
Abstract
Omics approaches can monitor responses and alterations of biological pathways at a genome scale, which are useful to predict potential adverse effects from environmental toxicants. However, high-throughput application of transcriptomics in chemical assessment is limited due to the high cost and lack of “standardized” toxicogenomic methods. Here, we have developed a reduced transcriptome approach as an alternative strategy to facilitate testing a wide range of chemical concentrations, which targets a reduced set of genes to focus on key toxic response genes and associated pathways. The reduced transcriptomic approach allows full dose range testing of hundreds of chemicals or mixtures using human cells or zebrafish embryos. Points of departure of genes and pathways can be used for potency ranking and to classify chemicals by disrupted biological pathways. It is anticipated that reduced transcriptomic approaches will significantly advance pathway-based high-throughput screening of potentially toxic substances.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


1 Introduction
A major challenge in regards to prioritizing environmental chemicals and/or assessing the hazard of complex mixtures is the lack of sufficient toxicological information for thousands of chemicals and endless possibility of mixtures (Zhang et al. 2018). Toxicity pathway profiling could help to predict potential apical toxicity and prioritize and guide subsequent testing of the chemicals. To support chemical risk assessment, analytical frameworks such as the use of adverse outcome pathways (AOPs) have been adopted to describe cascading chains of causal events occurring at different levels of biological organization that result in a measurable ecotoxicological effect (Conolly et al. 2017). In particular, the AOP framework has gained traction in regulatory science as it offers an efficient and effective means for linking toxicological mechanisms with the standardized toxicity end points required for regulatory assessments, increasing their relevance as predictors of ecosystem effects. Nevertheless, among the many challenges and limitations that must be addressed to realize the full potential of the AOP framework in regulatory decision-making, one prominent task is the development of appropriate in vitro bioassays to capture all possible molecular initiating events (MIEs) and/or key events (KEs) that could be generated by thousands of untested chemicals (Knapen et al. 2018).
Traditionally, monitoring and assessment of mixtures have relied on chemistry analyses. Although high-throughput targeted and nontargeted analytical methods have been developed for the detection of hundreds of chemicals present in complex environmental samples, chemical-focused analyses cannot detect contaminants with unknown structure, and cannot explain the cumulative toxicity of mixtures (Altenburger et al. 2015). Effect-based approaches such as high content screening can provide assessments of biological activity of environmental mixture. However, most current cell-based HTS assays are limited in their coverage of biological pathways, and subsequently their ability to predict a wide range of potential adverse outcomes (Escher et al. 2014).
Integrating genomic dose-response modeling into the hazard characterization with wide-range doses has shown to be valuable in risk assessment, particularly when applied to lower, more environmentally relevant doses. Omics technologies have the ability to provide a global view of the cellular processes of an individual in response to chemical exposure, and to do so in a high-throughput manner with the advancement of bioinformatics (Zhang et al. 2011). Therefore, the widespread adoption of omics can increase the efficacy, efficiency, and timeliness of chemical assessment, and generate new knowledge on the underlying mechanisms contributing to adverse effects (Zhang et al. 2018). Genomic studies on wide-range doses could help to determine new biomarkers and to derive points of departure for chemical risk assessment. For instance, certain endocrine-disrupting chemicals have been reported to alter gene expression in a nonmonotonic manner at low doses, which indicates a potential novel molecular mechanism. In addition, application of multiple doses with single replicate using human cells or zebrafish embryo has been shown to effectively identify vulnerable genes and pathways (Hermsen et al. 2012). Concentration-dependent bioactivity of chemicals or mixtures could indicate potential early responses. Pathway analysis based on the active values of differentially concentration-dependent genes implicates the potential bioactivity of samples, which can be used in diagnostic analysis of chemical profiles (Wang et al. 2018). However, utilization of biological-pathway responses derived from concentration-dependent genomic data is still limited in hazard characterization.
The development of reproducible, dose-dependent omics protocols for chemical testing is urgently needed to support the incorporation of omics technologies into chemical risk assessment (Zhang et al. 2018). While omics have been widely used to investigate whole-genome alteration for MOA prediction and classification of chemicals, consistent protocols for generation, processing, and interpretation of omics data should be established before such methodologies are incorporated into regulatory assessment. Great efforts have been made on the standardization of transcriptomics to profile genome expression. For instance, the MicroArray Quality Control (MAQC) project has been launched to evaluate the reproducibility of inter- and intraplatform microarray technologies. In ecotoxicology, a few studies have highlighted standardized transcriptomic protocols, ranging from RNA extracts to full bioinformatic pipelines, potentially improving interlaboratory comparability. One suggested advantage of concentration-dependent transcriptomic data is the generation of point of departure (POD) values. We also note that some concentration-dependent transcriptomics studies have been conducted using inconsistent bioinformatic methods for data filtering, concentration–response modeling, and quantitative characterization of genes and pathways. This makes comparison across studies problematic, and as such, we recommend that future studies consider the development of standardized protocols for concentration-dependent transcriptomic characterization of chemicals (Zhang et al. 2018).
As an alternative strategy to sequencing of the whole transcriptome, reduced transcriptome analysis targets a reduced set of genes to focus on key toxic response genes and associated pathways to facilitate testing a wide range of chemical concentrations (Xia et al. 2017). A key supporting principle is that a subset of representative genes in a network may function as surrogates for all genes of that network. The use of reduced transcriptomes has been proposed to measure a subset of genes to focus on toxicologically relevant genes; this reduces the complexity of such studies, which in turn supports extending the range of chemical concentrations being tested—a key statistical consideration. The principle supporting the use of reduced transcriptomes is that the expression of key genes can provide a proxy for expression of all genes in networks or pathways of interest. Recent examples of the use of gene subsets include the library of integrated network-based cellular signatures (LINCS) project, which has designed a key gene set of 978 human genes, and the National Institute of Environmental Health Sciences (NIEHS) has proposed the S1500 gene set consisting of 1500–3000 human genes, which were computationally selected from thousands of gene expression data sets in Gene Expression Omnibus (GEO) to be representative of the whole human transcriptome.
2 Development of Reduced Transcriptome for Human and Zebrafish
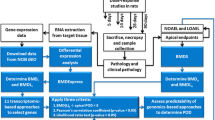
We have recently developed streamlined reduced transcriptome approaches using human cells (RHT) and zebrafish embryos (RZT) for the assessment of toxic substances (Fig. 1) (Xia et al. 2017; Wang et al. 2018). Two principles were employed when selecting genes for reduced transcriptomes: (1) maximal coverage of biological pathways and (2) toxicological relevance. Firstly, to cover comprehensive biological pathways, we selected all genes from existing biological-pathway databases such as the Kyoto Encyclopedia of Genes and Genomes (KEGG) or Gene Ontology (GO), followed by computational inference of a small set of genes playing central roles in entire gene networks. Secondly, toxicologically relevant genes were retrieved from existing toxicology testing databases, including all gene end points tested in ToxCast (https://www.epa.gov/chemical-research/toxicity-forecaster-toxcasttm-data) and all genes associated with MIEs and KEs in AOP-Wiki (https://aopwiki.org/aops). The above steps generated RHT and RZT gene sets consisted of 1200 human genes and 1637 zebrafish genes, respectively (Tables 1 and 2, Tables S1 and S2). In silico evaluation was performed to validate the coverage of biological pathways by RHT and RZT gene sets, which showed >90% KEGG and GO pathways were covered by at least one gene in each gene set. Further, the ability of RHT and RZT to represent the entire transcriptome was validated using existing transcriptome data sets, where reduced transcriptomes were seen to faithfully represent clustering patterns in entire transcriptomes. Finally, genes in reduced transcriptomes were multiplexed and PCR-amplified, followed by simultaneous measurement of amplicon abundance (RNA-ampliseq). Below RZT was used as an example to illustrate the development procedure for reduced transcriptome.

