stages in the first black box are used to control the relative time intervals between pulses. M = monochromator. D = detector
where ωt is the Fourier frequency conjugated with detection time t. In (1.19), |ELO(ωt)|2 contributes to the measured spectral interferogram as a constant independent from the two delay times τ and T so that it can be removed from the measured intensity. The signal field intensity, which is often referred to as the homodyne signal, |Es(ωt, T, τ)|2, is negligibly small compared to the last interference term because of the inequality |ELO(ωt)|≫|Es(ωt, T, τ)|. Thus, the measured heterodyne-detected signal that is needed to obtain the 2D spectrum is
To retrieve the complex signal electric field, i.e., both the real and imaginary parts of the signal field, from the spectral interferogram, it should first be Fourier transformed to the time domain data. The positive time component of the time-domain signal is only taken into consideration for the subsequent inverse Fourier transformation back to the frequency (ωt) domain. Thus, the obtained spectrum is a function of τ, T, and ωt, which is denoted as \( \bar{S}_{{\text{het}}} (\omega_{t} ,T,\tau ) \). Finally, the Fourier transformation of \( \bar{S}_{{\text{het}}} (\omega_{t} ,T,\tau ) \) with respect to τ produces the 2D spectrum S2D(ωt, T, ωτ). The absorptive component of the signal is measured when the signal is in phase with that of the LO field, whereas the dispersive component is obtained when the signal is 90° out of phase with respect to the LO. In practice, the dispersive and absorptive components of the heterodyned signal can be obtained using phase cycling methods such as the dual phase scan method.
To obtain purely absorptive 2D spectra, the non-rephasing and rephasing signals are measured independently using two different pulse time sequences at a fixed waiting time T. These signals are Fourier transformed with respect to t and τ and are added together to obtain the absorptive 2D spectrum:
In experiments, the time resolution is determined by the duration of the incident pulses, whereas the spectral resolution is determined by (1) the characteristics of the monochromator (e.g., the grating and focal length), (2) the pixel size of the CCD or IR array detector, and (3) the scanning range of the delay time τ between the first and second pulses.
The measured 2D spectra are then displayed with respect to the excitation (pump) frequency ωτ and the emission (probe) frequency ωt for varying waiting times T. The horizontal axis labeled as ωτ provides information on the molecular transition frequencies of an ensemble of chromophores, whereas the peak positions along the vertical probe frequency (ωt) axis provide information on the quantum states involved in the created superposition state of the system after the waiting time T. Thus, coherent 2D spectroscopy is composed of photon-labeling (i.e., writing), waiting, and photon-detecting (i.e., reading) steps. A molecule labeled (i.e., excited) by the first two field-matter interactions spontaneously undergoes a variety of relaxations, transitions, or reactions, which essentially result in the change in the transition frequency of the labeled molecule. The 2D optical spectrum can therefore be considered a 2D frequency correlation map between the initial and final frequencies or states, which provides quantitative information on the dynamics of molecules.
The overall experimental layout shown in Fig. 1.3 is also similar to the Mach-Zehnder interferometer. Usually, the local oscillator field does not need to pass through the optical sample. In practice, the amplitude ratio of |Es|/|ELO| is deliberately varied to optimize the signal-to-noise ratio. One of the most difficult issues in measuring the 2D spectrum is to keep the relative phases of the pulses stable and constant throughout the experimental time. The relative phase of the signal field with respect to that of the LO (reference) field is determined by the relative phases of incident pulses:
Here, the phase differences \( \phi_{{{\mathbf{E}}_{1} }} - \phi_{{{\mathbf{E}}_{2} }} \) and \( \phi_{\text{LO}} - \phi_{{{\mathbf{E}}_{3} }} \) fluctuate in time because the time delays between E1 and E2 and between E3 and ELO, respectively, fluctuate. Usually, it is quite challenging to precisely control the relative phase between the signal and LO fields. The heterodyne-detected signal inevitably consists of both absorptive and dispersive contributions. Thus, to extract the purely absorptive spectra from the measured spectral interferograms, the relative time delay errors between the incident pulses and the chirps on the incident pulses need to be numerically corrected afterward. This numerical procedure is often referred to as phasing . The most successful and widely used method is based on the pump-probe projection theorem. This theorem holds that the integrated spectrum of S2D(ωt, T, ωτ) over frequency ωτ should, in principle, be identical to the pump-probe spectrum with respect to the probe frequency ωt. Therefore, the phase factor by which the complex 2D spectrum is multiplied should be adjusted to ensure the 2D spectrum projected onto the ωt-axis better matches the pump-probe spectrum, thus completing phasing correction.
