
Abstract
Heart failure (HF) remains a major cause of disability, suffering, and death worldwide. The prevalence of HF increases with age and at an alarming pace in the elderly population aged 65 years or more. Importantly, the increase in HF prevalence, first seen in developed countries and currently in developing countries as well, has taken place despite tremendous advances in HF therapy and efforts to encourage implementation of management guidelines. The magnitude of this HF pandemic is staggering, affecting nearly 26 million people across the world. There are several reasons for this continued increase in HF prevalence despite optimal therapy; of these, two that stand out include (i) the aging-induced cardiovascular (CV) remodeling that modifies disease expression and response to therapy and aging-related increase in reactive oxygen species (ROS) and oxidative stress (OXS) that augment adverse left ventricular remodeling after myocardial injury; ii) the lifelong exposure to CV disease (CVD) risk factors that increase ROS and OXS, as well as inflammation. Other pathways and mechanisms leading to HF that are yet to be addressed may also involve OXS and inflammation. This chapter focuses on the evidence for ROS-induced myocardial damage during HF progression and some potential pharmacological interventions and strategies for reducing the damage. In addition, some key issues facing translation of experimental successes with antioxidant therapy into successes in clinical practice on the real-world stage are addressed.
The authors have nothing to disclose.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Aging
- Healing
- Infarct size
- Hypertrophy
- Heart failure with preserved ejection fraction
- Heart failure with reduced ejection fraction
- Hypertension
- Inflammation
- Myocardial infarction
- Mitochondria
- Oxidative stress
- Prevention
- Remodeling
- Reperfusion injury
1 Introduction
In this second decade of the twenty-first century, heart failure (HF) remains a major cause of disability, suffering, and death worldwide [1,2,3,4,5,6,7,8,9,10,11,12,13]. The prevalence of HF increases with age and reaches alarming proportions in elderly people aged ≥65 years, a group that is growing steadily [14,15,16,17,18,19]. Over the last two decades alone, HF has grown into a serious pandemic, affecting nearly 26 million people in developed and developing countries across the world [3,4,5,6]. In Europe, both the prevalence and risk of HF in the adult and aging population are significant; it is reported that HF occurs in about 1–2% of adults and rises to more than 10% in those who are older than 70 years [1]; the lifetime risk of HF in people aged 55 years is reported at 33% for men and 28% for women [20]. In the United States, HF prevalence in people aged ≥20 years is reported to have increased from 5.7 million over 2009–2012 to 6.5 million over 2011–2014, with a projected increase by 46% over 2012–2030, which equates to over 8 million people aged ≥18 years with HF [6]; HF incidence in people aged >65 years approached 21 per 1000 or 2.1%, and HF risk was highest among African Americans [6]; the lifetime risk of HF was 20–45% for people aged 45–95 years and was higher for people with hypertension (HTN) and obesity irrespective of age [6]. Across Asia, estimates of HF prevalence ranged between 1.26 and 6.7% [6, 21]. Taken together, the global burden of HF in the aging population is clearly and undeniably staggering [3,4,5,6, 14,15,16, 22, 23]. Importantly, this increase in HF prevalence, first in developed countries and currently in developing countries as well, has taken place despite tremendous advances in HF therapy and efforts to encourage implementation of management guidelines [1, 2, 7,8,9,10,11,12,13].
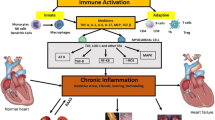
There are many causes for HF (Fig. 11.1) and these have been extensively reviewed [1, 8]. New pathophysiological mechanisms leading to HF continue to be elucidated, and potential targets are being identified for therapeutic intervention [24, 25]. The two leading causes of HF remain myocardial infarction (MI) and HTN [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19]. In his 2015 Lancet lecture, Braunwald boldly stated that it is “time to declare war on HF” and nicely underpinned several targets for urgent action [26]. However, as depicted in Fig. 11.2, one additional target that needs urgent action concerns the lifelong onslaught from reactive oxygen species (ROS), oxygen free radicals (OFRs), and oxidative stress (OXS) associated with exposure to cardiovascular disease (CVD) risk factors, such as hyperlipidemia, obesity, type 2 diabetes mellitus (DM2), HTN, and aging; although OXS has been recognized as an important mechanism of HF and a major contributor to both aging [16, 18] and HF progression [27, 28], definitive therapy for quenching increased levels of ROS/OFRs, and thereby limiting OXS and its potential contribution to excess morbidity and mortality in the aging population with HF, is still lacking [22]. Another target that needs urgent action is inflammation, which is also related to lifelong exposure to the same CVD risk factors and is exacerbated by acute triggers such as ischemia, ischemia-reperfusion (I/R), and MI, and appears to interact synergistically with OXS to intensify myocardial damage and HF progression [16, 18, 19, 28], as depicted in Fig. 11.3. In that context, inflammation can be viewed as the fuse that ignites the smouldering background fire of OXS into the raging flames of a wildfire that exacerbates HF progression.


The roles of ROS/OXS and inflammation in the cardiovascular disease continuum and the progression to heart failure. Adapted and modified from Jugdutt (2014) [18, 19]
∗Cardiovascular risk factors tied to ↑ ROS, OXS, and inflammation. Other abbreviations as in text: ECM extracellular matrix, HFpEF heart failure with preserved ejection fraction, HFrEF heart failure with reduced ejection fraction, I/R ischemia-reperfusion, LV left ventricular, OXS oxidative stress, PPCI primary percutaneous coronary intervention, ROS reactive oxygen species, STEMI ST-segment elevation myocardial infarction

Schematic of putative interactions between CVD risk factors and aging, comorbidities, increased ROS and OXS, increased inflammation, fibrosis, vascular, and LV dysfunction in the march to HFpEF
Abbreviations as in text: CAD coronary artery disease, CVD cardiovascular disease, ECM extracellular matrix, HDL high-density lipoprotein, HFpEF heart failure with preserved ejection fraction, HFrEF heart failure with reduced ejection fraction, I/R ischemia-reperfusion, LDL low-density lipoprotein, LV left ventricular, MI myocardial infarction, OXS oxidative stress, ROS reactive oxygen species
Extensive research over the last four decades has elucidated the underlying cellular, subcellular, and molecular mechanisms involved in the generation of ROS/OFRs and OXS as well as the central role of mitochondria [29, 30] and identified several key signaling pathways that could serve as potential targets for pharmacological intervention [31,32,33]. An in-depth review of all the mechanisms and pathways illustrating the role of OXS in HF would be too lengthy for this one chapter, and several of them are addressed in other chapters of this book. The chapter here focuses on some potential future pharmacological interventions aimed at reducing ROS-induced damage during HF progression in the clinical setting. In addition, some key issues facing the translation of experimental successes with antioxidant therapy into successes in clinical practice on the real-world stage are addressed, and the importance of proper validation in carefully designed randomized clinical trials (RCTs) is discussed.
2 Pathophysiology of Progressive Remodeling in Heart Failure
Six points about the pathophysiology of the progressive adverse remodeling leading to progression in HF severity need emphasis.
2.1 Multiple Causes of the HF Syndrome
It is important to appreciate that multiple etiologies (Fig. 11.1), via diverse pathophysiological mechanisms, converge to produce the final clinical syndrome of HF [1, 8]. In the final analysis, HF represents a failure of homeostasis on several fronts; simply put, the left heart fails to pump enough oxygenated blood forward and maintain optimal perfusion of the tissues in the systemic circuit, whereas the right heart fails to pump all venous blood returned from the tissues to the lungs for reoxygenation in the pulmonary circuit; the congested lungs in turn fail to optimally reoxygenate the venous blood and return it all to the left heart to maintain circulatory flow. The reduced cardiac output generated by the failing heart can no longer match the metabolic demands of normal activities of daily life. The net effect of these failures at the level of the heart, systemic and pulmonary circulatory circuits, and the backup of blood in the lungs and tissues manifest themselves in the patient’s typical complaints of generalized weakness and fatigue, shortness of breath, and swelling of the ankles and legs; on examination, the patients show characteristic signs such as prominent neck veins with elevated jugular venous pressure, congestion of the lungs with typical crackles on auscultation, evidence of edema in the extremities, hemodynamic evidence of reduced cardiac output and/or elevated intracardiac pressures at rest or with exercise stress, and bedside two-dimensional (2D) or three-dimensional (3D) echocardiographic evidence of left ventricular (LV) global and regional systolic dysfunction with reduced LV ejection fraction (LVEF), as well as evidence of LV diastolic dysfunction and remodeling of LV structure and shape [35,36,37,38,39,40,41,42,43,44,45,46]. Laboratory test panels often display metabolic abnormalities related to the severity and duration of stress on different organs and tissues, such as reduced mixed venous oxygen saturation and arteriovenous oxygen difference, metabolic and respiratory acidosis with elevated lactate levels, altered blood profile with anemia, electrolyte imbalance with changes in serum sodium (Na+) and potassium (K+) levels, changes in serum creatinine (kidney stress), serum albumin and liver enzymes (liver stress), and biomarkers such as N-terminal B-type natriuretic peptide (BNP) and NT-proBNP (cardiac stress) and C-reactive protein (CRP; for inflammation). Metabolic panels reveal evidence of neurohormonal activations such as the sympathetic nervous system, renin-angiotensin-aldosterone system (RAAS), and endothelin (ET) system. Evidence of hypothyroidism is often present in the blood test. Other tests, such as electrocardiogram (ECG), chest X-ray, multigated acquisition (MUGA) scan, nuclear stress test, magnetic resonance imaging (MRI) or cardiac magnetic resonance (CMR), contrast echo with 2D and 3D imaging, pharmacologic stress test, and cardiac catheterization with angiography, provide clues regarding the precise etiology. No routine blood tests are currently done to screen for OXS.
2.2 Two Main Subsets of HF with Divergent LV Remodeling
Notwithstanding the multiplicity of etiologies leading to HF (Fig. 11.1), the two commonest aforementioned causes are MI and HTN, and they account for the majority of cases of HF seen in clinical practice and show nearly equal distribution (about 50% with MI and about 50% with HTN) [1, 2, 7,8,9,10,11,12,13]. The type and degree of adverse cardiac remodeling in HF depend on the underlying cause; dilative and eccentric remodeling with eccentric LV hypertrophy develops after ST-elevation MI (STEMI) and volume overload conditions; and concentric remodeling with concentric LV hypertrophy develops in HTN and other pressure overload conditions (Fig. 11.2). The progressive, adaptive, and maladaptive remodeling of cardiac structure, geometric shape, and function that occurs over time in survivors of the different types of CVD insults takes different paths, as illustrated by MI [34,35,36,37,38,39,40,41,42,43,44] and HTN [45, 46], as well as various cardiomyopathies, including those associated with DM2 and obesity.
2.3 Different Subsets of HF Based on LVEF
Stratification of HF on the basis of LVEF has unmasked three categories with distinct phenotypes that are now recognized in the latest HF management guidelines [1, 2]: (i) HF with reduced LVEF <40% (HFrEF); (ii) HF with preserved LVEF ≥50% (HFpEF); and (iii) most recently, an intermediate group with midrange LVEF 40–49% (HFmrEF). Besides the difference in LVEFs, the criteria for diagnosis of HFpEF and HFmrEF both include the presence of elevated BNP levels and either LV diastolic dysfunction or abnormal structure reflected in LV hypertrophy and/or left atrial enlargement [1]. In survivors of the acute phase of the insults, MRI or CMR can be used, where available, not only to quantify ventricular volumes, LVEF, and LV mass but also to assess fibrosis and scar size, and provide clues as to the precise etiology in various cardiomyopathies [1, 2]. In the context of ROS and OXS, the most data currently available is for these two main categories of HF, namely, HFrEF and HFpEF.
2.4 Divergent Types of Remodeling in the Two Subsets of HF Based on LVEF
Over the last four decades, the underlying structural, biochemical, cellular, subcellular, molecular, and metabolic derangements in HF following MI and HTN have been extensively researched, and key pathways and molecules have been identified and targeted by therapeutic interventions that have undisputedly reduced the number of patients dying and suffering from this debilitating chronic disease [1, 2, 25,26,27,28,29,30,31,32,33,34,35]. It is well appreciated that the nature of the insults and rates of progression of remodeling after MI and HTN are very different (Figs. 11.2 and 11.3). Typically, remodeling after anterior transmural MI or STEMI has two components; an early, dramatic, and rapidly developing regional expansion of the infarct zone with thinning and dilation (infarct expansion), followed by more gradual dilative global LV remodeling of both the IZ and NIZ during the healing and repair phases. Both these two components of remodeling are associated with HFrEF and poor outcome [34, 39,40,41,42,43, 52,53,54,55,56,57,58], and cumulative evidence suggests that both OXS and inflammation play distinct pathophysiologic roles in the two components of remodeling during the development and progression of HFrEF [27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44, 48,49,50,51,52,53,54,55,56,57,58]. In contrast, remodeling in HTN progresses at a much slower pace, in parallel with progression of the hypertensive disease and increasing blood pressure; this results in progressive concentric LV remodeling with development of HFpEF and poor outcome [45, 46]. Cumulative evidence over the last two decades indicates that OXS and inflammation also play important pathophysiologic roles in the development and progression of HFpEF, often with significant contribution from various comorbidities [27,28,29,30,31,32,33, 59,60,61,62,63,64,65,66,67,68,69]. These current concepts are summarized in Figs. 11.3 and 11.4. Furthermore, research has shed light on the various mechanisms leading to OXS and inflammation and their roles in the pathophysiology of CVD and the progression to HF via the chains of ischemia-MI-I/R-/HFrEF and HTN-HFpEF, as illustrated in Figs. 11.2, 11.3, and 11.4. Research has also suggested novel potential therapeutic targets.

Schematic showing steps in myocardial remodeling in HFpEF versus HFrEF. Adapted from Paulus and Tschӧpe (2013) [46]
Abbreviations: as in text
2.5 Evidence on the Roles of OXS and Inflammation in HFrEF and HFpEF
Evidence for the roles of OXS and/or inflammation in the progression of HFrEF and HFpEF were addressed in several studies, and some key reports are summarized below.
2.5.1 Multiple Biomarkers
In a clinical study of the different biological pathways that characterize HFrEF and HFpEF, Tromp et al. [59] analyzed 92 biomarkers in a cohort of 804 elderly HF patients (47% HFrEF, 27% HFpEF; mean age 74 years); they found that key markers of HFrEF were BNP, growth differentiation factor-15 (GDF-15), IL-1 receptor-like 1, and activating transcription factor 2, whereas key markers in HFpEF were integrin subunit beta-2 and catenin beta-1. They concluded that biomarker profiles in HFrEF are related to regulation of sequence-specific DNA-binding transcription, smooth muscle cell proliferation, and nitric oxide (NO) biosynthesis and metabolism, whereas those for HFpEF were related to cell adhesion, leucocyte migration, cytokine response and inflammation, neutrophil degranulation, and ECM organization, and the profile for HFmrEF was intermediate between those of HFrEF and HFpEF [59]. However, OXS was not addressed.
2.5.2 Multiple Comorbidities
In a small experimental study of multiple comorbidities in swine with chronic streptozotocin-induced diabetes, high-fat diet, and HTN induced by renal artery embolization over 6 months, Sorop et al. [60] documented that increased blood glucose and triglyceride, kidney dysfunction, and HTN were associated with evidence of systemic inflammation (increased IL-6 and tumor necrosis factor-α [TNF-α]), myocardial OXS (with increased superoxide or O2•−, NADPH oxidase or NOX activity, and endothelial NO synthase or eNOS uncoupling), coronary microvascular dysfunction (with decreased NO and impaired endothelial-dependent vasodilation), as well as increased myocardial collagen, decreased capillary/fiber ratio, with hemodynamic evidence of increased passive myocardial stiffness, LV diastolic dysfunction, and HFpEF [60]. However, OXS was not directly addressed in that study.
2.5.3 Microvascular Dysfunction
In a small clinicopathologic study of LV biopsies in 3 groups of patients (HFpEF, n = 36; aortic stenosis, n = 67; and HFrEF, n = 43), van Heerebeek et al. [65] found that protein kinase G (PKG) activity was lower in patients with HFpEF than in patients with aortic stenosis or HFrEF; importantly, the lower PKG level was associated with lower cyclic guanosine monophosphate (cGMP) concentration and higher nitrosative-OXS as well as a higher cardiomyocyte resting tension which was normalized by in vitro exogenous PKG administration [65]. They also noted that higher nitrosative-OXS, reflected in higher myocardial nitrotyrosine content in HFpEF than in HFrEF (and aortic stenosis) and known to impair NO-cGMP-PKG signaling, might result from the higher prevalence of associated comorbidities that increase OXS and inflammation (such as HTN, obesity, and DM2) in the HFpEF group. The patients in that study were older adults with a mean age of 60–65 years [65]. The overall findings suggested that microvascular dysfunction and PKG might be useful targets in HFpEF, but these need validation.
