
Abstract
Fungi are well known for their ability to produce a multitude of secondary metabolites (SMs) which act as a weapon of defense to protect themselves against parasites and predators. A variety of fungal SMs have proved to serve as an important factor for decades. The synthesis of SMs in fungi is a complex, multi-step process and is stage-specific under specialized conditions. SMs are primarily synthesized by non-ribosomal peptide synthetase (NRPS) or polyketide synthase (PKS) enzymes. The genes encoded for SM synthesis are often located in the cluster form at the sub-telomere region. The SM biosynthetic gene cluster comprises of genes encoding for NRPS/PKS, a transcription factor, and other accessory genes essential for assembly and maturation of SM. The regulation of SM synthesis in fungi can be achieved by pathway-specific (in-clustered transcription factor), global regulatory proteins and chromatin remodeling. The regulatory protein-encoding gene present in each gene cluster is considered to be a crucial regulatory circuit of the SM biosynthetic pathway. Moreover, the regulation of fungal SM biosynthesis is also guided by global regulatory proteins responsive to pH, carbon, nitrogen, light/dark, and other environmental cues. Histone modifications by methylation and acetylation often altered the chromatin structure; as a result, these changes repress or express the genes of SM biosynthetic pathway. Taken together, we conclude that fungal SM ability as antibiotics to toxins is useful to mankind.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
2.1 Introduction
The enormous variety and diversity of fungi stand a prime place in the natural world. The geographical diversity of India is the most suitable to fungi that contributes more than half of the total fungal species of the world. However, the flora of the fungal kingdom is still undiscovered; thus it needs to be explored for detection of novel therapeutics for the treatment of a plethora of pathogens. Fungi are well known for their ability to produce a multitude of secondary metabolites (SMs) as products. SMs are small organic molecules (<4 kDa) which are unlikely to be involved in cell growth and development but have a pivotal role in self-defense, niche adaptation, and communication to the environment (Kamei and Watanabe 2005; Keller et al. 2005). SMs isolated from fungi have been considered the most resourceful natural product as they have been exploited by humankind as antimicrobial, antitumor, enzyme inhibitor, immunosuppressive, anticholesterol, and antiparasitic agents (Agathos et al. 1987; Demain 1999; Ramawat and Mérillon 2013). Despite their clinical importance, fungi also synthesize some toxic SM substances which are sometimes deleterious to human beings as they can cause systemic infections (Demain and Fang 2000; García-Estrada et al. 2011). A range of SMs with their effectiveness and producing organisms are listed in Table 2.1. Characteristically, synthesis of SMs in fungi is stage-specific as they are produced by a specific organism/group of organisms under certain growth/environmental conditions such as media, pH, temperature, and nitrogenous sources (Jensen et al. 2007; Fox and Howlett 2008; Brakhage 2013; Keller 2015). SM production most often occurs at the late log phase or stationary phase (Bu’Lock 1961). Earlier observation revealed that environmental conditions for both SM production and spore formation were found to be common, but they were more stringent than vegetative growth (Bu’Lock 1961; Sekiguchi and Gaucher 1977). Therefore, it is believed that synthesis of SMs is essential for sporulation. For example, the dark-brown melanin pigments synthesized by oxidative polymerization of phenolic compounds which are deposited in the cell wall during cell sporulation protect the spores from UV light and are also involved in the virulence (Calvo et al. 2002). Aside from melanins, zearalenone produced in F. graminearum also induces the sporulation (Wolf and Mirocha 1973). The fungal SM production is also linked to the asexual to sexual development. The switching from primary to secondary metabolism or asexual to sexual development depends on the psi factor or endogenous ratio of oleic acid- and linoleic acid-derived molecules (Champe and El-Zayat 1989; Calvo et al. 2001). It is well known that complex and diverse SMs derived from fungi are synthesized by sequential assembly of primary metabolic intermediate precursors molecule, i.e., acetyl coenzyme A (acetyl-CoA), 1-deoxyxylulose 5-phosphate, and mevalonic and shikimic acid (Dewick 2002). Structurally, fungal SMs are classified as non-ribosomal peptides, polyketides, indole alkaloids, and terpenes (Keller et al. 2005). The synthesis of SMs is a multi-step process which is initiated by the action of various classes of enzymes. However, a variety of tailoring enzymes, viz., transporters, oxygenases, hydroxylases, and regulatory proteins, are also involved in the biosynthesis of fungal SMs, and their encoded genes are often arranged in a cluster (Fox and Howlett 2008; Keller 2015). The reason why SM biosynthetic genes present together as clusters is still unanswered. It is speculated that all the essential genes for an SM synthesis are transcriptionally regulated by an intrinsic transcription factor (TF) under certain environmental conditions (Fox and Howlett 2008; Keller 2015).
2.2 Different Classes of Fungal Secondary Metabolites
2.2.1 Non-ribosomal Peptides
Non-ribosomal peptides are an enormous class of SMs which are derived from both proteinogenic and non-proteinogenic amino acids. SMs belonging to this class are synthesized by the large multifunctional and multi-modular enzymes called non-ribosomal peptide synthetases (NRPS) and synthesized independently to ribosomes (Stein et al. 1996). NRPS are organized in the modules, which are highly specific to a particular amino acid and also considered as an assembly unit (Fig. 2.1a). The number of modules depends on the length of the peptides. NRPS module characteristically consists of three basic subunits named as adenylation (A), peptidyl carrier protein or thiolation (PCP or T), and condensation (C) domains (Fig. 2.1b). Adenylation domain tends to activate the amino acid to amino-acyl adenylate by reacting with ATP (Fischbach and Walsh 2006). This activated amino acid is then transferred to the PCP domain which forms a thioester bond with the cysteine-thiol group of the PCP domain (Strieker et al. 2010). However, the condensation domain (C) catalyzes the peptide bond formation between amino acid bonded to the thiolation domain and nascent peptide chain on the preceding module. In addition to the minimal core module, NRPS enzymes also contain associatory domains such as cyclization (Cy), epimerization (E), oxidation (Ox), thioesterase (TE), and methyltransferase (MT) domains essential for maturation of SM molecule (Hoffmeister and Keller 2007; Strieker et al. 2010; Evans et al. 2011). The cyclization domain (Cy) is a variant of the classical C domain which introduces thiazoline or oxazoline ring on β-heteroatom-bearing amino acids such as threonine, serine, and cysteine. The heterocyclization of participatory amino acids of SM prevents them from the action of cellular protease (Marshall et al. 2001; Kelly et al. 2005).

Organization of multi-modular non-ribosomal peptide synthetase (NRPS). (a). Arrangement of different enzyme modules of NRPS. Each module contains adenylation (A), peptidyl carrier protein or thiolation (PCP or T), and condensation (C) domains. Amino acids are sequentially assembled by NRPS enzyme. Initiation module lacking C domain and termination module contains thioesterase domain (TE) which is involved in the cleavage. (b). Specified domains catalyze the amino acid to the peptides. Adenylation activates amino acid to amino-acyl adenylate by reacting with ATP, and it attaches to the PCP. Condensation domain catalyzes peptide bond formation between amino acid bonded to the thiolation domain and nascent peptide chain on preceding module
Initiation modules usually lacking C domain and termination modules usually possess an essential thioesterase (TE) domain at their C terminal. TE domain catalyzes product hydrolytic cleavage or complex macro-cyclization and releases the peptide molecule (Fig. 2.1b) (Strieker et al. 2010). A range of non-ribosomal peptides are methylated by MT domain which contain C-methyl or N-methyl groups containing amino acids using S-adenosyl methionine (SAM) as co-substrate (Walsh et al. 2001). Epimerization (E) domain is meant for the conversion of L-amino acids to the corresponding D-isomer of the peptides (Linne et al. 2001). Penicillin, cephalosporin, cyclosporine, echinocandin B, cryptocandin, and gliotoxin are the common examples of NRPS-mediated SMs (Cacho et al. 2012).