1.4.4 Phasing
As emphasized above, the important step for successful heterodyne-detected 2D spectroscopy is to measure the amplitude and phase of the signal field Es with respect to the local oscillator field ELO. The period of electromagnetic waves in the UV-visible range is just a few femtoseconds per cycle, so it is difficult to accurately and repeatedly measure the absorptive component of the third-order signals with fixed relative phases. Suppose that the phases of incident pulses vary in time due to fluctuations in the incident beam paths. The relative phase \( \phi (t) \) can be written as the sum of the time-averaged constant phase angle \( \phi_{0} \) and the fluctuating component \( \delta \phi_{f} (t) \) as \( \phi (t) = \phi_{0} - \delta \phi_{f} (t) \). If the relative phase between the signal and LO fields varies randomly over the timescale of the experimental data collection, the measured signal that represents the average of the fluctuating phases would be distorted or even vanish depending on the amplitude of the phase fluctuation.
To stabilize the relative phase, an active phase-locking scheme has been developed using an additional interferometer to monitor the phase errors. A feedback loop is then implemented to control the position of each individual optic component on the incident beam path. Another approach is to use diffractive optics for passive phase-locking. A diffractive optical element acts like a beam splitter. If two beams propagating along different directions are focused onto the diffractive optical element, each beam is split into two (±) first-order beams. The waiting time T is controlled before the diffractive optics, but the other delay times are scanned with a pair of sliding glass wedges. Thus, the four generated beams are reflected by the same optical element, e.g., mirrors, and are focused onto the sample. Therefore, the phase noise of the incident beams cancels out. This passive phase-locking technique based on diffractive optics can be easily implemented within a coherent 2D spectrometer without requiring additional interferometer or feedback electronics. A 2D optical spectrometer based on diffractive optics is compact and has been found to be more effective for 2D electronic spectroscopy in the visible frequency domain.
1.4.5 Frequency-Scanning 2D Pump-Probe Spectroscopy
Unlike coherent 2D spectroscopy that utilizes three pulses with different propagation directions, coherent 2D spectroscopy based on a pump-probe geometry has a relatively simple experimental configuration. Typical pump-probe spectroscopy is performed with two pulses propagating non-collinearly. The pump beam Epu(kpu) with frequency ωτ excites only those chromophores that are resonant with the ωτ field. After a finite delay time of T, the probe beam Epr(kpr) is used to monitor the relaxation processes of those excited molecules. In this case, the pump-pump-probe electric field-matter interactions create the corresponding third-order polarization P(3)(t), which in turn generates the pump-probe signal electric field E(3)(t), whose wave vector is ks = −kpu + kpu + kpr. This pump-probe signal field propagates along the same direction as the probe beam, and the spectrum of the interference between the signal and probe fields is measured with a monochromator and an array detector. Because no additional local oscillator (reference) field is used to detect the amplitude and phase of the pump-probe signal field, there is no need to precisely control the relative phase. This is why it is known as self-heterodyne detection spectroscopy. Note that the pump-probe signal field is naturally in phase with the probe beam, which allows the absorptive component of the 2D spectrum to be measured.
This pump-probe scheme has been used to obtain 2D spectra by scanning the pump frequency. The excitation frequency ωτ of the pump can be selected using, for example, a Fabry-Pérot interferometer. To obtain the 2D spectrum SPP(ωt, T, ωτ), the excitation frequency ωτ needs to be scanned over the molecular transition band. The frequency resolution is therefore determined by the spectral bandwidth of the frequency-selected pump beam. The temporal resolution of frequency-scanning 2D pump-probe spectroscopy is determined by the convolution of the frequency-selected pump beam and the probe beam at the sample position. The narrow spectral bandwidth of the pump beam unfortunately generates a 2D spectrum with poor time resolution. In other words, there is a trade off between the temporal resolution and the spectral resolution. Therefore, this frequency-scanning 2D pump-probe measurement method has been used to study molecular systems with relatively slow relaxation processes and chemical or biological reactions.