2.5.4 Coronary Flow Reserve and Microvascular Dysfunction
In a prospective observational study of coronary flow reserve in 202 patients with HFpEF and without unvascularized large vessel CAD, Shah et al. [62] documented a high prevalence of coronary microvascular dysfunction reflected in endothelial dysfunction (low reactive hyperemic index), higher albumin/creatinine ratio, NT-proBNP, and right ventricular dysfunction [62]. The patients in that study were elderly (mean age 72–75 years) with several comorbidities (such as HTN, obesity, DM2, hyperlipidemia, chronic kidney disease [CKD], cigarette smoking) known to increase OXS and inflammation.
In another study of coronary flow reserve using phase contrast cine-MRI of the coronary sinus to assess flow as an index of LV microvascular function in elderly patients (25 HFpEF, mean age 73 years; 13 hypertensive LV hypertrophy, age 67; 18 controls, age 65), Kato et al. documented that coronary flow reserve was lower in 76% of the HFpEF patients compared to that in patients with hypertensive LV hypertrophy and the controls; in addition, they found that coronary flow reserve correlated with serum BNP levels [64]. The findings suggested that impairment of coronary flow reserve might be related to the severity of HFpEF [64]. The patients in that study had several comorbidities (such as HTN, DM2, hyperlipidemia, cigarette smoking) that are known to increase OXS and inflammation.
2.5.5 OXS Markers
Despite these compelling reports of the role of OXS and inflammation in HFpEF, controversy exists. In one small study of 50 patients with and without HFpEF, Negi et al. [69] measured various OXS markers (such as derivatives of reactive oxidative metabolites, F2-isoprostanes, ratios of oxidized to reduced glutathione and cysteine) and angiotensin-converting-enzyme (ACE) levels and activity; while they found an association between HFpEF and male gender and higher body mass index (BMI), they did not find significant evidence of systemic renin-angiotensin system (RAS) activation or OXS, leading them to conclude that their finding may explain the failure of RAS inhibitors to alter outcomes in HFpEF [69].
2.5.6 Unexplained Mode of Death in HFpEF
In a systematic review of 1608 papers on HFpEF from 1985 to 2015, the authors found that about 25% of deaths were sudden, calling for a longitudinal multicenter global registry [70]. Whether there is an arrhythmic and/or ischemic contribution to the mode of death in HFpEF is not currently established; however, this appears very likely in view of the increased OXS [60,61,62,63, 65, 67,68,69], inflammation [59,60,61, 63], ECM and fibrosis [59,60,61,62,63,64,65,66,67,68,69], microvascular disease [62, 63], impaired coronary flow reserve [64, 66], and impaired bioenergetics [67], as depicted in Fig. 11.3.
2.5.7 Merits of RCT Versus Observational Data in HF
From another review of electronic databases on observational nonrandomized studies versus those in RCTs until the end of 2017 for the association between drug therapy and mortality in HF patients, the authors concluded that “treatment effects cannot be estimated from observational data” [71]. That finding supports the need for RCTs to guide therapy.
In summary, cumulative evidence supports the role of CVD risk factors in promoting OXS and inflammation (Figs. 11.2, 11.3, and 11.4), thereby leading to coronary microvascular dysfunction, subendocardial ischemia, regional and diffuse fibrosis, cardiac steatosis, vascular stiffness, and adverse LV remodeling.
2.6 Impact of Multiple Etiologies and Comorbidities on HF Therapy
The distinction between the two HF categories based on severity of systolic dysfunction is logical from a treatment perspective because the specific category might dictate the specific therapeutic approach to be recommended in compliance with the management guidelines [1, 2, 10,11,12,13]. Since the two main categories of HF (HFrEF and HFpEF) based on LVEF are characterized by two divergent types of LV remodeling (Fig. 11.2), it is not surprising that therapies recommended for the management of the two distinct categories of HF in the updated published guidelines should be quite different [1, 2, 10,11,12,13], as reflected in the summaries shown in Tables 11.1, 11.2, and 11.3. However, it should be noted that, whereas therapies for HFrEF are fairly well-defined (Tables 11.1 and 11.2), those for HFpEF still remain to be defined (Table 11.3); this is partly due to the heterogeneous causations and the presence of multiple CV comorbidities in HFpEF [66], such as HTN, obesity, metabolic syndrome, DM2, hyperlipidemias, CAD, atrial fibrillation, ventricular arrhythmias, renal dysfunction, and pulmonary hypertension (Fig. 11.3), and these require different specific therapies (Table 11.3). They also have several non-CV comorbidities, including osteoarthritis, hypothyroidism, chronic obstructive lung disease, sleep apnea, CKD, iron-deficiency anemia, anxiety, and depression, that require separate therapies. In addition, RCTs have not distinguished between HFpEF and HFmrEF because the phenotypes are still being characterized [72]; as a result, the recommendations have tended to lump the two categories into a single HFpEF category at this time [1, 2]. Furthermore and as mentioned before, at least 50% of HF patients have HFpEF, and they tend to be elderly, and aging is an important CV risk factor as shown in Figs. 11.3, 11.4, and 11.5.

Some aging-related cardiovascular and noncardiovascular physiological and pathophysiological changes and associated comorbidities
Adapted from Jugdutt [14,15,16,17,18,19, 34,35,36, 49]
Abbreviations: ↑ increased, enhanced, ↓ decreased, AV atrioventricular, CV cardiovascular, GI gastrointestinal, HF heart failure, LV left ventricular
Other abbreviations as in text
2.7 Management Guidelines for HF Therapy and HF Pathophysiology
It should also be noted that management guidelines that are updated by the major CV societies worldwide represent the consensus opinion of experts based on available evidence mainly from RCTs, are meant to guide therapy, and do not address all pertinent issues [1, 2]. Guideline-driven management of HF centers around improving clinical status, functional capacity, and quality of life and reducing hospitalization, morbidity, and mortality [1, 2]. It is recognized that several drugs that show efficacy in the short term may prove to be harmful in the long term [1, 2].
In HFrEF, pharmacotherapy consists of diuretics and neurohumoral antagonists such as ACE inhibitors (ACEIs), mineralocorticoid receptor antagonists (MRAs), and beta-blockers in the absence of contraindications or intolerance [Table 11.2]. Based on the results of a single RCT showing that the new compound LCZ696, which combines moieties of the angiotensin II (Ang II) type 1 receptor blocker (ARB) valsartan and the neprilysin (NEP) inhibitor sacubitril in a single molecule that is an angiotensin receptor neprilysin inhibitor (ARNI) and acts on both the RAAS and the neutral endopeptidase system, was superior to the ACEI enalapril in reducing HF mortality and hospitalization [73], it was recommended to replace ACEIs in HFrEF patients who continue to be symptomatic despite optimal therapy [1, 2, 74,75,76]. Several reports supported the replacement of ACEI with sacubitril/valsartan in patients with HFrEF [75, 76]. Moreover, since ARBs do not consistently reduce mortality, they are only used in ACEI-intolerant patients [1, 2]. The If-channel blocker ivabradine is used to control elevated heart rate above 70 beats per minute [1, 2].
Some safety issues with the ARNI sacubitril/valsartan include hypotension and angioedema [1, 2]. Recent reports in 2018 of contamination with human carcinogens N-nitrosodiethylamine (NDEA) in irbesartan and both N-nitrosodimethylamine (NDMA) and NDEA in valsartan supplied by certain pharmaceutical firms have raised additional concern that needs to be addressed. Of note, the recently updated recall list of the Food and Drug Administration (FDA) in the United States includes the ARB losartan for the same reason.
In the absence of firm and specific recommendations for HFpEF in the 2016 management guidelines [1, 2], several studies have been evaluating the efficacy of LCZ696 in HFpEF; while the final RCT results are still pending, there is considerable enthusiasm and hope of a positive outcome [76,77,78,79,80,81]. While awaiting RCT results in HFpEF, physicians in current clinical practice have tended to use therapies recommended for HFrEF to treat the HF component, including diuretics, beta-blockers, MRAs, ACEIs, and ARBs [1, 2], in addition to other specific therapies for common comorbidities such as HTN and DM2, an approach that has resulted in partial benefit [1, 2]. As summarized in Table 11.3, newer therapies, such as the sodium glucose cotransporter-2 (SGLT2) inhibitor empagliflozin and the glucagon-like peptide/receptor agonist (GLP-1RA) liraglutide, which were recently shown to improve control of DM2 as well as CV events and CV risks [80, 82, 83], are being implemented. However, there have been several negative studies; a large RCT of spironolactone in HFpEF failed to show benefit [84]. Although in patients with HFpEF an RCT with the phosphodiesterase-5 inhibitor sildenafil on exercise capacity [85] and another randomized double-blind crossover study with isosorbide mononitrate on daily activity [86] were negative, a subsequent meta-analysis of RCTs showed the benefit of exercise in HFpEF [87].
2.8 Studies on the Role of Inflammation in the Pathophysiology of HF Progression
The critical roles of acute and chronic inflammation during acute MI and the subsequent healing processes, and the progressive remodeling that spans these processes during remote MI and well beyond over years into the chronic HF stage, have been well documented, and various anti-inflammatory strategies have been proposed over the last five decades [34,35,36, 39, 41,42,43,44, 48,49,50,51,52,53,54,55, 58, 88,89,90,91,92,93,94,95,96,97,98,99,100,101,102]. These studies and others have underscored several pertinent points that need consideration when developing therapy for HFrEF after MI. The main points include the following.
2.8.1 Timing of Events Post-MI
The events that follow an acute MI all take place in tandem fashion (Table 11.4); early damage of muscle, matrix, and microvasculature triggers the healing process, which through a timed sequence of acute and chronic inflammation and associated biochemical, molecular, cellular, and subcellular reactions lead to formation of a fibrotic scar in the IZ, followed by fibrosis and hypertrophy in the NIZ and significant remodeling of structure, shape, and function [34,35,36, 41, 42, 49, 51,52,53,54,55, 101, 103]. Considering a single factor, such as the time interval needed for collagen deposition to reach a plateau during healing and repair after MI, this varies from weeks to months depending on the species, infarct size, reperfusion, and other factors; it usually takes a few days in mice, about 1 week in rats, 6 weeks in dogs, and 3 to 6 months in humans [51]. As reviewed before [51], this timing of the various events during the progression of fibrosis after MI is clearly important when deciding on timing and duration of therapies for HFrEF post-MI [34, 42, 99].
2.8.2 Multiple Cellular and Molecular Processes
An additional consideration with respect to therapy for HFrEF post-MI is the diversity and multiplicity of the cellular and molecular processes, cell types, and changes involved in the four main stages after STEMI or reperfused STEMI [50], as summarized in Table 11.4. Briefly, the early infarction phase over the first few hours involves damage to cardiomyocytes by apoptosis and necrosis, extracellular matrix (ECM) by matrix metalloproteinases (MMPs), and vascular cells by apoptosis and necrosis; the early healing phase with inflammation over days involves neutrophils, monocytes, macrophages, and mast cells; late healing with proliferation over weeks involves fibroblasts, myofibroblasts, collagen/ECM deposition, ECM remodeling, angiogenesis and vascular remodeling, and maturation with further ECM remodeling by cross-link formation, and scar formation, structural remodeling of the IZ and NIZ, and LV systolic and diastolic dysfunction [50]. The key modulators and mediators involved during the healing, repair, and fibrosis phases after MI and in the march to HFrEF, several of which can be targeted, have been reviewed before [51, 103] and are summarized in Table 11.5. Over all the phases leading to HFrEF, the three neurohumoral systems (RAAS, ET, and adrenergic) and OXS exert important modulating effects.
2.8.3 Persistent Inflammation Post-MI
The trickle of evidence over the last three decades indicates that low-grade inflammation and ECM remodeling both continue beyond the MI and collagen plateau phases during healing into the later phase of progression to the chronic HF phase, as reviewed elsewhere [101,102,103,104,105,106,107,108]; this is also an important consideration for timing and duration of therapy. During healing post-MI, the release of key factors that modulate healing, such as chemokines, cytokines, matrikines, growth factors including transforming growth factor-β (TGF-β), MMPs, and other matrix proteins, is quite precisely timed to orchestrate the sequence of acute and chronic inflammation with formation of granulation tissue, tissue repair with proliferation of fibroblasts, deposition of ECM, formation of myofibroblasts and scars, structural and functional remodeling of IZ and NIZ myocardium with cardiomyocyte hypertrophy and very little regeneration, and some angiogenesis [34, 51, 101,102,103, 108].
2.8.4 Timing of Remodeling Post-MI
The remodeling in post-MI survivors also occurs in a timed sequence, is progressive, and spans the infarction (first 24–48 h in humans) and healing (6 weeks to 3 months in humans) phases and far beyond (months to years) [34, 35, 40,41,42,43,44, 49, 51]; it is also modulated by multiple factors that orchestrate post-MI remodeling of myocardium, vascular tissue, ECM, and over time other cardiac chambers, tissues, cells, and molecules, resulting in a vicious cycle leading to end-stage HF. The additional role of exposure to CV risk factors that exacerbate OXS and inflammation in aging post-MI survivors during the march to HFrEF, just as in aging HTN victims in the march toward HFpEF as depicted in Figs. 11.2 and 11.3, in the progression of adverse remodeling needs to be recognized and addressed. Furthermore, the added contribution of non-CV risk factors in exacerbating OXS and inflammation and thereby promoting progressive adverse remodeling in the marches to both HFrEF and HFpEF need to be addressed.
2.8.5 Addressing Residual Inflammation in the Progression of Atherosclerosis Post-MI
It is well-known that (i) inflammation plays major roles in the initiation as well as the progression of atherosclerosis, as indicated by increased levels of the marker of inflammation, namely, high-sensitivity CRP (hs-CRP); and (ii) statins, through a pleotropic effect, reduce the residual inflammatory risk after MI [109,110,111,112,113]. However, the importance of “residual inflammation” in the progression of atherosclerosis in post-MI patients was only underscored in two recent RCTs [114, 115]. In the first report of the Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS) group, anti-inflammatory therapy with canakinumab to target IL-1β in 10,061 stable post-MI patients with evidence of residual inflammation, assessed by elevated hs-CRP levels, resulted in reducing the hs-CRP level and recurrence of CV events, independent of lipid-lowering [114]. In the second report of the CANTOS group, canakinumab therapy in 4833 patients with atherosclerosis showed that those patients in whom the IL-6 levels were lowered benefited from a reduction in major adverse CV events (MACE), hospitalization for unstable angina, and lower CV mortality as well as lower all-cause mortality independent of lipid-lowering. These findings suggested that modulation of the IL-6 proinflammatory pathway is beneficial for limiting vascular and CV events [115]. In the previous cholesterol and recurrent events (CARE) trial in MI survivors, levels of CRP increased over 5 years, indicating an increase in residual inflammation. Importantly, in that study, while therapy with the lipid-lowering agent pravastatin prevented the increase in CRP levels after MI, this benefit was not related to the extent of lipid-lowering, suggesting that a pleiotropic effect of the drug was involved [113]. In the more recent report of a cohort of 385 patients with hospitalization for HF from the original 10,061 patients with prior MI and elevated hs-CRP in CANTOS, the IL-1β inhibitor canakinumab showed a dose-dependent reduction in HF hospitalization and the composite endpoint of hospitalization for HF or HF-related mortality [116].
2.8.6 Addressing Inflammation in the ACS: Shift in Focus to Prevention of MI
In the last two decades, the thinking relating to the role of inflammation in acute coronary syndromes (ACS) and MI has shifted from the focus on treatment of the atherosclerotic plaque and epicardial coronary artery thrombosis, and the use of statins for lipid reduction and CV prevention, to the use of anti-inflammatory strategies for atherosclerosis and other strategies for ACS without thrombosis [117]. The CANTOS group continues to study IL-1 blockade with the IL-1β inhibitor canakinumab for interrupting the IL-1/IL-6 cascade and thereby reducing inflammatory risk in different settings [114, 115, 118, 119]. A small phase II RCT of 182 patients with non-ST elevation ACS showed that the recombinant IL-1 receptor antagonist (IL-1ra) anakinra reduced the elevated CRP, suggesting that IL-1 may be driving CRP elevation in ACS [120]. Interestingly, in that study, the CRP level rose again 16 days after treatment was stopped [120], suggesting that the persistent inflammation requires longer-term therapy. Furthermore, although the patients in CANTOS and the other studies with interleukin inhibitors were mostly older with mean ages of 60–64 years [114,115,116, 118, 120], and several factors in aging hearts are known to lead to increased ROS, O2•−, and myocardial Ang II which in turn trigger increased proinflammatory cytokines, MMPs, and OXS markers and thereby modulate post-MI healing and repair and progression of HF, OXS markers were not measured in those studies.
2.8.7 Role of IL-8 in Enhanced Damage After STEMI
While the canine aging study of Jugdutt et al. drew attention to the role of IL-6 as one important mediator of adverse LV remodeling after STEMI [58], other chemokines and proinflammatory cytokines may also be involved [121]. In a recent study of 258 patients with STEMI undergoing percutaneous coronary intervention (PCI) and who were followed for a median of 70 months, high levels of IL-8 in serially drawn blood samples were associated with large infarct size, impaired LV functional recovery, and adverse clinical outcome [122]. Interestingly, in that study, levels of IL-8 remained higher in nonsurvivors compared to survivors at 4 months [122]. While that study supports targeting of IL-8 for suppressing post-MI inflammation [122], the authors did not measure markers of OXS. Of note, the patients in that study were older adults, with an average age of 60 years (range 53–66 years) [122].