A streamlined workflow for reduced transcriptomic analysis of toxic substance by dose-response modeling. (1) Determination of point of departure of genes (PODgene); (2) Derivation of point of departure of pathways (PODpath) and development of dose-dependent response of pathways by ranking of PODpath; (3) Screening chemical by ranking of potency and chemical classification by disrupted biological pathways
Design of a Gene Set for Reduced Zebrafish Transcriptome. The RZT gene set was selected to represent the key biological pathways and toxicologically relevant processes in zebrafish (Danio rerio) genome. First, a list of genes associated with key biological pathways (in Entrez ID formats) was curated from three databases, including Kyoto Encyclopedia of Genes and Genomes (KEGG), zebrafish orthologs of L1000 landmark genes and zebrafish orthologs of pathway reporter genes (Table 2). The centrality values of genes were calculated using CentiScaPe in Cytoscape software. Centrality values are node parameters demonstrating the relevant position of nodes in a whole network. Higher centrality value suggests more central roles of a gene in biological pathways. Then the numbers of significantly enriched KEGG pathways and GO terms (adjusted p-value < 0.05) were calculated in clusterProfiler by walking down the list of curated genes from high to low centrality values by adding 100 genes each time. We selected a minimum number of genes playing central roles in biological pathways, which may significantly represent the maximum number of biological pathways. Additionally, a list of toxicology-relevant genes was curated to include the following: (1) genes measured as end points in ToxCast, (2) genes corresponding to molecular initiating events (MIEs) and key events (KEs) associated with adverse outcome pathways (AOPs) described in the AOP-Wiki (https://aopwiki.org/wiki/index.php/Main_Page), (3) genes listed in graphical model of the fish hypothalamic–pituitary–gonadal (HPG) axis, and (4) a set of manually retrieved genes associated with development. Finally, to avoid potential amplification bias during mRNA quantification, the combined genes were submitted for online multiplex primers designed by Ion Ampliseq Designer, where the genes with high transcript abundance across zebrafish transcriptome were removed from the RZT gene set.
In Silico Validation of RZT. The numbers of KEGG pathways or GO terms covered by genes in the RZT gene set were calculated to evaluate the biological coverage of RZT. Additionally, the numbers of significantly enriched KEGG pathways and GO terms associated with the RZT gene set were compared to a randomly selected gene set (repeated N = 1000) using clusterProfiler. Furthermore, to evaluate the representation of RZT gene set on the global expression patterns and sensitivity of whole zebrafish transcriptome, five microarray data sets of transcriptomic experiments were used during in silico simulation analysis, which covers five distinct life stages of zebrafish (Table 3). The global expression patterns of the previous study were simulated with principal component analysis (PCA) by using genes from RZT gene set and whole genome using edgeR. The sensitivities of zebrafish transcriptome to toxicants in two concentration-dependent whole transcriptome data sets were evaluated by transcriptional point of departure (PODt) calculated by ten previously reported approaches using RZT gene set and whole transcriptome. The PODt values estimated for the RZT gene set were compared with those for the whole transcriptome and the lowest observed adverse effect level (LOAEL) of apical end points.
Methods for estimating transcriptional point of departure (PODt). The values of PODt based on RZT gene set or whole transcriptome were derived from data of chemical toxicity testing on zebrafish embryo by ten approaches previously described by Farmahin1. In approach 1, 2, 3, 4, and 5, transcriptional benchmark dose (BMDt) values were derived from pathway level. In approach 6, 7, 8, 9, and 10, BMDt values were derived from gene level. The BMDt values derived from each approach were used for estimating PODt. The details are as following:
Calculation of BMDt of genes. BMDExpress2 was used for dose-response modeling and calculation of BMDt of each gene. First, raw counts of transcriptomic data were submitted to one-way ANOVA analysis in BMDExpress to identify genes significantly regulated in at least one treatment group compared to the vehicle control group. Then a best-fit model (Hill, Power, Linear, Polynomial 2°, or Polynomial 3°) for each gene was identified by the default parameters with slight changes as following: the Hill model was fagged if the “k” parameter was <1/3 of the lowest positive dose. In that situation, Hill model was excluded from the best-fit model selection of that gene.
Calculation of fold changes of genes. In approach 7, the 20 genes with the largest fold changes relative to controls were used. The normalized raw counts of genes were submitted to R package, edgeR3 to calculate fold change of each gene. 20 genes with the largest fold changes across all treatment were selected for further analysis.
Calculation of BMDt of pathways. Genes with calculated BMDt were submitted to R package, clusterProfiler4 to identify pathways of Gene Ontology Biological Process (GO BP) terms. A pathway enriched by at least three genes was assigned with the mean BMDt of genes matched to that pathway.
Estimating PODt by each approach. Bootstrap was used to present the distribution of BMDt estimated by each approach. For each approach, BMDt of genes or pathways was randomly sampled with replacement. Mean BMDt of genes (in terms of approach 6, 7, 8, 9, and 10) or mean BMDt of pathways (in terms of approach 1, 2, 3, 4, and 5) were used for each bootstrap. 2000 bootstraps were used to simulate the distribution of BMDt for each of the ten approaches. Finally, the distribution of BMDt for each approach was presented by boxplot, and the mean values were used as the PODt for each approach (except approach 7 that used median values).
The developed RZT gene set consists of 1637 zebrafish Entrez ID genes, including a list of 1000 genes with greatest pathway centrality scores and a list of 724 toxicology-relevant genes. The 1000 pathway-central genes were shown to be the minimum number of genes representing the maximum biological pathways in terms of GO BP terms and KEGG pathways (Fig. 2a). Toxicology-relevant genes (n = 724) were selected to provide linkages between molecular mechanism and apical end points (Table 2). Then 44 genes were removed by the online designer either because their background expression was too high or too low, or because effective multiplexed primers could not be designed. This resulted in 1637 genes as the final RZT gene set.

In silico evaluation of RZT gene set. a Investigation of minimal number of candidate genes for representing the maximal biological pathways including KEGG pathways and GO BP terms. The red dash line means the cutoff of 1000, where the number of top-ranked candidate genes is low enough for representing maximal biological pathways. The percentage of biological pathways coverage of b KEGG pathways and c GO BP terms by 1637 genes from RZT gene set. d, e Comparison on the distributions of transcriptional point of departure (PODt) estimated by RZT gene set (blue boxes) and the whole transcriptome (yellow boxes) using data from previously published studies (EMTAB-832 and GSE55618, respectively). The black bold lines within boxes represent PODt. The number above boxes represents the ratio of PODt between by RZT gene set and whole genome (larger value to smaller value). In plot (d), the solid lines in red and green represent LOAEL (13.5 μM) for pericardial edema (green line), and LOAEL (28 μM) for malformed heart (red line) induced by flusilazole in zebrafish embryo at 24 hpf. The dash lines in red and green represent threefold ranges of corresponding LOAELs. In plot (e), the red solid and dash lines represent 1/3 and 1/10 values of LOAEL (8 mM) for liver damage induced by isoniazid in zebrafish embryo reported
The RZT gene set showed a broad coverage of biological pathways, where 95% KEGG pathways and 94% GO BP terms were represented by at least one gene in RZT gene set (Fig. 2b and c). The uncovered pathways were mainly associated with basic metabolic processes. Furthermore, the RZT gene set of 1637 genes were significantly enriched in 29 KEGG pathways and 839 GO BP terms (adjusted p < 0.05) respectively, which was a 48-fold and 17-fold more than the average number of KEGG pathways and GO BP terms enriched by randomly selected genes from zebrafish transcriptome.
The RZT gene set could faithfully represent the global expression patterns and sensitivities of a zebrafish’s whole transcriptome to toxicants. The similar clustering patterns of samples were revealed by PCA analysis using the RZT gene set and whole genome on five transcriptomic studies across 24 hpf to adult zebrafish. Furthermore, the RZT gene set quantitatively represented the sensitivity of whole transcriptome for estimating PODt in response of toxicants. The PODt estimated by the RZT gene set was similar to that of whole transcriptome, where the overall ratios of PODt between RZT to whole genome were less than 1.5 more than 80% of the time (Fig. 2d and e). For the data set with recorded LOAEL, the overall PODt values calculated by RZT gene set were within threefold of whole transcriptomic LOAEL (Fig. 2d). For data sets without LOAEL, the overall PODt values calculated by the RZT gene set were still within tenfold ranges of LOAEL retrieved from other literature (Fig. 2e). The PODt values calculated from pathway-based approaches (approach 1, 2, 3, and 5) showed robust consistency between whole transcriptome and the RZT gene set, suggesting that pathway-based approaches may be applicable for estimating PODt by RZT.
3 Pipeline of Dose-Dependent Transcriptomes for High-Throughput Chemical Testing
A standardized pipeline for quantitative characterization of chemicals by dose-dependent reduced transcriptomes was developed (Fig. 1). Our study design employed wide-range doses (eight serial dilutions) with single biological replicates in human cells or zebrafish embryo for a specified exposure period, followed by RNA extraction for Ampliseq RNA library and HTS analysis. For example, the bioinformatics protocol for RHT contains four steps: (1) filtering genes which are unresponsive (counts < 5) to toxic substance exposure; (2) identifying the best-fit model for each gene to calculate POD values of genes (PODgene); (3) interpreting genes in the context of the biological pathways they influence; and (4) deriving POD values of pathways (PODpath) to support sample-based quantitative assessment. In subsequent dose–response modeling, the transcriptional expression of filtered genes against concentrations are fitted to nonlinear models (e.g., parabolic, linear, sigmoid) by assigning a best-fit model (least akaike information criterion (AIC) value) for each gene. PODgene values are determined according to the best-fit models (Fig. 3, Table 4). The PODpath value for a specific biological pathway can then be derived from the average value of PODgene according to genes assigned to that pathway.

