
Abstract
Epigenetics has a fascinating and convoluted history steeped within the fields of embryology and genetics. Here, we introduce genetics and epigenetics to the non-expert reader and give an account of the pivotal discoveries that have helped shape these fields. The significance of major discoveries will be explained and their impact on our understanding of epigenetic mechanisms in health and disease will be discussed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1.1 The Early Origins of Genetics
Long before the term ‘genetics’ was conceived, the basis of inheritance had been extensively debated. It is difficult to choose a specific point in history where the true basis of inheritance began to take shape; however, much can be said of the contributions of the mathematician and scientific philosopher Pierre Louis de Maupertuis. In 1751, he hypothesised that both parents contributed equally to their offspring and proposed a particulate basis of heredity. The concept of particulate inheritance was opposed to the beliefs of other philosophers of the time who argued that the characteristics of each parent were blended together in the next generation. Instead, his theory of particulate inheritance proposed that it had a physical basis caused by discrete particles (now known as genes) that are not diluted or diminished in the next generation. Shortly after, in 1753, Maupertuis was the first to apply probability estimates to predict disease risk in his study of a family with polydactyly and is credited with striking insights into the possibility of natural selection (Stubbe 1972). In his book Essai de Cosmologie published in 1751 (de Maupertuis 1751), he argued that variation in animals and plants arose spontaneously but that only a small proportion of individuals showed fitness and survival.
Decades later in the early nineteenth century, Jean-Baptiste Lamarck proposed the theory of inheritance of acquired characteristics, in which he suggested that species gradually developed characteristics that suited the physical conditions of life. In his publications Recherches sur l’organisation des corps vivants (de Monet de Lamarck 1802), and later in Philosophie Zoologique (de Monet de Lamarck 1809), Lamarck argued that a key driving force of his theory of inheritance was the effects of use and disuse and that the memory of these acquired characteristics were passed to future generations and therefore perpetuated. He used the example of the blind mole rat to illustrate loss of function through disuse. Though this theory is now widely disregarded as a major contributor to inheritance, it was nevertheless one of the first attempts to provide a tangible theory for biological evolution. Lamarck also asserted in his book Philosophie Zoologique that species, including man, are descended from other species.
The theory of natural selection is of course attributed to Charles Darwin in his book On the Origin of Species by Means of Natural Selection or the Preservation of Favoured Races in the Struggle for Life (often referred to simply as Origin of Species), published in 1859 (Darwin 1859). In the introduction of Origin of Species, two sentences perfectly summarise his theory: ‘As many more individuals of each species are born than can possibly survive; and as, consequently, there is a frequently recurring struggle for existence, it follows that any being, if it vary however slightly in any manner profitable to itself, under the complex and sometimes varying conditions of life, will have a better chance of surviving, and thus be naturally selected. From the strong principle of inheritance, any selected variety will tend to propagate its new and modified form’ (Darwin 1859). Origin of Species challenged the popular belief at the time that species were immutable, static in nature and as God, the Creator, had designed. Darwin himself grappled with the implications of his theories of natural selection and evolution, as he had also believed that life was created in its present form. However, Origin of Species presented the objective evidence, collected by himself and others over many decades, that particular species had adapted to their environments over many generations.
By Darwin’s own admission, his theory of evolution was imperfect; however, Darwin did not try to hide the faults in his theory and in fact discussed them extensively in his book. He clearly communicated that evolution and inheritance are inextricably linked but also acknowledged that his greatest problem was the absence of a mechanism by which traits were inherited from parent to offspring. This came in Gregor Mendel’s solution to the problem of inheritance in pea plants in 1865–1866 (Mendel 1866); however, throughout his lifetime, Darwin remained unaware of Gregor Mendel and his work. Darwin continued to publish updated editions of Origin of Species, with the sixth and final edition published in 1872, and published his work on human evolution in The Descent of Man and Selection in Relation to Sex in 1872 (Darwin 1872).
Gregor Mendel is now widely regarded as the founder of the modern science of genetics, with the rules he established on the basis of heredity now referred to as Mendelian inheritance. Initially overlooked, Mendel’s work on the inheritance pattern of pea plant characteristics was rediscovered in 1900, which lead to the establishment of genetics as a scientific field of study. The term genetics was coined by William Bateson in 1905, with the word originating from the Greek γεννώ (gennó), which translates as ‘to give birth’. He proposed the term should be used to describe the study of heredity, or how characteristics are transferred from parent to offspring.
1.2 Discovery of DNA
Deoxyribonucleic acid was first isolated from the nuclei of pus cells from surgical bandages in 1869 by the Swiss biologist Friedrich Meischer. Meischer was a mentee of the German scientist Felix Hoppe-Seyler, who is widely regarded as the founder of the disciplines of biochemistry and molecular biology. Meischer showed that the substance he had isolated was acidic, abundant in phosphorous and resistant to enzymes that degrade proteins. Given that he had isolated this substance from nuclei, he named it ‘nuclein’. Meischer submitted his work describing nuclein to Hoppe-Seyler, who was also editor of the journal Medizinisch-chemische Untersuchungen (Medical and Chemical Analysis). The unusual properties of nuclein described by Meischer were so unlike anything Hoppe-Seyler had seen previously that he decided to delay publication for 2 years whilst he repeated Meischer’s experiments with the help of two students Pal Plósz and Nikolai Lübavin. Once repeated, and confident of the accuracy of Meischer’s results, he finally published the original paper in 1871, another from Meischer describing a similar substance isolated from egg yolk and work from his own lab showing the isolation of nuclein from yeast and other cells. Neither Meischer nor Hoppe-Seyler appreciated the importance of nuclein in heredity; however, they recognised that its abundance and unusual chemical properties suggested it was important to the biology of the cell nucleus.
1.3 Early Characterisation of DNA
From 1878, the biochemist Albrecht Kossel, also a mentee of Hoppe-Seyler, began efforts to identify and characterise the chemical composition of the nucleus in greater detail. Kossel’s first major discovery in 1884 was the isolation of a type of protein (which he named histone) from the red blood corpuscles of birds that purified with nucleic acid (Kossel 1884). Following Meischer’s discovery of nuclein, many considered it chemically non-distinct from protein or closely related. This was due to methodological limitations at the time that prevented the isolation of pure protein-free nuclein. In the 1880s and 1890s, methods developed by Kossel and others (including Richard Altmann) allowed the isolation of protein-free nuclein and definition of its chemical composition. In 1889, following the discoveryFootnote 1 that protein-free nuclein was acidic, Altmann proposed the term nucleic acid (Altmann 1889). Efforts then turned to defining the building blocks of nucleic acid by characterising the products produced by breaking the chemical down into its constituent parts in a process called hydrolysis. Between 1885 and 1901, Kossel’s laboratory identified the nucleobases present in nucleic acid, beginning with adenine (A) in 1885 and later thymine (T), cytosine (C), guanine (G) and uracil (U).Footnote 2 These nucleobases, known as pyrimidines (cytosine, thymine and uracil) and purines (adenine and guanine), combine with phosphate and sugar in the form of ribose or deoxyribose. By the beginning of the twentieth century, Kossel had therefore helped define the basic building blocks of nucleic acids, eventually leading to the terms deoxyribonucleic acid (DNA) and ribonucleic acid (RNA).
1.4 Discovering That Genes Are Made of DNA
Based on its chemical properties, Meischer initially considered that DNA functioned to store phosphorous within the cell. Although its abundance in sperm lead Meischer and others to speculate a role in heredity, this was considered improbable due to its limited chemical composition (Friedrich 1874). In this section, we will describe the scientific discoveries that lead to the realisation that genes are made of DNA.
An appropriate starting point is the work of embryologist Thomas Morgan and his students Alfred Sturtevant, Calvin Bridges and Hermann Muller whose work in the early years of the twentieth century first demonstrated that inherited characteristics within the model organism Drosophila melanogaster were determined by physical units carried within chromosomes. Morgan’s team were the first to describe the role of chromosomes in sex-linked inheritance, to create the first genetic linkage map, to fully describe crossing over (now known as recombination) and to describe chromosomal abnormalities including non-disjunction, duplications and translocations (Morgan 2018). These were all monumental milestones that firmly asserted the importance of chromosomes in inheritance and that the linear order of genes on a chromosome could be defined.
Throughout the first half of the twentieth century, the term gene was used without an understanding of its chemical basis. In fact, most biologists thought that proteins, which can be composed of up to 20 different amino acids, were much more likely to carry hereditary information (McCarty 2003). Part of the reason for this was the tetranucleotide hypothesis proposed by the Russian biochemist Phoebus Levene in ~1910, which suggested that DNA was composed of equal amounts of guanine, cytosine, thymine and adenine in a repeating ring structure (Levene and Mandel 1908). This simple repeating configuration of DNA was to remain the accepted model of DNA structure until the 1940s and for many years contributed to the perception that DNA was far too simple in structure to be responsible for the molecular basis of inheritance. However, in the 1940s, it was discovered that the proportions of nucleobases in DNA can be different across species (Chargaff et al. 1949). This challenged the tetranucleotide hypothesis and began resurgence in research to define the molecular structure of DNA, which we shall return to later in this chapter.
In 1928, the British bacteriologist Frederick Griffith described the first widely accepted example of bacterial transformationFootnote 3 (Avery 1941). Griffith was a meticulous scientist who painstakingly characterised the different types of bacteria isolated from patients with pneumonia (the leading cause of death at the time). What particularly intrigued Griffith was that some strains of these bacteria could switch from virulent to non-virulent forms and vice versa. In crucial work, often referred to as Griffith’s Experiment, he injected mice with heat-inactivated preparations of the virulent strain and showed they did not develop pneumonia; however, when he injected both non-virulent and heat-inactivated virulent pneumococci, the mice developed pneumonia. This experiment suggested that a transforming factor persisted after heat-inactivation of virulent bacteria and that this transmitted virulence to the non-virulent strain (Griffith 1928). Though Griffith’s Experiment did not identify DNA as the transforming factor, his work showed that the characteristics of bacteria, including virulence, could be transformed and inspired others to identify the transforming factor, including Oswald Avery, Colin MacLeod and Maclyn McCarty at the Rockefeller Institute for Medical Research in New York City. The Avery-MacLeod-McCarty experiment isolated or enzymatically destroyed the different components of bacteria to identify the chemical that retained this transforming power. They first killed bacteria with heat, isolated the saline soluble components that contained protein, polysaccharides and DNA, then purified DNA using alcohol precipitation. To test whether DNA was the transforming factor, they treated extracts with enzymes that selectively destroyed protein or DNA. The transforming power of the extract was lost only after DNA was destroyed, thereby providing the definitive proofFootnote 4 that the function of DNA was to carry genetic information (Avery et al. 1944).
1.5 The Birth and Evolution of Epigenetics
In the late 1930s and early 1940s, the term epigenetics was coined by the British embryologist Conrad Waddington (Waddington 1939, 1940, 2012) to describe ‘the branch of biology that studies the causal interactions between genes and their products which bring the phenotype into being’ (Dupont et al. 2009; Waddington 1942). Waddington used the term broadly to describe the interaction between genetic mutations and cell differentiation (epigenesis); he was interested in how embryonic development unfolded at the molecular level. The term was further defined by David Nanney in the 1950s to include the concept of persistent homeostasis in the absence of genetic influence (Nanney 1958). This represented the first recognition of the importance of cellular memory in maintaining cell lineage and tissue type. To underscore the significance of epigenetics in developmental processes, Nanney highlighted some important cellular phenomenon that helped guide the use of the term (Nanney 1958), specifically:
-
1.
Cells with the same genetic material may manifest different phenotypes.
-
2.
Cellular properties are determined by the activity of an integrated set of genes.
-
3.
Specific patterns of gene activity can be induced.
-
4.
Epigenetic systems show stability (i.e. are heritable).
-
5.
Epigenetic control systems are localised in the nucleus of the cell.Footnote 5
These formed the basic tenets of epigenetics; however, the precise mechanisms that underpinned these cellular properties were not identified until decades later.
In 1975, two papers from Robin Holliday, John Pugh and Arthur Riggs, inextricably linked epigenetics with DNA methylation. Holliday, Pugh and Riggs proposed that DNA methylation was an epigenetic modification that regulated X-inactivation and gene expression (Holliday and Pugh 1975; Riggs 1975). For the first time, scientists could link cellular and phenotypic properties with differences at the molecular level. Consequently, the use of the term began to change from a description of cellular properties to one of molecular properties, beginning with DNA methylation and expanding to include all chromatin and DNA modifications that alter DNA function. The precise definition of the term epigenetics and its use (and misuse) have been extensively debated over many years (examples include Deans and Maggert 2015; Greally 2018; Haig 2004; Ptashne 2007). Throughout this book, the term epigenetics is used to refer to heritable chemical or structural modifications to chromatin that alter the function of DNA (see the Preface of this book for a more detailed description of the definition of epigenetics).