2.8.8 Surge of ROS and OXS After Reperfused STEMI
It is known that after STEMI, the extent of myocardial injury is massive and with reperfusion after a time delay; both the intensity of the inflammatory response and the extent of damage caused by the initial MI and the subsequent delayed reperfusion can be significant [48, 58]. Myocardial damage after STEMI is further exacerbated by the burst of OFR release and OXS with reperfusion as shown by Bolli et al. [123]. In that study, Bolli et al. showed that the levels of ROS, measured by electron paramagnetic resonance (EPR) and a spin trap, also called electron spin resonance (ESR), spectroscopy in the venous effluent from the stunned zone of myocardium in the dog model and sampled via a catheter positioned in the anterior interventricular vein, increased significantly to a peak over the first 20 minutes and persisted for several hours after reperfusion [116].
2.8.9 Role of Dysregulation of Immune Pathways in Adverse Post-MI Remodeling and HF
In a review on the topic, Prabhu and Frangogiannis [124] summarized the innate immune mechanisms involved in the four main steps of the cascade between MI and scar formation: (i) danger signals from necrotic cells in MI lead to activation of innate immune pathways that trigger inflammation; (ii) increased expression of proinflammatory cytokines (such as IL-1 and TNF-α) and chemokines (such as monocyte chemoattractant protein-1/CCL2) in response to stimulation of Toll-like receptor (TLR) signaling and complement promotes adhesive interactions between leukocytes and endothelial cells that lead to extravasation of neutrophils and monocytes; (iii) activation of repair mechanisms with suppression of inflammatory response cells leads to fibroblast proliferation and differentiation into myofibroblasts (driven by the RAAS and TGF-β), increase in ECM proteins, and scar formation; and (iv) scar maturation follows, with cross-linking of the collagen matrix, and removal of granulation tissue by apoptosis. The authors suggested that the combination of dysregulation of the immune pathways, impaired suppression or resolution of post-MI inflammation, failure of spatial containment of the inflammatory response, and excessive fibrosis contributes to adverse post-MI remodeling and HF [124]; however, they did not mention the possible contributions of persistent OXS, residual inflammation, the interaction between OXS, and inflammation in the progression from MI to HFrEF. While there is a vast body of compelling research evidence that supports therapeutic modulation of the inflammatory and repair responses after MI [124], Granger and Kochar pointed out that targeting inflammation in acute MI with a specific inflammatory agent might be “an elusive goal” [125]. In fact, the management guidelines up to 2016 recommend that anti-inflammatory agents such as steroids and nonsteroidal anti-inflammatory agents should be avoided after STEMI [1, 2, 109, 110].
2.8.10 Role of Different Monocyte Subsets in Post-MI Healing and HF
In another provocative review of innate immune mechanisms during healing after MI, Nahrendorf et al. [102] underscored the importance of two populations of monocytes involved in post-MI healing and found in both mice and humans [102, 126]; briefly, they noted that there is a biphasic response post-MI in the mouse, with Ly-6Chigh monocytes (resembling CD16− monocytes in humans) that are dominant in the early inflammatory phase and Ly-6Clow monocytes (resembling CD16+ monocytes in humans) that are dominant in the subsequent reparative phase [102]. They also noted that monocyte numbers are increased in atherosclerosis and increased recruitment of Ly-6Chigh monocytes after plaque rupture impairs healing of MI and promotes HF via a cascade of increased chemokines (such as TNF-α), increased protease activity (such as MMPs, cathepsins), resolution of inflammation (with decreased TGF-β), and decreased collagen synthesis, based on previous findings in apoE−/− mice [102, 127]; importantly, they postulated a bell-shaped parabolic relation between monocyte numbers and healing after MI, in which both too little or too many of the monocytes can impair healing [102]. The authors proposed “shifting the monocyte response to a hypothetical vertex that denotes ‘optimal’ healing” as a goal of therapy for preventing HF [102]. They also suggested tailoring therapy to modulate the recruitment of monocyte subsets [102].
In a more recent study, Ruparelia et al. found that the patterns of gene expression associated with monocytes in inflammation and proliferation are switched on before they infiltrate the injured myocardium, suggesting that early therapy might be beneficial [126]. Of note, in that study, the average ages of the control and STEMI groups of human subjects were 63 and 60 years, respectively [126]. It should also be noted that during the early inflammatory response, the recruited neutrophils, macrophages, and monocytes release OFRs and contribute to OXS.
Together, these studies underscore the need to address the possible contributions of continued inflammation and OXS as well as ECM remodeling during healing after MI and the progression to HFrEF in post-MI survivors.
2.9 Role of Aging on the Pathophysiology of HF Progression
The importance of aging in the progression of adverse ventricular remodeling and HF (Fig. 11.4) is slowly becoming appreciated since the publication of several critical reviews and books on the topic [1,2,3,4, 14,15,16,17,18,19, 128]. The main points are summarized below.
2.9.1 Aging-Related Changes and HFpEF
As reviewed before [14, 15, 18, 19, 46,47,48,49,50,51, 103, 128], the progressive physiological, biological, and structural changes that characterize CV aging lead to increased ECM deposition and fibrosis (driven by increased Ang II, aldosterone, and TGFβ) in the heart, thereby increasing ventricular-arterial stiffening and LV diastolic dysfunction resulting in HFpEF (Fig. 11.5). In addition, increased ECM deposition and fibrosis plus decreased elastin (associated with increased MMP-9, MMP-12, cysteine proteinases cathepsins S, K, L, and serine proteinase neutrophil elastase from inflammatory cells) lead to increased aortic stiffness which in turn leads to the chain of increased aortic pulse wave velocity, systolic blood pressure, and afterload, which results in systolic HTN, LVH, and fibrosis (driven by increased Ang II, aldosterone, and TGFβ). As pulse pressure widens, a decrease in diastolic blood pressure below 70 mmHg can lead to the J-curve effect and thereby decrease diastolic myocardial perfusion, which in turn can trigger subendocardial ischemia and increase CV risk [128]. In that construct, the physiological changes during aging predispose the subject to HFpEF (Fig. 11.4); the added insults from HTN and the associated combined effects of LV pressure overload, LV hypertrophy, excess LV and aortic ECM and fibrosis, and decrease in aortic elastin collaborate to drive the concentric LV remodeling in the initial stage of HFpEF. Exposure to other CVD risk factors cooperates in exacerbating OXS, inflammation, and microvascular dysfunction in the survivors, thereby contributing to the progressive adverse remodeling and the march toward increasing severity of HFpEF progression (Figs. 11.2, 11.3, 11.4, and 11.5). Later, as a result of acute ischemia or other aggravating factors, a mixture of the two types of LV remodeling may develop and lead to HFrEF superimposed on HFpEF, thereby accelerating the march toward end-stage disease (Figs. 11.2, 11.3, and 11.4).
2.9.2 Aging-Related Changes and HFrEF
In the aging patient who develops acute STEMI, the background physiological changes of CV aging that predispose the patient to HFpEF alter and augment the responses to acute injury; the combination of enhanced damage to the myocardium, ECM, microcirculation, and endothelium with STEMI drives the development of dilative LV remodeling and the shift toward HFrEF (Fig. 11.4). The continued exposure to risk factors that exacerbate OXS and inflammation in the survivor contributes to progressive adverse remodeling (Figs. 11.2, 11.3, and 11.4). Furthermore, reperfusion therapy with restoration of coronary blood flow in the infarct-related artery (IRA) after STEMI leads to an acute surge in OXS, which, in combination with the intense acute inflammation, exacerbates the damage [19, 31, 34, 48,49,50,51,52, 58, 101, 102].
2.9.3 Aging-Related Changes in Healing and Post-STEMI Remodeling
As mentioned before, progressive adverse remodeling that takes place during the subsequent healing, repair, and beyond the healing/repair phases in survivors of STEMI spurs on the progression to HFrEF (Fig. 11.2). Expanding on that general theme, it is now established that with aging (Fig. 11.5), adverse post-MI remodeling is more severe [14,15,16,17,18,19]. This is due to impaired healing and repair of the damaged tissue resulting from a dysregulation of the pathways that are involved [15, 18]. The augmented adverse remodeling affects the entire left ventricle and, in time, leads to remodeling of the left atrium and right ventricle as well (Fig. 11.3). Of note, cardiomyocyte hypertrophy and fibrosis after STEMI develop in both the spared myocardium within the IZ and the myocardium in the NIZ [51]. In the aging mouse model of reperfused MI, Bujak et al. showed that enhanced adverse remodeling was associated with suppression of the inflammatory response, delayed granulation tissue formation, and reduced collagen deposition [48]. In the aging canine model of reperfused STEMI, with reperfusion after 90 minutes of ischemia after coronary occlusion, Jugdutt et al. [58] showed age-dependent early increases in markers of damage (increased ischemic injury, infarct size, cardiomyocyte apoptosis, blood flow impairment and no-reflow), structural remodeling (increased LV dilation and dysfunction) and matrix remodeling (increased expression of secretory leucocyte protease inhibitor [SLPI], secreted protein acidic and rich in cysteine [SPARC], osteopontin [OPN], a disintegrin and metalloproteinase [ADAM]-10 and ADAM-17, and MMP-9 and MMP-2), and inflammation (with increased inducible NO synthase [iNOS], proinflammatory cytokines IL-6 and TNF-α, and TGF-β1, and decreased anti-inflammatory cytokine IL-10). Importantly in that study, early therapy with the ARB candesartan, initiated at the time of reperfusion, attenuated these adverse age-dependent changes [58].
As reviewed previously [50], the main changes in molecular and cellular responses during healing and repair after STEMI/reperfused STEMI in older subjects include (i) increased RAAS, Ang II, and ROS activity; (ii) impaired or defective healing, with amplification of damage in the infarction phase (reflected in increased apoptosis, necrosis, and ECM degradation), dysregulation of inflammation in the early inflammation phase (with decreased antioxidant response), and defective healing and repair in the late phase (reflected in decreased myofibroblasts, collagen/ECM, angiogenesis, and telomere lengths) resulting in a defective scar. The ECM changes during repair include increased collagen type III (more elastic and pliable) and decreased collagen type I (more rigid) and cross-linking [50].
2.9.3.1 Aging-Related Changes in Mitochondrial OXS and Inflammation During HF Progression
The roles of aging, mitochondrial OXS, inflammation, and CVD risk factors during HF progression have been underlined here in Figs. 11.2, 11.3, 11.4, 11.5, 11.6 and 11.7, and the pertinent advances recently reviewed by Paneni et al. [128] are summarized below under 14 subheadings.

Schematic of main sources of oxidants, superoxide, and peroxynitrite. Adapted from Bartesaghi and Radi (2018) [98]
Peroxynitrite (ONOO-) is cytotoxic and enhances cell damage during reperfusion following ischemia and can cause protein tyrosine nitration which can serve as a biomarker of oxidative stress. The nitrite radical NO2• is a strong oxidizing and nitrating agent. Protein tyrosine nitration is pertinent in reperfusion and inflammation. The carbonate radical CO3•- is involved mainly in nitro-oxidative damage
Abbreviations: NOX NADPH oxidases, NOS nitric oxide synthases

Schematic depicting the postulated temporal evolution of changes in myocardial damage, extracellular matrix, inflammation, and oxidative stress after myocardial infarction during progression of HFrEF
Data based on studies in the canine model by Jugdutt BI et al. using histopathology for myocardial damage (necrosis and apoptosis), inflammation, collagen and fibrosis, and biochemistry for collagen, MMP, and TIMP [18, 34,35,36, 42, 44, 49,50,51, 55, 58, 92, 94, 98, 103, 128, 129] and Bolli et al. using electron paramagnetic resonance and a spin trap for reactive oxygen species [123], and a review of inflammation and repair postinfarction by Nahrendorf et al. based initially on data in mouse [102]
Abbreviations: MMP matrix metalloproteinase, TIMP tissue inhibitor of metalloproteinase
Other Abbreviations as in text
2.9.3.2 The Problem of Changing Demographics of HF with Aging
Expanding on previous reviews on aging and HF [14,15,16,17,18,19, 50, 51], Paneni et al. reemphasized the impact of lifelong vascular remodeling on the development of CVD risk and HF [128]. Updating previous demographic data on aging [14], they underline the projected doubling of the population aged >65 years from 12% in 2010 to 22% in 2040 [128]. Importantly, they point out that the prevalence of CVD is increasing in people aged >65 years, more so in those aged >80 years, and will increase by 10% over the next 20 years [128]. More alarming, the projected increases between 2010 and 2030 are for 27 million more people with HTN, 8 million more with CHD, 4 million more with stroke, and 3 million more with HF, and these will be mainly in the expanding elderly group [128]. They also underscore several important points that are pertinent for therapeutic interventions [128] and are outlined below.
2.9.3.3 Molecular Mechanisms in Aging-Related Vascular Remodeling
The evidence for how the changes associated with aging-related vascular remodeling contribute to CVD risk and adverse CV events and what key cellular mechanisms and pathways are involved in endothelial dysfunction [128] are as follows: (i) decreased cofactor tetrahydrobiopterin (BH4), in eNOS-mediated generation of NO from L-arginine, leads to eNOS uncoupling with decreased NO release and increased formation of the pro-oxidant superoxide anion (O2•−); (ii) increased arginase activity leads to reduced L-arginine and decreased NO production and bioavailability; (iii) increased ROS can also increase NO degradation, which is mediated in part by chronic inflammation, and thereby results in NO depletion [128]; (iv) increased TNF-α and NADPH oxidase lead to increased superoxide (O2•−) that reacts with NO to form peroxynitrite (NOO−), which nitrosylates eNOS and antioxidant enzymes; (v) increased RAAS and Ang II activities contribute to NO inactivation; (vi) increased RAAS and Ang II activities also activate NADPH oxidase, thereby increasing ROS which, in turn, promotes vascular inflammation; (vii) increased H2O2 activates NF-kB, which in turn leads to release of proinflammatory cytokines (such as IL-6), chemokines (such as TNF-α), and adhesion molecules that are known to mediate atherogenesis; (viii) increased ET-1 levels in the blood and aortic wall promote vasoconstriction and impair endothelium-dependent dilatation; ix) increased cyclooxygenase (COX)-derived eicosanoids (such as prostaglandin [PG]-H2, thromboxane [Tx]-A2, and PGF2α) that are known to promote vasoconstriction and thrombosis, combined with decreased prostacyclin (PG-I2) that is known to prevent these effects, result in increased vasoconstrictor tone and thrombogenicity; (x) increased collagen (from increased collagen deposition and decreased breakdown, as well as increased advanced glycation end-products [AGEs]) and decreased elastin (via the aforementioned increase in MMP-9, MMP-12, cysteine proteinases cathepsins S, K, L, and serine proteinase neutrophil elastase from inflammatory cells with elastolytic activity) lead to increased arterial stiffness (especially the thoracic aorta) and reduced distensibility of large elastic arteries; and (xi) increased TGF-β activity, which induces increased synthesis of interstitial collagen by the adjacent vascular smooth muscle cells (VSMCs) and increased RAAS, Ang II, and TGFβ, which increase both synthesis of collagen and lysis of elastin in the arterial wall, contribute further to arterial stiffness [128]. Taken together, vascular aging is associated with arterial stiffening, endothelial dysfunction with decreased endothelial-dependent vasodilation and antithrombotic property, and increased OXS and proinflammatory cytokines, which act in concert to increase the predisposition to atherosclerosis, thrombosis, and CVD [128].
2.9.3.4 Impact of Lack of Evidence-Based Therapy for HFpEF in the Elderly
As mentioned before, whereas aging-related adverse myocardial and vascular remodeling are known to lead to HTN and HFpEF in the elderly, the lack of evidence-based effective therapies for HFpEF further predisposes the patients to increased risk of myocardial ischemia, MI, ischemic cardiomyopathy, and a switch to HFrEF [128]. Furthermore, while aging is associated with decreased skeletal calcium, the elderly develop increased risk of calcific aortic stenosis that is partly due to increased inflammation and can contribute to increase in afterload, LVH, and subendocardial ischemia. Paradoxically, statins appear to hasten coronary artery calcification [128]. In addition, aging-related frailty with sarcopenia (i.e., loss of muscle mass and function) results in increased sensitivity to drugs used for treating HTN in the elderly [15, 19, 128]. The diagnosis of frailty is complicated by the associated osteoporosis and obesity (that is partly due to the proinflammatory state) [128]. Frailty also appears to interact with the increased prevalence of CVD in the elderly, thereby resulting in increased vulnerability to stressors and OXS.
2.9.3.5 Aging-Related Increased Incidence of Cardiac Amyloidosis
OXS and inflammation may play a role in cardiac amyloidosis. Increase in light-chain amyloidosis is associated with the increase in multiple myeloma with aging, whereas cardiac amyloidosis is associated with wild-type transthyretin (wtTTR) that is more common in older men. Imaging detected wtTTR cardiac amyloidosis in about 13% of patients with HFpEF aged ≥60 years, whereas autopsy showed it in about 20% of people who died at age >80 years [128]. Although there is no proven treatment for wtTTR cardiac amyloidosis, several trials are under way. Chemotherapy is used in light-chain amyloidosis.