2.2.2 Polyketides
Polyketides are the structurally diverse class of fungal secondary metabolites which are primarily synthesized from the monoketide units (acetyl-CoA, methylmalonyl-CoA, and malonyl-CoA) derived from the primary metabolite pool (Keller et al. 2005; Wakimoto et al. 2011). Fungal polyketides are synthesized or assembled by polyketide synthases (PKS) which are closely related to the fatty acid synthases (FAS) (Hutchinson 2003). Similar to NRPS, PKS are also multi-modular enzymes, having ketoacyl CoA synthase (KS), acyltransferase (AT) and acyl carrier (ACP), and ketone-reducing enoyl reductase (ER), dehydratase (DH), and ketoreductase (KR) domains (Fig. 2.2) (Keller et al. 2005). PKS can be classified into three classes, PKS I, II, and III, which have identical enzymatic functions, but varying chain initiation and termination steps (Gallo et al. 2013). PKS I are complex and the most versatile in nature which can be sub-categorized into iterative and non-iterative type.

Domain organization of the PKS. The functional domains are represented by box. Loading module lacking KS domain and terminal module has a TE domain. Each module is responsible for the specific polymerization of single monoketide unit, and the specific action of respective domains synthesizes the polyketide molecule. Domains are represented as AT acyltransferase, ACP acyl carrier protein, KS ketosynthase, KR ketoreductase, DH dehydratase, ER enoyl reductase, TE thioesterase, and CoA coenzyme A
Iterative PKS I are multi-enzymes which produce non-reducing (NR), partially reducing (PR), and highly reducing (HR) polyketides. Non-iterative PKS I are organized into modules having multiple domains acting once and catalyze one cycle of polyketide chain elongation. The antibacterials orsellinic acid and erythromycin A and cholesterol-lowering agent squalestatin S1 are the known examples of iterative PKS I (Weissman 2009). PKS II are the aggregated complex of mono-functional proteins mainly comprising of ketosynthase (KS) α and β and acyl carrier protein (ACP). Each protein has a specific role in the synthesis and acts as iteratively. The iterative action of these proteins led to polyketone synthesis via chain extension to the defined number of cycles. PKS III, also known as chalcone synthases, are the homo-dimeric enzymes, which act iteratively. PKS III have simple, versatile, and broad substrate specificity and are very less abundant in fungi. They mostly utilize CoA thioesters as a substrate for decarboxylative condensation and cyclization reaction (Calne et al. 1989; Staunton and Weissman 2001; Shen 2003; Weissman 2009). The tri- and tetra-ketide pyrones, the penta- and hexa-ketide resorcylic acids, griseofulvin, bikaverin, and the penta-ketide resorcinols are such examples (Hashimoto et al. 2014; Liu et al. 2015).
2.2.3 Terpenoids
Terpenoids or terpenes are the odoriferous SMs which are mostly derived from plants (turpentine and camphor) and less commonly found in fungi (carotenoids, aristolochenes, trichothecenes, gibberellins, and indole-diterpenes). The basic unit of terpene is monomer isoprene (C5H8) which undergoes polymerization catalyzed by terpene synthases. The empirical formula of terpenes is (C5H8)n, where n represents the number of isoprene units. Structurally, terpenes can be cyclic or linear and unsaturated or saturated. Two abundant building blocks of terpenoids are isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP), derived from acetyl-CoA by glyceraldehyde 3-phosphate and pyruvate or mevalonic acid (Fig. 2.3) (Eisenreich et al. 2004).

Terpene biosynthetic pathway. Isopentenyl diphosphate and dimethylallyl diphosphate, the products of the mevalonate pathway, are the precursors of steroids, carotenoids, and coenzyme Q in many species
Terpenes are classified into three groups: monoterpenes (derived from geranyl pyrophosphate, also known as geranyl diphosphate), sesquiterpenes (derived from farnesyl pyrophosphate, also known as farnesyl diphosphate), and diterpenes/carotenoids (derived from geranyl-geranyl pyrophosphate, also known as geranyl-geranyl diphosphate) (Keller et al. 2005). Most common fungal terpenoids are carotenoids, gibberellins, and trichothecenes which act as a free-radical scavenger, plant hormone, and mycotoxin, respectively (Echavarri-Erasun and Johnson 2002; Alexander et al. 2009; Bömke and Tudzynski 2009).
2.2.4 Alkaloids
Alkaloids set up a diverse group of natural products; around 12,000 alkaloids have been identified that are most commonly produced in plants, animals, bacteria, and fungi. In the case of fungi, Ascomycota and Basidiomycota have been extensively producing the bioactive alkaloids (Guirimand et al. 2010; Xu et al. 2014). Alkaloids share the basic organic substances that contain at least one nitrogen atom in the heterocyclic ring (Mahmood et al. 2010). Alkaloids are bitter in taste and used in medical science against a diverse range of diseases and toxicants. The first microbial-derived alkaloid is ergot synthesized in Claviceps purpurea which causes an ergot of rye disease upon infection to the plant ovaries (Esser and Tudzynski 1978). Alkaloids accurately cannot be classified into families due to their diverse structure and precursor molecules, but based on structural similarities, the precursor molecule, and their source (Waller and Burström 1969; Anaya et al. 2006; Evans 2009; Roberts 2013; Wansi et al. 2013; Aniszewski 2015), alkaloid are separated in different types as shown in Table 2.2.
Indole alkaloids are most commonly produced in fungi which are derived from tryptophan and an isoprenoid precursor (dimethylallyl pyrophosphate). However, intermediate compounds of the shikimic acid pathway such as anthranilate, indole-3-glycerol-phosphate, 4-dimethylallyl tryptophan (4-DMAT), and tryptamine act as a precursor (Fig. 2.4) (Byrne et al. 2002; Xu et al. 2014). The synthesis of indole alkaloids is achieved by NRPS enzymes; adenylation (A) domain of NRPS activates the individual amino acids to aminoacyl-ADP, transfers to thiolation (T) domain, and forms aminoacyl thioesters. A variety of other enzymes, i.e., geranyl-geranyl pyrophosphate synthases, monooxygenases, methyltransferases, tryptophan decarboxylase, anthranilate synthase, chorismate mutase, strictosidine synthase, and tyrosine decarboxylase, are also involved in indole synthesis (Ziegler and Facchini 2008; Dubouzet et al. 2013; Xu et al. 2014). There are larger NRPS products that contain tryptophan, such as the tetrapeptide apicidin isolated from Fusarium semitectum that displays histone deacetylase inhibition activity (Darkin-Rattray et al. 1996; Jin et al. 2010; Xu et al. 2014).

Fungal indole alkaloid biosynthetic pathway. Paxilline biosynthesis from indole-3-glycerol phosphate in P. paxilli with the action of paxilline biosynthetic genes (Scott et al. 2013). Tryptoquialanine and meleagrin biosynthesis from L-tryptophan in P. aethiopicum and P. chrysogenum by respective biosynthetic genes (Gao et al. 2011; García-Estrada et al. 2011). Communesin F biosynthesis from L-tryptophan, tryptamine, and 4-dimethylallyl-L-tryptophan in P. expansum with respective biosynthetic genes (Lin et al. 2015)
2.3 Genetics of Secondary Metabolite Synthesis
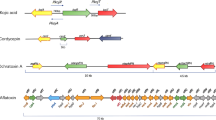
The fungal genome sequencing projects gave insight to the SM research, thus achieving the milestone for biosynthetic gene exploration and regulation (Galagan et al. 2005). Fungal SMs are synthesized by the action of a set of enzymes which are encoded by different genes. It has been reported that the SM gene clusters are randomly distributed in the genome but most often located at the sub-telomeric regions (Fig 2.5a) (Palmer et al. 2010; Cairns and Meyer 2017). The SM biosynthetic genes in fungi have special fetchers which are typically clustered in the genome and pooled to a single genetic locus (Fig 2.5b). The gene cluster encoded for SM is comprised of various functional genes. For example, in A. nidulans, each cluster contains 6–22 genes with a size ranging from 20 to 60 kb (William et al. 2005). It is expected that genes located in cluster could simplify the horizontal gene transfer and encourage the SM traits in nature (Khaldi and Wolfe 2011). It is also postulated that gene clustering could facilitate co-regulation of cluster’s genes, specifically those located in the sub-telomeric regions (Palmer and Keller 2010). The cluster genes encode for NRPS/PKS, regulatory transcription factors, MFS/ABC transporters for translocation, and hydrolases and oxidases in particular for maturation of SM. Two complex proteins either non-ribosomal peptides (NRPS) or polyketides (PKS) are involved in building the general scaffolds of most SMs as described elsewhere (Palmer and Keller 2010; Brakhage 2013).