1.6 The Double Helix Structure of DNA
In 1951, Erwin Chargaff and others built upon Levene’s tetranucleotide hypothesis by showing that the relative amounts of A, T, C and G can differ between species and that, despite this diversity, the amount of adenine always equalled the amount of thymine and the amount of cytosine always equalled the amount of guanine (Chargaff 1951; Chargaff et al. 1949). Chargaff’s findings were a pivotal piece of information that complemented crystallography studies by Rosalind Franklin, Maurice Wilkins and others, and collectively, these findings enabled the chemical structure of DNA to be deduced in 1953 (Watson and Crick 1953b; Wilkins et al. 1953). Their model described the double helix structure of DNA containing two antiparallel strands in which adenine on one strand pairs with thymine on the other and cytosine pairs with guanine. This pairing occurs through hydrogen bonds that can be broken, thereby allowing the two molecules to unwind and separate. This structure reconciled Franklin’s crystallography observations of a symmetrical molecule and Chargaff’s observations that the amount of guanine is equal to cytosine and the amount of adenine is equal to thymine. The double helix structure is hugely significant in genetics—but why? Firstly, it finally dispelled any doubts that genes were made of DNA by casting aside the argument that DNA was too simple in structure to encode something as complex as inheritance. The helical structure of DNA is remarkably elegant and has an immense capacity for complexity. As the length of a DNA molecule increases, the amount of information it can contain increases exponentially. At any given position on one strand of a DNA molecule, there are four possible letters, A, T, C or G. A two-base-pair-long piece of DNA has 16 possible combinations (AA, TT, CC, GG, AT, AC, AG, TA, TC, TG, CA, CT, CG, GA, GT, GC or 4 × 4 combinations). A three-base-pair-long piece of DNA has 64 possible combinations (4 × 4 × 4), four base pairs gives 256 combination, five base pairs has 1024 combinations and so on. At 150 base pairs long, a piece of DNA has more possible combinations than there are atoms in the observable universe (which is currently estimated to be between 1078 and 1082). The true content of information is doubled when one considers that there is also a second strand within the double helix structure that contains a different sequence of complementary letters. This new model therefore made it abundantly clear that DNA had the requisite complexity to encode the instructions for life. Secondly, the double helix structure provided the answer to the molecular basis of inheritance by explaining how a DNA molecule can be copied for transfer between generations. As Watson and Crick explained in one of their 1953 papers, the DNA molecule ‘is, in effect a pair of templates, each of which is complementary to the other. We imagine that prior to duplication the hydrogen bonds are broken down and the two chains unwind and separate. Each chain then acts as a template for the formation onto itself of a new companion chain so that eventually we shall have two pairs of chains, where we only had one before […]. Moreover the sequence of pairs of bases will have been duplicated exactly’ (Watson and Crick 1953a). The chemical structure of DNA is described in greater detail in Chap. 2.
1.7 The Discovery of DNA Methylation
One might anticipate that the discovery of DNA methylation occurred after the chemical structure of DNA was deduced; however, the existence of 5-methylcytosine was recognised long before.
Between 1898 and 1910, collaboration of the US chemists Henry Wheeler and Treat Johnson at Yale University helped to characterise the chemical natures of the nucleobases.Footnote 6 In 1904, Wheeler and Johnson hypothesised the existence of 5-methylcytosine and synthesised this artificially to characterise its chemical properties (Wheeler and Johnson 1904). Several years later in 1925, Johnson and Robert Coghill confirmed the presence of 5-methylcytosine as a natural constituent of DNA from the Mycobacterium tuberculosis (Johnson and Coghill 1925). However, the existence of 5-methylcytosine in mammalian cells was a serendipitous discovery of the American biochemist Rollin Hotchkiss. In one of his papers, Hotchkiss described a small amount of an additional base that separated from cytosine. This ‘minor constituent’ of calf thymus DNA was distinct from cytosine yet shared similar chemical characteristics suggesting a modified form, which he referred to as ‘epicytosine’ (Hotchkiss 1948). He correctly deduced this base was not uracil and that it was pre-existing in the nucleic acid before extraction from cells and therefore present naturally. He also noted that the properties of this additional base could be distinguished from cytosine in a similar way that 5-methyluracil (otherwise known as thymine) could be distinguished from uracil. Though he referred to 5-methylcytosine and the earlier work of Johnson and Coghill, he was cautious in his conclusions noting that, ‘More than this cannot be said until further study of epicytosine has been made’ (Hotchkiss 1948). This discovery did not precipitate an immediate impact; however, studies over the next two decades would find 5-methylcytosine within DNA from all vertebrate and plant species (Hall 1971). In mammalian DNA, approximately 2–7% of cytosine is converted to 5-methylcytosine (Vanyushin et al. 1970). This ubiquity suggested an important function and several hypotheses were proposed, including the regulation of protein-DNA interactions, gene regulation and differentiation, protection from eukaryotic restriction enzymes, the regulation of DNA replication, chromosome folding, packing and sorting, and recombination. However, its functional significance would remain unclear until the 1980s.
1.8 The X-Chromosome and Its Unique Place in Genetics and Epigenetics
The story of the X-chromosome is unique in human genetics and, as we will see, has a particular place in the history of epigenetics. Its unique properties have enabled several conceptual leaps by providing the chromosomal basis of sex determination, X-linked inheritance and X-inactivation, as well as precedents for dosage compensation and the role of DNA methylation in gene activity.
The X-chromosome was first identified in 1891 by the German cytologist Hermann Henking. Henking studied the firebug Pyrrhocoris apterus and was intrigued by a heavily stained chromatin body at the periphery of the nucleus that seemed to distinguish cells from males versus females. Females contained 24 chromosomes that arranged into 12 pairs, whereas males had 11 pairs and one solitary chromosome. In one of the figures of his papers, he labelled this body of chromatin with an X and referred to it as the X-element, primarily because he was unsure if it was a chromosome.Footnote 7 Henking observed that sperm cells from Pyrrhocoris were of two kinds: those with the X-element and those without, in approximately equal numbers. This observable difference between the chromatin of sperm cells and the growing evidence at the time that it was the chromatin of cells that contained the hereditary information, led Clarence McClung to propose a chromosomal basis of sex determination in 1902 (McClung 1902). McClung’s theory was significant because it provided early support for the chromosomal theory of inheritance, which was proposed the same year by Walter Sutton and Theodor Boveri. This introduced the concept that the physical characteristics of chromosomes (e.g. the number or type of chromosome) can drastically alter the physical characteristics of an organism, including something as fundamental as sex. Though McClung had identified the specific chromosome that appeared to be associated with sex determination he had mistakenly proposed that it was the number of chromosomes that determines sex. However, this was soon to be corrected in 1905 by the American cytologist Nettie Stevens, whose work helped solve the basis of what would become known as the XY sex determination system (Brush 1978). As a former student of Thomas Morgan, she had studied the fruit fly Drosophila several years before Morgan adopted this species as a model organism. In her studies of more than 50 species of beetles and flies, she investigated the chromosomal basis of sex determination. In the mealworm beetle Tenebrio molitor, she showed that males produced two kinds of sperm that contained either a small chromosome or a large chromosome, and that offspring that inherit the small chromosome were invariably male whereas those that inherited the large chromosome were invariably female. From this, she deduced that the chromosomal basis of sex depended on the presence or absence of the smaller chromosome. Though this mechanism of sex determination would prove relevant for many other species, including humans, she found that this was not true for all species. Around the same time, and independently, Edmund Wilson also confirmed that sex determination was much more complex and varied, depending on which species of insect was studied. Further contributions by both Stevens and Wilson showed that in the context of XY sex determination, females inherit two copies of the larger chromosome (and were referred to as XX), whilst males have one small and one large chromosome (and were referred to as XY). Therefore, it is the presence or absence of the Y-chromosome, rather than the number of chromosomes, that determines sex. As we now know, a consequence of this is ‘sex-related’ inheritance, which describes the inheritance of specific traits preferentially in one sex. In 1910, Morgan published his observations of the inheritance patterns of several traits in Drosophila, including the inheritance of white eyes instead of the normal red eyes. He first noted that white eyes occurred exclusively in males, but on further crosses, white-eyed females could be observed. From this he implied a physical relationship between the X-chromosome and the white-eyed trait. Morgan’s paper was therefore the first to propose X-linked inheritance, and in doing so, the field of genetics made an important conceptual leap by showing that genes were physical entities that reside on chromosomes. At this point, the concept of the gene ceased to be a theoretical term with no physical evidence and the modern theory of the gene was born. This modern theory posited that genes were located on chromosomes, that they could be studied and experimentally manipulated, and that the linear order of genes on any given chromosome could be mapped, which Morgan and his team (in particular Alfred Sturtevant) did to great effect in the years that followed. Finally, in 1911, Wilson published a famous review in which he described the XY sex determination system and predicted its consequences for human X-linked inheritance in the context of haemophilia and colour blindness (Kingsland 2007; Wilson 1911). Prior to this, the patterns of inheritance of these disorders and their predominance in males had puzzled geneticists.
In humans, and other species where sex is determined by the presence or absence of a Y-chromosome,Footnote 8 females contain two X-chromosomes, whereas males contain only one. Therefore, female cells contain one extra copy of all X-linked genes that could, theoretically, lead to a profound imbalance in the expression of those genes between males and females. To overcome this, all somatic cells in female eutherian mammalsFootnote 9 adopt a mechanism of dosage compensation, where one of the X-chromosomes is inactivated. In 1948, the Canadian researcher and physician Murray Barr and his student Ewart Bertram reported that cells from male and female cats could be distinguished by simply staining chromatin and viewing using a compound microscope. They discovered that somatic, non-dividing cells (specifically nerve cells) of male and female cats could be distinguished by the presence or absence of densely staining chromatin at the periphery of the nucleolus (Barr and Bertram 1949). This densely staining chromatin was, at the time, referred to as ‘sex chromatin’, and later as the ‘Barr body’. Barr and Bertram demonstrated that this was present in all female somatic cells, much like the structure described by Henking in insects over 50 years earlier. In their paper, they postulated that the Barr body was most likely comprised of the X-chromosomes and that in female cells, the presence of two X-chromosomes explained its visibility. For the next decade, the theory persisted that the Barr body represented the tight pairing of the two X-chromosomes; however, its precise nature remained a topic of intense investigation. The next major breakthrough was offered by Susumu Ohno and Theodore Hauschka in 1960 whose investigation of the Barr body in cells from female mice suggested it was comprised of a single X-chromosome (Ohno and Hauschka 1960).Footnote 10 This raised an interesting question: was it the paternal X or the maternal X? A theory favoured by Ohno and Hauschka was that with each cell division this ‘alternate[d] between the two X’s ... regardless of their parental derivation’ (Ohno and Hauschka 1960). However, this was soon superseded by a radical theory that was to have major implications for the field of epigenetics.
Mary Lyon was a visionary British geneticist whose work had a profound impact in clinical genetics. In 1961, soon after Ohno and Hauschka had proposed the theory that the Barr body was comprised of a single X-chromosome, Lyon proposed that this X-chromosome ‘can be either paternal or maternal in origin, in different cells of the same animal’, and that ‘it is genetically inactivated’ (Lyon 1961). Lyon’s hypothesis was derived from her years of experience in mouse genetics and mouse cytology and was based on two key pieces of genetic evidence:
-
1.
That mice with only one X-chromosome and no Y-chromosome (known as XO) are normal, fertile females (Welshons and Russell 1959)Footnote 11 and show no evidence of a Barr body, which shows that only one active X-chromosome is necessary for normal development of the female mouse.
-
2.
Specific X-linked traits in female animals, including the patchy appearance of different coat colours in female cats, suggested different X-chromosomes were active in different cells of the same animal—also known as genetic mosaicism.Footnote 12
Lyon’s theory was significant and far reaching in its implications. It proposed the concept of X-inactivation, a form of dosage compensation that renders one of the two X-chromosomes in XX females genetically inert. By indicating that the inactive X could be either paternal or maternal in origin, and that this could differ in different cells of the same animal (selected at random), Lyon’s theory also further explained the unusual characteristics of X-linked traits and diseases. These included colour blindness and haemophilia, traits that are rarely seen in females due to the presence of two X-chromosomes and the random inactivation of one, which prevents their penetrance.Footnote 13 Importantly, X-inactivation is a clear example of gene activity that is heritable through cell division because once one of the X-chromosomes is inactivated, this ‘choice’ is then fixed in all daughter cells thereafter. As an example, in cats, coat colour is determined by a gene on the X-chromosome and the pigment cells in each patch are descended from one cell with inactivation of a specific X-chromosome. Lyon was exacting in her hypothesis, going so far as stating, ‘Patches of intermediate color would arise by cell mingling in development, and the shape of the patches would depend on cell movement during growth’ and that this would ‘vary from one individual to another by chance’ (Lyon 1962). Interestingly, Lyon also showed that when genes from other chromosomes are abnormally translocated to the X-chromosome they too can become inactivated. This showed that the mechanism of X-inactivation was chromosome-wide and occurred independently of DNA sequence. At the time, it was unclear at what stage of embryonic development X-inactivation occurred, but it was known to be an early event that was already established by the late blastocyst stage in humans (Austin and Amoroso 1957; Park 1957). In honour of Lyon’s contribution, X-inactivation was for many years referred to as Lyonisation, though this term is no longer used.Footnote 14
The concept of X-inactivation was initially met with some resistance, with sceptics of the theory arguing that if surplus X-chromosomes are completely inert due to inactivation then why would sex chromosome aneuploidyFootnote 15 result in any clinical symptoms? These include females with a single X, known as Turner syndrome (referred to by cytogeneticists as 45,XO), who show developmental delay, infertility and short stature with extra folds of skin on the neck and low set ears. Males with XXY, known as Klinefelter syndrome (47,XXY) are characterised by low muscle tone, underdeveloped testicles, infertility and the development of breast tissue (gynaecomastia). There are various other described combinations (47,XXX, 48,XXXX, etc.), with each exhibiting variable clinical symptoms. In 45,XO cells, no Barr body is observed, whereas in cells containing 47,XXY a single Barr body is observed, and in cells with more than two X-chromosomes, multiple Barr bodies are found. During the 1960s, various explanations were proposed including incomplete inactivation and the effects of abnormal dosage in early development prior to inactivation (Lyon 1963). However, it was soon realised that specific genes on the X-chromosome that are involved in development escape inactivation, thereby resulting in an imbalance of some genes in individuals with sex chromosome aneuploidy (Burch and Burwell 1963).Footnote 16
X-inactivation posed a molecular puzzle; how are most of the thousands of genes located on only one of the X-chromosomes coordinately inactivated? Two landmark papers published independently by Arthur Riggs (1975), Robin Holliday and John Pugh (1975) would provide the stimulus for several conceptual advances toward answering this question.