2.9.3.6 Aging-Related Telomere Shortening During Cellular Senescence
As previously reviewed [19], evidence suggests that telomere shortening is associated with increased risk of CVD during aging. It is well established that aging-related increase in senescent cells in the vascular wall and heart contributes to adverse remodeling [128]. Telomeres consist of repetitive nucleotide sequences (TTAGGG on one strand and AATCCC on the other strand) at the ends of mammalian chromosomes. These act as caps that preserve chromosome stability and integrity by preventing deterioration or fusion with neighboring chromosomes; every cell division shortens telomeric DNA. When a critical length is reached, the capping function is lost, leading to DNA damage and apoptosis. Studies have shown an association between decreased leukocyte telomere length (TL) and vascular cell senescence, aortic stenosis, CV risk factors (such as HTN, DM2, obesity, and smoking), and risk of atherothrombotic events. Other studies showed correlations between leukocyte TL and atherosclerosis, ischemic and hemorrhagic stroke, and between reduced leukocyte TL and risk of plaque and its progression [128]. A meta-analysis (43,725 participants; CVD, 8400) showed that patients with the shortest leukocyte TL had a higher risk of coronary heart disease and cerebrovascular disease [129].
2.9.3.7 Mitochondrial Oxidative Stress and Cardiovascular Aging: Role of the p66Shc Gene
The molecular events in CV aging, mitochondrial OXS, chromatin remodeling, and genomic instability may all be linked [128]. Six lines of evidence from translational studies support the idea that mitochondrial OXS contributes to cellular senescence through a chain of O2•− or H2O2 formation, ROS overload, senescence, DNA damage, inflammation, and cell death pathways including apoptosis [128]. The evidence supporting the role of the mitochondrial adaptor p66Shc gene in this chain of cellular events is as follows: (i) cells lacking p66Shc show reduced intracellular ROS, whereas mice lacking p66Shc have reduced ROS upon exposure to high OXS, and mice with p66Shc deletion show increased longevity; (ii) aging p66Shc-deficient mice have reduced ROS and preserved NO bioavailability and are protected from both systemic and cerebral endothelial dysfunctions; (iii) p66Shc-deficient mice with brain injury from I/R show reduced ROS production in the brain and reduced stroke size, and in vivo postischemic silencing of p66Shc prevents I/R brain injury in mice; (iv) increased p66Shc expression found in stroke patients correlates with neurological deficits; (v) increased p66Shc is present in peripheral blood mononuclear cells of patients with ACS and DM2; and (vi) p66Shc protein activation is found in patients with CV risk factors, including hyperglycemia, oxidized low-density lipoprotein, smoking, and HTN. Together, these findings suggest that p66Shc may be a potential therapeutic target for age-related CVD and increased mitochondrial ROS [128].
2.9.3.8 Mitochondrial Oxidative Stress and Cardiovascular Aging: Role of JunD
Evidence suggests that the activated protein-1 (AP-1) transcription factor JunD mediates aging-related OXS [128]. JunD, which is formed from dimeric complexes from three main families of DNA-binding proteins (Jun, Fos, and ATF/CREB), is involved in regulation of cell growth and survival, as well as in protection against OXS by modulating genes involved in antioxidant defense and ROS production [128]. Five lines of evidence that support the role of JunD are as follows: (i) JunD levels are lower in the aorta of the aging mouse and in peripheral blood mononuclear cells from old compared to those in young healthy humans; (ii) young mice lacking JunD show endothelial dysfunction and vascular senescence similar to that found in old wild-type mice; (iii) JunD null mice show increased aging markers p53 and p16INK4a, reduced telomerase activity, and mitochondrial DNA damage in their aortas, whereas overexpression of JunD rescues vascular aging features in old mice; (iv) the age-associated decrease in JunD leads to an imbalance between the oxidant NADPH oxidase and scavenger enzymes manganese superoxide dismutase (MnSOD) and aldehyde dehydrogenase 2, which results in early redox changes, mitochondrial dysfunction, and vascular senescence; and (v) mice lacking JunD develop less hypertrophy after mechanical pressure overload whereas mice with cardiomyocyte-specific expression of JunD develop LV dilatation and contractile dysfunction; moreover, fra-1 transgenic mice overexpressing the AP-1 member fos-related antigen and lacking JunD develop dilated cardiomyopathy associated with defective mitochondria and increased cardiomyocyte apoptosis; the findings suggested that JunD promotes adaptive or maladaptive hypertrophy depending on its level [130].
2.9.3.9 Mitochondrial Oxidative Stress and Cardiovascular Aging: Role of Sirtuin-1
The evidence supporting the beneficial role of the sirtuins (silent information regulator [SIR] genes), which are NAD+-dependent enzymes of the big nicotinamide adenine dinucleotide (NAD)-dependent protein family, in human aging [128] is as follows: (i) endogenous sirtuin-1 (SIRT1) expression in VSMCs correlates inversely with donor age, and age-related loss of SIRT1 correlates with reduced stress response and increased senescence; (ii) endothelial-specific SIRT1 overexpression or chronic exposure to a SIRT1 activator in hypercholesterolemic mice decreases atherogenesis, whereas reduced SIRT1 increases atherosclerosis; (iii) inhibition of SIRT1 by immunosuppressant drugs (such as sirolimus and everolimus) leads to endothelial senescence; (iv) inhibition of SIRT1 with sirtinol impairs eNOS function, while activation of SIRT1 improves endothelial NO availability; (v) inhibition of SIRT1 with the endogenous inhibitor, microRNA-217, suppresses SIRT1-dependent eNOS function and promotes endothelial senescence; (vi) SIRT1 has been shown to regulate p66Shc transcription, whereas reduced SIRT1 leads to NF-kB p65 acetylation, resulting in increased inflammatory genes; (vii) SIRT1 has been shown to repress pathways of arterial aging, thereby preventing DNA damage, cell cycle arrest, and oxidative stress; and (vii) SIRT1 has been shown to activate the energy regulator enzyme 5 ́-adenosine monophosphate (AMP)-activated protein kinase involved in glucose homeostasis, thereby maintaining cellular ATP levels and maintaining endothelial integrity via regulation of eNOS activity and autophagy [128]. Together, the findings suggest that activation of SIRT1 may preserve endothelial function during aging and contribute to prevention of CVD and progression to HF.
2.9.3.10 Mitochondrial Oxidative Stress and Cardiovascular Aging: Role of Klotho
Recent evidence suggests that Klotho is an important antiaging gene and Klotho protein acts as a circulating hormone, which binds to a cell-surface receptor and thereby suppresses intracellular signals of insulin and IGF-1that are known to favor longevity [128]. Studies show that (i) Klotho deletion in mice induces premature aging and reduces life span, whereas overexpression increases life span and protects against age-related CV and kidney dysfunction; (ii) high plasma levels of Klotho in patients are associated with a reduced risk of CVD, whereas low serum Klotho concentrations predict CAD and arterial stiffness [128]. Together, these findings suggest that Klotho may be a useful biomarker and a potential target for limiting age-related CVD and stalling progression to HF.
2.9.3.11 Aging-Related Dysregulation of DNA Repair and Damaged DNA Overload
Evidence suggests that the buildup of damaged genetic material throughout life and defects in the DNA repair mechanism after damage (caused by chemicals, mutations, and epigenetic alterations [i.e., “change in gene activity without change in DNA sequence”]) might result in genomic instability and cellular senescence and thereby promote CV aging and dysfunction [128]. Studies have shown that (i) extensive nuclear DNA damage in the Hutchinson-Gilford progeria syndrome is associated with premature atherosclerosis and CVD that lead to early MI or stroke; (ii) mice with genomic instability from defective repair genes develop premature aging that is associated with endothelial cell senescence, vascular stiffness, and HTN, thought to be due to dysregulation of eNOS and sirtuin and to increased NADPH oxidase; and (iii) humans develop sporadic genomic mutations, as supported by findings of (a) DNA damage in circulating cells and plaques of patients with atherosclerosis, (b) chromosomal damage and mitochondrial DNA deletions in peripheral blood mononuclear cells of patients with coronary heart disease that correlate with severity of the disease, (c) association between variation in nucleotide excision repair components and carotid-femoral pulse wave velocity, and (d) increased phosphodiesterase type 1 (PDE1A) expression and impaired NO-cGMP signaling and endothelial dysfunction in senescent VSMCs, and association between PDE1A polymorphisms and diastolic blood pressure and carotid intima-media thickness in patients [128]. Together, these findings suggest that preserving genome stability may limit age-related CVD that contributes to HF progression.
2.9.3.12 Aging-Related Defects in Angiogenesis
Evidence suggests that defects in angiogenesis during CV aging lead to (i) increased stroke, peripheral artery disease, and MI in elderly, with worse outcomes compared to younger patients; (ii) increased mortality and rate of limb amputation after acute limb ischemia; (iii) reduced capillary density, associated with microvascular disease, defective eNOS functionality, and impaired insulin sensitivity in elderly men; (iv) decreased proliferative capacity, telomerase activity, and production of angiogenic growth factors such as VEGF-A in senescent endothelial cells; (v) impaired endothelial migration leading to reduced tube formation; (vi) reduced hypoxia-inducible factor 1α (HIF1α) activity, due mainly to increased degradation and reduced nuclear translocation by importin α; (vii) reduced activity of the transcriptional coactivator PGC-1α that is associated with hypoxia-driven angiogenesis; and (viii) dysregulation of angiogenic pathways associated with an age-dependent decrease in the number and function of stem and progenitor cells [128]. These findings are pertinent for the development of therapies for the elderly.
2.9.3.13 Aging-Related Vascular Remodeling via Nongenomic Regulation of Changes in Chromatin
Recent evidence suggests that, besides the well-recognized gene-driven regulation of aging, nongenomic regulation of aging via epigenetic modifications of transcription programs in OXS, inflammation, angiogenesis, and metabolism can also promote maladaptive pathways leading to vascular aging [128]. The epigenetic modifications that are acquired throughout life appear to be quite stable and explain how environmental factors may interact with genomic DNA and change gene expression. Importantly, epigenetic changes may be transmitted down or inherited and thereby contribute to senescent traits and CVD in young adults [128]. Epigenetic processes that modify chromatin include DNA methylation, acetylation, phosphorylation, ubiquitination, and sumoylation. Studies have shown that (i) DNA methylation, which is the most widely studied, decreases with age, whereas the rate of demethylation correlates inversely with age in mice and humans; (ii) unmethylated or partially methylated CpG (cytosine-guanine) islands are present in atherosclerotic plaques and leukocytes from patients and atherosclerosis-prone mice; (iii) changes in DNA methylation localize at promoter sites of several regulatory genes, including NOS and the vascular endothelial growth factor receptor, that are involved in atherosclerosis and aging; (iv) histone methylation has been implicated in regulating life span and vascular homeostasis that involves endothelial NF-kB, SIRT1, and other genes, including methyltransferase Set7 expression which is increased in peripheral blood mononuclear cells from DM2 patients and correlates with NF-kB-mediated inflammation, OXS, and endothelial dysfunction; (v) histone deacetylation by SIRT1 may influence age-related CVD and transgenic overexpression of SIRT1 was shown to improve metabolic efficiency and endothelial function in old mice; and (vi) SIRT6 can prevent endothelial dysfunction and atherosclerosis by epigenetic modulation of multiple atherosclerosis-related genes, including the pro-atherogenic gene of the TNF family [128]. Together, these findings implicate chromatin modifications in aging-related CVD.
2.9.3.14 Aging-Related Changes and Stem Cell-Based Therapies for Vascular Repair in the Elderly
With regard to the status of vascular repair and of autologous bone marrow-derived stem cell transplantation in the elderly, the conflicting results of RCTs of stem cell-based therapies in MI may be explained by the fact that, in most cases, the aged bone marrow cells have been down the senescence pathways and the substrate in the elderly involved activation of pathways that may impair the healing ability of transplanted stem cells. Therapies that can potentially modulate epigenetic modifications of chromatin and thereby restore gene expression may be useful [128]. Another consideration is the supplementation with adjunctive therapy for targeting both increased OXS and inflammation in elderly patients. Other aging-related molecular remodeling including impaired adrenergic signaling and calcium handling are discussed in detail elsewhere [16, 19].
2.9.4 Aging-Related Increase in Mitochondrial ROS and Autophagy
A prevalent theory is that aging is general, including CV aging, and involves increased generation of ROS from dysfunctional mitochondria leading to ROS-induced cumulative cellular damage [131, 132]; the dysfunctional mitochondria are thought to accumulate during cellular aging due to decreased autophagy [131, 133]. Autophagy, which performs cellular housekeeping and maintains homeostasis in the cell by renewing or recycling cytoplasmic materials and organelles (including mitochondria), removing toxic protein aggregates and harmful ROS, and providing essential energy and biomolecules to cells, is a cytoprotective function that has been shown to decline with aging [133, 134]. With aging, evidence for both the increase in basal ROS levels [135] and the increase in dysfunctional mitochondria from reduced or dysregulated autophagy [134] have been documented. Since cardiomyocytes are not frequently replaced, they are subjected to OXS and oxidative damage during aging [135]. Dai et al. have reviewed the interactions among mitochondrial ROS, redox, and other cellular signaling pathways as well as various therapeutic strategies and drugs (such as mitochondrial antioxidants MitoQ, SkQ1, and the mitochondrial protective peptide SS-31) that can potentially improve mitochondrial function during aging [132].
Evidence shows that homeostasis by autophagy is maintained through cues from “danger-associated molecular patterns” (DAMPs), such as ROS, mitochondrial DNA, and extracellular ATP that are detected by “pattern recognition receptors” (PRRs) comprised of several families, including “Toll-like receptors” (TLRs) and “NOD-like receptors” (NLRs), which control autophagy and are also involved in innate and adaptive immune response [136]. DAMPs activate the NLRP3 inflammasome in response to ROS and other stimuli, such as extracellular ATP and lysosomal disruption [136]. Evidence suggests that autophagy and inflammation are interdependent [137]. As mentioned before, impaired autophagy is associated with an increase in defective mitochondria and levels of ROS and leads to increased inflammatory cytokine levels; evidence suggests that increased ROS activates inflammasomes which process IL-1β.
While preclinical studies are consistent with decreased autophagy during aging (Fig. 11.5) and autophagy-driven regulation of the degree of inflammation, whether the decrease in autophagy with aging leads to increased basal levels of inflammation and OXS in the human heart or the CV system during HF needs further study. The studies reviewed by Linton et al. did not specifically address this question but the results were interesting [136]. In one small study of 9 HF patients with idiopathic ischemic cardiomyopathy maintained on an LV assist device (LVAD), LV biopsies showed a downregulation of the autophagy gene mRNA and protein [138]. In another study of 170 patients undergoing coronary artery bypass surgery (CABG), 22% who developed postoperative atrial fibrillation showed evidence of impaired autophagy in their atrial biopsies, while levels of hs-CRP, inflammation, fibrosis, and other markers were similar for patients with or without postoperative atrial fibrillation [139]. In a further study of 19 patients undergoing surgery with cardiopulmonary bypass (CPB), the test results suggested depletion of autophagy proteins [140]. In another study of patients undergoing heart surgery with I/R, biopsies of the right atrial appendage showed evidence of upregulation of autophagy-related genes in 13% and downregulation in 4% [141]. A small study of patients undergoing CABG and remote ischemic preconditioning (RIPC) failed to show changes in autophagy in LV biopsies [142], probably due to issues with study design [136]. In a cohort of older cardiac surgery patients (mean age 62 years, range 33–87 years) who had right atrial biopsies before and after CPB, Linton’s group found evidence for a “robust” autophagic response to the ischemic stress that was age-independent, albeit as assessed by lipidation of the autophagy protein LC3 which is somewhat controversial [143]. Another pertinent finding with respect to exposure to comorbidities is that animals with metabolic syndrome and DM2 show suppression of autophagy; in a cohort of their surgical patients, Linton’s group found evidence for impaired autophagy in those DM2 patients with poor glycemic control and HbA1C level > 7% [136].
Several studies addressed the role of autophagy in postremodeling and HF; in one study in young mice with chronic MI, the autophagy inhibitor bafilomycin A1 worsened remodeling, whereas the autophagy enhancer rapamycin attenuated remodeling, suggesting that autophagy might protect against adverse post-MI remodeling [144]; another more recent study in young mice with chronic MI showed that TLR3 upregulation contributes to persistent autophagy which promotes HF and death, whereas TLR3 deletion inhibits autophagy, reduces MI size, attenuates HF, and improves survival, and the autophagy inducer rapamycin abolishes these benefits [145].