Location, arrangement, and pathway-specific regulation of fungal SM biosynthetic genes. (a) Sub-telomeric arrangement of A. nidulans SM biosynthetic genes in the respective chromosomes. ORS orsellinic acid, ST sterigmatocystin, APD aspyridone, AUS austinol, PN penicillin, ES emericellamide, MDP monodictyphenone. (b) Physical arrangement of SM biosynthetic genes in the cluster along with NRPS/PKS. (c) Pathway-specific regulation of SM biosynthetic genes by the activation of in-cluster transcription factor
The architecture and arrangement of SM biosynthetic genes raise a question on their clustered form and sub-telomeric location. For example, there are 24 SM biosynthetic gene clusters present in the A. fumigatus genome. Out of 24 gene clusters, 8 were located in the sub-telomeric regions (William et al. 2005). Similarly, Zymoseptoria tritici contains 34 gene clusters; among them 12 are located at sub-telomeric regions (Cairns and Meyer 2017). In contrast, the sub-telomeric arrangement of the gene cluster may also influence another level of regulation incurred by telomeric positioning effect (TPE). TPE is a phenomenon in which telomeric or sub-telomeric region genes are transcriptionally repressed due to heterochromatin formation (Gottschling et al. 1990; Castaño et al. 2005; Rosas-Hernández et al. 2008; Smith et al. 2008). TPE covers up to 30-kb region of the telomere in A. nidulans. The expression for aflatoxin biosynthetic gene cluster is affected by TPE (Palmer and Keller 2010). The sub-telomeric occurrence of the gene cluster is also a signal for multi-step controlled regulation and production of SMs after stopping the primary metabolism or growth. Heterochromatin starts to turn into euchromatin at stationary phase, and SM gene clusters are more amenable to transcribe and turn on the SM biosynthesis.
2.4 Regulation of Fungal Secondary Metabolites (SMs)
The SM biosynthetic genes are often silent in the initial growth phase; SM genes are transcribed continuously when cells enter in the late exponential stage to the initial decline phase. The fungal SM biosynthetic genes are arranged in a cluster form with a size up to several hundred kilo base pairs (kb) long. The regulation of SM synthesis is either governed by a single or multiple transcriptional factors (TFs). Regulation of SM synthesis in fungi occurs either at pathway-specific level or global regulation level. The in-clustered TF gene located in a pathway-specific gene cluster plays a pivotal role in the SM regulation. AfoAAflR, SirZ, and GliZ are the examples of pathway-specific regulators (Chang et al. 1995; Bok et al. 2006a; Fox et al. 2008). On the other hand, the regulatory factors localized in the genome also respond to different environmental cues for SM synthesis. VeA, PacC, LaeA, and MeaB are the known fungal global regulators that monitor the expression of SM synthesis (Yin and Keller 2011; Brakhage 2013; Knox and Keller 2015).
2.4.1 Pathway-Specific Regulatory Proteins
Pathway-specific TFs are mostly present within the SM biosynthetic gene clusters, and they tend to regulate the expression of their own biosynthetic genes (Fig. 2.5c). Pathway-specific TFs are mostly Zn2Cys6 type which are abundantly present in fungal SM biosynthetic gene clusters (MacPherson et al. 2006; Chung et al. 2013). Nonetheless, TFs belonging to Cys2His2, bZIP, and winged helix are less common (Trapp et al. 1998; Kihara et al. 2008; Chang and Ehrlich 2013; Schmitt and Kück 2000; Hong et al. 2013). AflR, belonging to Zn2Cys6 family TF, specifically interacts with palindromic sequence (TCGN5CGR) of the promoters and activates aflatoxin and sterigmatocystin production in Aspergillus spp. (Ehrlich et al. 1999). Similarly, other pathway-specific regulators such as GliZSirZ, ApdR, CtnA, LovE, and AfoA also interact with the promoter sequence for their activation. Gliotoxin produced from Aspergillus, Trichoderma, and Penicillium spp. was shown to be regulated by an in-clustered TF GliZ in Aspergillus fumigatus (Bok et al. 2006a; Fox and Howlett 2008). Deletion of GliZ resulted in the loss of gliotoxin biosynthesis; however, its induced expression accelerates the gliotoxin level (Bok et al. 2006a). The SM sirodesmin PL produced from Leptosphaeria maculans is regulated by in-clustered TF SirZ (Fox et al. 2008). Moreover, ApdR, CtnA, and LovE regulate the biosynthesis of cytotoxic metabolite aspyridone in A. nidulans (Bergmann et al. 2007), citrinin in Monascus purpureus (Shimizu et al. 2007), and lovastatin in A. terreus, respectively (Van Den Berg and Hans 2011). An anti-cancerous drug asperfuranone produced from A. nidulans was regulated by AfoA (Chiang et al. 2009).
Tri6 belonging to Cys2His2 zinc finger TF has a role in the regulation of trichothecene biosynthesis in Fusarium spp. (Proctor et al. 1995). The TFs Bmr1, Cmr1, Cmr1p, and Pig1p, present in Bipolaris oryzae, Cochliobolus heterostrophus, Colletotrichum lagenarium, and Magnaporthe grisea, respectively, belong to the Cys2His2 class that regulates the melanin biosynthesis (Tsuji et al. 2000; Kihara et al. 2008). Despite this, bZIP TFs were also reported in pathway-specific regulation in fungi. These TFs are characterized by the presence of basic and leucine zipper regions. The basic region comprises of basic amino acids (arginine and lysine) or hydrophobic residues involved in the sequence-specific DNA binding; this region is unstructured in the absence of DNA but forms an α-helical structure upon binding the DNA target site and is also considered as an essential domain. The leucine zipper (heptad repeats of leucine residues) region mediates dimerization of the protein (Amoutzias et al. 2006). TOXE, a unique TF present in the HC toxin gene cluster, regulates the HC toxin biosynthesis by interacting with TOX2 gene promoters in Cochliobolus carbonum (Pedley and Walton 2001).
2.4.2 Global Regulatory Proteins
It was well documented that different regulatory proteins, other than in-clustered TFs, are also involved in the regulation of fungal SM biosynthesis in response to environmental cues. These cues include carbon and nitrogen source, light, pH, and temperature (Fig. 2.6). These factors are triggered in response to SM production (Brakhage 2013; Macheleidt et al. 2016). For example, CreA, AreA, PacC, LaeA, and HapX are triggered in response to environmental carbon, nitrogen, pH, light, and iron starvation condition, respectively (Dowzer and Kelly 1991; Eisendle et al. 2004; Bayram et al. 2008; Schönig et al. 2008; Schrettl et al. 2010; Brakhage 2013).