Riggs was a researcher in the department of Susumu Ohno, one of the discoverers of X-inactivation, at the City of Hope Medical Centre, Los Angeles and had a keen interest in understanding gene regulation in E.coli. During a short visit to Herbert Boyer’s laboratory at the University of California, San Francisco, Riggs learned about a group of proteins known as restriction enzymes. One enzyme in particular, named EcoK, was a methyltransferase in E.coli and had a preference for hemimethylated DNA. Riggs realised a similar enzyme in mammalian cells could provide a mechanism of cellular heredity by maintaining patterns of methylation following DNA replication. In his paper, he proposed that DNA methylation was important for the X-inactivation process and that there existed a hitherto unrecognised information-coding system based on methylation patterns. His theory referred specifically to the X-inactivation process but clearly had broader implications for gene regulation. Interestingly, Riggs’ paper was promptly rejected by the first journal and Ohno had to convince him to persist by submitting his theory for publication elsewhere (Riggs 1988). Independently, Robin Holliday and his student John Pugh were based at the National Institute for Medical Research in London. Similar to Riggs, Pugh and Holliday had concluded that methylation of DNA could be a mechanism for gene regulation, but also that gene expression patterns could be stably inherited during cell division. Moreover, Holliday and Pugh clearly conveyed the concept that methylation might also switch genes on and off during development. At the time, it was well appreciated that 5-methylcytosine was abundant in higher organisms (Doskocil and Sorm 1962; Wyatt 1951); however, its functional effects, if any, were unclear. An additional concept discussed in the paper by Holliday and Pugh was that DNA mutations could be caused by the deamination of 5-methylcytosine to generate thymine, which had earlier been proposed by Eduardo Scarano (1971). We now know that the methylation of cytosine is an important cause of transition to thymine and a major mutation mechanism in cancer.
How the cell carries out X-inactivation is an extremely complex process that is still under investigation, despite decades of research. The precise mechanisms of X-inactivation likely differ between species (Migeon 2017). The critical role of DNA methylation was confirmed by experiments showing that inhibition of methylation with the drug 5-azacytidine resulted in the reactivation of genes on the inactive X-chromosome (Mohandas et al. 1981). 1991 saw a major breakthrough with several publications in the journal Nature from laboratories in the UK and the USA (Borsani et al. 1991; Brockdorff et al. 1991; Brown et al. 1991a, b). These showed that X-inactivation was dependent on a region on the X-chromosome, designated the X-inactivation centre (XIC), from which the inactivation signal is initiated and spreads throughout the rest of the chromosome. DNA elements within the XIC regulate the different aspects of X-inactivation, namely the counting of X-chromosomes, choosing one for silencing, initiating the silencing and finally maintenance of the inactive state (Lu et al. 2017). Within the XIC, the gene XIST is expressed specifically from the inactive X-chromosome. The RNA produced from this gene does not encode protein and is known as a long non-coding RNA (lncRNA). Instead, the XIST lncRNA coats the X-chromosome and, through interactions with a range of different proteins, results in exclusion of the transcriptional machinery, chromatin modifications and tethering of the future inactive X to the inner nuclear membrane where most of the genes on the chromosome are silenced by DNA methylation.Footnote 17 The importance of the XIST transcript is clear from observations that mutations within the promoter of the gene that cause changes to XIST expression are associated with skewed X-inactivation (Plenge et al. 1997) whereby one of the X-chromosomes is preferentially inactivated.
Up to 200 different proteins interact with XIST through repetitive sequences in the lncRNA (Chu et al. 2015; Lu et al. 2017; Minajigi et al. 2015). One important interacting protein is known as LBR (laminin B receptor), which serves as a bridge to tether the XIST-coated X-chromosome to the inner nuclear membrane. Other interacting proteins include various histone modifying enzymes, which prompt the condensation of the X-chromosome and extinguish gene expression. Recent research suggests the existence of additional genes on some autosomesFootnote 18 that regulate X-inactivation. Evidence for this theory includes the fact that in diploidFootnote 19 cells with three X-chromosomes (47,XXX), all but one is subject to X-inactivation, whereas triploid cells (69,XXX and 69,XXY) contain two active X’s (Migeon et al. 1979, 2008; Weaver and Gartler 1975). Maintenance of more than one active X-chromosome suggests that ploidy (the number of sets of autosomes) is important as a counting mechanism. This has led some to suggest that in human cells there exists a dosage-sensitive XIST repressor, encoded by an autosomal gene (Migeon et al. 2008). In this respect, one or more of several genes on chromosome 19 may be important (Migeon 2017).
1.9 Heritability of DNA Methylation
DNA methylation helps to determine cellular identity by controlling the complement of genes that are expressed in a cell. As such, the heritability of DNA methylation across cell division has major biological implications for ensuring maintenance of this identity and gene expression patterns in daughter cells. How the levels and precise location of DNA methylation are maintained remained unclear until relatively recently; however, a famous experiment from 1958 would later provide a crucial piece of this puzzle.
Following the discovery of the structure of DNA, there were three hypotheses for the mechanism by which replication might occur. The conservative hypothesis proposed that an entirely new DNA molecule was formed using the original as a template. The dispersive hypothesis, proposed by Max Delbrück, was a complicated combination of DNA unwinding, double-stranded DNA breaks, copying and the end-to-end ligation to form two copies comprised of both newly synthesised and original template DNA. Finally, in the semi-conservative hypothesis, proposed by Watson and Crick, the two strands of a DNA molecule separate and act as the template for the synthesis of complementary strands. This generates two DNA molecules each consisting of one original template strand and one newly synthesised strand. The Meselson-Stahl experiment, which illuminated which of these hypotheses was correct, has been called ‘the most beautiful experiment in biology’ (Judson 1979). Meselson and Stahl cultured E.coli for several generations in the presence of the nitrogen isotope 15N (‘heavy’ nitrogen), which contains an additional neutron when compared with the naturally abundant 14N. During cell division, this isotope became incorporated into newly synthesised DNA, which would later allow it to be distinguished by virtue of molecular weight using a method known as density centrifugation. E.coli cultured in the presence of 14N contained ‘light’ DNA, whereas in the presence of 15N the DNA became ‘heavy’. They then transferred the E.coli back to 14N and observed that after one cell division the DNA showed an intermediate weight. Though this result excluded conservative replication, which would have given equal amounts of heavy and light DNA, it was consistent with both the semi-conservative and dispersive hypotheses. However, after two rounds of cell division equal amounts of intermediate and light DNA were observed, which is consistent only with semi-conservative DNA replication; dispersive replication would have resulted in DNA with a weight between intermediate and light, since the heavier 15N DNA would have been further diluted by 14N DNA and would be evenly distributed throughout all DNA molecules. Meselson and Stahl had managed to distil the complex process of DNA replication into a simple readout (the presence or absence of DNA at a specific weight) with predictable results.
Many years later in the 1990s and 2000s, the model of semi-conservative replication was to help answer the question of how DNA methylation was maintained during cell division. However, another crucial aspect of DNA methylation was yet to be discovered, specifically, how DNA methylation is established; in other words, how does cytosine become 5-methylcytosine? In 1959, the American biochemist Arthur Kornberg, who in the same year won the Nobel Prize for helping to decipher the mechanisms of DNA and RNA synthesis, suggested the possibility of an enzymatic mechanism for the methylation of DNA as a pre-formed polymer (Kornberg et al. 1959). In this model, DNA is firstly synthesised then an enzyme methylates specific sites along the DNA polymer. In 1963, this model proved accurate when the first descriptions of the enzymatic methylation of DNA in bacteria emerged (Gold et al. 1963) and by 1965 it was realised that bacteria contained at least two enzymes: those that methylate adenine to generate 6-methyladenine and those that methylate cytosine to generate 5-methylcytosine (Fujimoto et al. 1965). These enzymes became known as DNA methyltransferases, and though much of the early research of these enzymes focused on those found in bacterial species, it was anticipated that similar enzymes would exist in human cells.
In the 1980s, Timothy Bestor and colleagues at the Massachusetts Institute of Technology (MIT), Cambridge, USA, identified, cloned and characterised the first mammalian DNA methyltransferase from mouse cells, now known as DNMT1 (DNA methyltransferase 1), and showed it had a marked preference for DNA when only one strand is methylated (Bestor et al. 1988). Referred to as hemimethylated DNA, this describes DNA molecules that contain methylated cytosine on only one DNA strand (hence ‘hemi’, or ‘half’, methylated). The 1980s, 1990s and 2000s saw huge strides in our understanding of the cellular machinery that regulates DNA methylation and the identification of a family of DNA and RNA methyltransferases (described further in Chaps. 2 and 3).
We now know that during the cell cycle, DNMT1 expression dramatically increases and becomes localised to sites of DNA replication where it binds to newly synthesised DNA molecules (Leonhardt et al. 1992; Szyf et al. 1985). Due to the semi-conservative nature of DNA replication, each newly formed DNA molecule contains one original DNA strand that is modified by a specific pattern of DNA methylation, whereas the newly synthesised strand is unmethylated. In 2004, Albert Jeltsch and colleagues confirmed Bestor’s earlier findings that DNMT1 preferentially methylates cytosine at hemimethylated targets sites (Hermann et al. 2004). DNMT1 uses the original methylated DNA strand as a template to replicate the pattern of methylation on the newly synthesised DNA strand. If no methylation is present on the original strand then DNMT1 will not methylated the newly synthesised strand. The essential role of DNMT1 in maintaining DNA methylation has led to its description as the maintenance methyltransferase.
1.10 Genomic Imprinting
Each somatic cell in our body contains two sets of autosomes, one set that is maternally inherited and one set that is paternally inherited, and therefore each parent contributes a copy of every gene.Footnote 20 For some time, it was therefore assumed that the genetic contribution of each parent was equal. However, we now know some genes show parental-specific expression and, as a result, the contribution of each parent is different. Genomic imprinting is an epigenetic mechanism that restricts the expression of a gene to one of the two parental chromosomes (Barlow and Bartolomei 2014). Mechanistically, genomic imprinting is a complex phenomenon and, as with many pioneering discoveries, its existence was met with a large degree of scepticism for many years.
During the 1920s and 1930s, the laboratory of Charles Metz at the Carnegie Institution of Washington in Baltimore published several studies describing the embryonic development of Sciara, a type of Diptera (two-winged fly) that was amenable to genetic and cytological study at the time. As an embryologist, Metz was particularly interested in the unusual behaviour of chromosomes during development and spermatogenesis in this species, and the role this plays in sex determination (Metz 1938). During spermatogenesis all paternal chromosomes are selectively eliminated, but two identical copies of the maternal X-chromosome are retained. Therefore, the sperm contributes two X-chromosomes to the zygote whereas the oocyte contributes one X-chromosome and a complete set of autosomes. During development of the embryo, one or both of the X-chromosomes derived from the sperm are selectively eliminated so either one remains (XO) to derive a male or two remain (XX) to derive a female (Crouse 1960). This selective elimination of paternal chromosomes is dependent on a remarkable feature; the ability of the cell to somehow distinguish the parental origin of each chromosome. Helen Crouse, a student of Metz, realised that the mechanism by which the cell achieves this must be erasable, so that chromosomes that are recognised as paternal in origin in one generation may be recognised as maternal in the next, and vice versa. This unorthodox behaviour of chromosomes, driven by their parental origin, seemed to violate apparently well-established principles of heredity and revealed a new mechanism of inheritance. It was Crouse who first used the term ‘imprint’ to describe this unusual behaviour of chromosomes, commenting: ‘…the dramatic chromosome unorthodoxies in Sciara are clearly unrelated to the genic make-up of the chromosomes: a chromosome which passes through the male germ line acquires an “imprint” which will result in behavior exactly opposite to the “imprint” conferred on the same chromosome by the female germ line. In other words, the “imprint” a chromosome bears is unrelated to the genic constitution of the chromosome and is determined only by the sex of the germ line through which the chromosome has been inherited’ (Crouse 1960). Though the exact nature of the ‘imprint’ remained mysterious, Crouse’s own research had suggested that a specific segment of the X-chromosome, which she termed the ‘controlling element’, might be important in this process (Crouse 1960). This early work in Sciara is significant because it foreshadowed the discovery of similar mechanisms of inheritance in other species, including humans.
Chromosomal imprinting in mammals was first recognised in 1971 when it was shown that the paternal X-chromosome was inactivated in all cells of female marsupials and in extra-embryonic tissuesFootnote 21 during mouse development (Cooper et al. 1971). Further work during the 1970s described specific traits in mice when they were engineered to contain two maternal or paternal copies of specific chromosomes (Barlow and Bartolomei 2014). These experiments suggested that the expression of only the paternal or the maternal copy of specific genes was important for normal mouse development (Searle and Beechey 1978).
A major breakthrough came in the 1980s when two papers were published showing that mouse zygotesFootnote 22 generated from entirely paternal or maternal DNA were not viable (McGrath and Solter 1984; Surani and Barton 1983). These studies confirmed that both parental genomes are required for normal mouse development. The experiments involved taking fertilised zygotes containing maternal and paternal pronucleiFootnote 23 and using nuclear transfer techniques to engineer zygotes containing either two maternal pronuclei, two paternal pronuclei or one of each. Embryos derived from zygotes with both a maternal and paternal pronuclei survived, as expected. However, embryos derived from only maternal pronuclei were defective in extraembryonic tissues, whereas embryos derived from only paternal pronuclei were defective in embryonic tissue. This suggested that the development of extraembryonic tissues requires genes specifically expressed in the paternal genome, whereas development of the embryo requires genes specifically expressed in the maternal genome (Barton et al. 1984). The definitive proof of the existence of genomic imprinting in mammals came in 1991 when several papers published in the journals Cell and Nature identified three genes (Igf2, Igf2r and H19) that were expressed specifically from either the maternal or paternal chromosomes in mice (Barlow et al. 1991; Bartolomei et al. 1991; DeChiara et al. 1991; Ferguson-Smith et al. 1991).