1.4.6 Time-Scanning 2D Pump-Probe Spectroscopy
Unlike frequency-scanning 2D pump-probe spectroscopy, time-scanning 2D pump-probe spectroscopy utilizes three broadband femtosecond pulses in the pump-probe geometry. In contrast to the three-pulse scattering geometry (Fig. 1.3), the first two incident pulses (Epu1 and Epu2), which are separated by τ in time, propagate collinearly along the direction set by the same wave vector kpu. The pump 1/pump 2/probe interactions with the chromophores generate the pump-probe signal field along the probe direction due to the phase-matching condition of ks = −kpu + kpu + kpr = kpr. The generated signal electric field E(3)(t) interferes with the probe field itself and the spectrum of this interference term is measured using a monochromator and an array detector (Fig. 1.4). In contrast to pump frequency-scanning 2D pump-probe spectroscopy, temporal interferograms are collected with respect to τ at detection frequencies (ωt). Therefore, additional Fourier transformation of the τ-dependent data provides additional frequency information for the nonlinear response function along the ωτ axis. One of the advantages of this form of spectroscopy is that the absorptive 2D spectra can be obtained without sacrificing the temporal resolution. However, the desired 2D signal is measured together with the stronger pump-probe signals originating from the pump 2/pump 2/probe interactions with the chromophores, thus the noise inherently present in the two-beam pump-probe signals contributes to the pump 1/pump 2/probe 2D signal. These strong two-beam pump-probe signals act like an additional time-dependent local oscillator field, which causes the spectral distortion of the 2D signal. Furthermore, this time-scanning 2D pump-probe method still requires phasing correction to obtain the 2D spectrum, even though the number of phasing parameters is reduced. The timing error between Epu1 and Epu2 and the unbalanced chirp also give rise to the spectral distortion of the measured 2D spectrum. In real experiments, the two beams E1 and E2 are usually combined by a 50:50 beam combiner, which means that a loss of almost 50% in the intensity of each beam is unavoidable. To overcome some of these problems, pulse-shaping technology has thus been employed.

Schematic representation of time-scanning 2D pump-probe spectroscopy. The time delay between pump 1 and pump 2 is controlled with a translational stage (or sometimes two sliding wedged glasses). The probe field is delayed from pump 2 by waiting time T. The generated third-order pump-probe signal field interferes with the probe beam itself. The spectrum of the interference signal is measured by a monochromator (M) and an array detector (D)
1.4.7 2D Spectroscopy with a Pulse Shaper
With the development of pulse-shaping technology, it has become possible to carry out coherent 2D pump-probe spectroscopy experiments with commercially available pulse shapers. In principle, this approach is similar to time-scanning 2D pump-probe spectroscopy because a pair of collinearly propagating pump pulses are used to excite molecules and a time-delayed probe pulse is used to monitor the relaxation processes of the excited molecules in condensed phases. Here, it is the pulse shaper that generates the pair of femtosecond pump pulses that are temporally separated in time by τ (Fig. 1.5). The underlying principle of a pulse shaper is well-known. Suppose that an incident pulse electric field Ein(t) has the spectral distribution Ein(ω). To obtain time-separated twin pulses, the pulse shaper modulates the spectral distribution of the incident beam, which is achieved with the use of a mask in the frequency domain, producing the shaped spectral pulse electric field Eout(ω) as \( E_{\text{out}} (\omega ) = M(\omega )E_{\text{in}} (\omega ) \). Here, the spectrum of the acousto-optic modulator (AOM) is in general given by \( M(\omega ) = A(\omega )\exp [i\varphi (\omega )] \), where the spectral amplitude of the mask is denoted as \( A(\omega ) \) and its spectral phase is \( \varphi (\omega ) \). The time profile of the generated electric field Eout(t) is given by the inverse Fourier transformation of the output field spectrum Eout(ω). In principle, the phase and chirp of the shaped pulse or pulses can be controlled by changing the mask spectrum. To generate a pair of femtosecond pulses with a time delay of τ using the acousto-optic modulator, the static acoustic wave needs to be electronically generated so that the mask spectrum is given by \( M(\omega ) = \cos (\omega \tau /2)\exp [i\omega \tau /2] \). The time delay between the two pulses is inversely proportional to the fringe spacing (wavelength) of the acoustic wave created by the AOM. The maximum time delay is therefore limited by the resolution of the AOM. In addition to the time delay between the two pulses, the relative phase and chirp of the generated pulses can also be controlled using the pulse shaper. Essentially, the throughput of the pulse shaper is determined by the efficiency of the grating as well as the AOM. Often, a significant loss in the intensity of the input beam when it passes through the pulse shaper cannot be avoided.