2.9.5 Role of AGEs and RAGEs in OXS and Heart Failure
AGEs are condensates of glucose that nonenzymatically form cross-links between collagen molecules, thereby rendering them resistant to enzymatic degradation [128]. AGEs are known to accumulate in vessel walls and contribute to both microvascular and macrovascular complications through formation of cross-links. They exert adverse effects through either receptor or nonreceptor mechanisms; in the receptor mechanism, they bind to specific cell-surface receptors for AGEs (RAGEs) thereby activating RAGE and leading to a chain of NF-kB upregulation, proinflammatory cytokine (such as TNF-α, IL-1β) activation, growth factors (such as PDGF, IGF-1), adhesion molecules (such as VCAM-1), and increased ROS which converge on tissue damage. While soluble AGEs activate monocytes, basement membrane AGEs inhibit monocytes; AGE-bound RAGE increases endothelial permeability; AGEs block NO activity and induce ROS production; AGEs have also been implicated in diabetic vascular injury [146, 147]. Prasad et al. [147] referred to the adverse effects of AGE and AGE-RAGE as “AGE-RAGE stress” and its endogenous defense mechanisms as “antistressors”; the endogenous antistressor mechanism involves enzyme-mediated (through glyoxalase 1 and 2) and AGE receptor-mediated (AGER-1 and AGER-2) degradation of AGE, and increased soluble receptor of AGE (sRAGE); exogenous defense strategies include decreasing AGE consumption, preventing AGE formation, and downregulating RAGE. Pharmacological agents known to reduce AGE formation include those used to treat HFrEF and HTN (ACEIs, ARBs), DM2 (anti-DM2 drugs; aminoguanidine) and CVD risk (statins), and others [147]. Drugs that elevate sRAGE include ACEIs, statins, and antidiabetic drugs [147].
2.9.6 Role of Synergism Between OXS and Inflammation in HF Progression
The topic of stress and disease has raised concern since ancient times. Selye studied the role of stress in health and disease, focusing mainly on its role in the brain and related disorders [148]. Interest in the role of OXS as an important mediator of CVD was rekindled in the 1980s [31,32,33]. The vast literature on the biology, physiology, and biochemistry of OFRs, ROS, and OXS that stemmed from extensive basic, translational, and clinical research over nearly 5 decades since the 1970s has been recently reviewed elsewhere [31,32,33]. This has improved our understanding of the pathobiology, biochemistry, and pharmacology of OFRs (Fig. 11.6) and unveiled the mechanisms leading to OXS and its contribution to the pathophysiology of CVD, including myocardial ischemia, I/R and MI, and subsequent initiation and progression to HFrEF [27, 28, 31,32,33, 128]. In addition, the realization that both OXS and inflammation contribute to the progression of HFpEF and HFrEF has perked interest in the critical roles of OXS and inflammation in HF progression and underscored the importance of OXS and inflammation as potential targets for stalling HF progression [28,29,30,31,32,33]. The additional contributions of CVD risk factors in increasing both OXS and inflammation have also been underscored [31,32,33]. With the passage of time during aging, OXS and inflammation appear to propel the progression of HF forward toward end-stage heart disease (Figs. 11.2 and 11.4). However, more research is needed to unravel the mechanisms of how exactly CVD risk factors lead to increased OXS and inflammation and thereby mediate HF progression.
We hypothesize that the synergism between OXS and inflammation is an important factor in accelerating HF progression toward end-stage heart disease, and both OXS and inflammation need to be targeted for optimal results.
2.9.6.1 OXS and Inflammation in Post-STEMI Remodeling and HF Progression
The OXS-inflammation interaction during post-STEMI cardiac remodeling and its contribution during HF progression have been a topical issue for the last two decades (Figs. 11.2, 11.3, and 11.4). Although persistent inflammation has been addressed recently in several aforementioned RCTs showing the benefit of some anti-inflammatory strategies during post-MI HF progression [114, 115], RCTs that target persistent OXS alone or the OXS-inflammation interaction have not been launched.
2.9.6.2 Development of ROS Overload and OXS Post-STEMI and HF
The development of ROS overload and OXS in HF represents a breakdown of homeostasis that occurs whenever the production of OFRs far exceeds the capacity of endogenous antioxidant defense mechanisms to neutralize them; when that happens, the result is damage to the myocardium and other CV tissues. It is known that during aerobic respiration, molecular oxygen (O2) acts as the final acceptor of electrons in the ETC, thereby leading to generation of adenosine triphosphate (ATP) through the Krebs cycle in the mitochondria. The ATP supplies the energy needed for maintaining contractile function, cardiac output, and organ perfusion. Under physiological conditions, oxygen molecules are reduced to the ROS superoxide (O2•−) which is converted to hydrogen peroxide (H2O2) by the endogenous antioxidant superoxide dismutase (SOD); H2O2 is then broken down into H2O by other antioxidants (such as catalase or glutathione peroxidase), as shown in Fig. 11.6. However, under pathological conditions (such as myocardial ischemia, I/R, MI, and HF), the formation of ROS or OFRs including O2•−, H2O2, hydroxyl radical (•OH), hypochlorite (HOCl), and NO-derived peroxynitrite (NOO−) increases; the scavenging ability of the antioxidants is exceeded, thereby resulting in ROS overload and OXS (Fig. 11.6) [30, 31, 149]. H2O2 is converted to the reactive (•OH) via the Fenton or Haber-Weiss reactions (Fig. 11.6). Increased NO from inducible NOS (iNOS) leads to increase in the reactive NOO− (Fig. 11.6). Whereas most ROSs are lipid insoluble and remain within the cell, H2O2 can cross cell membranes and thereby cause damage at remote regions [149]. Endogenous antioxidants include SOD, glutathione peroxidases, catalase, and peroxiredoxins [149] (Fig. 11.6). Sources of ROS include enzymes in mitochondrial ETC, plasma membrane, peroxisomes, endoplasmic reticulum and nuclear membrane, and other enzymes such as xanthine oxidase, MPO, and cytochrome P450 enzymes (monooxygenases), some of which are found in macrophages and neutrophils that accompany inflammation, NADPH oxidases, and soluble heme proteins [149]. ROS produced by cardiomyocytes and infiltrating inflammatory cells leads to cellular damage through disruption of membranes, proteins, and nucleic acids and activation of cell death pathways that trigger apoptosis [150, 151] (Fig. 11.6). During postischemic reperfusion after MI, ROS may be produced by both enzymatic and nonenzymatic systems and in both cardiomyocytes and infiltrating inflammatory cells. Reperfusion stimulates neutrophil activation and lipid peroxidation in membranes which result in increased ROS. Although NO is nonreactive, iNOS generates reactive •NO from NO; NOXs generate reactive superoxide (O2•−) from O2; the •NO and O2•− interact to yield highly reactive peroxynitrite (NOO−) (Fig. 11.6). Evidence suggests that peroxynitrite leads to the formation of hydroxyl, nitrite, and carbonate radicals, which mediate cell damage (Fig. 11.6), and tyrosine nitration which serves as a biomarker of OXS [149]. Besides protein nitration, increased ROS and NOO− also lead to lipid peroxidation, single-strand nucleic acid breaks, and chromosomal changes [149]. Some pertinent harmful effects of ROS overload during I/R include (i) stimulation of platelets to release platelet activating factor, thereby attracting more neutrophils that exacerbate damage; (ii) attenuation of NO function, thereby enhancing endothelial dysfunction through NOO− formation and augmenting damage; and (iii) blunting of endothelium-dependent vasodilation and enhanced ET-1-induced vasoconstriction, which together aggravate the decrease in reflow.
2.9.6.3 Reasons for Failure to Reduce ROS Overload and OXS Post-STEMI and HF in Patients
Despite experimental animal studies suggesting that administration of exogenous antioxidants including SOD, xanthine oxidase inhibitors (such as allopurinol), N-acetyl cysteine, vitamin E, and vitamin C during after I/R and MI yields favorable results, overall clinical experience has been disappointing [1, 2].
There are several possible reasons for the lack of clinical benefit of antioxidants during and after I/R and MI at the bedside. These include the following: (i) in the setting of I/R and MI, the presence of intense inflammation can have many harmful effects as mentioned before, including increased ROS and activation of platelets which aggregate and provide the matrix scaffold for thrombi to form and plug blood vessels, thereby augmenting cardiomyocyte damage; (ii) the acute inflammation after STEMI also induces vascular damage, which (as mentioned before) is augmented by reperfusion, and involves sequential activation of resident macrophages, release of proinflammatory cytokines (such as IL-1 and TNF-α), upregulation of vascular adhesion molecules (such as selectins, integrins, and ICAM-1) and CXC chemokines (such as IL-8, macrophage inflammatory protein-2 [MIP-2]), neutrophil adhesion and transmigration, damage to endothelial cells, basement membrane and matrix, and increased permeability with microvascular hemorrhage; importantly, the intensity of the inflammatory response is regulated by the balance between proinflammatory cytokines (such as TNF-α, IL-1, and CXC chemokines) and anti-inflammatory cytokines (such as IL-10 and IL-13); (iii) the intense inflammatory response with I/R and reperfused STEMI appears to collaborate in the early surge in ROS in the first 2–10 min and the persistence for several hours thereafter (Figs. 11.7 and 11.8) [123]; since inflammation extends far beyond reperfusion and generates ROS as discussed before (Fig. 11.7), it can be expected to contribute to the ROS pool far beyond the early postreperfusion phase into the subsequent phase of healing and repair, with obvious pathophysiological and therapeutic implications; of note, SOD plus catalase, given over 15 min before and continued for 30 min after reflow, blocked ROS production in the stunned zone, and improved functional recovery in the dog model [152]; (iv) intracellular remodeling after I/R injury is associated with increased proteolytic enzyme activity and alterations in gene expression and translation mechanisms which can have persistent effects [153]; (v) humans with STEMI are middle-aged or older adults, or elderly, with a host of aging-related issues as discussed before (Fig. 11.5); for example, aging may blunt the response to therapy, as found with an ARB after reperfused STEMI in dogs [58] and with postischemic conditioning in mice [97]; aging may also modify inflammation, healing, and repair [48, 58, 124]; (vi) in the context of aging, age equivalence should be considered when assessing therapies in animal models with the goal of translation to humans. For example, a 6-week-old mouse or rat would be equivalent to a young human child by age, and not even to a young adult [99]; (vii) persistent ROS production over the healing phases of acute and chronic inflammation [48, 50, 97, 101,102,103], and far beyond, during progressive remodeling and HF as discussed below; (viii) the rate of reperfusion differs between animal models and humans, being usually more abrupt in most animal models than in humans who undergo PCI and/or thrombolysis after STEMI [97]; (ix) besides SOD and catalase, other protective endogenous mechanisms against ROS include adenosine, opening of ATP-sensitive potassium (K-ATP) channels, and release of NO [154]; (x) ROS overload and OXS-induced damage after reperfusion of STEMI and thereafter in humans may be enhanced by comorbidities such as hyperlipidemia, HTN, and DM2 (Figs. 11.2 and 11.3) that aggravate endothelial dysfunction and lead to adverse vascular remodeling [110, 111, 128]; (xi) targeting I/R injury and OXS in humans is further complicated by the multiple factors, players, mechanisms, mediators, signaling pathways, background drugs, pathologies, approaches, and timings involved [109,110,111] (Figs. 11.3, 11.4, 11.5, 11.6, and 11.7); and xii) despite successful primary percutaneous coronary intervention (PPCI) after STEMI and achieving TIMI grade 3 flow in IRAs, reperfusion at the tissue level is often incomplete in as many as 9–15 % of patients due to a combination of microvascular damage and distal embolization of bits of thrombi and debris from atherosclerotic plaques [155, 156]. Taken together, these points should be considered in designing RCTs to demonstrate the benefit of therapies targeting ROS overload and alleviate OXS in HFrEF and HFpEF. Importantly, rather than targeting ROS alone, both ROS and inflammation need to be targeted.

Schematic depicting concept of synergism between persistent ROS and inflammation in heart failure progression
↑ increases, ↓ decreases,  stimulation of
stimulation of
Abbreviations: as in text
2.9.6.4 ROS-Driven Progression of Early and Late Remodeling Post-STEMI and HF in Patients
The mechanisms that modulate post-STEMI cardiac remodeling have been addressed above and elsewhere before [34, 39,40,41,42,43, 52,53,54,55,56,57,58], as has the contribution of inflammation [27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44, 48,49,50,51,52,53,54,55,56,57,58, 102, 124, 126]. However, the molecular and cellular mechanisms that modulate ROS-driven progression of early and late remodeling after STEMI and thereby contribute to chronic HF were not addressed. Grieve et al. very nicely underlined some of the evidence implicating ROS, OXS, and redox signaling in the progression of early and late remodeling after STEMI [157]. They noted that under physiological conditions, ROS can (i) act as second messengers in several cellular functions, including reacting with cysteine residues in sulfhydryl groups of proteins, and thereby induce conformational changes that mediate signal transduction; (ii) stimulate DNA synthesis and induce expression of growth-related genes (such as c-fos, c-jun, and c-myc); and (iii) alter the activity of redox-sensitive phosphatases and kinases such as mitogen-activated protein kinases (MAPK), thereby leading to changes in gene transcription and cell phenotype. Under pathophysiological conditions, they suggested a chain of increased intracellular ROS, ROS-induced activation of kinases (such as p38MAPK, extracellular signal-related kinase [ERK]-1/2, c-Jun N-terminal kinase [JNK], protein kinase B/C, and protein tyrosine phosphatase/tyrosine protein kinase c-Src), redox-sensitive gene transcription, and the development and progression of early and late ventricular remodeling [157].
Increased ROS and OXS have been documented in animal models of IR, MI, and HF and in patients with chronic HF in early studies almost two decades ago [27, 123, 152, 158,159,160,161,162]. Hill et al. showed that rats with MI have marginal increases in SOD, glutathione peroxidase and catalase activities, and vitamin E levels during the nonfailure stage at 1 week but develop increased redox state (reduced/oxidized glutathione ratio) and lipid peroxidation associated with decreased SOD, glutathione peroxidase and catalase activities, and vitamin E levels during moderate to severe HF at 16 weeks, suggesting that HF post-MI is associated with an antioxidant-deficit and increased OXS [158]. Kinugawa et al. showed, using ESR spectroscopy in mice with MI, that the hydroxyl radical (•OH) scavenger dimethylthiourea (DMTU) attenuated the increase in •OH as well as MMP-2 in the NIZ [159]. Singh et al. suggested that antioxidant vitamins reduced OXS in patients with suspected MI [160]. Dhalla et al. showed that aortic banding in guinea pigs induced hypertrophy associated with decreased OXS (increased redox state) at 10 weeks followed by HF with increased OXS at 20 weeks; importantly, chronic vitamin E therapy improved the reserve of endogenous vitamin E, reduced OXS (decreased glutathione/oxidized glutathione ratio), and ultrastructural damage at 20 weeks, suggesting that long-term antioxidant therapy can potentially attenuate or prevent HF [161]. Ide et al. showed, using the dog model of congestive HF induced by 4 weeks of rapid ventricular pacing, that ROS (using ESR spectroscopy for measuring ROS superoxide anion) increased nearly threefold in HF; this was due to a functional block of electron transport at complex I (reflected in decreased complex I enzymatic activity) and uncoupling of the respiratory chain, leading to increased production of mitochondrial ROS and resulting in contractile dysfunction and structural damage to the myocardium during HF [162]. Kim et al. showed, using adenoviral-mediated gene transfer of a rac1 gene product in VSMCs in culture, that inhibition of rac1 blocks the release of ROS upon reoxygenation (as with reperfusion) and protects against reoxygenation-induced cell damage [163]. Talukder et al. used cardiomyocyte-specific overexpression of active rac to show that increased myocardial rac leads to increased injury, contractile dysfunction, and MI after I/R [164].
It is clear that there are multiple sources of ROS during HF, including inflammatory cells such as neutrophils and monocytes [49, 51, 157], cardiomyocyte mitochondria [29,30,31, 162, 165], xanthine oxidases [157], phagocytic-type NADPH oxidases [157, 166, 167], and dysfunctional NO synthase [157, 168, 169]. Vascular injury can occur not only during the initial MI but also with recurrent ischemia, I/R, and MI during progression of both HFrEF and HFpEF (Figs. 11.3 and 11.7). As mentioned before, acute inflammation appears to be regulated by anti-inflammatory mediators (such as IL-10, IL-13, and SLPI) that in turn can regulate NF-kB activation and dampen the release of proinflammatory mediators (such as TNFα and IL-1), thereby attenuating the damaging effects of oxidants and proteases derived from recruited neutrophils [170]. Lentsch et al. suggested that therapy to limit vascular injury should be based on anti-inflammatory mediators [170]. Many other studies have implicated other specific pathways involved in ROS overload and potential signaling pathways and molecules that could be targeted to quench ROS overload and reduce OXS during the adverse remodeling and progression to HF after MI [27,28,29,30,31,32,33].