Global regulatory proteins involved in the regulation of secondary metabolism. The NRPS- or PKS-mediated SMs are regulated in the different environmental cues such as light, pH carbohydrates, and carbon and nitrogenous source. The stimulus-responsive regulatory factors (global regulators) may also able to activate or repress the biosynthetic gene cluster of the SMs
2.4.2.1 Regulation by Nitrogen Source
Nitrogen is a crucial nutrient, required for growth and development. Microbes have the ability to metabolize a diverse range of nitrogen sources to colonize at diverse environmental niches and survive under nutrient limitations. Fungi most preferably utilize glutamine and ammonium, as favored nitrogen sources. In absence of these sources, non-preferred or complex nitrogen sources such as nitrate, uric acid, urea, amines, purines, pyrimidines, and amides may also be consumed (Wong et al. 2007). The selective utilization of complex nitrogenous sources is a state called nitrogen metabolite repression which activates the global regulatory circuit leading to transcriptional activation of genes involved in the uptake and metabolism and ensures degradation (Marzluf 1997; Fraser et al. 2001; Magasanik and Kaiser 2002). The nitrogen metabolite repression is mediated by the GATA family TFs. Both the key regulatory genes named as AreA and nit-2 in A. nidulans and N. crassa, respectively, are similar in sequence and closely related to each other. They contain Cys2/Cys2-zinc finger motifs, which preferentially bind to 5′HGATAR3′ DNA sequence (Kudla et al. 1990; Ravagnani et al. 1997). The activity of AreA is regulated by both extracellular and intracellular nitrogen levels (Caddick et al. 2006). At nitrogen-limiting conditions, the level of AreA gets up-regulated, while it was down-regulated at nitrogen-sufficient conditions (Tudzynski 2014). In another level of regulation, AreA was down-regulated when it interacts with co-repressor NmrA/Nmr1, whereas NmrA/Nmr1 is regulated by a bZIP TF MeaB in A. nidulans, N. crassa, and F. fujikuroi (Fig. 2.6) (Wong et al. 2007; Tudzynski 2014). AreB and NreB are also nitrogen-responsive regulators present in A. nidulans and P. chrysogenum, respectively. AreA/NIT2 is responsible for activation of non-preferred nitrogen-metabolizing genes, whereas AreB/NreB acts antagonistically and represses the same set of genes, most probably via DNA-binding competition (Haas et al. 1997; Wong et al. 2009; Tudzynski 2014).
In F. fujikuroi, the production of growth hormone gibberellic acid and red pigment bikaverin induced under nitrogen-limiting conditions is positively regulated by AreA (Bu’Lock et al. 1974; Mihlan et al. 2003). In F. verticillioides, fumonisins are regulated at nitrogen-repressing condition mediated by the AreA (Kim and Woloshuk 2008; Picot et al. 2010). In F. graminearum, mycotoxins deoxynivalenol (DON), zearalenone, and fusarielin H are produced at nitrogen-repressing condition under the control of AreA, whereas they are slightly negatively regulated by NmrA (Giese et al. 2013). Production of aflatoxin is mediated by nitrate repression in A. parasiticus, whereas in A. flavus it is mediated by ammonium induction with response to AreA (Feng and Leonard 1998; Ehrlich and Cotty 2002).
2.4.2.2 Regulation by Carbon Source
Carbon is an essential nutrient which drastically affects the growth and development of microorganisms. Fungi preferentially utilize simple sugars such as D-glucose, sucrose, D-xylose, and acetate. Moreover, other complex carbon sources such as fructose, cellulose, xylan, pectins, alcohols, amino acids, and glycerol were also utilized under non-availability of the simple sugars (Ruijter and Visser 1997). Cells grown with the simple sugars create a carbon catabolic repression which tends to repress the transcription of a gene, involved in the complex carbon catabolism to ensure preferential utilization of glucose.
CreA/Cre1 is a C2H2 type of key regulatory TF protein present in A. nidulans and A. niger (Tudzynski et al. 2000) which specifically binds to the 5′-SYGGRG-3′ DNA sequence in the promoters of glucose-repressible genes or complex carbon-catabolizing genes, leading to switching off of the expression in the presence of glucose (Dowzer and Kelly 1989; Flipphi et al. 2003). In A. nidulans, expression of xylanase (xlnR), cellulase, and arabinose encoding genes is also controlled by CreA (Ruijter and Visser 1997; Tamayo et al. 2008). Moreover, CreA also has a repressive role in SM biosynthesis in fungi (Fig. 2.6). The synthesis of penicillin was found to be repressed under sufficient glucose in the media, and also in response to CreA, it negatively regulates the transcription of pcbAB, pcbC, and penDE in Penicillium chrysogenum (Gutiérrez et al. 1999), not in A. nidulans (Espeso et al. 1993). The antibiotic pleuromutilin production was repressed in the presence of glucose, whereas it was enhanced by soybean oil in Pleurotus mutilus (Hu et al. 2009). Negative regulation of glucose on other SMs such as cephalosporin and lovastatin was also reported in A. chrysogenum and A. terreus, respectively (Jekosch and Kück 2000; Lai et al. 2007). However, unlike other SMs, AF production was induced by glucose (Wiseman and Buchanan 1987).
2.4.2.3 pH-Mediated Regulation
Extracellular pH is an important factor which influences the cell growth by alteration of the expression level of a variety of genes in fungi. The modulation of gene expression over a wide range of pH is controlled by the genetic regulatory system which is mediated by a global regulatory protein PacC (Caddick et al. 1986). The zinc finger PacC is constituently expressing protein, but full-length protein is inactive and unable to activate the responsive genes. The activation of PacC is mediated by the six pal gene products at alkaline ambient pH (Fig. 2.6). In acidic conditions, the full-length PacC (72-kD) protein predominantly is in a closed conformation in A. nidulans, but at alkaline to neutral pH, it goes through two rounds of proteolytic cleavage and yields a functional PacC 27 (27-kDa) TF. This PacC 27 activates the alkaline-expressed genes and represses the acid-expressed genes (Díez et al. 2002). A variety of SM biosynthetic genes are controlled by pH-mediated regulation (Fig. 2.6). In Trichoderma virens, 5% of the transcriptomes are pH-dependent; among these around 25% are PacC dependent, whereas secondary metabolism-related genes are predominantly regulated by PacC (Trushina et al. 2013).
SMs are mostly produced under acidic pH conditions, which are negatively regulated by PacC (Fig. 2.6). For example, a mycotoxin, fumonisin, is negatively regulated by Pac1, and its production was repressed under alkaline conditions in F. verticillioides (Flaherty et al. 2003). The mycotoxin, ochratoxin A, synthesized by A. ochraceus is another example which is synthesized under acidic pH conditions (O’Callaghan et al. 2006). On that note, the mycotoxins, aflatoxin and sterigmatocystin, induced their production by fivefold at pH 4–5 as compared to pH 8 in Aspergillus spp. (Keller et al. 1997). The mycotoxin deoxynivalenol (DON) regulates the production at low pH in F. graminearum (Gardiner et al. 2009). In contrast, PacC also positively regulates the SM production in few cases, such as penicillin biosynthesis at alkaline pH (Espeso et al. 1993).