2D pump-probe spectroscopy with a pulse shaper. A pair of twin pump pulses that are separated by τ in time are generated by a pulse shaper on the lower path. The time (T) delayed probe is used to generate the third-order pump-probe signal field and to produce the interference signal detected by the monochromator and the array detector
In this form of 2D pump-probe spectroscopy, the pulse shaper can modulate the relative phase between the pump 1 and pump 2 pulses, i.e., \( \Delta \phi = \phi_{{{\text{E}}_{ 1} }} - \phi_{{{\text{E}}_{ 2} }} = 0 \) and \( \Delta \phi = \pi \), and the absorptive 2D spectrum can be directly obtained by the phase-cycling method. A pulse shaper with a laser system operating at a repetition rate of 1 kHz can produce 500 data points per second. Therefore, a complete 2D spectrum at fixed waiting time T can be collected within a few seconds. However, a drawback of this method is that it cannot control the polarization states of the three beams, which limits its use in exploring all fourth-rank tensor properties of the nonlinear response function of molecules in condensed phases.
Over the past decade, a variety of coherent 2D spectroscopic techniques have been developed and demonstrated. Using a two-dimensional array detector (CCD) working in the visible frequency domain, it has been shown that single-shot 2D electronic and IR spectroscopic measurements are feasible [39, 40]. Using combined spherical and cylindrical lenses, the different spectral components of a broadband pulse can be encoded onto different positions of the sample in space and different pixels on a 2D array detector are used to record the different spectral components of coherent 2D signals. Another possibility is using a wedge-shaped material to induce a pump-to-pulse time delay gradient along the wedge axis. This approach is advantageous because neither a mechanical translational stage nor a pair of sliding wedged glasses is needed to control the time delay between the pulses. Nevertheless, the principles behind these techniques do not differ from those discussed above.
1.5 Perspectives and Concluding Remarks
1.5.1 Coherent Multidimensional Spectroscopy with Mixed IR and Visible Beams
Nonlinear IR (e.g., near-IR, mid-IR, far-IR, and THz) spectroscopy provides critical information on molecular structure and dynamics because vibrational properties are highly sensitive to chemical structure and intermolecular interactions. Although the theory and experimental feasibility of mixed IR-vis 4WM spectroscopy was proposed in 1998 and demonstrated in 1999, only recently have fully time-resolved mixed IR-vis 4WM experiments been performed. They are an IR-optical analogue of heteronuclear 2D NMR because there are two different kinds of oscillating charged particles that are resonant with external fields: electrons and nuclei. Because mixed IR-vis 2D spectroscopy can be used in the analysis of vibration-electronic coupling, it is especially useful for investigating electronic transition-induced changes in molecular structures and the long-lived electronic states or vibrational coherences of photo-excited molecules during chemical reactions.
Another important development in CMS is fluorescence-detected coherent 2D electronic or vibrational spectroscopy [41]. Instead of measuring third-order signal electric fields using spectral interferometric detection, the population of excited state molecules can be measured by detecting the fluorescence intensity after the molecular system interacts with a sequence of optical or IR pulses whose relative phases are accurately controlled. This form of fluorescence-detected 2D spectroscopy can be extended to coherent 2D microspectroscopy by combining it with a microscope system.
1.5.2 Coherent Multidimensional Spectroscopy with More Than One Phase-Stabilized Mode-Locked Laser
Most CMS techniques developed and used over the past two decades use a single mode-locked laser. Although it produces a train of femtosecond pulses, each CMS signal field is generated by considering one pulse at a time. More specifically, a single laser pulse is split into two or more pulses and their relative delay times are controlled with mechanical delay devices (Fig. 1.6a). The generated daughter pulses thus originate from the same mother pulse, meaning they should remain coherent. However, in practice, this is not always the case because the beam paths fluctuate due to the environmental vibrations of optical mounts and chaotic air flow, which results in the decoherence of pulsed electric fields. However, as long as this noise is actively or passively controlled, each individual signal measurement is performed with one pulse from the laser at a time. The 2D spectrum is thus the average of the signals obtained from many thousands of laser pulses.