1.11 Why Do Genes Become Imprinted?
Since the initial discovery of genomic imprinting in mammals, more than 100 imprinted genes have been identified. Most of these genes have a known role in embryonic and neonatal development and regulate the growth of embryonic or extra-embryonic tissues. Inquiries in different species show that imprinting occurs in placental mammals and marsupials, but not in egg-laying mammals. A distinguishing feature between these species is dependence on maternal nutrition during gestation. In placental mammals, a developing embryo can access maternal resources, whereas embryos developing within an egg cannot. Interestingly, some egg-laying animals, for example, some lizards, can undergo asexual reproduction whereby the embryo develops from a single oocyte following duplication of the maternal genome. This suggests that in egg-laying animals there is no prerequisite for both parental genomes, and therefore no imprinting, again indicating that the dependence of mammals on maternal resources during gestation may be key to understanding why imprinting exists. In 1991, Tom Moore and David Haig proposed the ‘parental conflict’ theory to explain the emergence of imprinting during evolution (Moore and Haig 1991). In this theory, Moore and Haig suggested: ‘imprinting has evolved in mammals because of the conflicting interests of maternal and paternal genes in relation to the transfer of nutrients from the mother to her offspring’ (Moore and Haig 1991). This theory posits that paternal imprints activate or repress the expression of genes to promote the uptake of nutrients from the mother, whereas maternal imprints limit this uptake. Another theory, named the ‘trophoblast defense’ theory, was proposed in 1994 by Susannah Varmuza and Mellissa Mann. This theory suggests that imprinting arose to protect females from spontaneous pregnancy and malignant trophoblast disease (Varmuza and Mann 1994). However, neither theory fully explains the imprinting of all genes, since some are specifically involved in neuronal development during the neonatal period.Footnote 24
1.12 How Do Genes Become Imprinted?
How is a cell able to distinguish the maternal and paternal copies of the same gene and what is the precise nature of the ‘imprint’? During gamete formation, the maternal and paternal chromosomes are separate, thereby providing a window of opportunity to establish parental imprints before fertilisation. Once fertilisation has occurred, these imprints allow the cell to distinguish the maternal and paternal copies of the same chromosome. Therefore, it is thought that the maternally and paternally derived genomes are already differently marked (imprinted) before the two genomes combine to form the zygote. After the discovery of imprinted genes in mammals, scientists began to focus on the mechanisms responsible. It was concluded that it must involve epigenetic modification of DNA because imprinting had been observed in inbred strains of mice wherein the two parental chromosomes contained identical DNA sequences. Crouse’s description of the switching of parental identity of the same chromosome in successive generations added weight to this hypothesis (see above). As a molecular imprint, DNA methylation fulfils several necessary criteria. Firstly, de novo methyltransferases present in the sperm or oocyte can methylate specific regionsFootnote 25 thereby providing a means to establish an imprint. Secondly, the maintenance methyltransferase can maintain an imprint following cell division and throughout development. Finally, DNA can also be demethylated (described further in Chap. 3), thereby allowing the imprint to be erased in the germline and reset in the next generation. To qualify as an imprint, the epigenetic mark must be present in only one of the gametes, persist after fertilisation and be maintained throughout development. Scientists therefore suspected the mechanism was cis-acting,Footnote 26 thereby allowing targeting of only one chromosome. The role of DNA methylation in X-inactivation and the availability of methods to measure its presence or absence provided a clear and testable candidate.
DNA methylation, it turned out, was a key factor governing genomic imprinting. A crucial insight was the realisation that imprinted genes are often found in clusters. In fact, around 80% of imprinted genes are clustered together within imprinted domains (Reik and Walter 2001), which provides strong evidence that they are coordinately regulated. Within imprinted gene clusters that have been well characterised, a common theme is methylation of at least one, and in some instances two, regions specifically on one of the parental chromosomes within the gametes, which regulates the imprinted expression of the entire cluster. These differentially methylated regions are maintained after fertilisation and in all cells of the developing embryo. The necessity of these differentially methylated regions is clear from experiments that show imprinting is lost following their deletion. For the majority of clusters, it is the maternal chromosome that contains the methylation imprint. Another common theme is the presence of at least one lncRNA within the imprinted cluster, which is also expressed specifically from one of the parental chromosomes (lncRNAs have been observed in all but one of the well-characterised clusters). Regions that are essential for establishing imprinting within an imprinted gene cluster are known as imprint control elements. Though DNA methylation of imprint control elements plays a critical role in imprinting, the precise mechanisms at play differ depending on the cluster. In several instances, the imprint control element overlaps with the promoter of the lncRNA within the cluster, and methylation silences its expression. Importantly, the expression of the lncRNA within an imprinted domain is crucial to establishing imprinted gene expression patterns. The specific mechanisms involved in imprinting at several important regions and the relevance of disorders of imprinting in human disease are described in Chap. 9.
1.13 Histones, Nucleosomes and Chromatin Structure
Despite knowledge from the 1800s that chromatin was composed of histones and DNA, the fine structure and three-dimensional arrangement of chromatin within the cell nucleus remained an enigma until the 1970s. As late as 1972, the prevailing hypothesis was of a superhelical structure in which DNA was coated with a layer of histones (Olins and Olins 2003). However, the 1960s and 70s saw a period of discovery that revolutionised our understanding of chromatin structure, and ultimately how this regulated DNA function. These discoveries included the demonstration that chromosomes are uninemic, i.e. that they represent a single DNA molecule running from end to end (Gall 1963); the fractionation, purification and characterisation of the different histones (Johns 1969); the recognition that modification of histones (acetylation and methylation) may regulate gene expression (Allfrey et al. 1964); improvements to methods for preparing and visualising chromatin using the transmission electron microscope (Zubay and Doty 1959); and the demonstration that approximately 50% of the DNA in chromatin was accessible to enzymes that degrade DNA and therefore not covered with proteins (Clark and Felsenfeld 1971; Itzhaki 1971; Mirsky 1971). However, Ada and Donald Olins, and Christopher Woodcock, who in 1973 independently visualised DNA and spherical histone particles in a repeating unit like beads on a string (Fig. 1.1a–c), made by far the biggest conceptual leap in this productive period (Olins and Olins 1973, 1974; Woodcock 1973; Woodcock et al. 1976). A year later Roger Kornberg and Jean Thomas described the chromatin subunit model (R. D. Kornberg 1974; Kornberg and Thomas 1974), in which DNA wraps around an octamer of histone proteins containing one histone (H3–H4)2 tetramer and two histone H2A–H2B dimers. This model was supported by independent evidence showing interactions between histones (D’Anna and Isenberg 1974; Roark et al. 1974) and by biochemical data showing that chromatin structure is a repeating unit (Oudet et al. 1975). In 1975, this repeating unit of chromatin was named the nucleosome (Oudet et al. 1975).

Chromatin structure. (a) An isolated metaphase chromatid pair from a mouse cell. (b) Transmission electron microscopy image of chromatin prepared from chicken erythrocytes. (c) A closer look at chromatin from the box in panel b. Black arrow heads indicate nucleosome core particles. White arrow heads indicate linker DNA between nucleosomes. Black brackets indicate nucleosomes and linker DNA. (d) The crystal structure of the nucleosome core particle consisting of H2A (yellow), H2B (red), H3 (blue) and H4 (green) core histones, and DNA. (e) Schematic of the N-terminal tails of Histone H3 showing the amino acid positions that can be methylated or acetylated. (f) Nuclear Magnetic Resonance (NMR) images of the binding of methyl-CpG binding domain (MBD) proteins bound to methylated DNA. Left, MBD1; centre, MBD2; right, MeCP2. (g) Interaction of MBD1 and methylated DNA at the atomic level. Left, hydrogen bonding between the H- and N-atoms (black line) and the H- and O-atoms (red line) in arginine 22 (ARG22) and guanine at position 107 (GUA107). Right, hydrogen bonding between the H- and N-atoms in the ARG44 and GUA119 (green line), H- and O-atoms in the ARG44-GUA119 pair (blue line). Images from Cell Image Library (http://www.cellimagelibrary.org) using the following accession numbers: Chromosome (panel A), CIL:40682, chromatin (panels B and C), CIL:709 (provided by Christopher Woodcock). Image of nucleosome core particle in panel D taken from https://en.wikipedia.org/wiki/Nucleosome. NMR images in panels F and G taken from Zou et al. (2012)
The nucleosome is the basic functional unit of chromatin and its discovery revolutionised the perception of how DNA function is regulated. As described by Ada and Donald Olins: ‘Higher-order packaging of chromosomal DNA and DNA-based processes, such as transcription, replication and repair, were now all viewed through a different lens. DNA was no longer seen as being coated by histones (superhelical models), but conceived as being coiled on the outside of a globular histone core, which is accessible to the binding of other nuclear proteins. The nucleosome became the ‘quantum’ of chromatin structure, the fundamental unit for the modulation of chromatin function’ (Olins and Olins 2003).
Over the next two decades the crystal structure of the nucleosome core particle would be solved with ever increasing resolution. In 1997, stunning high-resolution images of the nucleosome revealed the orientation of histones at the core and the histone amino-terminal tails that can be chemically modified (Fig. 1.1d, Luger et al. 1997). These protein tails emanate from the nucleosome core particle where they are accessible to a range of enzymes capable of adding or removing a wide range of post-translational modifications, including acetylation and methylation (Fig. 1.1e). The next challenges included defining the histone code and its relationship with gene expression, nucleosome occupancy, nucleosome positioning and DNA methylation (Chaps. 4 and 5 describe these aspects of epigenetics in greater detail) and deciphering the three-dimensional organisation of chromatin in the nucleus and gene regulation through long-range chromatin interactions.
1.14 Cancer Epigenetics
Two non-exclusive hypotheses have been proposed for the origin of cancer: (1) alterations to stem cellsFootnote 27 that result in loss of controlled proliferation and (2) the dedifferentiation of mature cells with specialised functions to stem-like cells that retain the ability to proliferate. In either case, cells must undergo genetic and epigenetic changes that enable these altered behaviours. Cancer can therefore be considered as a disease of altered differentiation.
By the early 1980s, reports that DNA methylation played a role in differentiation were beginning to emerge (Ehrlich et al. 1982; Jones and Taylor 1980; Mandel and Chambon 1979; McGhee and Ginder 1979; Razin and Riggs 1980; Shen and Maniatis 1980; Taylor and Jones 1979; van der Ploeg and Flavell 1980). These studies showed that methylation patterns were tissue-specific, heritable following cell division, and that inhibition of the enzymes that methylate DNA (DNA methyltransferases) disrupts methylation patterns and can alter the differentiation of cells. Given that cancer can be considered a disease of altered differentiation it seemed a natural progression to question whether DNA methylation was altered between normal and cancer tissues. In 1983, two papers from Andrew Feinberg, Bert Vogelstein and the laboratory of Melanie Ehrlich showed that tumours had lower levels of DNA methylation than matched normal tissue (Feinberg and Vogelstein 1983; Gama-Sosa et al. 1983). This hypomethylation was universal across tumour types and was evident in both benign and malignant tumours, suggesting it was an early event in the development of tumours (Feinberg et al. 1988; Goelz et al. 1985). It was also clear by this point that there was a relationship between methylation levels and gene activity; in normal tissues, active (expressed) genes were always less methylated (Razin and Riggs 1980). Accordingly, reports began to emerge of genes showing hypomethylation and overexpression (relative to normal tissue) or hypermethylation and transcriptional silencing (Feinberg and Tycko 2004). In 1989, the first example of epigenetic inactivationFootnote 28 of a known tumour suppressor was described for the retinoblastoma (RB1) gene (Greger et al. 1989; Sakai et al. 1991). This led to widespread acceptance that epigenetic alterations can contribute to the development of cancer and the subsequent description of epigenetic activation or inactivation of many cancer-related genes (Baylin and Jones 2016; see Chap. 7 for a detailed review of the role DNA methylation changes in cancer).
1.15 A Molecular Definition of the Term ‘Gene’
As discussed above, the term ‘gene’ was conceived long before its molecular attributes were known. The term itself has evolved over many years and has been reviewed previously (Portin and Wilkins 2017). In this section, we will briefly describe this evolution and offer a contemporary molecular definition.
When proposed by Wilhelm Johannsen in 1909, the word gene was an abstract term used to refer to a ‘unit of inheritance’. Thomas Morgan and his team confirmed that genes were physical entities in the early twentieth century by demonstrating that they reside on chromosomes in a linear order. However, the term was still somewhat abstract in that it had now become a dimensionless point on a chromosome (Portin and Wilkins 2017). In 1941, George Beadle and Edward Tatum showed that each gene directs the synthesis of a protein, also known as the one gene, one enzyme hypothesis (Beadle and Tatum 1941). Confirmation by Avery, MacLeod and McCarty that DNA was the hereditary molecule in 1944 and the discovery of the structure of DNA in 1953 gave the gene its chemical identity. In the 1950s and 60s, the central dogma of molecular biology was formulated primarily by Francis Crick to describe the unidirectional transfer of information from DNA to messenger RNA (mRNA) and protein during processes known as transcription (DNA to mRNA) and translation (mRNA to protein, (F. Crick 1970; F. H. Crick 1958)). Inherent to this model is the genetic code, whereby triplets (codons) of nucleobases composed of A, T, C and G encode information in the form of amino acids or stop codons. During the 1960s, Robert Holley, Har Gobind Khorana and Marshall Nirenberg cracked the genetic code by deciphering which codons encoded which amino acids, for which they received the Nobel Prize in 1968. By this time it was thought that a gene was a contiguous segment of DNA that encodes a protein.
In the late 1970s, it was realised that genes can contain many exons (coding DNA) interrupted by introns [non-coding DNA, Fig. 1.2 (Berget and Sharp 1977; Berk and Sharp 1977; Chow et al. 1977)]. Before the discovery of introns, it was thought that mRNA molecules were faithful copies of the DNA sequence from the genome; however, on average, around 90% of a gene is comprised of intronic sequence which is spliced out to generate mature mRNA. This discovery changed the way scientists thought about the architecture of the human genome and revealed that genes can have multiple isoforms due to alternative splicingFootnote 29 and the use of different transcription initiation sites. In humans, around 95% of genes with more than one exon are alternatively spliced (Pan et al. 2008). Alternative splicing can vary across tissue types, developmental stages or disease states and represents a mechanism of increasing the functionality encoded within a single gene.