Overview of three types of concentration–effect curves for calculating effect concentration of DEGs identified by reduced transcriptomic analysis. The red symbols stand for effective concentrations (ECs) used for each type of concentration–effect curve. EC50, ECFC=1.5, and ECMax stand for median effective concentration, concentration causing 1.5-fold change and concentration inducing maximum effect, respectively
The PODpath estimate can characterize and quantify biological pathways potentially disrupted by toxic substances by identifying potentially sensitive pathways. This knowledge can inform understanding of responses at the molecular level in terms of molecular initiating events (MIEs) or key events (KEs) of chemical toxicity. The potency values (PODgene or PODpath) each provide critical information on the bioactivity of the chemicals. Moreover, altered pathways identified by this process can provide insight into substance mode of action, which can be used to predict likely adverse outcomes, guiding chemical cross read. In an RZT analysis of zebrafish embryos following 8–32 h of exposure post fertilization (hpf) to 10E–5–10 μM bisphenol A (BPA), the most sensitive pathways (PODpath < 0.001 μM) identified were those involved in neurogenesis-related processes (e.g., central nervous system development, nervous system development, locomotion), which was concordant with a previously reported adverse effect of hyperactivity under BPA exposure.
Furthermore, the altered pathways and their corresponding PODpath value by reduced transcriptomics can be used to prioritize chemicals or environmental mixtures based on biological activity. RHT in two human cell lines (HepG2 and MCF7) and RZT in zebrafish embryos have been applied to a set of water samples ranging from wastewater to drinking water using a concentration-dependent transcriptomics protocol. Both RHT and RZT approaches were responsive, identifying a wide spectrum of biological activities associated with water-extracts exposure. Moreover, the most sensitive biological pathways were successfully identified, and were linked to adverse reproductive, genotoxic, and developmental outcomes. In this way, water quality was benchmarked by the sensitivity distribution curve of biological pathways, where the PODpath values of different samples can be ranked to discriminate polluted and clean samples. Overall, RHT and RZT approaches provided efficient and cost-effective tools to prioritize toxic substances based on the responsiveness of biological pathways.
Although used as a representative approach to whole transcriptomes, reduced transcriptomes may lose signals of unmeasured genes or pathways covered by only few genes. Computational methods have been developed to infer the remainder of whole transcriptomes, but regions of poor inference remain due to limitations of existing knowledge and differences among cell types. Currently, the reduced transcriptome approach can only be applied successfully for organisms with well-annotated genomes. In the future, however, reduced transcriptome approaches will be extended to other species as knowledge improves. It is also worth adding that the genes curated in reduced transcriptomes should be optimized, updating toxicology databases such as AOP-Wiki, when both existing and new AOPs are under development.
4 Benchmarking Water Quality from Wastewater to Drinking Waters Using Reduced Transcriptome of Human Cells
One of the major challenges in environmental science is monitoring and assessing the risk of complex environmental mixtures. In vitro bioassays with limited key toxicological end points have been shown to be suitable to evaluate mixtures of organic pollutants in wastewater and recycled water. Omics approaches such as transcriptomics can monitor biological effects at the genome scale. However, few studies have applied omics approaches in the assessment of mixtures of organic micropollutants. Here, an omics approach was presented to profile the biological activity of water samples in human cells by RHT approach and dose-response models. A pair of widely used in vitro cell models in human toxicology, human hepatoma (HepG2), and human mammary cancer (MCF7) cells was used to assess the transcriptomic response induced by the mixtures. Cells were exposed to eight serial dilutions of each sample, and the transcriptional expression of 1200 selected genes was quantified by an RNA amplicon-seq technology. To evaluate the performance of RHT for benchmarking water mixtures, the samples tested in this study were a set of ten water extracts that has been previously characterized by a battery of in vitro assays and chemical analysis. The objectives of the study were (1) to identify the mRNA expression profiles of the 1200 RHT genes in the HepG2 and MCF7 cells exposed to water samples in serial dilutions, (2) to evaluate the ability of RHT to assess biological activity of water samples in comparison with in vitro bioassays, and (3) to compare the concentration-dependent distribution of biological activity by mixture to that of chemical profiles.
Methods
Water samples. The ten water samples (Fig. 4) were collected and extracted with solid-phase extraction (SPE) as previously described (Escher et al. 2014). The dried extracts were stored in −80 ℃ until analysis. Samples were dissolved in DMSO as stock solutions with relative enrichment factor (REF) of 10 000. The REF represents the concentrations of water samples (e.g., a REF of ten means tenfold concentrated sample; a REF of 0.1 means a tenfold diluted sample). All the ten samples have been tested by 103 in vitro bioassays covering the relevant steps of cellular toxicity pathways, including the activation of nuclear receptors, disruption of hormone synthesis, genotoxicity, adaptive stress response, and cytotoxicity.

Description of ten water samples
Cell Culture and Cytotoxicity Assay. HepG2 and MCF7 (ATCC) cells were maintained in DMEM medium with 10% fetal bovine serum (FBS) in a humidified atmosphere of 5% CO2 at 37 °C. After 24 h of incubation, cells seeded into 96 well plates with 1 × 105 cells/mL were dosed with twofold serial dilutions of water samples (DMSO = 0.1%). After 24 h of exposure, cell viability was measured using 96 Aqueous One Solution Reagent (Promega, Madison, WI) according to the manufacturer’s instruction.
Transcriptome Analysis Using RHT. The transcriptional expression of 1200 selected genes was measured by an amplicon-seq technology (Fig. 5, Table 1, and Table S1).

Workflow of reduced human transcriptome (RHT) analysis of water samples. AIC, Akaike’s Information Criterion; EC, effect concentration; DEGs, differentially expressed genes; REF, relative enrichment factor; GO, Gene Ontology
RHT Experiment. Cells in 12 well plates with 1 × 105 cells/mL were treated with 8 5-fold noncytotoxic dilutions (from 10 to 0.000 128 REF for all ten samples in MCF7; from 2 to 0.000 0256 REF for sample Eff2, and from 10 to 0.000 128 REF in the other nine samples in HepG2) of water samples with a single replicate, in addition to two vehicle control. After 24 h of dosing, cells were harvested for total RNA isolation using an RNeasy Mini Kit (Qiagen, Hilden, Germany). A total of 164 RNA samples (80 treatments and 2 vehicle controls for HepG2 and MCF7, respectively) were stored at −80 ℃ until used. RNA concentrations were measured using QuBit fluorometer 2.0 (Thermo Fisher Scientific, Waltham, MA) with Quant-iT RNA HS Assay Kit according to the manufacturer’s procedure. Libraries were prepared from 10 ng of RNA of each sample using Ion AmpliSeq Library Kit 2.0 and Ion AmpliSeq custom panels (Thermo Fisher Scientific, Waltham, MA), followed by high-throughput sequencing of RHT panel on Ion Torrent Proton (Thermo Fisher Scientific, Waltham, MA). Briefly, the 1200 human genes of RHT panel were multiplex-amplified, and then the counts of genes were quantified using the coverageAnalysis plugin on Ion Torrent Service. To examine the suitable sequence depth of RHT for each sample, Monte Carlo simulations were performed on the samples with maximum counts in HepG2 and MCF7, respectively (Text S1). Genes whose counts were not detected in vehicle control groups were removed, followed by normalization of counts between libraries using the R package edgeR. Fold changes of genes in each treatment were calculated by dividing the counts in treatment by the mean of counts in two vehicle controls.
Concentration Effect Analysis. For each gene, the fold change against concentration was first subjected to linear regression analysis using the R function cor.test (method “Pearson”). Next, the significant genes (p-value of <0.05) were subjected to automatic concentration–effect curves fitting analysis using drc and DoseFinding. Briefly, log2-transformed fold changes of each gene in log10-transformed concentrations were fitted with any of the nine concentration–effect models (Table 4). The model with the least akaike’s information criterion (AIC) value was identified as the best-fit model for that gene. Genes whose best-fit models showed significant curve-fitting performance (p-value of <0.05) were identified as differentially expressed genes (DEGs). The best-fit models of DEGs were used to derive the effect concentrations (ECs) of DEGs (Fig. 1). Briefly, for sigmoidal curves in which the maximum response could be defined, the concentrations causing 50% maximum effect were used as the ECs. For linear concentration–effect curves in which no maximum response can be defined, the concentrations causing absolute 1.5-fold change were used as the ECs of the gene. For the gene with U-shaped concentration–effect curves, its EC was assigned by the concentration causing the maximum response. A value of 100 REF was an inflection point of all ranked EC values, above which a small portion of DEGs (EC ≥ 100 REF) were defined as extrapolation artifacts and removed from further analysis. Finally, concentration-dependent sequences of molecular events were investigated by network analysis of DEGs using stringApp in Cytoscape.
Pathway Analysis. An unsupervised gene ontology (GO) analysis was performed to investigate potential bioactivity of each sample identified by RHT. First, DEGs of each sample were matched to their corresponding GO terms. GO terms with less than three DEGs were removed because three was the minimum number to calculate mean and standard deviation (SD). Then the EC of each GO term was calculated as the geometric mean of the ECs of matched DEGs. A previous study has used the mean of benchmark dose of DEGs for GO analysis, in which a narrow range of concentrations across only one or two orders of magnitude was assessed. However, a wide range of concentrations across six orders of magnitude was used here. The geometric mean rather than mean value of ECs of DEGs was used to represent the EC of each GO term to avoid heavy influence by DEGs with high EC values. Principal component analysis (PCA) was performed on ECs of GO in all samples using FactoMineR.
Representation of In Vitro Pathways. A supervised approach was used to assess the RHT representation of the previous in vitro bioassay. First, gene sets associated with cellular toxicity pathways tested by in vitro bioassays were manually curated from WikiPathways and Gene Ontology. Next, the EC of each pathway was calculated by the geometric mean of the ECs of matched DEGs. To be consistent with in vitro bioassays, the EC values of in vitro pathways of >30 REF were all assigned with 30 REF. For the in vitro pathways matched by single DEG, only the single DEG with acceptably high efficacy (the maximum absolute fold changes across all treatment being >1.5) was considered to be robust to represent the perturbation of its matched pathway(s). For in vitro pathways that were not matched by any DEGs, their ECs were assigned with 30 REF. Finally, the EC values of in vitro pathways identified by RHT analysis in each cell line were presented as a heatmap using the R package gplot. For biological end points tested by multiple bioassays, the geometric mean of EC values from multiple bioassays was calculated to provide a single integrated EC value for the corresponding pathways. The patterns of hierarchical clusters of water samples identified by RHT analysis were compared with the results of in vitro bioassays.
Comparison of Bioactivity Potency with Chemical Profiles. Chemical profiles in water samples may indicate biological effects of mixture, although there may be a lack of explanation for overall bioactivity. A total of 54 chemicals were previously characterized in 6 of 10 water samples at concentrations above the limit of detection (LOD). The RHT profiles were compared with chemical profiles of water samples. The overall biological potency of each sample was characterized by fitting proportionally ranked EC values of GO into a four-parameter dose–response curve using GraphPad (GraphPad Prism 5.0 software, San Diego, CA), which was defined as biological potency distribution curve (BDC). BDC was not fitted for samples with GO terms of <20, which provided too few dots for fitting meaningful distribution curves. The overall chemical profile of each sample was characterized by fitting proportionally ranked concentrations of detected chemicals into a four-parameter dose–response curve defined as chemical concentrations distribution curve (CDC). Furthermore, relative biological potency (REP) and relative chemical contamination (REC) of each sample were calculated according to BDC and CDC using Formulas 1 and 2, respectively:
MBref and MBi are the median values calculated from BDC of the reference sample and sample i, respectively:
MCref and MCi are the median values calculated from CDC of reference sample and sample i, respectively. Sample Eff2 showed the highest biological activity and chemical contamination among all water samples and thus was selected as the reference sample for calculation of REP and REC. Finally, the REP of each sample was compared to its REC.
Results and Discussions
Analysis of RHT by Mixture Samples. Only sample Eff2 showed cytotoxicity at REF > 2 in HepG2, while none of the samples showed any cytotoxicity at REF ≤ 10 in MCF7. The RHT experiment on serial noncytotoxic dilutions of total 164 samples showed sequence counts that ranged from 327 000 to 8 327 170 in HepG2 and from 158 025 to 3 025 355 in MCF7 (Fig. 6a, b). Monte Carlo simulations revealed that sequence depth of 300,000 reads is needed for detective signals (counts of >5) of at least 750 genes. For the only five samples sequenced with counts <300 000, even the sample with the lowest counts of 158 025 (the sixth dilution of O3/BAC in MCF7) still showed coverage of >100 counts for each gene, suggesting that the sequence depth of each sample was adequate for further data analysis. Out of the RHT 1200 gene list, 756 and 767 genes were expressed in HepG2 and MCF7, respectively, with 667 common genes (Fig. 7). The 95% percentile ranges of log2-fold changes across all ten samples in HepG2 was 0.03–9.51, relatively narrower than 0.02–163.14 in MCF7 (Fig. 8a). The sequence data was deposited in the NCBI BioProject database (accession no. PRJNA385238; https://www.ncbi.nlm.nih.gov/bioproject/).