a Conventional interferometric measurement of a sample’s linear or nonlinear optical response signal field. Here, only one mode-locked laser producing a train of femtosecond pulses is used. b Schematic representation of dual optical frequency comb (OFC) spectroscopy. The two frequency comb lasers are phased-locked with each other using one standard atomic clock radio frequency. Any pair of pulses from the two OFC lasers will thus have a fixed phase relationship, which means that the pulsed fields are coherent. The upper wave from OFC1 is used as the reference field, which is then allowed to interfere with the sample’s generated linear or nonlinear optical response signal field. By deliberately making the two repetition frequencies differ slightly from each other, automatic time scanning can be achieved, and the down-converted temporal interferogram can be measured with a single detector
Very recently, an interesting development in CMS has been reported, in which more than one mode-locked laser is used, where the phase-stabilized ultrafast laser is referred to as an optical frequency comb, which generates a train of pulses with highly stable repetition and carrier-envelop-offset frequencies. To understand how this differs from more conventional Ti:Sapphire lasers, it should be noted that the optical phase of a pulse from an ordinary 1 kHz Ti:Sapphire laser has no correlation with that of another pulse separated by 1 ms. On the other hand, there is a fixed phase relationship between any pair of pulses within the train of pulses from a highly stabilized frequency comb laser. This long-term coherence among the pulses is a key feature of the frequency comb. If two frequency comb lasers are stabilized simultaneously with respect to one standard frequency, e.g., a GPS-disciplined Rb atomic clock, the two trains of pulses from the two comb lasers will have a precisely defined phase relationship. Therefore, these two radiation sources together can be used to carry out coherent nonlinear spectroscopic measurements (Fig. 1.6b). This can be viewed as Young’s double slit interference, where the coherent waves from the two slits that will combine to produce an interference fringe pattern are replaced with two phase-stabilized comb lasers. Due to the fixed phase relationship between any pair of pulses from the two frequency comb lasers, the two waves are coherent with each other. One of the two trains of pulses can be used as the reference (i.e., local oscillator) field, whereas the other linearly or nonlinearly interacts with molecular systems. The signal field from the latter then interferes with the reference field to produce a time-domain interferogram. Its Fourier transformation provides information about the spectrum of molecular quantum eigenstates. Therefore, CMS signals can be obtained with a single detector [42]. It is anticipated that a variety of CMS techniques will be developed by combining optical frequency comb and IR frequency comb lasers in the future.
1.5.3 Nonlinear Microspectroscopy
A natural extension of nonlinear optical spectroscopy is to combine it with a microscope to develop novel microspectroscopy techniques. One of the most successful label-free imaging techniques is coherent Raman scattering microscopy. Since the first experimental demonstration of coherent anti-Stokes Raman scattering microscopy, a number of papers have reported a variety of coherent nonlinear optical or vibrational microspectroscopy techniques. For 2D electronic microspectroscopy, the detection of the fluorescence emitted by chromophores that are excited by multiple femtosecond laser pulses can be used, where the coherent 2D spectroscopic signal is selectively measured using phase-cycling techniques.
To achieve coherent 2D IR microspectroscopy, reflective objective lenses or upconversion techniques can be used to measure the 2D IR response with detectors in the visible frequency range.
1.5.4 Computational Spectroscopy
A number of computational methods for calculating 2D vibrational spectra have been developed and demonstrated to be of exceptional use in simulating and interpreting experimental results. In addition to pairwise couplings, diagonal and off-diagonal potential anharmonicities, and mode-mode frequency correlation, more information about multiple oscillator systems can be obtained from higher-order CMS methods generating multidimensional spectra. For example, 3D infrared spectroscopy has been explored experimentally and simulated based on classical or semiclassical methods. Nevertheless, it is still difficult to fully interpret these 2D spectra because of the increased number of couplings and the higher dimensionality of the relevant potential energy surface.