2.4.2.4 Light-Mediated Regulation
Light is an information carrier of nature; the cellular/molecular machinery converts its electromagnetic energy (photons) into the chemical language and transmits their dynamic signals to cellular functions. The plants used it as an energy source, whereas other organisms used it as a source of information to execute biological signal for the growth and development. In fungi, light is the essential signal for asexual sporulation and induces the conidiation at red light, whereas it is suppressed at far-red light (Mooney and Yager 1990; Corrochano 2007). The reception and transmission of the light signal are modulated by the complex proteins, consisting of LOV (light-oxygen-voltage) sensing domain (that senses blue light) and PAS (Per-Arnt-Sim) domain (involved in the dimerization of proteins), which form a complex regulatory network (Ballario et al. 1998). In N. crassa, two major light response regulatory proteins are White Collar-1 (WC-1) and White Collar-2 (WC-2) that contain photoreceptors which dimerize by the exposure of blue light and act together as a TF complex (white collar complex, WCC). The homolog of WC-1 and WC-2 in A. nidulans is named as LreA (light response) and LreB (Ballario et al. 1998; Purschwitz et al. 2008). These photoreceptors activate the clock-controlled genes and regulate the circadian clock (Ballario et al. 1996; Crosthwaite et al. 1997). The LOV domain of WC-1 acts as a photoreceptor, responds to blue light, and binds to the flavin molecule (FAD or FMN). The blue light-induced conformational change eventually transduced the light signal to downstream biochemical signals (Crosson and Moffat 2002; Crosson et al. 2003). The downstream signals eventually activate the targeted proteins such as histone acetyltransferase NGF-1/Gcn5p which acetylates the H3K14 residues of light-inducible gene promoter wrap under histone proteins at heterochromatic mark (Grimaldi et al. 2006; Brenna et al. 2012). In dark conditions, WCC forms “dark” WCC which consists of WC-1/WC-2 heterodimer and binds to light-responsive elements (LREs) in the promoters of light-responsive genes, leading to the transcriptional repression (Talora et al. 1999; Froehlich et al. 2002). After elongated exposure to blue light, another photoreceptor PAS protein VIVID (VVD) accumulates to the threshold level and disrupts the WCC complex proteins, leading to switching off of the light-dependent transcription (Schwerdtfeger and Linden 2001, 2003). VVD is a white collar-dependent blue light photoreceptor which represses the light responses and also deals with the changes in light intensities (Schwerdtfeger and Linden 2001, 2003). The disruption of WCC complex is mediated by the conformational change of WC-1 protein by phosphorylation reaction leading to switching off of the gene (Schwerdtfeger and Linden 2000; He and Liu 2005). Sterigmatocystin (ST) biosynthesis in A. nidulans was suppressed on exposure of blue or white light and induced on red light exposure greater than the dark condition (Purschwitz et al. 2008).
2.4.2.4.1 Velvet (VeA) Family of Light-Responsive Global Regulators
Proteins under velvet (VeA) family are abundantly distributed in ascomycetes and basidiomycetes and play a vital role in the secondary metabolism (Ni and Yu 2007). VeA is a light-dependent regulatory protein; exposure of light governs its localization within the cell. During dark conditions, VeA accumulates in the nucleus and upon exposure of light mobilizes it to the cytoplasm (Stinnett et al. 2007; Bayram et al. 2008). The truncated VeA mutant protein does not respond to the light signals and is effectively unable to synthesize mycotoxin ST and antibiotic penicillin (Kato et al. 2003). VeA also has a crucial role in conidia formation, which is induced by red light (Mooney and Yager 1990). The heterotrimeric velvet complex VelB/VeA/LaeA is also involved in the light-dependent regulation of cellular developmental and secondary metabolism in A. nidulans. Upon light exposure, the cellular VeA level and its translocation to the nucleus both get reduced (Fig. 2.6).
The VelB (another developmental regulator like VeA) forms the dimer with VeA (VelB-VeA) in the cytoplasm and transports to the nucleus. VeA acts as a bridge between VelB and a secondary metabolism nuclear master regulator, LaeA. The deletion of either VeA or VelB results in impaired sexual fruiting body formation and SM production (Fig. 2.6) (Bayram et al. 2008; Purschwitz et al. 2008). The similar trimeric complexes have also been reported in other fungal species such as F. oxysporum and P. chrysogenum (Kopke et al. 2013; López-Berges et al. 2013). Other than these, VosA protein also mediates asexual differentiation via heterodimer formation with VeA or VelB. The heterodimer (VosA-VelB) formation results in inhibition of asexual differentiation in the dark and induces trehalose biosynthesis in spores to protect from stress (Ni and Yu 2007). In A. nidulans a sexual development positive regulator, VelC, has also been recently characterized (Park et al. 2014). FphA is another light-responsive transcription regulatory protein which senses red light. It interacts with the VeA, induces the asexual spore formation, and represses the fruiting body formation (Blumenstein et al. 2005; Purschwitz et al. 2009). LreA and LreB act as activators of the sexual cycle and are negatively regulated by light with the action of FphA (Purschwitz et al. 2008).
2.4.2.5 Chromatin-Mediated Regulation
The eukaryotic gene expression and SM biosynthesis are also controlled by another hierarchical level of regulation known as chromatin-mediated regulation. This primarily depends on the degree of chromatin compaction or chromosomal organization of biosynthetic genes. The fungal SM biosynthetic gene clusters are most often present in the telomeric region and usually not expressed in the normal conditions (Reyes-Dominguez et al. 2010; Strauss and Reyes-Dominguez 2011). Telomeres of the chromosomes are normally heterochromatic in nature, which get relaxed into the euchromatic form by the chemical modifications of nucleosomal histone amino acids. These modifications at the epigenetic level dramatically monitor the expression of SM biosynthetic genes (Bok et al. 2009). The majority of chemical modifications are achieved at the basic amino acids of histone proteins located in the nucleosome.
2.4.2.5.1 Histone Modifications
The large eukaryotic genome (10–100,000 Mb) is systematically compacted to several thousand folds in the nucleus (Hodgkin 2001). This compaction is mediated by histone proteins which bind to the DNA molecule in a systematic manner and are organized in the chromatin structure (Kornberg 1974). Chromatin consists of a repetitive unit of nucleosome which comprises of 147 bp of DNA molecule, which is wrapped around an octameric histone protein (H3/H4 and H2A/H2B) and thereby forms an 11-nm fiber structure (Thomas and Kornberg 1975; Luger 2003). The thread of nucleosome is further condensed into a 30-nm fiber by linker histone H1 (Brosch et al. 2008). The various associated proteins further condense to higher hierarchical level and form compact chromatin structure. Certain segments of the entire chromosomes become more condensed than the rest of the sections; the condensed region is termed as heterochromatin and the relaxed region as euchromatin. The heterochromatic state is mostly present at the telomeric and centromeric regions of the chromosome, whereas the euchromatic state is present in the rest of the region. The heterochromatic state prevents the transcription of genes, whereas the euchromatic state facilitates to interact with TFs (Sterner and Berger 2000). These core histones (H2A, H2B, H3, and H4) are conserved throughout the eukaryotes; these consist of a globular domain and versatile N-terminal tail (Luger 2003). N-terminal tail can be easily modified by the action of various post-translation modification enzymes as acetylation, phosphorylation, methylation, ubiquitination, ADP-ribosylation, and SUMOylation (Verdone et al. 2006; Brosch et al. 2008; Martinez-Zamudio and Ha 2012; Rossetto et al. 2012; Wilson and Hochstrasser 2016). However, methylation and acetylation have been widely involved in the chromatin remodeling and secondary metabolite biosynthetic gene regulation (Gacek and Strauss 2012).
2.4.2.5.2 Histone Methylation
Histone methylation inferred the transfer of a methyl group from S-adenosyl-L-methionine (SAM) to substrate proteins or specific amino acid residue. Methylation of histone proteins is specifically restricted to lysine and arginine at different positions and represents a much more complex language as compared to acetylation. The methylation of some residues such as H3K9, H3K27, and H3K4 is best studied in SM production (Fig. 2.6). Acetylation and methylation events on lysine do not occur simultaneously on the same residue (Gacek and Strauss 2012). The H3K9 in A. nidulans is strongly methylated by methyltransferase ClrD; di- and trimethylation of H3K9 subsequently recruits HepA and forms the condensed heterochromatin (Bannister et al. 2001; Reyes-Dominguez et al. 2010). In contrast, deletion of ClrD in A. nidulans decreased the trimethylation state of H3K9 and subsequently resulted to loss of HepA recruitment in the ST cluster. Furthermore, deletion of HepA decreased the heterochromatin formation and increased the AflR expression (Reyes-Dominguez et al. 2010). In addition, H3K4 methylation is also associated with SM production; it occurred by CclA gene in A. nidulans. The deletion of CclA resulted in the reduction of H3K4 methylation along with H3K9 trimethylation in some SM gene promoters which increases the production of SMs F9775B and F9775A and expression of their associated genes (Fig. 2.6) (Bok et al. 2009). However, methyltransferase LaeA directly/indirectly induces the secondary metabolism and virulence in the A. nidulans and neutralizes the heterochromatic mark establishment. Though LaeA contains the methyltransferase domain, substrate or methyl-accepting residue has not been identified up to date, whereas it self-methylates methionine 207 in A. nidulans. Furthermore, self-methylation of methionine 207 did not show any role in its biological function (Patananan et al. 2013). The LaeA knockout strain of A. nidulans down-regulates the expression of ST, penicillin, and some indole alkaloid biosynthesis gene clusters in microarray experiments (Bok et al. 2006b; Yin and Keller 2011). Furthermore, LaeA knockout strain of A. fumigatus up-regulates the 22 secondary metabolism gene clusters and down-regulates the 13 SM gene clusters (Perrin et al. 2007).