Sequencing counts of ten water samples across eight fivefold dilutions and two DMSO controls in a HepG2 and b MCF7. The number 1–8 of water samples stands for dilutions of samples from highest concentrations to the lowest concentrations. The numbers 1 and 2 of DMSO mean two replicates of vehicle controls. The red lines stand for the sequencing depth of 300,000 for detecting at least 750 genes, which was calculated by Monte Carlo simulation of sequencing counts of c fifth dilution of AO in HepG2 d eighth-dilution of DW in MCF7

Venn diagram of RHT detective genes (counts >0 in both two vehicle control) in HepG2 and MCF7

a Log2-fold changes of all genes identified by RHT analysis across all ten water samples in HepG2 and MCF7, respectively. b Number of differentially expressed genes (DEGs) of ten water samples identified by RHT analysis in HepG2 and MCF7, respectively
The number of DEGs selected by the nine dose–response models across all ten samples ranged from 24 to 109 in HepG2 and from 7 to 157 in MCF7, respectively (Fig. 8b). The DEGs responsive at low dose range (ECs ≤ 1 REF) were mainly fitted with U-shaped models (Fig. 9a, b), which suggest that the mode of hormesis dominates the low dose response of transcriptome. Taking the DEGs of Eff2 in HepG2, for instance, (Fig. 10), CSF1R, SIRT3, and TEK were potentially early response genes (ECs ≤ 0.1 REF) associated with the regulation of ERK1/2 cascade, which were all fitted with the Gaussian model. It has been widely reported that the translocation of ERK1/2 was involved in early gene response. Secondary and adverse biological effects were identified by DEGs with higher EC values, including the regulation of signal transduction (such as CDKN2A, DUSP3, and FOXP1, 0.1 < EC ≤ 1 REF), regulation of cell proliferation (such as CSF1, FYN, and MAPK1, 1 < EC ≤ 10 REF), and regulation of apoptosis (all four DEGs including RTN4, CDKN1A, SRC, and HMOX1, EC > 10 REF). Genes responsive at high dose ranges (ECs > 10 REF) were mainly fitted with linear models, which may be due to severely secondary effects specifically induced at high doses. These results indicated that the dose–response profiles of DEGs identified by RHT analysis could differentiate low dose response from a wide spectrum of biological activities by water samples.

Histogram of the effective concentration (EC) values of differentially expressed genes (DEGs) across ten water samples in a HepG2 and b MCF7, respectively

Concentration-dependent gene network of DEGs identified by RHT analysis in HepG2 cells treated by Eff2. The EC of DEGs is expressed as the relative enrichment factor (REF) of the water sample
The RHT profiles in both HepG2 and MCF7 demonstrated significant responses to the polluted water samples. Most of the DEGs were identified in three polluted samples (Eff2, Eff1, and MF) in both HepG2 and MCF7. For all three polluted samples, 7 and 36 DEGs, mainly involved with cellular response to toxic substance, were identified by RHT in HepG2 and MCF7, respectively. Furthermore, three DEGs (ABCC3, CYP1A1, and KLF9) were identified in all the three polluted samples in both HepG2 and MCF7. CYP1A1 encodes a member of the cytochrome P450 superfamily of enzymes well known for metabolism of xenobiotics. ABCC3 encodes a member of the superfamily of ATP-binding cassette (ABC) transporter involved in multidrug resistance. KLF9 encodes a transcription factor has been widely reported to be involved in response to oxidative stress. The results suggested that polluted water samples prevalently induced a cellular stress response that could be commonly identified by HepG2 and MCF7. Moreover, 29, 23, and 13 DEGs were commonly identified by HepG2 and MCF7 for Eff2, Eff1, and MF, suggesting that HepG2 and MCF7 may be consistent in identification of positive responses in polluted samples.
Fewer DEGs were identified in blank sample in both HepG2 and MCF7 compared with the polluted water samples, suggesting that relatively low response was identified in HepG2 and MCF7 for blank sample. The DEGs of blank sample in HepG2 and MCF7 were mainly associated with nonspecific cellular response, but only four and nine DEGs of the blank sample in HepG2 and MCF7, respectively, showed an absolute fold change of ≥1.5 across all dilution treatments, suggesting very low biological effects induced by blank sample. The results are consistent with the previous results that the blank sample could induce slight nonspecific bioactivity, which may be due to the tiny impurities present in the solvent during SPE extract of samples. In addition, MGMT, encoding the O-6-methylguanine-DNA methyltransferase involved in DNA repair, showed an upregulation trend in HepG2, which may explain the observed genotoxicity previously reported in the blank sample.
The RHT profiles in both HepG2 and MCF7 may show specific response to water samples in middle or lower contaminated situation. The number of DEGs of DW in MCF7 was 103, which was significantly greater than the 25 DEGs of blank sample. However, only 24 DEGs of DW were identified in HepG2, which was equal to the DEGs number of blank sample. It is suggested that the RHT analysis of MCF7 was more sensitive in response to DW sample than that of HepG2. Previous studies have reported that the DW sample induced broad bioactivity of xenobiotic metabolism and genotoxicity. The formation of disinfection by-products increases the toxicity of DW as compared to RW. Disinfection by-products may become more electrophilic after metabolic activation, resulting in enhanced biological effects, such as genotoxicity. The relatively weak effects of DW in HepG2 may be due to the poor expression of drug-metabolizing enzymes in HepG2.
Unsupervised GO Analysis of RHT. GO analysis of RHT profiles may implicate distinct bioactivity between water samples. The number of identified GO terms was proportional to the number of identified DEGs for a sample. For HepG2, the fewest GO terms were identified at blank sample (4) and the most at MF (170). For MCF7, the fewest GO terms were identified at O3/BAC (0) and the most at Eff2 (288). A small number of GO terms enriched by the RHT profiles of blank sample in HepG2 (4) and MCF7 (6) suggest low biological activities of blank sample. Change of the identified GO terms suggests potential changes of bioactivity of water samples from initial to advanced treatment processes. A total of 60 GO terms identified in Eff1 diminished after MF treatment, while 89 GO terms were specifically identified in MF in HepG2 (Fig. 11a). The reduced 60 GO terms of Eff1 were mainly associated with basic cellular response such as cellular oxidant detoxification, while the newly introduced GO terms in MF were associated with more adverse responses such as programmed necrotic cell death. It is suggested that extra and severe bioactivity was produced after MF treatment, which may be explained as the process of chloramination during MF treatment. The remaining six GO terms after the advanced treatment process of AO may be explained as background effect introduced during sample extract, which was similar to the blank sample. The three remaining GO terms identified in all Eff1 (MF, RO, and AO) were annotated to biological processes including that of cellular response to DNA damage stimulus (GO: 0006974), suggesting potential remaining of genotoxicity, which was observed in the previous study. Furthermore, principal component analysis (PCA) of ECs of identified GO terms showed that polluted and clean samples were distinctly separated in both HepG2 and MCF7 cells (Fig. 11b, c), suggesting that GO analysis may distinguish polluted and clean samples by bioactivity. However, in MCF7 cells, DW, a presumed clean sample, was grouped with polluted samples Eff2 and MF, suggesting that HepG2 and MCF7 cells demonstrated cell-type-specific responses.