In the numerical integration of the Schrödinger equation method, an essential step is the modeling and parameterization of the multi-chromophore Hamiltonian from the electronic structure calculations of their static structures. In contrast, the classical mechanical approach relies on an accurate description of molecular vibrational properties in the context of classical dynamics. This often requires the use of sophisticated molecular mechanics potential models or quantum mechanical potential. In this regard, recently proposed ab initio theories of vibrational solvatochromism and direct QM/MM or full ab initio MD simulations of vibrational spectra can be fruitfully adapted for either approach to improve their efficiency and accuracy.
Two-dimensional vibrational spectra can be calculated using the exact hierarchical equation of motion (HEOM) approach. It should be noted, however, that this HEOM approach is based on the assumption that the spectral density description of the bath is valid and accurate. This means that the bath is modeled as a collection of independent harmonic oscillators, i.e., the Caldeira-Leggett quantum dissipative bath model, and the spectral distribution of solute-solvent coupling constants is well-known. Under this assumption, the quantum correlation between the system and the bath can be accounted for. However, this method scales rather unfavorably, limiting its application to relatively small systems even though a number of approximations have been developed to improve its efficiency. Another limitation is that the method requires the parameterization of the spectral density.
1.5.5 Summary
In this chapter, I have presented a brief introduction to the fundamental theory, computational methods, and experimental techniques that have been used to describe or measure a variety of coherent multidimensional spectroscopic signals. Theoretically, the formalism of the nonlinear response function has been developed to describe various CMS methods. For nonlinear vibrational spectroscopy, one of the widely used theoretical approaches is based on the approximation that the associated quantum mechanical nonlinear vibrational response function can be evaluated using the MD simulation trajectories of either equilibrium or non-equilibrium systems. Another popular method of choice is to solve the time-dependent vibrational Schrödinger equation of coupled oscillators where their frequencies, coupling constants, and anharmonicities, which fluctuate in time due to the system-bath interactions, can be obtained with independent computational methods and theoretical models. Experimentally, over the past decade, a variety of 2D vibrational and/or electronic spectroscopic techniques that utilize femtosecond IR, THz, and/or UV-visible pulses have been developed and used to study molecular structure and dynamics, protein conformation and folding-unfolding processes, large-scale delocalized modes in condensed phases, and ultrafast reaction dynamics during chemical and biological reactions. We have recently witnessed novel forms of coherent multidimensional spectroscopy, such as multiple frequency comb spectroscopy and nonlinear optical and vibrational microspectroscopy. There is no doubt that the theory and experimentation in coherent multidimensional spectroscopy and microspectroscopy will pave the way for the development of novel time and space resolved spectroscopy and microscopy tools that are useful for understanding the underlying principles, mechanisms, and functions of novel materials and synthetic and biological molecules in condensed phases.
References
G.R. Fleming, Chemical Applications of Ultrafast Spectroscopy. (Oxford University Press, 1986)
A.H. Zewail, Femtochemistry: atomic-scale dynamics of the chemical bond using ultrafast lasers (Nobel Lecture). Angew. Chem. Int. Ed. 39, 2586–2631 (2000). https://doi.org/10.1002/3527600183.ch1