2.4.2.5.3 Histone Acetylation
Acetylation of histone proteins under distinct physiological conditions also plays an important role in the regulation of secondary metabolism gene clusters. Histone acetylation marks are subjected to active transcription of genes at several positions; it occurs by histone acetyltransferase (HAT) enzymes, and these marks were removed by histone deacetylases (HDAC). The hyperacetylation of histone commonly responds to euchromatin formation, while hypoacetylation responds to heterochromatin formation and silences the gene transcription. Few histones involved in acetylation such as H3K9, H3K14, and H4K12 greatly influence the SM production and are acquired by multi-subunit Saga/Ada complexes or other associated histone acetyltransferases (Lan et al. 2016).
The GcnE and AdaB histone acetyltransferases are a part of SAGA-ADA complex and acetylate the H3K9 and H3K14 residue at the promoter region of orsellinic acid biosynthesis genes in fungal and bacterial interaction in A. nidulans. However, this acetylation mark is also detected in genes other than secondary metabolism gene clusters (Nützmann et al. 2011). In addition, the regulatory subunits of Saga/Ada, GcnE, and AdaB, respectively, are required for induction of conidiation in A. nidulans (Cánovas et al. 2014). Furthermore, the acetylation of H3K9 which enhances the orsellinic acid and sterigmatocystin biosynthesis gene clusters is triggered by S. rapamycinicus (Reyes-Dominguez et al. 2008). Targeted mutation of the potentially acetylated lysine residues 9, 14, 18, and 23 of histone H3 led to major changes in the transcriptional and metabolite levels of sterigmatocystin, orsellinic acid, and penicillin in A. nidulans (Nützmann et al. 2013). EsaA acetylates the H4K12 and enhances the sterigmatocystin, penicillin, terrequinone, and orsellinic acid production (Soukup et al. 2012). Furthermore the SM production is also enhanced by histone deacetylase (HDAC) inhibitor such as suberoylanilide hydroxamic acid (SAHA), trichostatin A, and 5-azacytidine (Fisch et al. 2009; Lim et al. 2012; Zutz et al. 2013; Albright et al. 2015), whereas SM production is blocked by histone acetyltransferase HAT inhibitor anacardic acid and curcumin (Netzker et al. 2015; Wee 2015).
For the activation of cryptic SM gene clusters, HDAC inhibitors (HDACi) have been widely used in fungi. These HDACi are valproic acid and trichostatin A (TSA), and suberoylanilide hydroxamic acid (SAHA) treatment effectively increases the SM production by chromatin modification in fungi (Henrikson et al. 2009; Zutz et al. 2013). In addition to the HDACi, histone demethylase inhibitor such as 5-azacytidine is also used in the SM modulation (Zutz et al. 2013).
References
Agathos S, Madhosingh C, Marshall J, Lee J (1987) The fungal production of cyclosporine A. Ann N Y Acad Sci 506:657–662
Albright JC et al (2015) Large-scale metabolomics reveals a complex response of Aspergillus nidulans to epigenetic perturbation. ACS Chem Biol 10:1535–1541
Alexander NJ, Proctor RH, McCormick SP (2009) Genes, gene clusters, and biosynthesis of trichothecenes and fumonisins in Fusarium. Toxin Rev 28:198–215
Amici AM, Minghetti A, Scotti T, Spalla C, Tognoli L (1967) Ergotamine production in submerged culture and physiology of Claviceps purpurea. Appl Microbiol 15:597–602
Amoutzias G et al (2006) One billion years of bZIP transcription factor evolution: conservation and change in dimerization and DNA-binding site specificity. Mol Biol Evol 24:827–835
Anaya AL, Cruz-Ortega R, Waller GR (2006) Metabolism and ecology of purine alkaloids. Front Biosci 11:2354
Aniszewski T (2015) Alkaloids: chemistry, biology, ecology, and applications. Elsevier, Amsterdam
Ballario P, Vittorioso P, Magrelli A, Talora C, Cabibbo A, Macino G (1996) White collar-1, a central regulator of blue light responses in Neurospora, is a zinc finger protein. EMBO J 15:1650–1657
Ballario P, Talora C, Galli D, Linden H, Macino G (1998) Roles in dimerization and blue light photoresponse of the PAS and LOV domains of Neurospora crassa white collar proteins. Mol Microbiol 29:719–729. https://doi.org/10.1046/j.1365-2958.1998.00955.x
Bannister AJ, Zegerman P, Partridge JF, Miska EA (2001) Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410:120
Bayram Ö et al (2008) VelB/VeA/LaeA complex coordinates light signal with fungal development and secondary metabolism. Science 320:1504–1506
Bergmann S, Schümann J, Scherlach K, Lange C, Brakhage AA, Hertweck C (2007) Genomics-driven discovery of PKS-NRPS hybrid metabolites from Aspergillus nidulans. Nat Chem Biol 3:nchembio869
Blumenstein A et al (2005) The Aspergillus nidulans phytochrome FphA represses sexual development in red light. Curr Biol 15:1833–1838
Bok JW et al (2006a) GliZ, a transcriptional regulator of gliotoxin biosynthesis, contributes to Aspergillus fumigatus virulence. Infect Immun 74:6761–6768
Bok JW, Hoffmeister D, Maggio-Hall LA, Murillo R, Glasner JD, Keller NP (2006b) Genomic mining for Aspergillus natural products. Chem Biol 13:31–37
Bok JW et al (2009) Chromatin-level regulation of biosynthetic gene clusters. Nat Chem Biol 5:462–464
Bömke C, Tudzynski B (2009) Diversity, regulation, and evolution of the gibberellin biosynthetic pathway in fungi compared to plants and bacteria. Phytochemistry 70:1876–1893
Brakhage AA (2013) Regulation of fungal secondary metabolism. Nat Rev Microbiol 11:21–32
Brenna A, Grimaldi B, Filetici P, Ballario P (2012) Physical association of the WC-1 photoreceptor and the histone acetyltransferase NGF-1 is required for blue light signal transduction in Neurospora crassa. Mol Biol Cell 23:3863–3872
Brosch G, Loidl P, Graessle S (2008) Histone modifications and chromatin dynamics: a focus on filamentous fungi. FEMS Microbiol Rev 32:409–439
Bu’Lock J (1961) Intermediary metabolism and antibiotic synthesis. Adv Appl Microbiol 3:293–342
Bu’Lock J, Detroy R, Hošťálek Z, Munim-Al-Shakarchi A (1974) Regulation of secondary biosynthesis in Gibberella fujikuroi. Trans Br Mycol Soc 62:377–389
Byrne KM, Smith SK, Ondeyka JG (2002) Biosynthesis of nodulisporic acid A: precursor studies. J Am Chem Soc 124:7055–7060
Cacho RA, Jiang W, Chooi Y-H, Walsh CT, Tang Y (2012) Identification and characterization of the echinocandin B biosynthetic gene cluster from Emericella rugulosa NRRL 11440. J Am Chem Soc 134:16781–16790
Caddick MX, Brownlee AG, Arst HN (1986) Regulation of gene expression by pH of the growth medium in Aspergillus nidulans. Mol Gen Genet MGG 203:346–353