a Venn diagram of GO terms identified by RHT analysis in Eff1, MF, RO, and AO in HepG2; Principle component analysis of the EC values of GO terms of ten water samples identified by RHT analysis in b HepG2 c MCF7, respectively
Cell-type-specific responses identified by GO analysis of RHT profiles in HepG2 and MCF7. GO terms associated with DNA damage such as mismatch repair of DW sample were identified by RHT analysis in MCF7. However, in HepG2, only nonspecific biological responses such as regulation of cell growth were identified by RHT analysis of DW. Moreover, the cell-type-specific responses may be explained by the GO terms with top lowest EC values, which may represent low dose effects. For instance, in Eff2, the GO with the top ten lowest EC values in HepG2 was mainly associated with immune response, such as transmembrane receptor protein tyrosine kinase signaling pathway, negative regulation of inflammatory response, and natural killer cell mediated cytotoxicity, but the GO terms with the top lowest EC values in MCF7 were only annotated to pathways associated with xenobiotic metabolism, such as canonical Wnt signaling pathway.
Supervised Analysis of RHT for Comparison with In Vitro Bioassays. The RHT profiles of water samples in HepG2 and MCF7 both showed similar patterns with in vitro bioassays, as biological effects were mainly identified in polluted samples (Fig. 12). For end points of xenobiotic metabolism, PXR, CAR, and AHR pathways in both two cell lines showed high responses in less-treated samples and nearly no response in clean samples, which was highly consistent with in vitro assays. Consistent with the results of in vitro assays, the overall activity of pathways associated with PPAR (PPARα, PPARδ, and PPARγ) across ten water samples identified by RHT analysis was relatively low compared with other xenobiotic metabolism pathways. The PPARα and PPARδ pathway showed slight activity (EC > 10 REF) only at RW in HepG2. The PPARγ pathway showed high responses (EC ≤ 10 REF) only at polluted samples (Eff1 and MF) in MCF7, partially accordant with in vitro assays. For specific MOA, the ACHE pathway showed no response at either of the two cell lines, indicating that HepG2 and MCF7 were not suitable for identification of neurotoxic response. The activity of ER and AR pathways was similar to in vitro assays. A slight difference was shown in HepG2, in which the activity of AR pathway was identified in clean samples O3/BAC and AO but at very high REF (EC ≥ 20). For other hormone receptors, the PR pathway showed responses only at polluted samples in MCF7, while no active response of PR was observed in HepG2. The responses of the GR pathway were similar between in vitro assays and RHT profiles in both HepG2 and MCF7, whereas GR activity was specifically shown in MCF7. TR activity was not identified by in vitro assays in any samples, while TR was identified to be activated at multiple samples by RHT analysis in both HepG2 and MCF7. No response of TR activity was observed in previous in vitro thyroid receptor gene bioassays, which has been explained as nonexistence of thyroid agonists and goitrogens in extraction of SPE. Nevertheless, RHT analysis in both HepG2 and MCF7 identified TR activity by multiple samples, which might be due to the “cross-talk effect” between molecular pathways or nongenomic modulation of thyroid hormone signaling pathways on transcript levels.

Heatmap of ECs of the 24 in vitro pathways calculated from RHT analysis in a HepG2, b MCF7, and c in vitro bioassays previously analyzed
For reactive MOA, activity of pathways associated with adaptive stress responses and genotoxicity was identified in HepG2 and MCF7 exposed to polluted samples, which was consistent with the results of in vitro assays. Specific end points associated with adaptive stress responses, such as the Nrf2 and AP-1 pathways in sample Eff2 showed higher activity in HepG2 than in MCF7, suggesting that HepG2 may be more sensitive in response to adaptive stress responses than MCF7. Although immune-related response was not identified in any samples by in vitro assays, obvious immune responses were identified in RHT analysis in polluted samples. Only THP1 cytokine assay in antagonist mode was conducted in a previous in vitro bioassay to measure immunotoxicity. It has previously reported a poor correlation between immunosuppressive chemicals and immunosuppressive activity identified by THP1 assay in water samples. RHT analysis could complement in vitro assays by providing measurement of a broader range of immune-related responses.
Overall, integration of the RHT profiles of HepG2 and MCF7 may provide a broad representation of bioactivity identified by the 103 in vitro assays. In the previous study, the 103 in vitro bioassays included multiple cell lines including 11 types of human cell lines, 2 types of rat cell lines and zebrafish embryos as well as lower organisms such as yeast and Escherichia coli, which were used to measure integrative end points across cellular toxicity pathway. Although the results might not be comparable between in vitro bioassays with different platform background on cell types and species, the RHT analysis of water samples with less cell lines, HepG2 and MCF7, was shown to be capable of reflecting patterns identified by in vitro bioassays. Furthermore, our results support the importance of the utilization of multiple cells in RHT analysis of water samples. HepG2 cell line was more specific in identification of adaptive stress response, and the MCF7 cell line was more specific in response to endocrine disruption effects. To assess the biological responses related to other toxicological end points, such as neurotoxic potential of water samples, other functional cell types, such as a human neuronal cell line, would be necessary.
Comparison of Chemical Profiles with Bioactivity Identified by RHT. Distribution curve of the EC values of GO is a novel approach to estimate the potency of overall biological activities by water samples. Although the RHT profiles in both HepG2 and MCF cells showed the decreasing potency of overall biological activities after serial water treatment processes, cell-type-specific signatures can be observed in the two cell lines (Fig. 13a, b). For the BDC in both HepG2 and MCF7, Eff2 showed the highest biological potency, followed by Eff1, and the clean samples such as SW and AO showed relatively low biological potency. MF and DW showed relatively high potency close to Eff1 in MCF7, and DW showed an even greater potency than its source water RW. However, in HepG2, both MF and DW showed lower potency than their upstream samples Eff1 and RW, respectively. This might be due to genotoxic and oxidative stress effects of disinfection by-products existing in water samples such as MF and DW. Endocrine tissue origin MCF7 cells may be more sensitive than hepatocyte HepG2 cells for identifying the bioactivity of water samples.