2.9.6.5 Role of ECM Remodeling in Progression of LV Remodeling in HFrEF After STEMI
The evidence for the role of the ECM in cardiac remodeling has been reviewed elsewhere [18, 34,35,36,37, 42, 44, 49,50,51,52,53,54,55,56,57,58]. Several points need emphasis: (i) dramatic ECM remodeling in early STEMI is driven by the sharp and early rise in MMP levels (Fig. 11.7), causing a chain of MMP/TIMP imbalance, ECM degradation, decreased collagen, adverse LV remodeling, LV dysfunction, HFrEF, and adverse outcome; (ii) the disturbed ECM homeostasis after acute injury continues during healing and repair phases, and beyond initial scar formation in survivors, and contributes to further progressive ECM and LV remodeling and HF progression; whereas the MMP and TIMP levels subside over weeks as inflammation subsides and the early rise in ROS falls (Fig. 11.7); chronically high MMP/TIMP ratios may promote continued ECM degradation and contribute to the commonly observed progressive LV dilation during healing and remote MI and progressive increase in severity of HFrEF; (iii) continued low-grade inflammation and low level of ROS production (Figs. 11.7 and 11.8), in part due to the continued inflammation itself, contribute further to the progression of HFrEF; (iv) in contrast to that construct, a chronically low MMP/TIMP ratio can contribute to increased ECM and fibrosis in both the IZ and NIZ and thereby lead to increased stiffness and diastolic dysfunction and aggravate LV systolic dysfunction over time; (v) defective ECM and fibrosis together with increased cross-linking can also augment adverse LV remodeling and systolic/diastolic dysfunction; in addition, decreased ECM deposition and fibrosis and/or defective ECM (with increased type III collagen and decreased or abnormal cross-linking) can further exacerbate adverse LV remodeling, LV systolic dysfunction, and LV rupture; (vi) whereas LV remodeling is a major mechanism for LV enlargement leading to HF after STEMI, it is the ECM disruption that mediates the initial step in LV dilatation and is the key mechanism underlying LV structural remodeling and Ang II (a primary effector molecule of the RAAS) that drives both ECM and LV remodeling [34, 35]; (vii) the aforementioned mix of cells (inflammatory cells, fibroblast, and vascular cells) together with a mix of molecules (growth factors, cytokines, chemokines, and matrikines) in the healing post-MI substrate act in concert to further modulate LV remodeling [36]; (viii) fibroblasts regulate ECM synthesis/deposition and mediate ECM degradation/turnover through MMP/TIMP balance, and various MMPs (including MMP-1, MMP-2, and MMP-9) modulate ECM remodeling; concurrently, growth factors (such as TGF-β) and proinflammatory cytokines (such as Ang II, IL-6, and TNF-α) that are released after MI modulate MMP/TIMP imbalance, ECM degradation or interstitial fibrosis, and remodeling [36]; (ix) diffusion of proteins and migration of cells from the IZ to borders and the NIZ may extend fibrotic remodeling to those areas, whereas MMP-9 can interact with inflammatory response proteins in the post-MI inflammatory response (such as activator protein-1, specificity protein-1 and NF-κB) and, in fact, shows correlation with various inflammatory markers (such as IL-6, hs-CRP, and fibrinogen), LV hypertrophy, adverse remodeling with LV dilation, dysfunction in HFrEF and HFpEF as well as CV mortality [49, 50, 96]; (x) TIMP-3 also regulates inflammation and inhibits ADAM-17 and ADAM-10, which in turn can alter integrins (cell-surface matrix receptors) and thereby disrupt cell-matrix interactions, degrade ECM, and contribute to LV dilation; xi) ADAMS also interact with inflammatory cytokines and alter MMPs and thereby impact LV remodeling and/or injury; these interactions between the matrix proteins and inflammatory cytokines may modulate ECM damage; (xii) since inflammation is a key modulator of healing/repair after MI and impacts both ECM and cardiac remodeling, tight regulation of the inflammatory response is essential not only for adequate healing/repair and scar formation but also during the subsequent progression to HF and beyond; it follows that dysregulation of the inflammatory response can result in defective scars, increased adverse ECM and LV remodeling, and more rapid progression to HFrEF and end-stage disease; (xiii) the inflammatory cells also release MMPs and oxidants; infiltrating neutrophils release MMPs such as MMP-9; activated neutrophils and monocytes produce MPO which enhances remodeling through generation of oxidants and MMP activation; the aforementioned Ly-6Chigh monocytes secrete inflammatory cytokines, ROS, and matrix-degrading proteases, whereas the Ly-6Clow monocytes trigger collagen/ECM synthesis by myofibroblasts and promote healthy infarct scar formation. Myofibroblasts persist in the infarct scar and maintain ECM; xiv) ROS modulates fibroblast proliferation and collagen synthesis and activates MMPs [171,172,173] which are redox-sensitive [172,173,174]; and various cytokines (such as Ang II, cytokines, and cyclic load) stimulate both ECM remodeling and intracellular ROS generation [175,176,177]; and (xv) ACEIs and ARBs attenuate both adverse post-MI remodeling and OXS [178,179,180,181,182]; chronic treatment with the ROS scavenger DMTU was shown to inhibit adverse LV remodeling and HF, and reduce collagen deposition, MMP-2 activity, LV hypertrophy, and dilatation in mice [159]; the antioxidant probucol was shown to improve LV function, prevent LV dilatation, and reduce cardiac fibrosis post-MI in rats [183]. Although a time-course analysis showed nuclear-mitochondrial cross-talk in global myocardial ischemia, the effect of HF therapies on the cross-talk needs further study [184].
Taken together, the findings support the idea that interactions between ROS and inflammation after STEMI contribute to adverse ECM and cardiac remodeling and progression to HFpEF.
3 Conclusion and Future Directions
The rising trend in disability and death from HF has persisted despite adherence to therapies recommended in the published management guidelines from major societies including the American Heart Association, American College of Cardiology, and European Society of Cardiology [1,2,3,4,5, 7]. Clearly, this trend burdens the available healthcare resources [9]. As mentioned before, HF prevalence in Europe is reported to be about 1–2% in adults, rising to over 10% in those older than 70 years [1], and the lifetime risk of HF in people aged 55 years is 33% in men and 28% for women [20]. In the United States, the lifetime risk of HF was 20–45% for people aged 45–95 years and was higher for people with HTN and obesity irrespective of age [6]. The recommended pharmacologic therapies in the guidelines for ST-segment elevation MI (STEMI) and HF are listed in Tables 11.1, 11.2, and 11.3, respectively. Whereas mortality and morbidity after ACS and MI have improved dramatically over the last four decades due to adherence to guideline-driven timely reperfusion and adjunctive therapies [1, 2, 74], there remains a 7% mortality and 22% morbidity in patients with STEMI at 1 year after prompt reperfusion and PPCI [1, 110, 111]. Although several pharmacological interventions were shown to be effective for preventing and limiting the ravages of ROS and OXS during myocardial ischemia, I/R, MI, and HF in animal models, they did not prove to be effective in patients [1, 2, 7, 12, 109, 110]. The guidelines therefore did not recommend the use of antioxidants in the setting of myocardial ischemia, I/R, MI, and HF [1, 2, 7,8,9,10, 12, 109, 110].
As is clear from the discussions in this chapter, the area of prevention of OXS on the clinical front is complex. While there are several reasons for the continued increase in HF prevalence despite optimal approved therapy, three reasons that stand out include (i) the aging-induced CV remodeling that modifies disease expression and response to therapy; (ii) the lifelong exposure to CVD risk factors that increase ROS and OXS as well as inflammation (Fig. 11.4); and (iii) the contribution of synergism between ROS overload/OXS and inflammation as proposed here (Fig. 11.8) and other yet to be addressed pathways and mechanisms leading to HF.
The multiplicity of CVD risk factors, comorbidities, and pathophysiological mechanisms and pathways involved in HFpEF, coupled with the lack of guideline-approved therapy for HFpEF in 2018, actually provides unique opportunities for drug development in this area. As shown in Fig. 11.8, whereas OXS and inflammation appear to show synergism in mediating damage, this aspect of the background mechanisms still remains to be fully characterized and addressed. Whereas there are recommended therapies for HFrEF, the persistence of background levels of inflammation as well as ROS and MMPs, as shown in Fig. 11.7, also provides unique opportunities for drug development in this area. Already, the CANTOS trial group has tested effective adjunctive anti-inflammatory therapy for limiting the residual inflammation on top of background therapies in post-STEMI/HFrEF patients [114, 115]. A similar approach can be used to test a carefully selected antioxidant or pathway in a well-designed RCT for HFrEF and HFpEF. It is also important to select a biomarker or panel of biomarkers for OXS that can be used to assess response to therapy and to predefine realistic endpoints.
In this war against OXS, the matter of background therapy is an important consideration when undertaking RCTs. Background drugs used by both HFrEF and HFpEF patients include ACEIs, ARBs, MRAs, beta-blockers, and other drugs that already decrease OXS. In addition, in spite of the lack of support for the use of antioxidants in the management guidelines, off-the-counter vitamins and other related drugs that are advertised to reduce OXS are widely used, especially in the elderly. Furthermore, the health-conscious patients are already on diet and exercise programs that reduce OXS.
Thus, the selection of the appropriate placebo arm of an OXS-RCT becomes a difficult task. It is essential for investigators designing the RCTs to limit OXS to have a clear appreciation of the key basic mechanisms and problems facing the translation of strategies that are found to be effective when tested in research studies carried out in appropriate animal models. Well-designed preliminary clinical studies in humans are also essential before launching equally well-designed RCTs and before moving to application at the bedside of patients suffering from HF. What, when, and exactly how best to target OXS and translate promising experimental therapy to the real-world by well-designed RCTs before application at the bedside by practicing cardiologists and physicians remains an unanswered question. Waging war on OXS is a worthy goal but it can be challenging.
Abbreviations
- ACS:
-
acute coronary syndrome
- ACE:
-
angiotensin-converting enzyme
- ACEIs:
-
angiotensin-converting-enzyme inhibitors
- ADAM:
-
a disintegrin and metalloproteinase
- AMP:
-
adenosine monophosphate
- Ang II:
-
angiotensin II
- ARB:
-
angiotensin II type 1 receptor blocker
- ARNI:
-
angiotensin receptor neprilysin inhibitor
- ATP:
-
adenosine triphosphate
- BH4:
-
tetrahydrobiopterin
- BNP:
-
N-terminal B-type natriuretic peptide
- CABG:
-
coronary artery bypass surgery
- CAD:
-
coronary artery disease
- CANTOS:
-
Canakinumab Anti-Inflammatory Thrombosis Outcomes Study
- CARE:
-
cholesterol and recurrent events
- cGMP:
-
cyclic guanosine monophosphate
- CKD:
-
chronic kidney disease
- CMR:
-
cardiac magnetic resonance
- CPB:
-
cardiopulmonary bypass
- CRP:
-
C-reactive protein
- CVD:
-
cardiovascular disease
- DM2:
-
type 2 diabetes mellitus
- ECG:
-
electrocardiogram
- ECM:
-
extracellular matrix
- eNOS:
-
endothelial nitric oxide synthase
- EPR:
-
electron paramagnetic resonance
- ESR:
-
electron spin resonance
- ET:
-
endothelin
- ETC:
-
electron transfer chain
- GDF:
-
growth differentiation factor
- GLP-1:
-
antidiabetic glucagon-like peptide-1
- GLP-1RA:
-
glucagon-like peptide/receptor agonist
- H2O2:
-
hydrogen peroxide
- HDL:
-
high-density lipoprotein
- HF:
-
heart failure
- HFmrEF:
-
heart failure with midrange ejection fraction
- HFpEF:
-
heart failure with preserved ejection fraction
- HFrEF:
-
heart failure with reduced ejection fraction
- hs-CRP:
-
high-sensitivity C-reactive protein
- IGF:
-
insulin-like growth factor
- IL:
-
interleukin
- IL-1ra:
-
recombinant IL-1 receptor antagonist
- iNOS:
-
inducible nitric oxide synthase
- I/R:
-
ischemia-reperfusion
- IRA:
-
infarct-related artery
- IZ:
-
infarct zone
- LDL:
-
low-density lipoprotein
- LV:
-
left ventricular
- LVAD:
-
LV assist device
- MACE:
-
major adverse cardiovascular events
- MI:
-
myocardial infarction
- MMP:
-
matrix metalloproteinase
- MPO:
-
myeloperoxidase
- MRA:
-
mineralocorticoid receptor antagonist
- MRI:
-
magnetic resonance imaging
- MUGA:
-
multigated acquisition scan
- NDEA:
-
N-nitrosodiethylamine
- NEP:
-
neprilysin
- NADPH:
-
nicotinamide adenine dinucleotide phosphate
- NDMA:
-
N-nitrosodimethylamine
- NIZ:
-
noninfarct zone
- •NO:
-
nitric oxide
- NOO−:
-
NO-derived peroxynitrite
- NOS:
-
nitric oxide synthase
- NOX:
-
NADPH oxidase
- NSTEMI:
-
non-ST-segment elevation MI
- O2:
-
oxygen
- OFRs:
-
oxygen free radicals
- OPN:
-
osteopontin
- OXS:
-
oxidative stress
- •OH:
-
hydroxyl radical
- PCI:
-
percutaneous coronary intervention
- PDGF:
-
platelet-derived growth factor
- PKG:
-
phosphokinase G
- PPCI:
-
primary PCI
- O2•−:
-
superoxide anion radical
- RAAS:
-
renin-angiotensin-aldosterone system
- RCT:
-
randomized clinical trial
- RIPC:
-
remote ischemic preconditioning
- ROS:
-
reactive oxygen species
- SGLT2:
-
sodium glucose cotransporter-2
- SLPI:
-
secretory leucocyte protease inhibitor
- SOD:
-
superoxide dismutase
- SPARC:
-
secreted protein acidic and rich in cysteine
- STEMI:
-
ST-segment elevation MI
- TIMP:
-
tissue inhibitor of metalloproteinase
- TGF:
-
transforming growth factor
- TNF:
-
tumor necrosis factor
- VSMC:
-
vascular smooth muscle cell
References
Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJS et al (2016) 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure. The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J 37:2129–2200
Yancy CW, Jessup M, Butler J, Casey DE, Colvin MM, Drazner MH et al (2016) 2016 ACC/AHA/HFSA focused update on new pharmacological therapy for heart failure: an update of the 2013 ACCF/AHA guideline for the management of heart failure. A report of the American College of Cardiology/American Heart Association Task force on clinical practice guidelines and the Heart Failure Society of America. Circulation 134:e282–e293
Lloyd-Jones D, Adams RJ, Brown TM, Carnethon MR, Dai S, de Simone G et al (2010) Heart disease and stroke statistics – 2010 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation 121:e46–e215
Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB et al (2012) Heart disease and stroke statistics-2012 update: a report from the American Heart Association. Circulation 125:e2–e220
Benjamin EJ, Blaha MJ, Chiuve SE, Cushman M, Das SR, Deo R et al (2017) Heart disease and stroke statistics-2017 update. A report from the American Heart Association. Circulation 35:e146–e603
Benjamin EJ, Virani SS, Callaway CW, Chamberlain AH, Chang AR, Cheng S et al (2018) Heart disease and stroke statistics- 2018 update. A report from the American Heart Association. Circulation 137:e67–e492
Hunt SA, Abraham WT, Chin MH, Feldman AM, Francis GS, Ganiats TG et al (2009) 2009 focused update incorporated into the ACC/AHA 2005 guidelines for the diagnosis and Management of Heart Failure in adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation 119:e391–e479
Dickstein K, Cohen-Solal A, Filippatos G et al (2008) ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2008: the Task Force for the diagnosis and treatment of acute and chronic heart failure 2008 of the European Society of Cardiology. Eur J Heart Fail 10:933–989
Jessup M, Abraham WT, Casey DE, Feldman AM, Francis GS, Ganiats TG et al (2009) Focused update: ACCF/AHA guidelines for the diagnosis and management of heart failure in adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 119:1977–2016
McMurray J, Adamopoulos S, Anker S, Auricchio A, Bӧhm M, Dickstein K et al (2012) ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2012- the task force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail 14:803–869
Butler J, Fonarow GC, Zile MR, Lam CS, Roessig L, Schelbert EB et al (2014) Developing therapies for heart failure with preserved ejection fraction: current state and future directions. JACC Heart Fail 2:97–112
Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Drazner MH et al (2013) 2013 ACCF/AHA guideline for the Management of Heart Failure: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 128:1810–1852
Filippatos G, Khan SS, Ambrosy AP, Cleland JGF, Collins SP, Lam CSP et al (2015) International registry to assess medical practice with longitudinal observation for treatment of heart failure (REPORT-HF): rationale for and design of a global registry. Eur J Heart Fail 17:527–533
Jugdutt BI (2010) Aging and heart failure: changing demographics and implications for therapy in the elderly. Heart Fail Rev 15:401–405
Jugdutt BI (2010) Heart failure in the elderly: advances and challenges. Expert Rev Cardiovasc Ther 8:695–715
Jugdutt BI (ed) (2014) Aging and heart failure: mechanisms and management. Springer, New York
Jugdutt BI (2014) Changing demographics of the aging population with heart failure and implications for therapy. Chapt 1. In: Jugdutt BI (ed) Aging and heart failure: mechanisms and management. Springer, New York, pp 1–14
Jugdutt BI (2014) Aging and remodeling of the RAS and RAAS and related pathways: implications for heart failure therapy. Chapt 18. In: Jugdutt BI (ed) Aging and heart failure: mechanisms and management. Springer, New York, pp 259–290
Jugdutt BI (2014) Biology of aging and implications for heart failure therapy and prevention. Chapt 2. In: Jugdutt BI (ed) Aging and heart failure: mechanisms and management. Springer, New York, pp 14–34
Bleumink GS, Knetsch AM, Sturkenboom MCJM, Straus SMJM, Hofman A, Deckers JW et al (2004) Quantifying the heart failure epidemic: prevalence, incidence rate, lifetime risk and prognosis of heart failure The Rotterdam Study. Eur Heart J 25:1614–1619
Sakata Y, Shimokawa H (2013) Epidemiology of heart failure in Asia. Circ J 77:2209–2217
Roger VL (2013) Epidemiology of heart failure. Circ Res 113:646–659
Savarese G, Lund LH (2017) Global public health burden of heart failure. Card Fail Rev 3:7–11
Braunwald E (2012) Paul Lichtlen lecture 2011: the rise of cardiovascular medicine. Eur Heart J 33:838–846
Braunwald E (2013) Research advances in heart failure. A compendium. Circ Res 113:633–645
Braunwald E (2015) The war against heart failure: the Lancet lecture. Lancet 385:812–824
Tsutsui H, Kinugawa S, Matsushima S (2011) Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol 301:H2181–H2190
Münzel T, Gori T, Keaney JF Jr, Maack C, Daiber A (2015) Pathophysiological role of oxidative stress in systolic and diastolic heart failure and its therapeutic implications. Eur Heart J 36:2555–2564
Murphy MP (2009) How mitochondria produce reactive oxygen species. Biochem J 417:1–13
Murphy E, Ardehali H, Balaban RS, DiLisa F, Dorn GW, Kitsis RN et al (2016) American Heart Association council on basic cardiovascular sciences, council on clinical cardiology, and council on functional genomics and translational biology. Mitochondrial function, biology, and role in disease: a scientific statement from the American Heart Association. Circ Res 118:1960–1991
Sack MN, Fyhrquist FY, Saijonmaa OJ, Fuster V, Kovacic JC (2017) Basic biology of oxidative stress and the cardiovascular system: part 1 of 3-part series. J Am Coll Cardiol 70:196–211
Münzel T, Camici GG, Maack C, Bonetti NR, Fuster V, Kovacic JC (2017) Impact of oxidative stress on the heart and vasculature: part 2 of a 3-part series. J Am Coll Cardiol 70:212–229
Niemann B, Rohrback S, Miller MR, Newby DE, Fuster V, Kovacic JC (2017) Oxidative stress and cardiovascular risk: obesity, diabetes, smoking, and pollution: part 3 of a 3-part series. J Am Coll Cardiol 70:230–251
Jugdutt BI (2003) Ventricular remodeling post-infarction and the extracellular collagen matrix. When is enough enough? Circulation 108:1395–1403
Jugdutt BI (2003) Remodeling of the myocardium and potential targets in the collagen degradation and synthesis pathways. Curr Drug Targets Cardiovasc Haematol Disord 3:1–30
Jugdutt BI (2008) Aging and remodeling during healing of the wounded heart: current therapies and novel drug targets. Curr Drug Targets 9:325–344
Jugdutt BI, Michorowski B (1987) Role of infarction expansion in rupture of the ventricular septum after acute myocardial infarction. A two-dimensional echocardiography study. Clin Cardiol 10:641–652
Jugdutt BI, Warnica JW (1988) Intravenous nitroglycerin therapy to limit myocardial infarct size, expansion and complications: effect of timing, dosage and infarct location. Circulation 78:906–919. Erratum in (1989) Circulation 79:1151
Jugdutt BI, Basualdo CA (1989) Myocardial infarct expansion during indomethacin and ibuprofen therapy for symptomatic post-infarction pericarditis: effect of other pharmacologic agents during early remodelling. Can J Cardiol 5:211–221
Jugdutt BI (1990) Identification of patients prone to infarct expansion by the degree of regional shape distortion on an early two-dimensional echocardiogram after myocardial infarction. Clin Cardiol 13:28–40
Pfeffer MA, Braunwald E (1990) Ventricular remodelling after myocardial infarction. Circulation 81:1161–1172
Jugdutt BI (1993) Prevention of ventricular remodelling post myocardial infarction: timing and duration of therapy. Can J Cardiol 9:103–114
Gaudron P, Eilles C, Kugler I, Ertl G (1993) Progressive left ventricular dysfunction and remodeling after myocardial infarction. Potential mechanisms and early predictors. Circulation 87:755–763
Jugdutt BI (1996) Prevention of ventricular remodeling after myocardial infarction and in congestive heart failure. Heart Fail Rev 1:115–129
Carabello BA (2002) Concentric versus eccentric remodeling. J Cardiac Fail 8(6 Suppl):S258–S263
Paulus WJ, Tschöpe C, Sanderson JE, Rusconi C, Flachskampf FA, Rademakers FE et al (2007) How to diagnose diastolic heart failure: a consensus statement on the diagnosis of heart failure with normal left ventricular ejection fraction by the Heart Failure and Echocardiography Associations of the European Society of Cardiology. Eur Heart J 28:2539–2550
Lakatta EG, Levy D (2003) Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises. Part I. Aging arteries: a “set up” for vascular disease. Circulation 107:139–146
Bujak M, Kweon HJ, Chatila K, Li N, Taffet G, Frangogiannis NG (2008) Aging-related defects are associated with adverse cardiac remodeling in a mouse model of reperfused myocardial infarction. J Am Coll Cardiol 51:1384–1392
Jugdutt BI (2011) Modulators of remodeling after myocardial infarction. In: Dhalla NS, Nagano M, Ostadal B (eds) Molecular defects in cardiovascular disease. Springer Media, Inc, New York, pp 231–242
Jugdutt BI, Jelani A (2013) Aging and markers of adverse remodeling after myocardial infarction, chapter 27. In: Jugdutt BI, Dhalla NS (eds) Cardiac remodeling: molecular mechanisms. Springer, New York, pp 487–512
Jugdutt BI (2013) Regulation of fibrosis after myocardial infarction: implications for ventricular remodeling, chapter 29. In: Jugdutt BI, Dhalla NS (eds) Cardiac remodeling. Molecular mechanisms. Springer, New York, pp 525–545
Jugdutt BI, Amy RW (1986) Healing after myocardial infarction in the dog: changes in infarct hydroxyproline and topography. J Am Coll Cardiol 7:91–102
Jugdutt BI, Khan MI (1992) Impact of increased infarct transmurality on remodeling and function during healing after anterior myocardial infarction in the dog. Can J Physiol Pharmacol 70:949–958
Jugdutt BI, Tang SB, Khan MI, Basulado CA (1992) Functional impact on remodeling during healing after non-Q-wave versus Q-wave anterior myocardial infarction in the dog. J Am Coll Cardiol 20:722–731
Jugdutt BI, Joljart MJ, Khan MI (1996) Rate of collagen deposition during healing after myocardial infarction in the rat and dog models: mechanistic insights into ventricular remodeling. Circulation 94:94–101
Spinale FG (2002) Matrix metalloproteinases: regulation and dysregulation in the failing heart. Circ Res 90:520–530
Spinale FG (2007) Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function. Physiol Rev 87:1285–1342
Jugdutt BI, Jelani A, Palaniyappan A, Idikio H, Uweira RE, Menon V et al (2010) Aging-related changes in markers of ventricular and matrix remodelling after reperfused ST-segment elevation myocardial infarction in the canine model. Effect of early therapy with an angiotensin II type 1 receptor blocker. Circulation 122:341–351
Tromp J, Westenbrink BD, Ouwerkerk W, van Veldhuisen DJ, Samani NJ, Ponikowski P et al (2018) Identifying pathophysiological mechanisms in heart failure with reduced versus preserved ejection fraction. J Am Coll Cardiol 72:1081–1090
Sorop O, Heinonen I, van Kranenburg M, van de Wouw J, de Beer VJ, Nguyen TN et al (2018) Multiple comorbidities produce left ventricular diastolic dysfunction associated with coronary microvascular dysfunction, oxidative stress, and myocardial stiffening. Cardiovasc Res 114:954–964
O’Gallagher K, Shah AM (2018) Modelling the complexity of heart failure with preserved ejection fraction. Cardiovasc Res 114:919–921
Shah SJ, Lam CSP, Svedlund S, Saraste A, Hage C, Tan R-S et al (2018) Prevalence and correlates of coronary microvascular dysfunction in heart failiure with preserved ejection fraction: PROMIS-HFpEF. Eur Heart J 39:3439–3450
Wei J, Nelson MD, Sharif B, Scufelt C, Merz CNB (2018) Why do we care about coronary microvascular dysfunction and heart failure with preserved ejection fraction: addressing knowledge gaps for evidence-based guidelines. Eur Heart J 39:3451–3453
Kato S, Saito N, Kirigaya H, Gyotoku D, Linuma N, Kusakawa Y et al (2016) Impairment of coronary flow reserve evaluated by phase contrast cine-magnetic resonance imaging in patients with heart failure with preserved ejection fraction. J Am Heart Assoc 5:e002649. https://doi.org/10.1161/JAHA.115.002649)
van Heerebeek L, Hamdani N, Falcão-Pires I, Leite-Moreira AF, Begieneman MP, Bronzwaer JG et al (2012) Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation 126:830–839
Paulus WJ, Tschöpe C et al (2013) A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol 62:263–271
Ter Maaten JM, Damman K, Verhaar MC, Paulus WJ, Duncker DJ, Cheng C et al (2016) Connecting heart failure with preserved ejection fraction and renal dysfunction: the role of endothelial dysfunction and inflammation. Eur J Heart Fail 18:588–598
Hiebert JB, Shen Q, Thimmesch A, Pierce J (2017) Impaired myocardial bioenergetics in HFpEF and the role of antioxidants. Open Cardiovasc Med J 10:158–162
Negi SI, Jeong EM, Shukrullah I, Veleder E, Jones DP, Fan TH et al (2015) Renin-angiotensin activation and oxidative stress in early heart failure with preserved ejection fraction. Biomed Res Int 2015:825027. https://doi.org/10.1155/2015/825027
Vaduganathan M, Patel RB, Michel A, Shah SJ, Senni MGheorgiade M et al (2017) Mode of death in heart failure with preserved ejection fraction. J Am Coll Cardiol 69:556–569
Rush CJ, Campbell RT, Jhund PS, Petrie MC, McMurray JJV (2018) Association is not causation: treatment effects cannot be estimated from observational data in heart failure. Eur Heart J 39:3417–3438
Shah SJ, Katz DH, Selvaraj S, Burke MA, Yancy CW, Gheorghiade M et al (2015) Phenomapping for novel classification of heart failure with preserved ejection fraction. Circulation 131:269–279
McMurray JJ, Packer M, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, PARADIGM-HF Investigators and Committees et al (2014) Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med 371:993–1004
Maggioni AP, Anker SD, Dahlstrӧm U, Filippatos G, Ponikowski P, Zannad F et al (2013) Are hospitalized or ambulatory patients with heart failure treated in accordance with European Society of Cardiology guidelines? Evidence from 12,440 patients of the ESC Heart Failure Long-Term Registry. Eur J Heart Fail 15:1173–1184
Mernendez JT (2016) The mechanism of action of LCZ696. Card Fail Rev 2:40–46
Hayman S, Atherton JJ (2016) Should angiotensin receptor neprilysin inhibitors replace angiotensin converting enzyme inhibitors in heart failure with a reduced ejection fraction? Card Fail Rev 2:47–50
Solomon SD, Zile M, Pieske B, Voors A, Shah A, Kraigher-Krainer E et al (2012) The angiotensin receptor neprilysin inhibitor LCZ696 in heart failure with preserved ejection fraction: a phase 2 double-blind randomised controlled trial. Lancet 380:1387–1395
Senni M, Paulus WJ, Gavazzi A, Fraser AG, Diez J, Solomon SD et al (2014) New strategies for heart failure with preserved ejection fraction: the importance of targeted therapies for heart failure phenotypes. Eur Heart J 35:2797–2815
Lim SL, Lam CSP (2016) Breakthrough in heart failure with preserved ejection fraction: are we there yet? Korean J Intern Med 31:1–14
Owens AT, Brozena SC, Jessup M (2016) New management strategies in heart failure. Circ Res 118:480–495
Ilieșiu AM, Hodorogea AS (2018) Treatment of heart failure with preserved ejection fraction. Adv Exp Med Biol 1067:67–87
Zheng SL, Roddick AJ, Aghar-Jaffar R, Shun-Shin MJ, Francis D, Oliver N et al (2018) Association between use of sodium-glucose cotransporter 2 inhibitors, glucagon-like peptide 1 agonists, and dipeptidyl peptidase 4 inhibitors with all-cause mortality in patients with type 2 diabetes: a systematic review and meta-analysis. JAMA 319:1580–1591
Das SR, Everett BM, Birtcher KK, Brown JM, Cefalu WT, Januzzi JL Jr et al (2018) 2018 ACC expert consensus decision pathway on novel therapies for cardiovascular risk reduction in patients with type 2 diabetes and atherosclerotic cardiovascular disease pathways. A report of the American College of Cardiology task force on expert consensus decision pathways Endorsed by the American Diabetes Association Writing Committee. J Am Coll Cardiol. https://doi.org/10.1016/j.jacc.2018.09.020
Pitt B, Pfeffer MA, Assmann SF, Boineau R, Anand IS, Claggett B (2014) Spironolactone for heart failure with preserved ejection fraction. N Engl J Med 370:1383–1392
Redfield MM, Chen HH, Borlaug BA, Semigran MJ, Lee KL, Lewis G et al (2013) Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA 309:1268–1277
Redfield MM, Anstrom KJ, Levine JA (2015) Isosorbide mononitrate in heart failure with preserved ejection fraction. N Engl J Med 373:2314–2324
Fukuta H, Goto T, Wakami K, Ohte N (2016) Effects of drug and exercise intervention on functional capacity and quality of life in heart failure with preserved ejection fraction: a meta-analysis of randomized controlled trials. Eur J Prev Cardiol 23:78–85
Jugdutt BI (1985) Delayed effects of early infarct-limiting therapies on healing after myocardial infarction. Circulation 72:907–914
Jugdutt BI (1996) Pharmacological intervention in post-infarction wound healing. EXS 76:501–512
Jugdutt BI, Lucas A, Khan MI (1997) Effect of angiotensin-converting enzyme inhibition on infarct collagen deposition and remodelling during healing after transmural canine myocardial infarction. Can J Cardiol 13:657–668
Jugdutt BI, Menon V (2002) Beneficial effects of therapy on the progression of structural remodeling during healing after reperfused and nonreperfused myocardial infarction: different effects on different parameters. J Cardiovasc Pharmacol Ther 7:95–107
Jugdutt BI, Palaniyappan A, Uwiera RR, Idikio H (2009) Role of healing-specific-matricellular proteins and matrix metalloproteinases in age-related enhanced early remodeling after reperfused STEMI in dogs. Mol Cell Biochem 322:25–36
Jugdutt BI (2008) Pleiotropic effects of cardiac drugs on healing post-MI. The good, bad, and ugly. Heart Fail Rev 13:439–452
Jugdutt BI, Idikio H, Uwiera RR (2007) Therapeutic drugs during healing after myocardial infarction modify infarct collagens and ventricular distensibility at elevated pressures. Mol Cell Biochem 304:79–91
Jugdutt BI (2007) Cyclooxygenase inhibition and adverse remodeling during healing after myocardial infarction. Circulation 115:288–291
Jugdutt BI (2006) Matrix metalloproteinases as markers of adverse remodeling after myocardial infarction. J Card Fail 12:73–76
Jugdutt BI, Jelani A (2008) Aging and defective healing, adverse remodeling, and blunted post-conditioning in the reperfused wounded heart. J Am Coll Cardiol 51:1399–1403
Palaniyappan A, Uwiera RR, Idikio H, Menon V, Jugdutt C, Jugdutt BI (2013) Attenuation of increased secretory leukocyte protease inhibitor, matricellular proteins and angiotensin II and left ventricular remodeling by candesartan and omapatrilat during healing after reperfused myocardial infarction. Mol Cell Biochem 376:175–188
Jugdutt BI (2011) Optimizing pharmacotherapy for limiting cardiovascular remodeling a matter of timing therapy to match biology. J Am Coll Cardiol 57:2029–2030