Caddick MX et al (2006) Opposing signals differentially regulate transcript stability in Aspergillus nidulans. Mol Microbiol 62:509–519
Cairns T, Meyer V (2017) In silico prediction and characterization of secondary metabolite biosynthetic gene clusters in the wheat pathogen Zymoseptoria tritici. BMC Genomics 18:631. https://doi.org/10.1186/s12864-017-3969-y
Calne R et al (1989) Rapamycin for immunosuppression in organ allografting. Lancet 334:227
Calvo AM, Gardner HW, Keller NP (2001) Genetic connection between fatty acid metabolism and sporulation in Aspergillus nidulans. J Biol Chem 276:25766–25774
Calvo AM, Wilson RA, Bok JW, Keller NP (2002) Relationship between secondary metabolism and fungal development. Microbiol Mol Biol Rev 66:447–459
Cánovas D et al (2014) The histone acetyltransferase GcnE (GCN5) plays a central role in the regulation of Aspergillus asexual development. Genetics 197:1175–1189
Castaño I, Pan SJ, Zupancic M, Hennequin C, Dujon B, Cormack BP (2005) Telomere length control and transcriptional regulation of subtelomeric adhesins in Candida glabrata. Mol Microbiol 55:1246–1258
Champe SP, El-Zayat A (1989) Isolation of a sexual sporulation hormone from Aspergillus nidulans. J Bacteriol 171:3982–3988
Chang P-K, Ehrlich KC (2013) Genome-wide analysis of the Zn(II)2Cys6 zinc cluster-encoding gene family in Aspergillus flavus. Appl Microbiol Biotechnol 97:4289–4300
Chang P-K, Ehrlich KC, Yu J, Bhatnagar D, Cleveland TE (1995) Increased expression of Aspergillus parasiticus aflR, encoding a sequence-specific DNA-binding protein, relieves nitrate inhibition of aflatoxin biosynthesis. Appl Environ Microbiol 61:2372–2377
Chiang Y-M, Szewczyk E, Davidson AD, Keller N, Oakley BR, Wang CCC (2009) A gene cluster containing two fungal polyketide synthases encodes the biosynthetic pathway for a polyketide, Asperfuranone, in Aspergillus nidulans. J Am Chem Soc 131:2965–2970. https://doi.org/10.1021/ja8088185
Chung H, Choi J, Park S-Y, Jeon J, Lee Y-H (2013) Two conidiation-related Zn(II)2Cys6 transcription factor genes in the rice blast fungus. Fungal Genet Biol 61:133–141
Corrochano LM (2007) Fungal photoreceptors: sensory molecules for fungal development and behaviour. Photochem Photobiol Sci 6:725–736
Crosson S, Moffat K (2002) Photoexcited structure of a plant photoreceptor domain reveals a light-driven molecular switch. Plant Cell 14:1067–1075
Crosson S, Rajagopal S, Moffat K (2003) The LOV domain family: photoresponsive signaling modules coupled to diverse output domains. Biochem(Mosc) 42:2–10
Crosthwaite SK, Dunlap JC, Loros JJ (1997) Neurospora wc-1 and wc-2: transcription, photoresponses, and the origins of circadian rhythmicity. Sci 276:763–769
Darkin-Rattray SJ et al (1996) Apicidin: a novel antiprotozoal agent that inhibits parasite histone deacetylase. Proc Natl Acad Sci 93:13143–13147
Degenkolb T et al (2000) Roseoferin, a new aminolipopeptide antibiotic complex from Mycogone rosea DSM 12973, structures and biological activities. J Antibiot 53:184–190
Demain A (1999) Pharmaceutically active secondary metabolites of microorganisms. Appl Microbiol Biotechnol 52:455–463
Demain AL, Fang A (2000) The natural functions of secondary metabolites. In: History of modern biotechnology I. Springer, New York, pp 1–39
Dewick PM (2002) Medicinal natural products: a biosynthetic approach. Wiley, Chichester
Díez E et al (2002) Activation of the Aspergillus PacC zinc finger transcription factor requires two proteolytic steps. EMBO J 21:1350–1359
Dorner JW, Cole RJ, Diener UL (1984) The relationship of Aspergillus flavus and Aspergillus parasiticus with reference to production of aflatoxins and cyclopiazonic acid. Mycopathologia 87:13–15
Dowzer CE, Kelly JM (1989) Cloning of the creA gene from Aspergillus nidulans: a gene involved in carbon catabolite repression. Curr Genet 15:457–459
Dowzer C, Kelly JM (1991) Analysis of the creA gene, a regulator of carbon catabolite repression in Aspergillus nidulans. Mol Cell Biol 11:5701–5709
Dubouzet JG, Matsuda F, Ishihara A, Miyagawa H, Wakasa K (2013) Production of indole alkaloids by metabolic engineering of the tryptophan pathway in rice. Plant Biotechnol J 11:1103–1111
Echavarri-Erasun C, Johnson EA (2002) Fungal carotenoids. In: Applied mycology and biotechnology. Elsevier, Amsterdam, pp 45–85
Ehrlich K, Cotty P (2002) Variability in nitrogen regulation of aflatoxin production by Aspergillus flavus strains. Appl Microbiol Biotechnol 60:174–178
Ehrlich K, Montalbano B, Cary J (1999) Binding of the C6-zinc cluster protein, AFLR, to the promoters of aflatoxin pathway biosynthesis genes in Aspergillus parasiticus. Gene 230:249–257
Eisendle M, Oberegger H, Buttinger R, Illmer P, Haas H (2004) Biosynthesis and uptake of siderophores is controlled by the PacC-mediated ambient-pH regulatory system in Aspergillus nidulans. Eukaryot Cell 3:561–563
Eisenreich W, Bacher A, Arigoni D, Rohdich F (2004) Biosynthesis of isoprenoids via the non-mevalonate pathway. Cell Mol Life Sci 61:1401–1426
Espeso EA, Tilburn J, Arst H Jr, Penalva M (1993) pH regulation is a major determinant in expression of a fungal penicillin biosynthetic gene. EMBO J 12:3947–3956
Esser K, Tudzynski P (1978) Genetics of the ergot fungus Claviceps purpurea. Theor Appl Genet 53:145–149
Evans WC (2009) Trease and Evans’ pharmacognosy e-book. Elsevier Health Sciences, New York
Evans BS, Robinson SJ, Kelleher NL (2011) Surveys of non-ribosomal peptide and polyketide assembly lines in fungi and prospects for their analysis in vitro and in vivo. Fungal Genet Biol 48:49–61
Feng GH, Leonard TJ (1998) Culture conditions control expression of the genes for aflatoxin and sterigmatocystin biosynthesis in Aspergillus parasiticus and A. nidulans. Appl Environ Microbiol 64:2275–2277
Fisch K et al (2009) Chemical induction of silent biosynthetic pathway transcription in Aspergillus niger. J Ind Microbiol Biotechnol 36:1199–1213
Fischbach MA, Walsh CT (2006) Assembly-line enzymology for polyketide and nonribosomal peptide antibiotics: logic, machinery, and mechanisms. Chem Rev 106:3468–3496
Flaherty JE, Pirttilä AM, Bluhm BH, Woloshuk CP (2003) PAC1, a pH-regulatory gene from Fusarium verticillioides. Appl Environ Microbiol 69:5222–5227. https://doi.org/10.1128/aem.69.9.5222-5227.2003
Flipphi M, Kocialkowska J, Felenbok B (2003) Relationships between the ethanol utilization (alc) pathway and unrelated catabolic pathways in Aspergillus nidulans. Eur J Biochem 270:3555–3564
Fox EM, Howlett BJ (2008) Biosynthetic gene clusters for epipolythiodioxopiperazines in filamentous fungi. Mycol Res 112:162–169