Distribution curves of EC values of GO of water samples identified by RHT analysis in a HepG2 and b MCF7 and c distribution curves of concentrations of chemicals detected in water samples, which were used to derive d REP and REC for comparison between biological potency and chemical contamination
The relative potencies of water samples estimated by RHT profile were consistent with chemical contamination level of the samples. A total of six water samples (Eff2, Eff1, O3/BAC, MF, SW, and RO) have been previously characterized by chemistry analysis. Although the concentration of a chemical does not necessarily reflect overall contamination level, the chemical profiles showed distinct separation between polluted samples (Eff2, Eff1, and MF) and clean samples (SW, O3/BAC, and RO) (Fig. 13c). The CDC showed that Eff2 was the most contaminated sample, followed by Eff1 and MF, which was highly accordant with the ranks of biological potency reflected by GO distribution. For clean samples, the CDC showed SW may be more contaminated than O3/BAC and RO, which was also consistent with the GO distribution and the results of previous in vitro bioassays, in which SW showed higher potency than O3/BAC and RO.
The relative biological potencies may quantitatively reflect the alteration of chemical contamination. The overall values of REPs and REC of the ten samples were mainly within one magnitude (Fig. 13d and Table 5). The REP of polluted samples, Eff1 and MF, in MCF7 was both similar to their REC values, implicating that RHT analysis using MCF7 may be sensitive for reflecting the relatively small alteration of chemical profiles between polluted or less-treated water samples. Although the REPs of polluted samples in HepG2 were quite inconsistent with REC, the REP of clean samples, O3/BAC and RO, in HepG2 was similar to their REC values. It is suggested that RHT analysis using HepG2 was more appropriate for quantitatively inferring the alteration of chemical status of clean samples. More samples and more comprehensive chemical profiles are needed to validate suitability of MCF7 and HepG2 cells in assessing specific biological response by water samples in future study.
The chemical profiles may help to explain the biological activity identified by RHT analysis. The occurrence of chemicals such as organophosphate pesticides (chlorpyrifos and diazinon) and steroids (17-β-Estradiol) in Eff2 might contribute to the activation of immune response pathway, which was also suggested by the results of GO terms analysis of Eff2. Chlorpyrifos and diazinon are two kinds of organophosphate pesticides and have been reported to induce immunotoxicity in human cells. 17-β-Estradiol has been widely reported to be able to induce immune response and may be synergistic with organophosphate pesticides. In a relatively clean sample SW, bisphenol A was the dominant chemical with the highest detected concentrations, suggesting that bisphenol A may partially contribute to the biological effects of SW. Bisphenol A is a well-known endocrine-disrupting chemical capable of affecting multiple nuclear receptors such as AR, ER, and AHR, which may explain the effects of AR, ER, and AHR identified by RHT analysis in SW sample. In a clean sample of O3/BAC, although few chemicals were detected, CYP1A1 was identified as DEG by RHT analysis in both HepG2 and MCF7, which may be related to the N, N-diethyl-meta-toluamide (DEET) detected in O3/BAC. Studies have reported that DEET may induce CYP1A1 enzyme in human liver cells. However, knowledge gaps exist in the mixture toxicology of environmental samples, such as response from unknown chemicals and a combined effect. To directly link the observed biological response with the measure chemicals, an effect-directed analysis (EDA) is necessary. The RHT analysis could provide a novel untargeted bioassay in EDA analysis of toxicants in the mixture.
Overall, the RHT method provides a highly dynamic approach to assess the biological response and to benchmark the potencies of water samples. The RHT profiles in two cell lines, HepG2 and MCF7, provided a comparable biological characterization on the water samples by the previous 103 in vitro bioassays, which could significantly increase the efficiency and throughput of assessment. Concentration-dependent bioactivity of water samples could be identified to indicate potential early responses. Pathway analysis based on the active values of differentially concentration-dependent genes implicates the potential bioactivity of samples, which can be used in diagnostic analysis of chemical profiles. The data analysis strategies developed here are also applicable to other omics approaches, such as metabolomics and proteomics, which have also been applied in mixture toxicology. Furthermore, our results highlight the value of the integration of multiple approaches, targeted in vitro assays, and untargeted omics approach, together with a higher resolution of chemical analysis in the study of mixtures. Finally, the selection of cell types that are relevant to toxicological end points is essential in the assessment of mixture by RHT approach. Future studies should explore the utility of other cell types, such as human primary cells and embryonic stem cells, to increase the coverage of biological activities and improve the implications for human health.
5 Assessing Environmental Toxicants Using Zebrafish Embryo Test
Here, a reduced zebrafish transcriptome (RZT) approach was developed to represent the whole transcriptome and to profile bioactivity of chemical and environmental mixtures in zebrafish embryo. RZT gene set of 1637 zebrafish Entrez genes was designed to cover a wide range of biological processes, and to faithfully capture gene-level and pathway-level changes by toxicants compared with the whole transcriptome. Concentration–response modeling was used to calculate the effect concentrations (ECs) of DEGs and corresponding molecular pathways. To validate the RZT approach, quantitative analysis of gene expression by RNA-ampliseq technology was used to identify differentially expressed genes (DEGs) at 32 hpf following exposure to seven serial dilutions of reference chemical BPA (10–10E–5 μM) or each of four water samples ranging from wastewater to drinking water (relative enrichment factors 10–6.4 × 10–4). The RZT-ampliseq-embryo approach was both sensitive and able to identify a wide spectrum of biological activities associated with BPA exposure. Water quality was benchmarked based on the sensitivity distribution curve of biological pathways detected using RZT-ampliseq-embryo. Finally, the most sensitive biological pathways were identified, including those linked with adverse reproductive outcomes, genotoxicity, and development outcomes. RZT-ampliseq-embryo approach provides an efficient and cost-effective tool to prioritize toxicants based on responsiveness of biological pathways.
The objectives of this study were threefold. The first was to curate a reduced gene list from zebrafish transcriptome (RZT) that can comprehensively represent biological pathways and toxicologically relevant processes, and be quantified by Ion Ampliseq Technology (RZT-ampliseq). Second, we aimed to develop a chemical test protocol integrating RZT-ampliseq and dose–response modeling in zebrafish embryo (RZT-ampliseq-embryo). Bisphenol A (BPA), a well-studied endocrine disruptor frequently detected in water samples, was selected as a reference chemical. Finally, we wanted to evaluate the performance of RZT-ampliseq-embryo for use in hazard assessment of environmental mixtures. The mixture samples tested in this study were a set of water extracts previously characterized by the RHT method.
Zebrafish Embryo Culture and Exposure. Embryos at 1 hpf were obtained from group spawns and incubated in buffered embryo medium at 28 ℃ until 8 hpf. Then, 1 mL of exposure solution was prepared in buffered embryo medium with a series of concentrations of single chemical or mixture (Table 5) with a final vehicle concentration of 0.1% dimethyl sulfoxide (DMSO; Sigma-Aldrich). Ten embryos were added to each well of 24 well microtiter plates. Plates were sealed with parafilm and incubated at 28 ℃ on a 14 h/10 h, light/dark cycle. At 32 hpf, the zebrafish embryos were collected for RNA isolation. The basal transcriptional expression of RZT gene set was evaluated on three replicates of 0.1% DMSO treatment sampled from six batches of culture.
Single Chemical. Stock solutions of BPA (Sigma-Aldrich) were prepared in DMSO. First, a single concentration experiment (0.1 and 10 μM BPA) with three replicates was conducted to validate the gene expression profiling of RZT-ampliseq platform by comparing with the whole transcriptome evaluated using microarray. Second, embryos were dosed to a serial tenfold dilutions of BPA (10–10E–5 μM) with a single replicate, in addition to three vehicle control (0.1% DMSO).
Environmental Mixtures. Four water extract samples previously characterized as representing high-, medium-, and low-toxic potencies (11) were tested by the RZT-ampliseq-embryo approach. The dried extracts were stored at −80 ℃ until analysis. Stock solutions of samples were prepared in DMSO with relative enrichment factor (REF) of 10 000, followed by 8–32 hpf embryo exposure. The REF represents the concentration in the ambient water samples (e.g., REF of ten means tenfold concentrated sample; REF of 0.1 means tenfold diluted sample).
mRNA Expression Profiling by Ampliseq. At 32 hpf, zebrafish embryos were collected for total RNA extraction using RNeasy mini kit (QIANGEN, GmbH, Hilden). RNA quantification was performed by using Agilent 2100 Bioanalyzer (Agilent technologies, Santa Clara, CA). Ten ng total RNA from each sample were reverse transcribed into cDNA by poly-A priming followed by PCR preamplification (15 cycles) according to the protocol supplied with the Ion AmpliSeq RNA Library Kit (Life Technologies, Carlsbad, CA). The library was amplified, purified, and stored at −20 ℃. Amplicon size and DNA concentration were measured using Agilent High Sensitivity DNA Kit (Agilent Technologies, Waldbronn, Germany) according to the manufacturer’s recommendation. The resulting libraries were sequenced by Ion Proton (Life Technologies).
Gene Expression Analysis Pipeline. Raw reads were automatically quantified by using Torrent Mapping Alignment Program. Then log2-fold change of each gene was calculated using edgeR package. Differentially expressed genes (DEGs) of a single BPA concentration were identified by the threshold of absolute fold change ≥1.5 and Benjamini–Hochberg adjusted p < 0.05. For full dose–response profiling, log2-fold change of genes against concentrations was submitted to dose–response modeling analysis using drc and DoseFinding as described previously with minor modification. Briefly, nine dose–response models (Table 4) were fitted for each gene. The best-fit model, with the lowest akaike’s information criterion (AIC) value, was used for calculation of effect concentration (EC) values for genes whose best-fit model showed significant curve-fitting performance (p-value < 0.05). Finally, genes whose EC ≤ LOEC (BPA) or ≤ 30 REF (water samples, NO effect concentration) were defined as DEGs.
Robustness Analysis of RZT-Ampliseq. The zebrafish embryos treated with 0.1% DMSO were used to evaluate the basal expression of RZT genes, since 0.1% DMSO is frequently used as a control in toxicological studies. To evaluate the optimum sequencing depth of RZT-ampliseq-embryo for consistently detecting most genes, mRNA sample of 32 hpf zebrafish embryo exposed to 0.1% DMSO with 300 0000 sequencing counts of RZT-ampliseq (coverage of ~200 counts for each gene) was used. The optimum sequencing depth of samples was determined by a Monte Carlo simulation conducted using R language. Each sequenced depth was repeated 100 times to calculate the detected number of genes (Reads > 0, Reads > 20) and the coefficient of variation (CV) (SD/Mean) of each gene’s expression abundance. For each gene, the relationship between CV and sequencing depth was fitted with loess model and then the minimum sequencing depth of ensuring CV < 15% was calculated. To evaluate the mRNA profiling performance of ampliseq on the RZT gene set, the number of detected and undetected genes, as well as each gene expression abundance measured by RZT-ampliseq was compared to that by microarray platform (GSE43186) and RNA-seq platform on 36 hpf zebrafish embryo. Correlations of the gene expression abundance between different technologies were calculated using number of reads per amplicon for RZT-ampliseq, RPKM (reads per kilo base per million reads) values for RNA-seq and signal intensity values for microarray technology. Finally, to evaluate the repeatability of RZT-ampliseq, the CV of RZT gene set in zebrafish embryos of 0.1% DMSO (n = 3) from six batches was analyzed using the edge package.
Pathway-Level and Biological Process Validation. For single dose experiment, functional enrichment analysis of identified DEGs was performed using a one-sided Fisher’s exact test on GO of biological process (BP), and KEGG pathways with RZT gene list (Table S2) as background. For full dose experiment, the EC values of GO terms and KEGG pathways were calculated as the geometric mean of EC values of matched DEGs. Only GO terms or KEGG pathways matched by at least three genes were included in EC calculation and further analysis. Finally, to analyze the overall biological potency of each sample, the proportionally ranked distribution of GO and KEGG of EC values was fitted with a four-parameter dose–response curve using GraphPad Prism 5.0 software (San Diego, CA).
The molecular responses profiling (DEGs, KEGG pathways of DEGs) of 0.1 μM BPA treatment by RZT-ampliseq were compared with whole transcriptome analysis of BPA archived in NCBI. To compare RZT-ampliseq-embryo approach with existing Toxcast high-throughput in vitro assays with regard to biological activities associated with BPA exposure, the responsive gene end points and molecular pathways (KEGG, GO BP terms) identified by both methods were evaluated. The responsive molecular gene end points were DEGs captured of dose–response model analysis of RZT-ampliseq-embryo. The responsive genes of Toxcast in vitro assay were downloaded from (https://www.epa.gov/chemical-research/toxicity-forecaster-toxcasttm-data). The responsive molecular end points were converted to zebrafish orthologous genes.
Comparison of RZT with In Vitro Bioassays and RHT Method on Mixtures. A supervised approach was used to assess the RZT representation of the previous in vitro bioassays. First, gene sets associated with cellular toxicity pathways tested by in vitro bioassays were manually curated from Wiki Pathways and Gene Ontology, KEGG. Then the EC of each pathway was calculated by the geometric mean of the ECs of matched DEGs. Pathway patterns identified by the RZT approach were shown by heatmap using gplot package. The hierarchical clusters of water samples identified by RZT analysis were compared with the results of in vitro bioassays.
To evaluate the sensitivity and specificity of RZT-ampliseq-embryo in identification of bioactivity of mixtures, the results of RZT-ampliseq-embryo were compared to that of RHT-ampliseq using human HepG2 and MCF7 cells on the same sample set. Briefly, the sensitivity of 50% biological potency of water samples identified by RZT was compared with those identified by RHT in HepG2 and MCF7 cells in terms of KEGG or GO. In addition, linear regression was conducted on values of 50% biological potency identified by RZT and RHT. Finally, the coverage of the most sensitive pathways (top 20 sensitive KEGG pathways) of Eff2, the sample with potential highest and broadest bioactivity was compared between RZT and RHT approaches.
RZT Assessment of a Classical Chemical: BPA. The RZT approach showed good repeatability for quantifying transcriptional response to chemical by zebrafish embryo. Common CV of 32 hpf embryo mRNA samples exposed to 0.1% DMSO from 8 to 32 hpf was 13% (biological replication within one batch), 14% (biological replication among six batches) for RZT-ampliseq-embryo. This variation was acceptably low when compared with other RNA profiling technology, such as qPCR (CV: 1 ~ 15%), microarray (CV:5 ~ 15%) or RNA-seq (CV:10 ∼ 15%). After exposure of two independent batches of embryos to a single dose of 10 μM BPA, 67, 45 DEGs (ANOVA, p < 0.05), respectively, were identified by RZT-ampliseq-embryo and 26 DEGs were common to both batches. Moreover, the fold change values of all expressed genes showed significant correlation between batches (R2 = 0.62) and the difference among the DEGs of two batches was nearly within twofold.
The full dose–response analysis of an RZT profile following exposure to BPA provided a distinct DEGs profile compared to that detected following single doses. Following 24-h exposure to 0.1 or 10 μM BPA (single dose), 67 and 58 DEGs, respectively, were captured by RZT-ampliseq-embryo and 31 DEGs were commonly expressed. Eight GO BP terms and five KEGG terms were shared between 10 and 0.1 μM BPA concentration groups. Transcriptional changes associated with DNA damage (DNA repair, cellular response to DNA damage stimulus) and central nervous system development were only captured after 0.1 μM BPA exposure. Oocyte meiosis was only identified after 10 μM BPA exposure. Moreover, there were no common DEGs in RZT-ampliseq-embryo and the published microarray data in embryo exposed to 8–32 hpf 0.1 μM BPA. Three common KEGG pathways (counts of mapped gene ≥3) were identified in two platforms, but these were only involved in fundamental apoptosis process (FoxO signaling pathway, p53 signaling pathway) and regulation of actin cytoskeleton. However, 98 DEGs were identified by dose–response analysis of the embryo exposed to the seven, tenfold, dilutions of BPA (10 ∼ 10–5 μM). Three and five of the DEGs identified by dose–response analysis were also detected as DEGs in embryos exposed to 10 and 0.1 μM, respectively.
One significant advantage of dose–response analysis by RZT-ampliseq is the sensitivity analysis of genes and biological pathways in response to chemicals, which could aid inference regarding the potentially sensitive apical end point effects. The responsive DEGs were mainly fitted with U-shaped models, which suggest that the mode of hormesis dominates the low dose response of transcriptome. However, there are alternative interpretations other than true hormesis. For example, the time course of dynamic transcriptional response may change at different doses; in higher doses, for example, the transcript abundance can peak earlier, but falls by 32 h. Alternatively, perhaps there is a developmental delay at the higher doses, associated with triggering more and more AOPs in the organism, thereby causing more and more disruption of normal development. What is effectively a monotonic response to the chemical may produce a nonmonotonic dose–response for a given snapshot in time. The response genes (EC < 0.001 μM) (foxa3, dhrs3a, src, rpa3, myb, csf1ra, flt4) were mainly fitted with Gaussian model (Fig. 14a) and were primarily associated with pathways of cell-based process, responses to external stimuli, immune system, and neurogenesis. The significant enrichment of neurogenesis-related processes (central nervous system development, nervous system development, locomotion) at very low concentrations of BPA (EC < 0.001 μM) was corroborated by hyperactivity behavior (0.0068 μM(35)) and increased neuronal development(35) observed in previous study. Moreover, hyperactivity behavior showed a nonmonotonic concentration-dependent response and was induced only at very low effect concentrations, which was consistent with the Gaussian model of the relevant DEGs. For the DEGs with EC between 0.001 and 0.01 μM, only apoptosis relevant FOXO signaling pathway (FDR = 0.057, covered four genes) was enriched. Previous research reported BPA could induce apoptosis of mice spermatocytes and zebrafish embryo. A larger number of pathways, which included liver development, regulation of nucleobase-containing compound metabolic process, sensory organ morphogenesis, and DNA binding, were associated with the DEGs with EC between 0.01 and 0.1 μM. The DEGs with EC (0.1–10 μM), close to the LOEC (10 μM), were mainly associated with carbohydrate metabolic process and blood vessel development process.