Molecular definition of the gene. (a) A gene can be defined as a DNA sequence (whose component segments do not need to be physically contiguous) that includes the regulatory sequences that can control expression and that produces one or more sequence-related RNAs/proteins. Depicted is an enhancer upstream of a multi-exon gene with two transcription initiation sites and a promoter overlapping a CpG island. Genes can reside on either the positive or the negative DNA strand. Cis-regulatory elements (CREs) such as enhancers and silencers usually operate irrespective of the orientation or strand relative to their target genes. One gene can have multiple CREs and the distance between them is highly variable. The positioning of a CRE may be upstream, downstream or within the gene body. A gene may have multiple promoters and transcription initiation sites. (b) CpG islands are stretches of DNA that are enriched in CpG dinucleotides (see section titled ‘CpG islands’). In approximately 72% of human genes the promoter region has a high CpG content. (c) A gene can give rise to multiple different mRNA transcript isoforms though differential promoter usage and alternative splicing
In addition, there are other non-coding DNA sequences that are important in gene function and should be included in the definition of a gene. These are known as cis-regulatory elements and include gene promoters, enhancers and silencers. Gene promoters are found near the transcription initiation site of a gene and contain specific DNA sequences that recruit proteins (transcription factors, transcriptional activators or repressors) that are important in the initiation of gene transcription. Enhancers and silencers are often many kilobases away from a target gene but can regulate their activity by physical interaction with the promoter via by chromatin looping, which bring two distant loci on the same chromosome into physical proximity. Identifying the enhancer for a specific gene can be extremely difficult because their positioning relative to each other can be highly variable. Also, one enhancer may interact with many genes, one gene may interact with multiple enhancers and these interactions may be tissue- or stimulus-specific (Fig. 1.3). The first enhancer was discovered at the immunoglobulin heavy chain locus (Banerji et al. 1983; Gillies et al. 1983; Mercola et al. 1983); however, it is now estimated that there are hundreds of thousands of enhancers scattered throughout the human genome (Pennacchio et al. 2013). A large part of the prolific discovery of these regulatory regions has been due to the development of methods for mapping three-dimensional chromatin structure, massively parallel sequencing technologies and international initiatives such as the Encyclopedia of DNA elements (ENCODE) and the International Human Epigenome Consortium (IHEC). These are described in greater detail later in this chapter.

Cis-regulatory elements. Cis-regulatory elements (CREs) include enhancers and silencers that can be located many kilobases from a target gene. Black boxes represent genes. Coloured boxes represent CREs. TF, transcription factor. (a) One CRE may target interact with multiple genes. (b) Multiple CREs may interact with one gene and this may depend on the tissue type or developmental stage. This may occur due to the expression of a specific transcription factor in a particular tissue type or stage of development. (c) The regulation of CREs may be altered by DNA sequence variants (red triangle), DNA methylation (black circles) or structural variants including copy number alterations. In either case, these can modify the binding of transcription factors to the CRE
To conclude, many modern definitions of the term gene have been proposed (Burian 2004; Griffiths and Stotz 2006; Keller and Harel 2007; Moss 2003; Pesole 2008; Portin and Wilkins 2017; Scherrer and Jost 2007; Stadler et al. 2009). Here, we use the term to refer to a DNA sequence (whose component segments do not need to be physically contiguous) that includes the regulatory sequences that can control expression of the gene and that produces one or more sequence-related RNAs/proteins. However, biology and genetics are seldom simple and some exceptions that challenge this general definition of a gene include RNA editing,Footnote 30 gene sharing,Footnote 31 gene fusions eventsFootnote 32 and pseudogenesFootnote 33 (Portin and Wilkins 2017).
1.16 CpG Islands
Levels of DNA methylation vary widely across the human genome, which is divided into heavily methylated and non-methylated domains. This is because the primary target of DNA methylation in human cells is cytosine that precedes guanine, also known as the CpG dinucleotide, which is not evenly distributed across the genome. The term CpG is used to distinguish the single-stranded linear sequence (where the ‘p’ represents the phosphate backbone of DNA) from the complementary base pairing of C and G on opposite strands. Approximately 70–80% of CpG cytosines are methylated in mammalian DNA (Jabbari and Bernardi 2004). Throughout mammalian genomes, CpG dinucleotides are under-represented and cluster within regions known as CpG islands. These islands are defined as stretches of DNA that are at least 200 bp in length and contain a GC percentage greater than 50% and an observed-to-expected CpG ratio greater than 60% (Gardiner-Garden and Frommer 1987). CpG islands are often found at the start of a gene, but many exist within repetitive DNA sequences spread throughout the genome, including more than one million copies of a repetitive sequence known as the Alu element (Szmulewicz et al. 1998). Importantly, CpG islands frequently overlap gene promoters (Fig. 1.2b). In the human genome, 72% of gene promoters contain a high CpG content (Saxonov et al. 2006). It has been known for some time that CpG islands overlapping gene promoters are protected from methylation (Bird et al. 1985). This ensures these promoters retain an open chromatin structure and that the underlying DNA sequence is accessible to the transcriptional machinery. However, hypermethylation of a CpG island promoter leads to transcriptional silencing of the linked gene.
1.17 How Does DNA Methylation Cause Transcriptional Silencing?
Although the functional effect of DNA methylation is context dependent, the hypermethylation of a CpG island promoter is usually associated with transcriptional silencing of the gene (Jones 2012). However, there are multiple interdependent layers within chromatin that regulate DNA function and gene expression, including nucleosome occupancy and positioning, post-translational histone modifications, and histone variants. Stable transcriptional repression of a gene involves the remodeling of chromatin structure, which renders the underlying promoter DNA sequence inaccessible to the transcriptional machinery (Ng and Bird 1999). In 1997, key experiments showed that DNA methylation directed a time-dependent repression of transcription (Kass et al. 1997). This was demonstrated by showing that naked methylated DNA displays equivalent expression than non-methylated DNA; however, as chromatin is assembled, the methylated template becomes transcriptionally silent and DNA becomes inaccessible. This indicated that transcriptional silencing by methylation involves a hierarchy of epigenetic events. A year later it was shown that methylated CpG dinucleotides serve as docking sites for the recruitment of a range of proteins containing methyl-CpG binding domains including MBD1 and MeCP2 (Fig. 1.1f and g, Chandler et al. 1999; Nan et al. 1998). MeCP2 interacts with a histone deacetylase complex that catalyses the removal of acetyl groups from histones thereby restoring a positive charge to lysine residues and increasing the affinity between histones and negatively charged DNA (Nan et al. 1998). These changes are accompanied by increased nucleosome occupancy and the addition and removal of a range of other histone modifications and histone variants. The chromatin structure of silent and active gene promoters are described in more detail in Chap. 4.
1.18 Epigenomics
Human genetics and epigenetics has been revolutionised by recent technological advances and by international efforts to democratise data. An example of this is the International Human Epigenome Consortium (IHEC)Footnote 34—an international effort to produce reference maps of at least 1000 human epigenomes from different cellular states, including cells from different tissues and diseases. Contributions are from leading scientists from the European Union, the USA, Canada, Australia, Japan, South Korea, Hong Kong and Singapore and encompasses the Encyclopedia of DNA elements (ENCODE)Footnote 35 project led by the National Institute of Health in the USA. This includes mapping DNA binding proteins (including transcription factors, histone modifications and histone variants), gene transcription, DNA accessibility, RNA binding proteins, DNA methylation, replication timing, three-dimensional chromatin structure and RNA structure. The profiling of genetic and epigenetic characteristics across tissues on this huge scale is possible only with the coordination of global expertise and with the use of massively parallel sequencing and microarray technologies. The types of chromatin modifications and the technologies used to investigate them are summarised in Fig. 1.4.

Data and technologies used by the Encyclopedia of DNA Elements (ENCODE) Consortium to discover and annotate functional DNA elements. Taken from the ENCODE website December 2018 (https://www.encodeproject.org/)
The value of these reference maps and the amount of information they contain is immense; they allow us understand the epigenetic marks that characterise healthy and disease states. They are a reference source that allows scientists to understand how the different layers of epigenetic information enable different interpretations of the same genome. This also allows identification of epigenetic differences that characterise healthy and diseased states. An example of how this information can be mined to understand epigenetic regulation in a specific region of the human genome is shown in Fig. 1.5. In this example, DNA methylation, histone modifications, gene expression, DNA accessibility, CTCF binding and DNA sequence conservation across a cluster of genes and several nearby enhancers is interpreted to convey some of the principles of epigenetic regulation in the human haemoglobin gene locus.

Visualising epigenetic characteristics: the human haemoglobin locus. Data shown are from the myeloid cell line K562 taken from ENCODE and visualised using the UCSC Human Genome Browser (https://genome.ucsc.edu). (a) The haemoglobin locus at human chr11:5,223,622-5,315,172 (GRCh37/hg19; February 2009 freeze). Shown are the locations of several haemoglobin genes (HBB, HBD, HBG1, HBG2 and HBE1), a pseudogene of HBB1 known as HBBP1 and a long non-coding RNA known as BGLT3. (b) A closer view of the HBG1 and HBG2 genes in the region chr11:5,267,292-5,277,485. Green arrows indicate the direction and gene transcription and the transcription start sites. (1) RNA data showing expression of three genes HBG1, HBG2 and HBE1 from the negative DNA strand. (2) RNA data from the positive DNA strand showing no detectable gene expression, as expected due to the fact that all genes in this region reside on the negative strand. (3) DNA methylation at specific sites across the region determined using Illumina Infinium Human Methylation 450 Bead Array technology. Black bars = methylated sites, dark grey bars = partially methylated sites and light grey bars = unmethylated sites. (4) Levels of trimethylation of lysine 4 on histone H3 (H3K4me3). The abundance of H3K4me3 immediately downstream of the transcription start site (as indicated by black arrows) is typical in actively transcribed genes. (5) Levels of monomethylation of lysine 4 on histone H3 (H3K4me1). H3K4me1 is abundant in regulatory regions known as enhancers and less abundant in actively transcribed genes. Note the abundance of H3K4me1 in the region indicated by black lines. This region is a known regulatory region containing several enhancer sites (as indicated by ENCODE GeneHancer data). (6) DNA sequence conservation across vertebrate species. Peaks above the line indicate sites showing evolutionary conservation of DNA sequence, peaks below the line indicate sites where DNA sequence is not conserved. DNA sequence conservation is greatest within the coding regions of genes (exons, see panel b), but conservation in non-coding DNA may indicate important gene regulatory regions such as enhancers. Note that the exons (coding DNA) of all genes across the region are conserved, as well as some sites within enhancer regions. (7) Sensitivity of DNA to cleavage by the enzyme DNaseI, which is evidence of DNA accessibility and an open chromatin structure. DNaseI accessibility can identify important regulatory regions and is often high at the transcription start sites of highly expression genes and at enhancers. Note that the highest levels of DNA accessibility are the transcription start sites of the two genes that are highly expressed (HGB1 and HGB2, see panel B), but also at several conserved sites within enhancer regions. (8) Sites of physical interaction between regions on the same chromosome (as indicated by ENCODE GeneHancer data). Note that several regions of high DNA accessibility physical interact. This includes the interaction of enhancers with several genes across the region including the highly expressed HGB1 gene. (9) The entire haemoglobin locus is bookended by two sites enriched for CTCF binding. CTCF is a protein that plays a key role in gene regulation by binding specific sites in the genome and forming boundaries that demarcate chromosome domains. This partitioning allows the independent regulation of different domains within a chromosome
1.19 Key Milestones in Genetics and Epigenetics
This introductory chapter has provided an overview of the history of genetic and epigenetic discoveries that have brought us to modern technologies. Below is a timeline of some of the key milestones in genetics and epigenetics with important terms underlined and milestones specific to epigenetics shown in italicised text. Due to the pace of discovery the timeline ends with the launch of The International Human Epigenome Consortium in 2010. An abridged version focused on epigenetics discoveries is shown in Fig. 1.6.