Manhenke C, Ueland T, Jugdutt BI, Godang K, Aukrust P, Dickstein K et al (2014) The relationship between markers of extracellular cardiac matrix turnover: infarct healing and left ventricular remodelling following primary PCI in patients with first-time STEMI. Eur Heart J 35:395–402
Frangogiannis NG, Smith CW, Entman ML (2002) The inflammatory response in myocardial infarction. Cardiovasc Res 53:31–47
Nahrendorf M, Pittet MJ, Swirski FK (2010) Monocytes: protagonists of infarct inflammation and repair after myocardial infarction. Circulation 121:2437–2445
Jugdutt BI (2015) Aging-related changes in cardiac extracellular matrix: implications for heart failure in older patients. J Cardiol Curr Res 3(3):00101. https://doi.org/10.15406/jccr.2015.03.00101
Weber KT (1997) Extracellular matrix remodeling in heart failure. A role for De Novo Angiotensin II generation. Circulation 96:4065–4082
Jugdutt BI (2005) Extracellular matrix and cardiac remodeling, chapter 3. In: Villareal FJ (ed) Interstitial fibrosis in heart failure. Springer, New York, pp 23–55
Deschamps AM, Spinale FG (2006) Pathways of matrix metalloproteinase induction in heart failure: bioactive molecules and transcriptional regulation. Cardiovasc Res 69:666–676
Spinale FG, Zile MR (2013) Integrating the myocardial matrix into heart failure recognition and management. Circ Res 113:300–309
Jugdutt BI (2014) Cardiac matrix remodeling and heart failure. In: Li R (ed) Cardiac regeneration and repair: pathology and therapies, Vol 1, Part 1. Woodhead Publishing Ltd, Oxford, pp 3–26
O’Gara PT, Kushner FG, Ascheim DD, Casey DE Jr, Chung MK, de Lemos JA et al (2013) 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: executive summary: a report of the American college of cardiology foundation/American heart association task force on practice guidelines. J Am Coll Cardiol 61:485–510
Ibanez B, James S, Agewall S, Antunes MJ, Bucciarelli-Ducci C, Bueno H et al (2018) 2017 ESC guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation: the task force for the management of acute myocardial infarction in patients presenting with ST-segment elevation of the European Society of Cardiology (ESC). Eur Heart J 39:119–177
Hausenloy DJ, Botker HE, Engstrom T, Erlinge D, Heusch G, Ibanez B et al (2017) Targeting reperfusion injury in patient with ST-segment elevation myocardial infarction: trials and tribulations. Eur Heart J 38:935–941
Libby P (2001) Current concepts of the pathogenesis of the acute coronary syndromes. Circulation 104:365–372
Ridker PM, Rifai N, Pfeffer MA, Sacks F, Braunwald E (1999) Long-term effects of pravastatin on plasma concentration of C-reactive protein: the cholesterol and recurrent events (CARE) investigators. Circulation 100:230–235
Ridker PM, Everett TT, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, for the CANTOS trial group et al (2017) Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med 377:1119–1131
Ridker PM, Libby P, MacFadyen JG, Thuren T, Ballantyne C, Fonseca F, on behalf of the CANTOS Trial Group et al (2018) Modulation of the interleukin-6 signalling pathway and incidence rates of atherosclerotic events and all-cause mortality: analyses from the canakinumab anti-inflammatory thrombosis outcomes study (CANTOS). Eur Heart J 39:3499–3507
Everett BM, Cornel JH, Lainscak M, Anker SD, Abbate A, Thuren T et al (2018) Anti-inflammatory therapy with canakinumab for the prevention of hospitalization for heart failure. Circulation. https://www.ahajournals.org/. https://doi.org/10.1161/CIRCULATIONAHA.118.038010
Crea F, Libby P (2017) Acute coronary syndromes. The way forward from mechanisms to precision treatment. Circulation 136:1155–1166
Ridker PM (2017) Canakinumab for residual inflammatory risk. Implications of CANTOS for clinical practice and drug development. Eur Heart J 38:3545–3548. https://doi.org/10.1093/eurheartj/ehx723
Abbate A, Dinarello CA (2015) Anti-inflammatory therapies in acute coronary syndromes: is IL-1 blockade a solution? Eur Heart J 36:337–339
Morton AC, Rothman AM, Greenwood JP, Gunn J, Chase A, Clarke B et al (2015) The effect of interleukin-1 receptor antagonist therapy on markers of inflammation in non-ST elevation acute coronary syndromes: the MRC-ILA Heart Study. Eur Heart J 36:377–384
Frangogiannis NG (2004) Chemokines in the ischemic myocardium: from inflammation to fibrosis. Inflamm Res 53:585–595
Shetelig C, Limalanathan S, Hoffmann P, Seljeflot I, Gran JM, Eritsland J et al (2018) Association of IL-8 with infarct size and clinical outcomes in patients with STEMI. J Am Coll Cardiol 72:187–198
Bolli R, Jeroudi MO, Patel BS, DuBose CM, Lai EK, Roberts R et al (1989) Direct evidence that oxygen-derived free radicals contribute to postischemic myocardial dysfunction in the intact dog. Proc Natl Acad Sci U S A 86:4695–4599
Prabhu SD, Frangogiannis NG (2016) The biological basis for cardiac repair after myocardial infarction. From inflammation to fibrosis. Circ Res 119:91–112
Granger CB, Kochar A (2018) Understanding and targeting inflammation in acute myocardial infarction. An elusive goal. Am Coll Cardiol 72:199–200
Ruparelia N, Godec J, Lee R, Chai JT, Dall’Armellina E, McAndrew D et al (2015) Acute myocardial infarction activates distinct inflammation and proliferation pathways in circulating monocytes, prior to recruitment, and identified through conserved transcriptional responses in mice and humans. Eur Heart J 36:1923–1934
Panizzi P, Swirski FK, Figueiredo JL, Waterman P, Sosnovik D, Aikawa E et al (2010) Impaired infarct healing in atherosclerotic mice with Ly-6Chi monocytosis. J Am Coll Cardiol 55:1629–1638
Paneni F, Diaz Cañestro C, Libby P, Lüscher TF, Camici GG (2017) The aging cardiovascular system: understanding it at the cellular and clinical levels. J Am Coll Cardiol 69:1952–1967
Haycock PC, Heydon EE, Kaptoge S, Butterworth AS, Thompson A, Willeit P et al (2014) Leucocyte telomere length and risk of cardiovascular disease: systematic review and meta-analysis. BMJ 349:g4227. https://doi.org/10.1136/bmj.g4227
Ricci R, Eriksson U, Oudit GY, Eferl R, Akhmedov A, Sumara I et al (2005) Distinct functions of junD in cardiac hypertrophy and heart failure. Genes Dev 19:208–213
Kroemer G, Marino G, Levine B (2010) Autophagy and the integrated stress response. Mol Cell 40:280–293
Dai DF, Chiao YA, Marcinek DJ, Szeto HH, Rabinovitch PS (2014) Mitochondrial oxidative stress in aging and healthspan. Longevity & Healthspan 3:6. https://doi.org/10.1186/2046-2395-3-6
Terman A, Brunk UT (2005) Autophagy in cardiac myocyte homeostasis, aging, and pathology. Cardiovasc Res 68:355–365
Terman A, Kurz T, Navratil M, Arriaga EA, Brunk UT (2010) Mitochondrial turnover and aging of long-lived postmitotic cells: the mitochondrial-lysosomal axis theory of aging. Antioxid Redox Signal 12:503–535
Judge S, Jang YM, Smith A, Hagen T, Leeuwenburgh C (2005) Age-associated increases in oxidative stress and antioxidant enzyme activities in cardiac interfibrillar mitochondria: implications for the mitochondrial theory of aging. FASEB J 19:419–421
Linton P-J, Gurney M, Sengstock D, Mentzer RM Jr, Gottlieb RA (2016) This old heart: cardiac aging and autophagy. J Mol Cell Cardiol 83:44–54
Levine B, Mizushima N, Virgin HW (2011) Autophagy in immunity and inflammation. Nature 469:323–335
Kassiotis C, Ballal K, Wellnitz K, Vela D, Gong M, Salazar R et al (2009) Markers of autophagy are downregulated in failing human heart after mechanical unloading. Circulation 120:S191–S197
Garcia L, Verdejo HE, Kuzmicic J, Zalaquett R, Gonzalez S, Lavandero S et al (2012) Impaired cardiac autophagy in patients developing postoperative atrial fibrillation. J Thorac Cardiovasc Surg 143:451–459
Jahania SM, Sengstock D, Vaitkevicius P, Andres A, Ito BR, Gottlieb RA et al (2013) Activation of the homeostatic intracellular repair response during cardiac surgery. J Am Coll Surg 216:719–726
Singh KK, Yanagawa B, Quan A, Wang R, Garg A, Khan R et al (2014) Autophagy gene fingerprint in human ischemia and reperfusion. J Thorac Cardiovasc Surg 147:1065–1072
Gedik N, Thielmann M, Kottenberg E, Peters J, Jakob H, Heusch G et al (2014) No evidence for activated autophagy in left ventricular myocardium at early reperfusion with protection by remote ischemic preconditioning in patients undergoing coronary artery bypass grafting. PLoS One 9:e96567
Sengstock D, Jahania S, Andres A, Ito BR, Gottlieb RA, Mentzer RM Jr (2014) Homeostatic intracellular repair response (HIR2) is increased in older adults and is upregulated by ischemia. J Am Geriatr Soc 62:S108
Kanamori H, Takemura G, Goto K, Maruyama R, Tsujimoto A, Ogino A et al (2011) The role of autophagy emerging in postinfarction cardiac remodeling. Cardiovasc Res 91:330–339
Gao T, Zhang S-P, Wang J-F, Liu L, Wang Y, Cao Z-Y et al (2018) TLR3 contributes to persistent autophagy and heart failure in mice after myocardial infarction. J Cell Mol Med 22:395–408
Goldin A, Beckman JA, Schmidt AM, Creager MA (2006) Advanced glycation end products. Sparking the development of diabetic vascular injury. Circulation 114:597–605
Prasad K, Mishra M (2018) AGE-RAGE stress, stressors, and antistressors in health and disease. Int J Angiol 27:1–12
Selye H (1976) Stress in health and disease, 1st edn. Butterworths, Boston, pp 762–807
Bartesagh S, Radi (2018) Fundamentals on the biochemistry of peroxynitrite and protein tyrosine nitration. Redox Biol 14:618–625
Kumar D, Jugdutt BI (2003) Apoptosis and oxidants in the heart. J Lab Clin Med 142:288–297
Jugdutt BI, Idikio HA (2005) Apoptosis and oncosis in acute coronary syndromes: assessment and implications. Mol Cell Biochem 270:177–200
Bolli R, Jeroudi M, Patel B, Aruoma O, Halliwell B, Lai E et al (1989) Marked reduction of free radical generation and contractile dysfunction by antioxidant therapy begun at the time of reperfusion. Evidence that myocardial “stunning” is a manifestation of reperfusion injury. Circ Res 65:607–622
Ducas A, Bartekova M, Dhalla NS (2015) Ischemia-reperfusion injury of the heart: moving forward with our knowledge. J Heart Health 1:1–10. https://doi.org/10.16966/2379-769X.110
Jugdutt BI (2002) Nitric oxide and cardioprotection during ischemia-reperfusion. Heart Fail Rev 7:391–405
Neumann F-J, Gick M (2018) Direct stenting in ST-segment elevation myocardial infarction: convenient, but not improving outcomes. Eur Heart J 39:2480–2483
Mahmood KO, Jolly SS, James S, Dzavik V, Cairns JA, Olivecrona CK et al (2018) Clinical impact of direct stenting and interaction with thrombus aspiration in patients with ST-segment elevation myocardial infarction undergoing percutaneous coronary intervention: thrombectomy trialists collaboration. Eur Heart J 39:2472–2479
Grieve DJ, Byrne JA, Cave AC, Shah A (2004) Role of oxidative stress in cardiac remodelling after myocardial infarction. Heart Lung Circ 13:132–138
Hill MF, Singal PK (1996) Antioxidant and oxidative stress changes during heart failure subsequent to myocardial infarction in rats. Am J Pathol 148:291–300
Kinugawa S, Tsutsui H, Hayashidani S, Ide T, Suematsu N, Satoh S et al (2000) Treatment with dimethylthiourea prevents left ventricular remodelling and failure after experimental myocardial infarction in mice: role of oxidative stress. Circ Res 87:392–398
Singh RB, Niaz MA, Rastogi SS, Rastogi S (1996) Usefulness of antioxidant vitamins in suspected acute myocardial infarction (the Indian experiment of infarct survival-3). Am J Cardiol 77:232–236
Dhalla AK, Hill MF, Singal PK (1996) Role of oxidative stress in transition of hypertrophy to heart failure. J Am Coll Cardiol 28:506–514
Ide T, Tsutsui H, Kinugawa S, Utsumi H, Kang D, Hattori N et al (1999) Mitochondrial electron transport complex I is a potential source of oxygen free radicals in the failing myocardium. Circ Res 85:357–363
Kim K-S, Takeda K, Sethi R, Pracyk JB, Tanaka K, Zhou YF et al (1998) Protection from reoxygenation injury by inhibition of rac1. J Clin Invest 101:1821–1826
Talukder MAH, Elnakish MT, Yang F, Nishijima Y, Alhai MA, Velayutham M et al (2013) Cardiomyocyte-specific overexpression of an active form of Rac predisposes the heart to increased myocardial stunning and ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 304:H294–H302
Ide T, Tsutsui H, Kinugawa S, Suematsu N, Hayashidani S, Ichikawa K et al (2000) Direct evidence for increased hydroxyl radicals originating from superoxide in the failing myocardium. Circ Res 86:152–157
Griendling KK, Sorescu D, Ushio-Fukai M (2000) NAD(P)H oxidase. Role in cardiovascular biology and disease. Circ Res 86:494–501
Heymes C, Bendall JK, Ratajczak P, Cave AC, Samuel JL, Hasenfuss G et al (2003) Increased myocardial NADPH oxidase activity in human heart failure. J Am Coll Cardiol 41:2164–2171
Bauersachs J, Bouloumie A, Fraccarollo D, Hu K, Busse R, Ertl G (1999) Endothelial dysfunction in chronic myocardial infarction despite increased vascular endothelial nitric oxide synthase and soluble guanylate cyclase expression: role of enhanced vascular superoxide production. Circulation 100:292–298
Saavedra WF, Paolocci N, St. John ME, Skaf MW, Stewart GC, Xie JS et al (2002) Imbalance between xanthine oxidase and nitric oxide synthase signaling pathways underlies mechanoenergetic uncoupling in the failing heart. Circ Res 90:297–304
Lentsch AB, Ward PA (2000) Regulation of inflammatory vascular damage. J Pathol 190:343–348
Irani K, Xia Y, Zweier JL, Sollott SJ, Der CJ, Fearon ER et al (1997) Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science 275:1649–1652
Arnold RS, Shi J, Murad E, Whalen AM, Sun CQ, Polavarapu R et al (2001) Hydrogen peroxide mediates the cell growth and transformation caused by the mitogenic oxidase Nox1. Proc Natl Acad Sci U S A 98:5550–5555
Siwik DA, Pagano PJ, Colucci WS (2001) Oxidative stress regulates collagen synthesis and matrix metalloproteinase activity in cardiac fibroblasts. Am J Phys 280:C53–C60
Morin I, Li WQ, Su S, Ahmad M, Zafarullah M (1999) Induction of stromelysin gene expression by tumor necrosis factor alpha is inhibited by dexamethasone, salicylate, and N-acetylcysteine in synovial fibroblasts. J Pharmacol Exp Ther 289:1634–1640
Sano M, Fukuda K, Sato T, Kawaguchi H, Suematsu M, Matsuda S et al (1999) ERK and p38 MAPK, but not NF-kB, are critically involved in reactive oxygen species-mediated induction of IL-6 by angiotensin II in cardiac fibroblasts. Circ Res 89:661–669
Kunsch C, Medford RM (1999) Oxidative stress as a regulator of gene expression in the vasculature. Circ Res 85:753–766
Campbell SE, Katwa LC (1997) Angiotensin II stimulated expression of transforming growth factor-beta1 in cardiac fibroblasts and myofibroblasts. J Mol Cell Cardiol 29:1947–1958
Anthonio RL, van Veldhuisen DJ, van Gilst WH (1998) Left ventricular dilatation after myocardial infarction: ACE inhibitors, beta-blockers, or both? J Cardiovasc Pharmacol 32(Suppl 1):S1–S8
Mankad S, d’Amato TA, Reichek N, McGregor WE, Lin J, Singh D et al (2001) Combined angiotensin II receptor antagonism and angiotensin-converting enzyme inhibition further attenuates postinfarction left ventricular remodelling. Circulation 103:2845–2850
Diez J, Querejeta R, Lopez B, Gonzalez A, Larman M, Martinez Ubago JL (2002) Losartan-dependent regression of myocardial fibrosis is associated with reduction of left ventricular chamber stiffness in hypertensive patients. Circulation 105:2512–2517
Kuno A, Miura T, Tsuchida A, Hasegawa T, Miki T, Nishino Y et al (2002) Blockade of angiotensin II type 1 receptors suppressed free radical production and preserved coronary endothelial function in the rabbit heart after myocardial infarction. J Cardiovasc Pharmacol 39:49–57
Moe G, Konig A, Liu P, Jugdutt BI (2008) Selective type 1 angiotensin II receptor blockade attenuates oxidative stress and regulates angiotensin II receptors in the canine failing heart. Mol Cell Biochem 17:97–104
Sia YT, Lapointe N, Parker TG, Tsoporis JN, Deschepper CF, Calderone A et al (2002) Beneficial effects of long-term use of the antioxidant probucol in heart failure in the rat. Circulation 105:2549–2555
Marín-García J, Damle S, Jugdutt BI, Moe GW (2012) Nuclear-mitochondrial cross-talk in global myocardial ischemia. A time-course analysis. Mol Cell Biochem 364:225–234
Acknowledgement
None.
Funding Sources
None.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Jugdutt, B.I., Jugdutt, B.A. (2019). Oxidative Stress and Heart Failure. In: Chakraborti, S., Dhalla, N., Dikshit, M., Ganguly, N. (eds) Modulation of Oxidative Stress in Heart Disease. Springer, Singapore. https://doi.org/10.1007/978-981-13-8946-7_11
Download citation
DOI: https://doi.org/10.1007/978-981-13-8946-7_11
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-8945-0
Online ISBN: 978-981-13-8946-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)