J. Zheng, K. Kwak, J. Xie, M.D. Fayer, Ultrafast carbon-carbon single-bond rotational isomerization in room-temperature solution. Science 313, 1951–1955 (2006). https://doi.org/10.1126/science.1132178
M. Maroncelli, J. Macinnis, G.R. Fleming, Polar solvent dynamics and electron-transfer reactions. Science 243, 1674–1681 (1989). https://doi.org/10.1126/science.243.4899.1674
G.R. Fleming, M. Cho, Chromophore-solvent dynamics. Annu. Rev. Phys. Chem. 47, 109–134 (1996). https://doi.org/10.1146/annurev.physchem.47.1.109
S. Mukamel, Principles of Nonlinear Optical Spectroscopy. (Oxford University Press, 1995)
M. Cho, Two-Dimensional Optical Spectroscopy. (CRC Press, 2009)
P. Hamm, M. Zanni, Concepts and Methods of 2D Infrared Spectroscopy. (Cambridge University Press, 2011)
H. Kim, M. Cho, Infrared probes for studying the structure and dynamics of biomolecules. Chem. Rev. 113, 5817–5847 (2013). https://doi.org/10.1021/cr3005185
J. Ma, I.M. Pazos, W. Zhang, R.M. Culik, F. Gai, Site-specific infrared probes of proteins. Annu. Rev. Phys. Chem. 66, 357–377 (2015). https://doi.org/10.1146/annurev-physchem-040214-121802
M. Cho, Coherent two-dimensional optical spectroscopy. Chem. Rev. 108, 1331–1418 (2008). https://doi.org/10.1021/Cr078377b
A.M. Weiner, S. De Silvestri, E.P. Ippen, Three-pulse scattering for femtosecond dephasing studies: theory and experiment. J. Opt. Soc. Am. B 2, 654–662 (1985). https://doi.org/10.1364/JOSAB.2.000654
D. Zimdars et al., Picosecond infrared vibrational photon echoes in a liquid and glass using a free electron laser. Phys. Rev. Lett. 70, 2718–2721 (1993). https://doi.org/10.1103/PhysRevLett.70.2718
Y. Tanimura, S. Mukamel, Two-dimensional femtosecond vibrational spectroscopy of liquids. J. Chem. Phys. 99, 9496–9511 (1993). https://doi.org/10.1063/1.465484
D.A. Blank, L.J. Kaufman, G.R. Fleming, Fifth-order two-dimensional Raman spectra of CS2 are dominated by third-order cascades. J. Chem. Phys. 111, 3105–3114 (1999). https://doi.org/10.1063/1.479591
M. Cho et al., Intrinsic cascading contributions to the fifth- and seventh-order electronically off-resonant Raman spectroscopies. J. Chem. Phys. 112, 2082–2094 (2000). https://doi.org/10.1063/1.480777
M. Cho, On the resonant coherent two-dimensional Raman scattering. J. Chem. Phys. 109, 5327–5337 (1998). https://doi.org/10.1063/1.477151
M. Cho, G.R. Fleming, Fifth-order three-pulse scattering spectroscopy: can we separate homogeneous and inhomogeneous contributions to optical spectra? J. Phys. Chem. 98, 3478–3485 (1994). https://doi.org/10.1021/j100064a033
J.D. Hybl, A.W. Albrecht, S.M. Gallagher Faeder, D.M. Jonas, Two-dimensional electronic spectroscopy. Chem. Phys. Lett. 297, 307–313 (1998). https://doi.org/10.1016/S0009-2614(98)01140-3
P. Hamm, M. Lim, R.M. Hochstrasser, Structure of the amide I band of peptides measured by femtosecond nonlinear-infrared spectroscopy. J. Phys. Chem. B 102, 6123–6138 (1998). https://doi.org/10.1021/jp9813286
K. Park, M. Cho, Time- and frequency-resolved coherent two-dimensional IR spectroscopy: Its complementary relationship with the coherent two-dimensional Raman scattering spectroscopy. J. Chem. Phys. 109, 10559–10569 (1998). https://doi.org/10.1063/1.477756
W. Zhao, J.C. Wright, Spectral simplification in vibrational spectroscopy using doubly vibrationally enhanced infrared four wave mixing. J. Am. Chem. Soc. 121, 10994–10998 (1999). https://doi.org/10.1021/ja9926414
S. Mukamel, Multidimensional femtosecond correlation spectroscopies of electronic and vibrational excitations. Annu. Rev. Phys. Chem. 51, 691–729 (2000)
T.L.C. Jansen, S. Saito, J. Jeon, M. Cho, Theory of two-dimensional vibrational spectroscopy. J. Chem. Phys. 150, 100901 (2019). https://doi.org/10.1063/1.5083966
S. Mukamel, V. Khidekel, V. Chernyak, Classical chaos and fluctuation-dissipation relations for nonlinear response. Phys. Rev. E 53, R1–R4 (1996). https://doi.org/10.1103/PhysRevE.53.R1