Timeline of epigenetic discoveries. Image sources: Homunculus, (Hartsoeker 1694); 5-methylcytosine, (Wheeler and Johnson 1904); Barr body, (http://glencoe.mcgraw-hill.com/sites/dl/free/0078664276/281029/ccq_ch13_q3.gif), Electron micrographs of chromatin, (Olins and Olins 2003); Crystal structure of the nucleosome core particle to 2.8 Angstrom resolution (Luger et al. 1997); Fractal globule of the three-dimensional structure of chromatin in the nucleus (Lieberman-Aiden et al. 2009)
1.20 Key Discoveries
- 1751:
Pierre Louis de Maupertuis hypothesises equal contribution of both parents to their offspring and a particulate basis of heredity
- 1800s:
Advances in microscopy results in epigenesis superseding preformationism as the preferred model of embryological development
- 1809:
Jean-Baptiste Lamarck proposes the model of inheritance of acquired characteristics
- 1814:
Physician and surgeon Joseph Adams classifies hereditary disorders as hereditary and congenital and distinguishes between predisposition and disposition
- 1859:
Charles Darwin Publishes Origin of Species
- 1866:
Gregor Mendel describes patterns of particulate inheritance in pea plants and introduces the terms dominant and recessive
- 1866:
Ernst Heinrich Haeckel proposes that the nucleus of a cell transmits its hereditary information
- 1869:
Charles Darwin publishes Variation in Animals and Plants
- 1871:
Friedrich Miescher isolates nuclein (DNA)
- 1871:
Charles Darwin publishes Descent of Man
- 1873:
First descriptions of cell division by Anton Schneider
- 1875:
Francis Galton uses twins to study characteristics
- 1875:
Oscar Hertwig recognises that fertilisation represents the union of the nuclei contributed by male and female germ cells
- 1876:
Francis Galton offers a statistical approach to heredity
- 1878:
Walther Flemming discovers a substance he calls chromatin and identifies it as a constituent of chromosomes
- 1882:
Walther Flemming introduces the term mitosis
- 1882:
Eduard Strasburger introduces the terms cytoplasm and nucleoplasm
- 1883:
Edouard van Beneden recognises that the sperm and egg contain fewer chromosomes and that chromosome number is combined after fertilisation
- 1883:
August Weismann makes the distinction between somatic cells and germ cells and proposes that only germ cells carry information that can be transmitted to offspring
- 1884:
Eduard Strasburger introduces the terms prophase, metaphase and anaphase to describe stages of cell division
- 1884:
Albrecht Kossel discovers histones and protamines
- 1885:
Hans Driesch clones the first animal (sea urchin) using a process known as embryo splitting
- 1887:
August Weismann deduces the existence of a reduction division (now known as meiosis) in all sexual organisms, which is observed during germ cell maturation by Edouard van Beneden later in the same year
- 1884–1888:
Oscar Hertwig, Eduard Strasburger, Albrecht von Kölliker and August Weismann show the basis of inheritance is contained within the cell nucleus
- 1888:
Heinrich Gottfried von Waldeyer-Hartz introduces the term chromosome
- 1888:
Theodor Boveri first studies chromosomes and suggests they are involved in heredity
- 1880–1890:
Walther Flemming, Eduard Strasburger, Edouard van Beneden and others fully describe cell division, including the equal separation of chromosomes to daughter cells
- 1890:
Theodor Boveri and Jean-Louis-Léon Guignard recognise that there is equal contribution of paternal and maternal chromosomes at fertilisation
- 1891:
Hermann Henking identifies the ‘X’ body in a proportion of germ cells in insects
- 1892:
August Weismann publishes Das Keimplasma (The Germ Plasm), which provided a framework to study development, evolution and heredity
- 1894:
William Bateson publishes his book Materials for the Study of Variation, which illustrates the significance of discontinuous characteristics (those that do not ‘blend’) for the understanding of heredity
- 1899:
Clarence McClung finds the ‘X body’ in locusts and identifies it as a chromosome
- 1899:
First International Congress of Genetics
- 1899:
Richard Altmann renames nuclein as nucleic acid
- 1900:
Hugo de Vries, Erich von Tschermak and Carl Correns rediscover Gregor Mendel’s work
- 1885–1901:
Albrecht Kossel isolates and names the constituents of the non-protein component of nucleic acids as adenine, cytosine, guanine, thymine and uracil
- 1901:
T. H. Montgomery recognises the pairing of maternal and paternal chromosomes during meiosis
- 1902:
First description of the theory of chromosomes independently by Theodor Boveri and Walter Sutton
- 1902:
William Bateson, Edith Saunders, William Castle and William Farabee first describe Mendelian inheritance in human disease (alkaptonuria, albinism and brachydactyly)
- 1902:
Clarence McClung proposes that particular chromosomes determine sex
- 1904:
William Bateson and Reginald Punnett first describe gene linkage
- 1904:
Hugo De Vries coins the term mutation in his book Species and Varieties: Their Origin by Mutation
- 1904:
Henry Wheeler and Treat Johnson artificially synthesise 5-methylcytosine
- 1905:
William Bateson proposes the term genetics to define the study of heredity
- 1905:
Nettie Stevens first identifies the chromosomal basis for sex determination in flies and beetles, later referred to as the XY sex-determination system
- 1908:
Godfrey Hardy and Wilhelm Weinberg propose the Hardy-Weinberg equilibrium to explain the mathematical relationship of genotype frequencies
- 1908:
Archibald Garrod publishes Inborn Errors of Metabolism and proposes they are determined by genetics
- 1909:
Willhelm Johannsen defines the terms gene, genotype and phenotype
- 1910:
Thomas Morgan describes sex-limited inheritance in drosophila using the white-eyed mutant
- 1910:
Albrecht Kossel is awarded the Nobel Prize in Physiology or Medicine for his research in cell biology, the chemical composition of the cell nucleus, and the isolation and description of nucleic acids
- 1910:
Phoebus Levene introduces the tetranucleotide hypothesis for the structure of DNA
- 1910–1911:
Thomas Morgan shows that chromosomes carry genes and describes the crossing over (recombination) theory of chromosomes
- 1911:
Edmund Wilson uses knowledge of the XY sex-determination system to predict consequences for X-linked inheritance for haemophilia and colour blindness
- 1913:
Alfred Sturtevant creates the first genetic linkage map
- 1914:
Calvin Bridges describes non-disjunction of sex chromosomes, thereby proving the chromosome theory of heredity
- 1915:
Morgan, Sturtevant, Muller and Bridges publish The Mechanism of Mendelian Heredity
- 1915:
Frederick Twort discovers the first bacteriophage
- 1917:
Independently of Twort, Felix Hubert D’Herelle discovers another virus capable of infecting and destroying bacteria and coins the term bacteriophage
- 1919:
Calvin Bridges discovers duplications within a chromosome in drosophila
- 1923:
Calvin Bridges discovers chromosome translocations in drosophila
- 1925:
Treat Johnson and Robert Coghill discover 5-methylcytosine in the Mycobacterium tuberculosis
- 1926:
Alfred Sturtevant discovers chromosome inversions in drosophila
- 1927:
Hermann Muller mutates genes using X-rays
- 1928:
Frederick Griffith’s bacterial transformation experiments provides initial evidence that DNA contains hereditary information
- 1920s–1930s:
Chromosomal imprinting described in insects by Charles Metz
- 1930:
First description of position effect variegation
- 1941:
George Beadle and Edward Tatum show that genes direct synthesis of proteins (one gene, one enzyme hypothesis)
- 1942:
Conrad Waddington introduces the term epigenetics
- 1944:
Demonstration that protein-free DNA carries genetic information (Oswald Avery, Colin MacLeod and Maclyn McCarty)
- 1946:
Hermann Muller receives Nobel Prize in Medicine for his work in radiation genetics
- 1948:
Rollin Hotchkiss discovers 5-methylcytosine in mammalian cells
- 1948:
Hermann Muller introduces the term dosage compensation
- 1949:
James Neel shows sickle cell anaemia is inherited in a Mendelian autosomal recessive manner
- 1950:
Erwin Chargaff shows the proportion of nucleobases differs in different species and that the amount of adenine equals the amount of thymine and the amount of cytosine equals the amount of guanine (A = T and G = C)
- 1937–1951:
X-ray diffraction of DNA reveals a regular repeating periodic structure (William Astbury, Maurice Wilkins, Rosalind Franklin and others)
- 1952:
Fred Sanger determines the sequence of amino acids in the protein insulin
- 1952:
Alfred Hershey and Martha Chase show that it is the DNA from bacteriophage that enters the host bacterium, thereby dispelling any remaining doubt that DNA contains the heredity information
- 1953:
The double helix structure of DNA is elucidated by Francis Crick and James Watson, using X-ray diffraction data from Rosalind Franklin
- 1955:
Definition of the human karyotype by Joe Hin Tjio
- 1956:
Arthur Kornberg crystallises DNA polymerase, the enzyme required for synthesising DNA
- 1957:
Francis Crick proposes the central dogma of molecular biology
- 1958:
Semi-conservative mechanism of DNA replication elucidated by Matthew Meselson and Franklin Stahl
- 1958:
George Beadle and Edward Tatum receive the Nobel Prize in Medicine ‘for their discovery that genes act by regulating definite chemical events’ with the other half to Joshua Lederberg ‘for his discoveries concerning genetic recombination and the organization of the genetic material of bacteria’
- 1958:
Fred Sanger receives Nobel Prize for Chemistry for his work on the structure of proteins, especially that of insulin
- 1959:
Arthur Pardee, Francois Jacob and Jacques Monod publish their study of the lactose operon in Escherichia coli
- 1959:
Trisomy 21 identified as the cause of Down syndrome by Jerome Lejeune, Martha Gautier and Raymond Turpin
- 1959:
The Nobel Prize in Physiology or Medicine is awarded jointly to Severo Ochoa and Arthur Kornberg ‘for their discovery of the mechanisms in the biological synthesis of ribonucleic acid and deoxyribonucleic acid’
- 1960:
Helen Crouse introduces the term imprint to describe the marking of chromosomes by parent-of-origin
- 1960s:
Fractionation of histones by E. W. Johns and others
- 1961:
Messenger RNA (mRNA) identified as the intermediate that carries information from DNA in the nucleus to the cytoplasm where protein is made (Sydney Brenner, Francois Jacob and Matthew Meselson)
- 1961:
Triplets of DNA bases proposed to code for one of the 20 amino acids by Sydney Brenner and Francis Crick
- 1961:
Discovery of X-inactivation by Mary Lyon
- 1962:
First patient diagnosed with a mitochondrial disease
- 1962:
The Nobel Prize in Physiology or Medicine is awarded jointly to Francis Crick, James Watson and Maurice Wilkins ‘for their discoveries concerning the molecular structure of nucleic acids and its significance for information transfer in living material’
- 1963:
Mitochondrial DNA identified
- 1964:
Vincent Allfrey, Robert Faulkner and Alfred Mirsky propose that modification of histones (acetylation and methylation) can regulate gene expression
- 1964:
Development of the DNA hypomethylating drug 5-azacytidine
- 1964:
Robin Holliday describes concept of gene conversion and the Holliday junction
- 1965:
The Nobel Prize in Physiology or Medicine is awarded jointly to François Jacob, André Lwoff and Jacques Monod ‘for their discoveries concerning genetic control of enzyme and virus synthesis’
- 1961–1966:
Marshall Nirenberg cracks the genetic code
- 1967:
Mary Weiss and Howard Green map human genes using somatic cell hybridisation
- 1968:
First therapeutic abortion performed entirely on the basis of a genetic test (Down syndrome)
- 1968:
The Nobel Prize in Physiology or Medicine is awarded jointly to Robert Holley, Har Gobind Khorana and Marshall Nirenberg ‘for their interpretation of the genetic code and its function in protein synthesis’
- 1969:
The Nobel Prize in Physiology or Medicine is awarded jointly to Max Delbrück, Alfred Hershey and Salvador Luria ‘for their discoveries concerning the replication mechanism and the genetic structure of viruses’
- 1970:
Ron Laskey and John Gurdon show that a somatic cell nucleus contains all the necessary information to direct embryogenesis when introduced into an enucleated egg. This represented definitive proof that cell differentiation and embryonic development was not driven by the loss of genetic material
- 1970:
Howard Temin and David Baltimore discovery reverse transcriptase, an enzyme that makes DNA from RNA
- 1971:
Alfred Knudson’s proposes his two-hit hypothesis of cancer
- 1971:
Chromosomal imprinting described in mammals (X-chromosome in the kangaroo)
- 1971:
Eduardo Scarano proposes that 5-methylcytosine can deamination to generate thymine
- 1972:
First recombinant DNA molecules
- 1973:
First images of higher-order chromatin structure by Ada and Donald Olins and Christopher Woodcock
- 1974:
The chromatin subunit model is proposed by Ada and Donald Olins, Roger Kornberg and Jean Thomas
- 1975:
The term nucleosome is proposed by Oudet, Gross-Bellard and Chambon
- 1975:
Robin Holliday, John Pugh and Arthur Riggs propose that DNA methylation controls gene expression, is heritable following cell division and is an epigenetic mechanism that explains X-chromosome inactivation
- 1977:
First DNA sequencing methods developed by Walter Gilbert, Alan Maxam and Fred Sanger
- 1977:
The enterobacteriaphage phiX174 becomes the first genome sequenced (Fred Sanger)
- 1977:
Discovery of introns and the concept of splicing
- 1978:
Methylation sensitive restriction endonucleases first used to detect DNA methylation
- 1978:
David Botstein uses restriction enzymes to map human genes and identify genetic differences between individuals
- 1980:
Concept of positional cloning of genes proposed by Ron Davis and David Botstein
- 1980:
The Nobel Prize in Chemistry is divided, one half awarded to Paul Berg ‘for his fundamental studies of the biochemistry of nucleic acids, with particular regard to recombinant-DNA’ and the other half jointly to Walter Gilbert and Frederick Sanger ‘for their contributions concerning the determination of base sequences in nucleic acids’
- 1981:
Sequencing of the mitochondrial genome
- 1982:
Melanie Ehrlich shows that DNA methylation content and distribution differs amongst different tissue and cell types
- 1983:
Invention of PCR by Kary Mullis
- 1983:
Discovery of the first enhancer within the immunoglobulin heavy chain locus
- 1983:
Andy Feinberg, Bert Vogelstein and Melanie Ehrlich demonstrate that DNA hypomethylation is a ubiquitous feature of human cancers
- 1984:
Solution of the crystal structure of the nucleosome core particle to 7 Angstrom resolution
- 1984:
Alec Jeffreys develops DNA fingerprinting
- 1984:
Demonstration that both maternal and paternal genomes are essential for normal mammalian embryonic development
- 1985:
CpG islands first recognised
- 1985:
Discovery of an imprinted region
- 1986:
Penny Jeggo and Robin Holliday introduce the term epimutation
- 1986:
First gene identified by positional cloning (chronic granulomatous disease)
- 1986:
Development of the first automated DNA sequencer using fluorescent dyes (Leroy Hood)
- 1986:
First descriptions of epigenetic inactivation of a tumour suppressor gene (RB1 in retinoblastoma)
- 1986–1990:
Human genome project launched
- 1980–1991:
Experiments showing that histone amino-terminal tails are essential in the regulation of gene expression
- 1987:
Robin Holliday proposes the inheritance of epigenetic defects
- 1990:
BRCA1 gene identified and linked to familial breast and ovarian cancer (Mary-Claire King and Mark Skolnick)
- 1991:
Abnormal hypermethylation of FMR1 recognised in Fragile X syndrome, representing the first description of a constitutional epimutation in humans
- 1991:
Imprinted genes first identified in mice by Denise Barlow, Anne Ferguson-Smith and others
- 1991:
Parental conflict theory proposed to explain the evolution of genomic imprinting
- 1992:
Mechanistic basis of the heritability of DNA methylation shown when it’s discovered that Dnmt1 is targeted to sites of DNA replication
- 1993:
Positional cloning of the gene mutated in Huntington’s disease by Nancy Wexler and James Gusella
- 1993:
Bryan Turner proposes that post-translational modifications of the amino-terminal tails of histones encodes epigenetic information
- 1993:
The Nobel Prize in Chemistry is awarded ‘for contributions to the developments of methods within DNA-based chemistry’ jointly with one half to Kary Mullis ‘for his invention of the polymerase chain reaction (PCR) method’ and with one half to Michael Smith ‘for his fundamental contributions to the establishment of oligonucleotide-based, site-directed mutagenesis and its development for protein studies’
- 1994:
Bisulphite DNA conversion method developed by Marianne Frommer and Susan Clark
- 1994:
Généthon publishes a map of the human genome based on microsatellite markers
- 1995:
Development of DNA microarrays
- 1995:
The genome of Haemophilus influenzae is sequenced (J. Craig Venter)
- 1995:
Map of the human genome based on Sequence Tagged Sites (STSs)
- 1995:
The Nobel Prize in Physiology or Medicine is awarded jointly to Edward Lewis, Christiane Nüsslein-Volhard and Eric Wieschaus ‘for their discoveries concerning the genetic control of early embryonic development’
- 1996:
Sequencing of the genome of Saccharomyces cerevisiae (baker’s yeast)
- 1996:
Dolly the sheep is cloned by Keith Campbell and Ian Wilmut
- 1997:
The crystal structure of the nucleosome core particle to 2.8 Angstrom resolution shows the histone amino terminal tails modified by post-translational modifications
- 1997:
The relationship between DNA methylation, chromatin structure and gene silencing becomes clearer when it is shown that methylated DNA only becomes transcriptionally inactive after being packaged into chromatin
- 1998:
Recognition that transposable and viral elements within the mammalian genome are hypermethylated
- 1998:
Discovery that DNA methylation increases with age
- 1999:
Recognition that DNA methyltransferase preferentially recognises hemimethylated DNA
- 2000:
CTCF identified as a key mediator of imprinting at the H19/Igf2 locus
- 2001:
First draft of the human genome published
- 2001:
The histone code is proposed by Thomas Jenuwein and C. David Allis
- 2002:
Development of method for capturing chromosome conformation and identifying long-range chromatin interactions
- 2003:
The National Human Genome Research Institute launches ENCODE (Encyclopedia of DNA Elements) to identify all functional elements in the human genome
- 2004:
First description of a constitutional epimutation predisposing to cancer by Robyn Ward and colleagues (MLH1 in Lynch syndrome)
- 2009:
First description of the three-dimensional architecture of a human genome
- 2010:
The International Human Epigenome Consortium is founded to lead efforts in understanding the human epigenome and to generate reference databases of healthy and disease-related cell types
Notes
- 1.