Concentration-dependent network of differentially expressed genes (DEGs) (p < 0.05) in 32 hpf zebrafish embryo treated by BPA (a). Coverage and sensitivity of biological pathways enriched by BPA in RZT-ampliseq-embryo (blue, log10 EC value) and Toxcast in vitro assays (orange, log10 AC50 value) at gene level (b), KEGG pathway level (c) and GO BP term (d). For KEGG pathways and GO BP terms, only those with counts of mapped genes ≥3 were included in this analysis. Common molecular end points were labeled by red triangle. Pathway scores (EC or AC50 value) were the geometric mean of the effect concentrations (ECs) (RZT-ampliseq-embryo) or AC50 (Toxcast) values of the relevant genes. The horizontal distance of 50% biological potency between RZT-ampliseq-embryo and Toxcast in vitro assays was labeled in red
The coverage of enriched pathways and the corresponding sensitivity detected following full dose–response profiling using RZT-ampliseq-embryo were compared with ToxCast in vitro results for BPA. Not only was there consistency with ToxCast in terms of the coverage of biological pathways, the RZT-ampliseq-embryo method appeared more sensitive (Fig. 14b–d). Bisphenol A was tested with regard to 821 ToxCast assay end points. Out of 96 genes that aligned with the 390 relevant assays, 78 genes had a corresponding orthologous zebrafish gene. In total, 5 KEGG pathways and 15 GO BP terms were commonly identified by Toxcast and RZT, although only five genes were identified by both approaches (Fig. 14b–d). Furthermore, the sensitivity rank of common genes (ptgre2a, tcf7l1b, src, xbpl, tfap2b) and KEGG pathways (metabolic pathways, MAPK signaling pathway, focal adhesion, endocytosis, adhere junction) was similar across two platforms (Fig. 14b, c). However, RZT-ampliseq-embryo was more sensitive than in vitro tests in Toxcast with 2.3, 2.9, and 2.8 magnitude difference at gene, KEGG, and GO BP level, respectively.
RZT Assessment of Mixtures in Zebrafish Embryo. The RZT-ampliseq-embryo was able to discriminate the relatively clean water samples from the polluted environmental samples by the relative potency of altered genes and molecular pathways. The DEGs responsive at low dose range (ECs ≤ 0.1 REF) were mainly fitted with U-shaped models (Fig. 15). For example, the DEGs of MF were mainly associated with altered metabolic process (ar, smad1, prpf40a, ak2, uqcrc2b, polr2gl, smc3, gtf2b, psmc1a, rpl7a, gars, ndufv2, htatsf1, rps24, u2af2b, cwc15, zgc:86599, nup85, psmc5, tcf7, hnrnpm, ikbkap, nup107), developmental process (such as kif1 bp, atp6v1e1b, flt4, sema3d, ctnnb1, smad1, raf1a, tgfbr2, ak2, rpl6, tgfb3, rps24, sumo1, tcf7, cyp26c1, plod3, ikbkap, hdac6), and cellular response to stimulus (such as ar, flt4, sema3d, smad1, raf1a, tgfbr2, smc3, ssr4, psmc1a, tgfb3, psmc5, tcf7, ephb2b). The number of DEGs selected by the nine dose–response models across all four samples ranged from 78 to 300 (SI Fig. 15). Although the number of DEGs did not directly correlate with decreasing pollution level, the sensitivity distribution curve of biological pathways indicated the decrease of pollution level (Eff2 > MF > RW > DW). The EC values of the most sensitive KEGG or GO pathways of DW samples were 1–2 orders of magnitude higher than those of effluent samples, suggesting relatively weak biological effects were induced by D.

a Venn diagram of DEGs by four water samples identified by RZT-ampliseq-embryo in 32 hpf. Histogram of log10 EC values of differentially expressed genes (DEGs) across total four water samples (b) and by each water sample (c), (d), (e), and (f)
The enriched pathways in RZT analysis could be used to prioritize potential biological end points for future assessment. Specifically, the most sensitive KEGG or GO BP pathways may be linked with adverse outcomes. All development relevant pathways (GO terms, each covered at least three DEGs suggested Eff2 and MF samples might induce potential development toxicity while RW and DW samples may not (Fig. 16a). The predicted adverse outcomes were corroborated by zebrafish embryo 48hpf lethality and 120hpf sublethal development experiment.