S. Mukamel, J.B. Maddox, All-forward semiclassical simulations of nonlinear response functions. J. Chem. Phys. 121, 36–43 (2004). https://doi.org/10.1063/1.1756582
T.I.C. Jansen, K. Duppen, J.G. Snijders, Close collisions in the two-dimensional Raman response of liquid carbon disulfide. Phys. Rev. B 67, 134206 (2003). https://doi.org/10.1103/physrevb.67.134206
T. Hasegawa, Y. Tanimura, Calculating fifth-order Raman signals for various molecular liquids by equilibrium and nonequilibrium hybrid molecular dynamics simulation algorithms. J. Chem. Phys. 125, 074512 (2006). https://doi.org/10.1063/1.2217947
A. Sakurai, Y. Tanimura, Does ℏ play a role in multidimensional spectroscopy? reduced hierarchy equations of motion approach to molecular vibrations. J. Phys. Chem. A 115, 4009–4022 (2011). https://doi.org/10.1021/jp1095618
M. Reppert, P. Brumer, Classical coherent two-dimensional vibrational spectroscopy. J. Chem. Phys. 148, 064101 (2018). https://doi.org/10.1063/1.5017985
T.I.C. Jansen, J. Knoester, Nonadiabatic effects in the two-Dimensional infrared spectra of peptides: application to alanine dipeptide. J. Phys. Chem. B 110, 22910–22916. https://doi.org/10.1021/jp064795t (2006)
B. Błasiak, C.H. Londergan, L.J. Webb, M. Cho, Vibrational probes: From small molecule solvatochromism theory and experiments to applications in complex systems. Acc. Chem. Res. 50, 968–976 (2017). https://doi.org/10.1021/acs.accounts.7b00002
J. Kim, J. Jeon, T.H. Yoon, M. Cho, Dual frequency-comb spectroscopy of chromophores in condensed phases. Chem. Phys. 520, 122–137 (2019). https://doi.org/10.1016/j.chemphys.2018.12.012
M.C. Asplund, M.T. Zanni, R.M. Hochstrasser, Two-dimensional infrared spectroscopy of peptides by phase-controlled femtosecond vibrational photon echoes. Proc. Natl. Acad. Sci. 97, 8219–8224 (2000). https://doi.org/10.1073/pnas.140227997
B. Lomsadze, S.T. Cundiff, Frequency combs enable rapid and high-resolution multidimensional coherent spectroscopy. Science 357, 1389–1391 (2017). https://doi.org/10.1126/science.aao1090
J. Kim, T.H. Yoon, M. Cho, Interferometric measurement of transient absorption and refraction spectra with dual frequency comb. J. Phys. Chem. B 122, 9775–9785 (2018). https://doi.org/10.1021/acs.jpcb.8b09262
I. Coddington, N. Newbury, W. Swann, Dual-comb spectroscopy. Optica 3, 414–426 (2016). https://doi.org/10.1364/optica.3.000414
T. Ideguchi, A. Poisson, G. Guelachvili, N. Picqué, T.W. Hänsch, Adaptive real-time dual-comb spectroscopy. Nat. Commun. 5, 3375 (2014). https://doi.org/10.1038/ncomms4375
M.F. DeCamp, L.P. DeFlores, K.C. Jones, A. Tokmakoff, Single-shot two-dimensional infrared spectroscopy. Opt. Express 15, 233–241 (2007). https://doi.org/10.1364/OE.15.000233
E. Harel, A.F. Fidler, G.S. Engel, Real-time mapping of electronic structure with single-shot two-dimensional electronic spectroscopy. Proc. Natl. Acad. Sci. 107, 16444–16447 (2010). https://doi.org/10.1073/pnas.1007579107
P.F. Tekavec, G.A. Lott, A.H. Marcus, Fluorescence-detected two-dimensional electronic coherence spectroscopy by acousto-optic phase modulation. J. Chem. Phys. 127, 214307 (2007). https://doi.org/10.1063/1.2800560
J. Kim, B. Cho, T.H. Yoon, M. Cho, Dual-frequency comb transient absorption: Broad dynamic range measurement of femtosecond to nanosecond relaxation processes. J. Phys. Chem. Lett. 9, 1866–1871 (2018). https://doi.org/10.1021/acs.jpclett.8b00886
Acknowledgements
This work was supported by IBS-R023-D1.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Cho, M. (2019). Introduction to Coherent Multidimensional Spectroscopy. In: Cho, M. (eds) Coherent Multidimensional Spectroscopy. Springer Series in Optical Sciences, vol 226. Springer, Singapore. https://doi.org/10.1007/978-981-13-9753-0_1
Download citation
DOI: https://doi.org/10.1007/978-981-13-9753-0_1
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-9752-3
Online ISBN: 978-981-13-9753-0
eBook Packages: Physics and AstronomyPhysics and Astronomy (R0)