Interestingly, the separation of nucleic acid from protein was so difficult at the time that when Richard Altmann achieved this, and described the acidic derivative (nucleic acid), he believed he was describing a novel subcomponent of nuclein. Friedrich Meischer had in fact defined nuclein as acidic in his original paper.
- 2.
Uracil is usually found only in RNA.
- 3.
The term transformation describes the uptake of genetic material by a cell from its surroundings. This can be measured by a change in the form and function of the cell, such as the transformation of a non-virulent strain of bacteria into a virulent strain.
- 4.
Though others quickly confirmed the findings of the Avery-MacLeod-McCarty experiment the conclusion that DNA was the genetic material was still met with resistance from those that believed genes were made of protein. Further work in 1948 by Rollin Hotchkiss, a mentee of Avery, would demonstrate that transformation was not the result of contaminating protein and that DNA was indeed responsible. Work by Alfred Hershey and Martha Chase in 1952 would provide the final confirmation by showing that it is the DNA from bacteriophage that enters the host bacterium.
- 5.
These cellular phenomena are not taken verbatim from Nanney’s original 1958 paper and have been adapted for clarity.
- 6.
A total of 48 research papers from this collaboration were published as a book titled Papers on Pyrimidines in 1910 and can be accessed freely at The Internet Archive at https://archive.org/details/papersonpyrimidi00wheerich.
- 7.
Work by Clarence McClung at the University of Kansas in 1902 showed the X-element was indeed a chromosome (though McClung referred to it as the accessory chromosome). Edmund Wilson and Nettie Stevens are credited with designating the names X-chromosome and Y-chromosome following the discovery of the XY sex determination system.
- 8.
In some species, it is the number of X chromosomes that determines sex.
- 9.
Eutherians include all placental mammals.
- 10.
In 1991, Barbara Migeon and colleagues used fluorescence in situ hybridisation to show that the Barr body is comprised of a single X-chromosome folded in two and attached at both ends to the periphery of the nucleus (Walker et al. 1991).
- 11.
Though XO female mice are phenotypically normal and fertile, in humans, individuals with only one X-chromosome are infertile.
- 12.
The term mosaicism is used in genetics to describe genetic differences between cells of the same organism.
- 13.
The term penetrance is used in genetics to describe the proportion of individuals carrying a particular variant (or allele) of a gene (the genotype) that also express an associated trait (the phenotype).
- 14.
In 2017, The Royal Society paid tribute to Mary Lyon with a series of review articles in the journal Philosophical Transactions of the Royal Society B (http://rstb.royalsocietypublishing.org/content/372/1733) and audio recordings (https://royalsociety.org/science-events-and-lectures/2016/10/x-chromosome-inactivation/), for readers interested in further details regarding X-inactivation.
- 15.
The term aneuploidy refers to an abnormal number of chromosomes.
- 16.
In humans, approximately 15% of X-linked genes escape X-inactivation, whereas in mice it is only 3% (Berletch et al. 2011), which explains why XO mice remain fertile. Interestingly, species comparison of genes on the X-chromosome suggests genes that escape X-inactivation once originated from autosomal chromosomes (chromosomes other than the sex chromosomes X or Y), which are not subject to dosage compensation (Ballabio and Willard 1992).
- 17.
Methylation of the inactive X chromosome is dependent on the gene SMCHD1 (Blewitt et al. 2008), which is an important regulator of DNA methylation genome-wide.
- 18.
The term autosome refers to a chromosome other than one of the sex chromosomes. In humans, there are 22 pairs of autosomes and two sex chromosomes.
- 19.
The term diploid is used to define a cell or nucleus containing two complete sets of chromosomes, one from each parent. In a triploid cell or nucleus, three complete sets of chromosomes are present.
- 20.
In the human genome, duplicated or deleted regions known as copy number variants can result in changes to the number of copies of specific genes. This may be responsible for genotypic and phenotypic variation between individuals. An example is the gene CYP2D6, which is responsible for the metabolism of a range of different drugs. The presence of more than two copies of this gene can cause individuals to metabolise some drugs more rapidly than individuals with two copies, which has implications for the dose of drug they should receive.
- 21.
Extra-embryonic tissues are those that contribute to the placenta.
- 22.
A zygote is a diploid cell that results from the fusion of two haploid gametes (i.e. a fertilised oocyte).
- 23.
The term pronucleus describes the structure within the oocyte or sperm that contains the maternal or paternal chromosomes, respectively. Around 12 h after fertilisation of the oocyte by a sperm the maternal and paternal pronuclei fuse to form the nucleus.
- 24.
It is possible that neuronal changes may affect behaviour and competition for maternal resources during the neonatal period, including the regulation of appetite for maternal milk, which would support the parental conflict model.
- 25.
It is currently unclear how a specific region is selected for methylation.
- 26.
The term cis in genetics refers to two genetic features on the same chromosome. In this context, the element controlling imprinting and the imprinted gene must be on the same chromosome.
- 27.
Stem cells have the ability to perpetually self-renew and can differentiate into specific cell types.
- 28.
In the context of cancer, epigenetic inactivation refers to the hypermethylation of the promoter of a gene and its transcriptional silencing in tumour tissue relative to matched normal tissue.
- 29.
Alternative mRNA splicing describes the joining together of different combinations of exons. This can vary the sequence of an mRNA, and thus protein. The discovery of alternative splicing earned Richard Roberts and Phil Sharp the Nobel Prize in 1993.
- 30.
Post-transcriptional alterations to RNA sequence.
- 31.
Gene sharing describes rare circumstances whereby the same protein sequence derived from the same gene can assume different conformations and functions in different cellular contexts.
- 32.
The physiological transcription of genes in tandem to produce hybrid mRNA molecules.
- 33.
A pseudogene is a non-functioning copy of another gene which may under some circumstances produce mRNA.
- 34.
Further information about IHEC can be accessed at http://ihec-epigenomes.org/.
- 35.
Further information about ENCODE can be accessed at https://www.encodeproject.org/.
References
Allfrey VG et al (1964) Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc Natl Acad Sci U S A 51:786–794
Altmann R (1889) Ueber Nucleinsäuren. Arch f Anatomie u Physiol:524–536
Austin CR, Amoroso EC (1957) Sex chromatin in early cat embryos. Exp Cell Res 13(2):419–421
Avery O (1941) Obituary: Frederick Griffith. Lancet:588–589
Avery OT et al (1944) Studies on the chemical nature of the substance inducing transformation of pneumococcal types: induction of transformation by a desoxyribonucleic acid fraction isolated from Pneumococcus type Iii. J Exp Med 79(2):137–158
Ballabio A, Willard HF (1992) Mammalian X-chromosome inactivation and the XIST gene. Curr Opin Genet Dev 2(3):439–447
Banerji J et al (1983) A lymphocyte-specific cellular enhancer is located downstream of the joining region in immunoglobulin heavy chain genes. Cell 33(3):729–740
Barlow DP, Bartolomei MS (2014) Genomic imprinting in mammals. Cold Spring Harb Perspect Biol 6(2):a018382
Barlow DP et al (1991) The mouse insulin-like growth factor type-2 receptor is imprinted and closely linked to the Tme locus. Nature 349(6304):84–87
Barr ML, Bertram EG (1949) A morphological distinction between neurones of the male and female, and the behaviour of the nucleolar satellite during accelerated nucleoprotein synthesis. Nature 163(4148):676
Bartolomei MS et al (1991) Parental imprinting of the mouse H19 gene. Nature 351(6322):153–155
Barton SC et al (1984) Role of paternal and maternal genomes in mouse development. Nature 311(5984):374–376
Baylin SB, Jones PA (2016) Epigenetic determinants of cancer. Cold Spring Harb Perspect Biol 8(9):a019505
Beadle GW, Tatum EL (1941) Genetic control of biochemical reactions in neurospora. Proc Natl Acad Sci U S A 27(11):499–506
Berget SM, Sharp PA (1977) A spliced sequence at the 5’-terminus of adenovirus late mRNA. Brookhaven Symp Biol (29):332–344
Berk AJ, Sharp PA (1977) Sizing and mapping of early adenovirus mRNAs by gel electrophoresis of S1 endonuclease-digested hybrids. Cell 12(3):721–732
Berletch JB et al (2011) Genes that escape from X inactivation. Hum Genet 130(2):237–245
Bestor T et al (1988) Cloning and sequencing of a cDNA encoding DNA methyltransferase of mouse cells. The carboxyl-terminal domain of the mammalian enzymes is related to bacterial restriction methyltransferases. J Mol Biol 203(4):971–983
Bird A et al (1985) A fraction of the mouse genome that is derived from islands of nonmethylated, CpG-rich DNA. Cell 40(1):91–99
Blewitt ME et al (2008) SmcHD1, containing a structural-maintenance-of-chromosomes hinge domain, has a critical role in X inactivation. Nat Genet 40(5):663–669
Borsani G et al (1991) Characterization of a murine gene expressed from the inactive X chromosome. Nature 351(6324):325–329
Brockdorff N et al (1991) Conservation of position and exclusive expression of mouse Xist from the inactive X chromosome. Nature 351(6324):329–331
Brown CJ et al (1991a) A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature 349(6304):38–44
Brown CJ et al (1991b) Localization of the X inactivation centre on the human X chromosome in Xq13. Nature 349(6304):82–84
Brush SG (1978) Nettie M. Stevens and the discovery of sex determination by chromosomes. Isis 69(247):163–172
Burch PR, Burwell RG (1963) Lyonisation of the X chromosome. Lancet 2(7319):1229
Burian RM (2004) Molecular epigenesis, molecular pleiotropy, and molecular gene definitions. Hist Philos Life Sci 26(1):59–80
Chandler SP et al (1999) The methyl-CpG binding transcriptional repressor MeCP2 stably associates with nucleosomal DNA. Biochemistry 38(22):7008–7018
Chargaff E (1951) Structure and function of nucleic acids as cell constituents. Fed Proc 10(3):654–659
Chargaff E et al (1949) The composition of the desoxypentose nucleic acids of thymus and spleen. J Biol Chem 177(1):405–416
Chow LT et al (1977) A map of cytoplasmic RNA transcripts from lytic adenovirus type 2, determined by electron microscopy of RNA: DNA hybrids. Cell 11(4):819–836
Chu C et al (2015) Systematic discovery of Xist RNA binding proteins. Cell 161(2):404–416
Clark RJ, Felsenfeld G (1971) Structure of chromatin. Nat New Biol 229(4):101–106
Cooper DW et al (1971) Phosphoglycerate kinase polymorphism in kangaroos provides further evidence for paternal X inactivation. Nat New Biol 230(13):155–157
Crick FH (1958) On protein synthesis. Symp Soc Exp Biol 12:138–163
Crick F (1970) Central dogma of molecular biology. Nature 227(5258):561–563
Crouse HV (1960) The controlling element in sex chromosome behavior in Sciara. Genetics 45(10):1429–1443
D’Anna JA Jr, Isenberg I (1974) A histone cross-complexing pattern. Biochemistry 13(24):4992–4997
Darwin C (1859) On the origin of species by means of natural selection: or the preservation of favoured races in the struggle for life. D. Appleton, New York
Darwin C (1872) The descent of man, and selection in relation to sex. D. Appleton, New York
de Maupertuis PLM (1751) Essai de Cosmologie. Sorli
de Monet de Lamarck JB (1802) Recherches sur l’organisation des corps vivants et particulièrement sur son origine, sur la cause de son développement et des progrès de sa composition ... Précédé du discours d’ouverture du cours de zoologie donné dans le Muséum d’histoire naturelle, l’an X de la République. Par J.-B. Lamarck: L’auteur
de Monet de Lamarck JBPA (1809) Philosophie zoologique: ou Exposition des considérations relative à l’histoire naturelle des animaux. Dentu, Paris
Deans C, Maggert KA (2015) What do you mean, “epigenetic”. Genetics 199(4):887–896
DeChiara TM et al (1991) Parental imprinting of the mouse insulin-like growth factor II gene. Cell 64(4):849–859
Doskocil J, Sorm F (1962) Distribution of 5-methylcytosine in pyrimidine sequences of deoxyribonucleic acids. Biochim Biophys Acta 55:953–959
Dupont C et al (2009) Epigenetics: definition, mechanisms and clinical perspective. Semin Reprod Med 27(5):351–357
Ehrlich M et al (1982) Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res 10(8):2709–2721
Feinberg AP, Tycko B (2004) The history of cancer epigenetics. Nat Rev Cancer 4(2):143–153
Feinberg AP, Vogelstein B (1983) Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature 301(5895):89–92
Feinberg AP et al (1988) Reduced genomic 5-methylcytosine content in human colonic neoplasia. Cancer Res 48(5):1159–1161