a Development-related biological processes affected by four water samples from RZT-ampliseq-embryo in 32 hpf. Plotted were log 10 EC values with a unit of relative enrichment factor (REF). b Venn diagram of top 20 sensitive KEGG pathways ranked by EC values identified by RZT, RHT in HepG2, and RHT in MCF7, respectively, for Eff2 sample. The labels of “zebrafish” in blue, “HepG2” in yellow, and “MCF7” in green stand for approaches of RZT, RHT in HepG2, and RHT in MCF7, respectively. The labels in red stand for the main function of KEGG pathways
Comparison between RZT-Embryo and RHT Cell Profiles of Water Samples. RZT-embryo also provided different profiles of altered genes and pathways of the four water samples from that by RHT approach, which might be due to the greater biological complexity represented by a fish embryo compared to a single cell type. The most sensitive pathways identified by RZT following exposure to the water samples were distinct with those by RHT in HepG2 and MCF7. Take Eff2 for example (Fig. 16b), only four KEGG pathways were overlapped between the 20 most sensitive pathways (with lowest EC values) identified by RZT-embryo and RHT in HepG2 and MCF7 cells. Nine of the 20 most sensitive pathways uniquely identified by RZT-embryo were associated with basic biological processes, which may suggest that rapidly developing and differentiating zebrafish embryos were more sensitive to alterations of basic processes, such as oxidative phosphorylation, than the single cell type in vitro system. Moreover, the most sensitive KEGG pathways identified by RHT in HepG2 and RHT in MCF7 showed cell-type responses, such as pathways involved in immune response and cellular communication, which were not among the most sensitive KEGG pathways by RZT-embryo assay. However, some cell-type-specific responses, including endocrine response in MCF7 and metabolism response in HepG2, were also identified by RZT as sensitive KEGG pathways responding to Eff2.
A distinct pathway sensitivity distribution in response to the MF sample was identified by RZT-embryo compared to RHT in HepG2 and MCF7 cells. Although the potency of the median sensitive pathway (50% biological potency) following exposure to MF was lower than that of RW and DW in RZT-embryo, MF was more potent than that of RW and DW at the most sensitive pathways which were profiled by the RZT-embryo. These highly sensitive biological responses induced by MF were primarily related to embryo development (e.g., heart jogging, embryo pattern specification, notochord development, determination of left/right symmetry) (Fig. 16a), which might be related to developmentally toxic pollutants present in the MF sample. The MF sample was water taken after microfiltration using filters disinfected by chlorination to avoid biofouling in a water reclamation plant, in which micropollutants such as carbamazepine, a teratogen, with the highest detected concentrations (1.9 μg/L) out of ten samples were present. Carbamazepine has been reported to disturb embryonic development with increasing hatching rate, body length, swim bladder appearance, and yolk sac absorption rate at 1 μg/L. However, knowledge gaps associated with unknown chemicals present in the mixtures and their potential combined effects still exist in toxicological assessment of these environmental samples. An effect-directed analysis (EDA) integrating extract fractionation and instrument analysis with the sensitive RZT approach may be used to identify the chemicals responsible for the observed effect in future.
In conclusion, we developed reduced transcriptome approaches (RHT and RZT) by integrating reduced transcriptome, RNA-ampliseq technology, and human cells or zebrafish embryo test to assess environmental toxicants. Firstly, the concentration-dependent transcriptomic approach could identify early molecular response and molecular mechanism of single chemical which would help to predict apical effect. The reduced transcriptome approach has potential to be used to evaluate and prioritize chemicals for further testing and potentially to predict adverse outcomes. These results demonstrate a promising and powerful tool for screening hundreds of chemicals or mixtures by potency ranking and to classify chemicals by the spectrum of disrupted biological pathways. The omics-based biological pathway strategy can also be used in the characterization of potential toxicity by environmental mixtures. It is anticipated that reduced transcriptomic approaches will significantly advance pathway-based high-throughput screening of potentially toxic substances.
References
Altenburger R, Ait-Aissa S, Antczak P, Backhaus T, Barcelo D, Seiler TB, Brion F, Busch W, Chipman K, de Alda ML, de Aragao Umbuzeiro G, Escher BI, Falciani F, Faust M, Focks A, Hilscherova K, Hollender J, Hollert H, Jager F, Jahnke A, Kortenkamp A, Krauss M, Lemkine GF, Munthe J, Neumann S, Schymanski EL, Scrimshaw M, Segner H, Slobodnik J, Smedes F, Kughathas S, Teodorovic I, Tindall AJ, Tollefsen KE, Walz KH, Williams TD, Van den Brink PJ, van Gils J, Vrana B, Zhang X, Brack W (2015) Future water quality monitoring–adapting tools to deal with mixtures of pollutants in water resource management. Sci Total Environ 512–513:540–551
Bluhm, K.; Otte, J. C.; Yang, L.; Zinsmeister, C.; Legradi, J.; Keiter, S.; Kosmehl, T.; Braunbeck, T.; Straehle, U.; Hollert, H., Impacts of Different Exposure Scenarios on Transcript Abundances in Danio rerio Embryos when Investigating the Toxicological Burden of Riverine Sediments. Plos One 2014, 9 (9)
Choi JS, Kim RO, Yoon S, Kim WK (2016) Developmental toxicity of zinc oxide nanoparticles to zebrafish (Danio rerio): a transcriptomic analysis. 11 (8), e0160763
Conolly RB, Ankley GT, Cheng W, Mayo ML, Miller DH, Perkins EJ, Villeneuve DL, Watanabe KH (2017) Quantitative adverse outcome pathways and their application to predictive toxicology. Environ Sci Technol 51(8):4661–4672
Driessen M, Vitins AP, Pennings JL, Kienhuis AS, Water B, van der Ven LT (2015) A transcriptomics-based hepatotoxicity comparison between the zebrafish embryo and established human and rodent in vitro and in vivo models using cyclosporine A, amiodarone and acetaminophen. Toxicol. Lett. 232(2), 403–412
Escher BI, Allinson M, Altenburger R, Bain PA, Balaguer P, Busch W, Crago J, Denslow ND, Dopp E, Hilscherova K, Humpage AR, Kumar A, Grimaldi M, Jayasinghe BS, Jarosova B, Jia A, Makarov S, Maruya KA, Medvedev A, Mehinto AC, Mendez JE, Poulsen A, Prochazka E, Richard J, Schifferli A, Schlenk D, Scholz S, Shiraishi F, Snyder S, Su G, Tang JY, van der Burg B, van der Linden SC, Werner I, Westerheide SD, Wong CK, Yang M, Yeung BH, Zhang X, Leusch FD (2014) Benchmarking organic micropollutants in wastewater, recycled water and drinking water with in vitro bioassays. Environ Sci Technol 48(3):1940–1956
Guiu J, Bergen DJM, De Pater E, Islam ABMMK, Ayllon V, Gama-Norton L, Ruiz-Herguido C, Gonzalez J, Lopez-Bigas N, Menendez P, Dzierzak E, Espinosa L, Bigas A (2014) Identification of Cdca7 as a novel Notch transcriptional target involved in hematopoietic stem cell emergence. J Exp Med 211(12):2411–2423
Haggard DE, Noyes PD, Waters KM, Tanguay RL (2016) Phenotypically anchored transcriptome profiling of developmental exposure to the antimicrobial agent, triclosan, reveals hepatotoxicity in embryonic zebrafish. Toxicol Appl Pharmacol 308:32–45
Hermsen SA, Pronk TE, van den Brandhof EJ, van der Ven LT, Piersma AH (2012) Concentration-response analysis of differential gene expression in the zebrafish embryotoxicity test following flusilazole exposure. Toxicol Sci 127(1):303–312
Hofsteen P, Mehta V, Kim MS, Peterson RE, Heideman W (2013) TCDD inhibits heart regeneration in adult zebrafish. Toxicological sciences: an official journal of the Society of Toxicology 132(1):211–221
Jiang J, Wu S, Wu C, An X, Cai L, Zhao X (2014) Embryonic exposure to carbendazim induces the transcription of genes related to apoptosis, immunotoxicity and endocrine disruption in zebrafish (Danio rerio). Fish Shellfish Immunol 41(2):493–500
Knapen D, Angrish MM, Fortin MC, Katsiadaki I, Leonard M, Margiotta-Casaluci L, Munn S, O’Brien JM, Pollesch N, Smith LC, Zhang X, Villeneuve DL (2018) Adverse outcome pathway networks I: development and applications. Environ Toxicol Chem 37(6):1723–1733
Lam SH, Hlaing MM, Zhang X, Yan C, Duan Z, Zhu L, Ung CY, Mathavan S, Ong CN, Gong Z (2011) Toxicogenomic and phenotypic analyses of bisphenol-A early-life exposure toxicity in zebrafish. PLoS ONE 6(12):e28273
Li Y, Qi X, Yang Y-W, Pan Y, Bian H-M (2014) Toxic effects of strychnine and strychnine N-oxide on zebrafish embryos. Chin J Nat Med 12(10):760–767
Maves L, Waskiewicz AJ, Paul B, Cao Y, Tyler A, Moens CB, Tapscott SJ (2007) Pbx homeodomain proteins direct Myod activity to promote fast-muscle differentiation. Development (Cambridge, England) 134(18):3371–3382
Verleyen D, Luyten FP, Tylzanowski P (2014) Orphan G-Protein Coupled Receptor 22 (Gpr22) Regulates cilia length and structure in the zebrafish kupffer’s vesicle. Plos One 9(10)
Wang P, Xia P, Yang J, Wang Z, Peng Y, Shi W, Villeneuve DL, Yu H, Zhang X (2018) A reduced transcriptome approach to assess environmental toxicants using zebrafish embryo test. Environ Sci Technol 52(2):821–830
Wanglar C, Takahashi J, Yabe T, Takada S (2014) Tbx protein level critical for clock-mediated somite positioning is regulated through interaction between Tbx and ripply. Plos One 9 (9)
Xia P, Zhang X, Zhang H, Wang P, Tian M, Yu H (2017) Benchmarking water quality from wastewater to drinking waters using reduced transcriptome of human cells. Environ Sci Technol 51(16):9318–9326
Xu M, Liu D, Dong Z, Wang X, Wang X, Liu Y, Baas PW, Liu M (2014a) Kinesin-12 influences axonal growth during zebrafish neural development. Cytoskeleton 71(10):555–563
Xu M, Liu D, Dong Z, Wang X, Wang X, Liu Y, Baas PW, Liu M (2014b) Kinesin-12 influences axonal growth during zebrafish neural development. Cytoskeleton 71(10):555–563
Zhang X, Wiseman S, Yu H, Liu H, Giesy JP, Hecker M (2011) Assessing the toxicity of naphthenic acids using a microbial genome wide live cell reporter array system. Environ Sci Technol 45(5):1984–1991
Zhang X, Xia P, Wang P, Yang J, Baird DJ (2018) Omics advances in ecotoxicology. Environ Sci Technol 52(7):3842–3851
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
1 Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Rights and permissions
Copyright information
© 2020 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Zhang, X., Wang, P., Xia, P. (2020). Dose-Dependent Transcriptomic Approach for Mechanistic Screening in Chemical Risk Assessment. In: Jiang, G., Li, X. (eds) A New Paradigm for Environmental Chemistry and Toxicology. Springer, Singapore. https://doi.org/10.1007/978-981-13-9447-8_4
Download citation
DOI: https://doi.org/10.1007/978-981-13-9447-8_4
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-9446-1
Online ISBN: 978-981-13-9447-8
eBook Packages: Earth and Environmental ScienceEarth and Environmental Science (R0)