Ferguson-Smith AC et al (1991) Embryological and molecular investigations of parental imprinting on mouse chromosome 7. Nature 351(6328):667–670
Friedrich M (1874) Die Spermatozoen einiger Wirbeltiere. Ein Beitrag zur Histochemie. Verhandlungen der naturforschenden Gesellschaft in Basel VI, pp 138–208
Fujimoto D et al (1965) On the nature of the deoxyribonucleic acid methylases. Biological evidence for the multiple nature of the enzymes. Biochemistry 4(12):2849–2855
Gall JG (1963) Kinetics of deoxyribonuclease action on chromosomes. Nature 198:36–38
Gama-Sosa MA et al (1983) The 5-methylcytosine content of DNA from human tumors. Nucleic Acids Res 11(19):6883–6894
Gardiner-Garden M, Frommer M (1987) CpG islands in vertebrate genomes. J Mol Biol 196(2):261–282
Gillies SD et al (1983) A tissue-specific transcription enhancer element is located in the major intron of a rearranged immunoglobulin heavy chain gene. Cell 33(3):717–728
Goelz SE et al (1985) Hypomethylation of DNA from benign and malignant human colon neoplasms. Science 228(4696):187–190
Gold M et al (1963) The enzymatic methylation of RNA and DNA, Ii. On the species specificity of the methylation enzymes. Proc Natl Acad Sci U S A 50(1):164–169
Greally JM (2018) A user’s guide to the ambiguous word ‘epigenetics’. Nat Rev Mol Cell Biol 19(4):207–208
Greger V et al (1989) Epigenetic changes may contribute to the formation and spontaneous regression of retinoblastoma. Hum Genet 83(2):155–158
Griffith F (1928) The significance of pneumococcal types. J Hyg (Lond) 27(2):113–159
Griffiths PE, Stotz K (2006) Genes in the postgenomic era. Theor Med Bioeth 27(6):499–521
Haig D (2004) The (dual) origin of epigenetics. Cold Spring Harb Symp Quant Biol 69:67–70
Hall RH (1971) The modified nucleosides in nucleic acids. Columbia University Press, New York
Hartsoeker N (1694) Essay de dioptrique. Kessinger, Whitefish
Hermann A et al (2004) The Dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA processively with high preference for hemimethylated target sites. J Biol Chem 279(46):48350–48359
Holliday R, Pugh JE (1975) DNA modification mechanisms and gene activity during development. Science 187(4173):226–232
Hotchkiss RD (1948) The quantitative separation of purines, pyrimidines, and nucleosides by paper chromatography. J Biol Chem 175(1):315–332
Itzhaki RF (1971) Studies on the accessibility of deoxyribonucleic acid in deoxyribonucleoprotein to cationic molecules. Biochem J 122(4):583–592
Jabbari K, Bernardi G (2004) Cytosine methylation and CpG, TpG (CpA) and TpA frequencies. Gene 333:143–149
Johns EW (1969) The histones, their interactions with DNA and some aspects of gene control. Churchill, London
Johnson TB, Coghill RD (1925) Researches on pyrimidines. C111. The discovery of 5-methyl-cytosine in tuberculinic acid, the nucleic acid of the tubercle bacillus. J Am Chem Soc 47(11):2838–2844
Jones PA (2012) Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 13(7):484–492. https://doi.org/10.1038/nrg3230
Jones PA, Taylor SM (1980) Cellular differentiation, cytidine analogs and DNA methylation. Cell 20(1):85–93
Judson HF (1979) The eighth day of creation: makers of the revolution in biology, n. edn. Cold Spring Harbor Laboratory Press, New York
Kass SU et al (1997) DNA methylation directs a time-dependent repression of transcription initiation. Curr Biol 7(3):157–165
Keller EF, Harel D (2007) Beyond the gene. PLoS One 2(11):e1231
Kingsland SE (2007) Maintaining continuity through a scientific revolution: a rereading of E. B. Wilson and T. H. Morgan on sex determination and Mendelism. Isis 98(3):468–488
Kornberg RD (1974) Chromatin structure: a repeating unit of histones and DNA. Science 184(4139):868–871
Kornberg RD, Thomas JO (1974) Chromatin structure; oligomers of the histones. Science 184(4139):865–868
Kornberg A et al (1959) Enzymatic synthesis of deoxyribonucleic acid. Influence of bacteriophage T2 on the synthetic pathway in host cells. Proc Natl Acad Sci U S A 45(6):772–785
Kossel A (1884) Ueber einen peptonartigen Bestandtheil des Zellkerns. Zschr Physiol Chem 8:511
Leonhardt H et al (1992) A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei. Cell 71(5):865–873
Levene PA, Mandel J-A (1908) Über die darstellung und analyse einiger nucleinsäuren. XIII. Über ein verfahren zur gewinnung der purinbasen. Biochem Z 10:215–220
Lieberman-Aiden E et al (2009) Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326(5950):289–293
Lu Z et al (2017) Mechanistic insights in X-chromosome inactivation. Philos Trans R Soc Lond Ser B Biol Sci 372(1733):20160356
Luger K et al (1997) Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389(6648):251–260
Lyon MF (1961) Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature 190:372–373
Lyon MF (1962) Sex chromatin and gene action in the mammalian X-chromosome. Am J Hum Genet 14:135–148
Lyon MF (1963) Lyonisation of the X Chromosome. Lancet 2(7317):1120–1121
Mandel JL, Chambon P (1979) DNA methylation: organ specific variations in the methylation pattern within and around ovalbumin and other chicken genes. Nucleic Acids Res 7(8):2081–2103
McCarty M (2003) Discovering genes are made of DNA. Nature 421(6921):406
McClung CE (1902) The accessory chromosome—Sex determinant? Biol Bull 3
McGhee JD, Ginder GD (1979) Specific DNA methylation sites in the vicinity of the chicken beta-globin genes. Nature 280(5721):419–420
McGrath J, Solter D (1984) Completion of mouse embryogenesis requires both the maternal and paternal genomes. Cell 37(1):179–183
Mendel G (1866) Versuche über Plflanzenhybriden. Verhandlungen des naturforschenden Vereines in Brünn, Bd. IV für das Jahr. Abhandlungen, 3–47
Mercola M et al (1983) Transcriptional enhancer elements in the mouse immunoglobulin heavy chain locus. Science 221(4611):663–665
Metz CW (1938) Chromosome behavior, inheritance and sex determination in Sciara. Am Nat 72(743):485–520
Migeon BR (2017) Choosing the active X: the human version of X inactivation. Trends Genet 33(12):899–909
Migeon BR et al (1979) Stability of the “two active X” phenotype in triploid somatic cells. Cell 18(3):637–641
Migeon BR et al (2008) X inactivation in triploidy and trisomy: the search for autosomal transfactors that choose the active X. Eur J Hum Genet 16(2):153–162
Minajigi A et al (2015) Chromosomes. A comprehensive Xist interactome reveals cohesin repulsion and an RNA-directed chromosome conformation. Science 349(6245):aab2276
Mirsky AE (1971) The structure of chromatin. Proc Natl Acad Sci U S A 68(12):2945–2948
Mohandas T et al (1981) Reactivation of an inactive human X chromosome: evidence for X inactivation by DNA methylation. Science 211(4480):393–396
Moore T, Haig D (1991) Genomic imprinting in mammalian development: a parental tug-of-war. Trends Genet 7(2):45–49
Morgan TH (2018) The theory of the gene, 2nd edn. Creative Media Partners, LLC, Sacramento, CA
Moss L (2003) What genes can’t do. MIT Press, Cambridge, MA
Nan X et al (1998) Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 393(6683):386–389
Nanney DL (1958) Epigenetic control systems. Proc Natl Acad Sci U S A 44(7):712–717
Ng HH, Bird A (1999) DNA methylation and chromatin modification. Curr Opin Genet Dev 9(2):158–163
Ohno S, Hauschka TS (1960) Allocycly of the X-chromosome in tumors and normal tissues. Cancer Res 20:541–545
Olins DE, Olins AL (1973) Spheroid chromatin units (v bodies). J Cell Biol 59:A252
Olins DE, Olins AL (1974) Spheroid chromatin units (v bodies). Science 183(4122):330–332
Olins DE, Olins AL (2003) Chromatin history: our view from the bridge. Nat Rev Mol Cell Biol 4(10):809–814
Oudet P et al (1975) Electron microscopic and biochemical evidence that chromatin structure is a repeating unit. Cell 4(4):281–300
Pan Q et al (2008) Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 40(12):1413–1415
Park WW (1957) The occurrence of sex chromatin in early human and macaque embryos. J Anat 91(3):369–373
Pennacchio LA et al (2013) Enhancers: five essential questions. Nat Rev Genet 14(4):288–295
Pesole G (2008) What is a gene? An updated operational definition. Gene 417(1–2):1–4
Plenge RM et al (1997) A promoter mutation in the XIST gene in two unrelated families with skewed X-chromosome inactivation. Nat Genet 17(3):353–356
Portin P, Wilkins A (2017) The evolving definition of the term “Gene”. Genetics 205(4):1353–1364
Ptashne M (2007) On the use of the word ‘epigenetic’. Curr Biol 17(7):R233–R236
Razin A, Riggs AD (1980) DNA methylation and gene function. Science 210(4470):604–610
Reik W, Walter J (2001) Genomic imprinting: parental influence on the genome. Nat Rev Genet 2(1):21–32
Riggs AD (1975) X inactivation, differentiation, and DNA methylation. Cytogenet Cell Genet 14(1):9–25
Riggs AD (1988) X-inactivation, differentiation, and DNA methylation. Curr Content 31(1):15
Roark DE et al (1974) A two-subunit histone complex from calf thymus. Biochem Biophys Res Commun 59(2):542–547
Sakai T et al (1991) Allele-specific hypermethylation of the retinoblastoma tumor-suppressor gene. Am J Hum Genet 48(5):880–888
Saxonov S et al (2006) A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc Natl Acad Sci U S A 103(5):1412–1417
Scarano E (1971) The control of gene function in cell differentiation and in embryogenesis. Adv Cytopharmacol 1:13–24
Scherrer K, Jost J (2007) Gene and genon concept: coding versus regulation. A conceptual and information-theoretic analysis of genetic storage and expression in the light of modern molecular biology. Theory Biosci 126(2–3):65–113
Searle AG, Beechey CV (1978) Complementation studies with mouse translocations. Cytogenet Cell Genet 20(1–6):282–303
Shen CK, Maniatis T (1980) Tissue-specific DNA methylation in a cluster of rabbit beta-like globin genes. Proc Natl Acad Sci U S A 77(11):6634–6638
Stadler PF et al (2009) Defining genes: a computational framework. Theory Biosci 128(3):165–170
Stubbe H (1972) History of genetics: from prehistoric times to the rediscovery of Mendel’s laws. MIT Press, Cambridge, MA
Surani MA, Barton SC (1983) Development of gynogenetic eggs in the mouse: implications for parthenogenetic embryos. Science 222(4627):1034–1036
Szmulewicz MN et al (1998) Effects of Alu insertions on gene function. Electrophoresis 19(8–9):1260–1264
Szyf M et al (1985) Cell cycle-dependent regulation of eukaryotic DNA methylase level. J Biol Chem 260(15):8653–8656
Taylor SM, Jones PA (1979) Multiple new phenotypes induced in 10T1/2 and 3T3 cells treated with 5-azacytidine. Cell 17(4):771–779
van der Ploeg LH, Flavell RA (1980) DNA methylation in the human gamma delta beta-globin locus in erythroid and nonerythroid tissues. Cell 19(4):947–958
Vanyushin BF et al (1970) Rare bases in animal DNA. Nature 225(5236):948–949
Varmuza S, Mann M (1994) Genomic imprinting—defusing the ovarian time bomb. Trends Genet 10(4):118–123
Waddington CH (1939) An introduction to modern genetics. Macmillan, New York
Waddington CH (1940) Organisers and genes. Cambridge University Press, Cambridge
Waddington CH (1942) The epigenotype. Endeavour 1:18
Waddington CH (2012) The epigenotype. 1942. Int J Epidemiol 41(1):10–13
Walker CL et al (1991) The Barr body is a looped X chromosome formed by telomere association. Proc Natl Acad Sci U S A 88(14):6191–6195
Watson JD, Crick FH (1953a) Genetical implications of the structure of deoxyribonucleic acid. Nature 171(4361):964–967
Watson JD, Crick FH (1953b) Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature 171(4356):737–738
Weaver DD, Gartler SM (1975) Evidence for two active X chromosomes in a human XXY triploid. Humangenetik 28(1):39–42
Welshons WJ, Russell LB (1959) The Y-chromosome as the bearer of male determining factors in the mouse. Proc Natl Acad Sci U S A 45(4):560–566
Wheeler HL, Johnson TB (1904) 5-methylcytosine. Am Chem J 31:591
Wilkins MH et al (1953) Molecular structure of deoxypentose nucleic acids. Nature 171(4356):738–740
Wilson EB (1911) The sex chromosomes. Archiv fu¨r mikroskopische Anatomie Pt. 2(77):249–271
Woodcock C (1973) Ultrastructure of inactive chromatin. J Cell Biol 59:A368
Woodcock C et al (1976) Structural repeating units in chromatin. I. Evidence for their general occurrence. Exp Cell Res 97:101–110
Wyatt GR (1951) The purine and pyrimidine composition of deoxypentose nucleic acids. Biochem J 48(5):584–590
Zou X et al (2012) Recognition of methylated DNA through methyl-CpG binding domain proteins. Nucleic Acids Res 40(6):2747–2758
Zubay G, Doty P (1959) The isolation and properties of deoxyribonucleoprotein particles containing single nucleic acid molecules. J Mol Biol 1:1–20
Author information
Authors and Affiliations
Corresponding authors
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

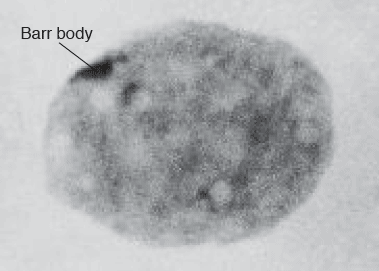{kind=link}
Cite this chapter
Hesson, L.B., Pritchard, A.L. (2019). Genetics and Epigenetics: A Historical Overview. In: Hesson, L., Pritchard, A. (eds) Clinical Epigenetics. Springer, Singapore. https://doi.org/10.1007/978-981-13-8958-0_1
Download citation
DOI: https://doi.org/10.1007/978-981-13-8958-0_1
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-8957-3
Online ISBN: 978-981-13-8958-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)