Fox EM, Gardiner DM, Keller NP, Howlett BJ (2008) A Zn(II)2Cys6 DNA binding protein regulates the sirodesmin PL biosynthetic gene cluster in Leptosphaeria maculans. Fungal Genet Biol 45:671–682. https://doi.org/10.1016/j.fgb.2007.10.005
Fraser JA, Davis MA, Hynes MJ (2001) The formamidase gene of Aspergillus nidulans: regulation by nitrogen metabolite repression and transcriptional interference by an overlapping upstream gene. Genetics 157:119–131
Froehlich AC, Liu Y, Loros JJ, Dunlap JC (2002) White Collar-1, a circadian blue light photoreceptor, binding to the frequency promoter. Science 297:815–819
Gacek A, Strauss J (2012) The chromatin code of fungal secondary metabolite gene clusters. Appl Microbiol Biotechnol 95:1389–1404
Galagan JE, Henn MR, Ma L-J, Cuomo CA, Birren B (2005) Genomics of the fungal kingdom: insights into eukaryotic biology. Genome Res 15:1620–1631. https://doi.org/10.1101/gr.3767105
Gallo A, Ferrara M, Perrone G (2013) Phylogenetic study of polyketide synthases and nonribosomal peptide synthetases involved in the biosynthesis of mycotoxins. Toxins 5:717–742
Gao X, Chooi Y-H, Ames BD, Wang P, Walsh CT, Tang Y (2011) Fungal indole alkaloid biosynthesis: genetic and biochemical investigation of the tryptoquialanine pathway in Penicillium aethiopicum. J Am Chem Soc 133:2729–2741
García-Estrada C et al (2011) A single cluster of coregulated genes encodes the biosynthesis of the mycotoxins roquefortine C and meleagrin in Penicillium chrysogenum. Chem Biol 18:1499–1512. https://doi.org/10.1016/j.chembiol.2011.08.012
Gardiner DM, Osborne S, Kazan K, Manners JM (2009) Low pH regulates the production of deoxynivalenol by Fusarium graminearum. Microbiology 155:3149–3156. https://doi.org/10.1099/mic.0.029546-0
Giese H, Sondergaard TE, Sørensen JL (2013) The AreA transcription factor in Fusarium graminearum regulates the use of some nonpreferred nitrogen sources and secondary metabolite production. Fungal Biol 117:814–821
Gottschling DE, Aparicio OM, Billington BL, Zakian VA (1990) Position effect at S. cerevisiae telomeres: reversible repression of Pol II transcription. Cell 63:751–762
Grimaldi B et al (2006) The Neurospora crassa White Collar-1 dependent blue light response requires acetylation of histone H3 lysine 14 by NGF-1. Mol Biol Cell 17:4576–4583
Grisham LM, Wilson L, BENSCH KG (1973) Antimitotic action of griseofulvin does not involve disruption of microtubules. Nature 244:294–296
Guirimand G, Courdavault V, St-Pierre B, Burlat V (2010) Biosynthesis and regulation of alkaloids. In: Plant developmental biology-biotechnological perspectives. Springer, Berlin, pp 139–160
Gutiérrez S et al (1999) Transcription of the pcbAB, pcbC and penDE genes of Penicillium chrysogenum AS-P-78 is repressed by glucose and the repression is not reversed by alkaline pHs. Microbiology 145:317–324
Haas H, Angermayr K, Zadra I, Stöffler G (1997) Overexpression of nreB, a new GATA factor-encoding gene of Penicillium chrysogenum, leads to repression of the nitrate assimilatory gene cluster. J Biol Chem 272:22576–22582
Harper JK et al (2003) Pestacin: a 1, 3-dihydro isobenzofuran from Pestalotiopsis microspora possessing antioxidant and antimycotic activities. Tetrahedron 59:2471–2476
Hashimoto M, Nonaka T, Fujii I (2014) Fungal type III polyketide synthases. Nat Prod Rep 31:1306–1317
Hayakawa S, Matsushima T, Kimura T, Minato H, Katagiri K (1968) Zygosporin A, a new antibiotic from Zygosporium masonii. J Antibiot 21:523–524
He Q, Liu Y (2005) Molecular mechanism of light responses in Neurospora: from light-induced transcription to photoadaptation. Genes Dev 19:2888–2899
Hellmig V, Grothe T, Mayer-Bartschmid A (2002) Altersetin, a new antibiotic from culture of endophytic Alternarla sp. taxonomy, fermentation, isolation, structure elucidation and biological activities. J Antibiot (Tokyo) 55:881–892
Henrikson JC, Hoover AR, Joyner PM, Cichewicz RH (2009) A chemical epigenetics approach for engineering the in situ biosynthesis of a cryptic natural product from Aspergillus niger. Org Biomol Chem 7:435–438
Hodgkin J (2001) Genome size. In: Brenner S, Miller JH (eds) Encyclopedia of genetics. Academic, New York, p 865
Hoffmeister D, Keller NP (2007) Natural products of filamentous fungi: enzymes, genes, and their regulation. Nat Prod Rep 24:393–416. https://doi.org/10.1039/b603084j
Hong S-Y, Roze LV, Linz JE (2013) Oxidative stress-related transcription factors in the regulation of secondary metabolism. Toxins 5:683–702
Hosokawa N et al (2000) New strobilurins O and P from a mushroom. J Antibiot 53:297–300
Hu C, Zou Y, Zhao W (2009) Effect of soybean oil on the production of mycelial biomass and pleuromutilin in the shake-flask culture of Pleurotus mutilus. World J Microbiol Biotechnol 25:1705–1711
Hu Y, Zhang W, Zhang P, Ruan W, Zhu X (2013) Nematicidal activity of Chaetoglobosin A poduced by Chaetomium globosum NK102 against Meloidogyne incognita. J Agric Food Chem 61:41–46. https://doi.org/10.1021/jf304314g
Hutchinson CR (2003) Polyketide and non-ribosomal peptide synthases: falling together by coming apart. Proc Natl Acad Sci 100:3010–3012
Igarashi Y et al (2000) Xanthoepocin, a new antibiotic from Penicillium simplicissimum IFO5762. J Antibiot 53:928–933
Jekosch K, Kück U (2000) Glucose dependent transcriptional expression of the cre1 gene in Acremonium chrysogenum strains showing different levels of cephalosporin C production. Curr Genet 37:388–395
Jensen PR, Williams PG, Oh D-C, Zeigler L, Fenical W (2007) Species-specific secondary metabolite production in marine actinomycetes of the genus Salinispora. Appl Environ Microbiol 73:1146–1152
Jin JM et al (2010) Functional characterization and manipulation of the apicidin biosynthetic pathway in Fusarium semitectum. Mol Microbiol 76:456–466
Kamei K, Watanabe A (2005) Aspergillus mycotoxins and their effect on the host. Med Mycol 43:95–99
Kato N, Brooks W, Calvo AM (2003) The expression of sterigmatocystin and penicillin genes in Aspergillus nidulans is controlled by veA, a gene required for sexual development. Eukaryot Cell 2:1178–1186
Keller NP (2015) Translating biosynthetic gene clusters into fungal armor and weaponry. Nat Chem Biol 11:671–677
Keller NP, Nesbitt C, Sarr B, Phillips TD, Burow GB (1997) pH regulation of sterigmatocystin and aflatoxin biosynthesis in Aspergillus spp. Phytopathology 87:643–648. https://doi.org/10.1094/phyto.1997.87.6.643
Keller NP, Turner G, Bennett JW (2005) Fungal secondary metabolism—from biochemistry to genomics. Nat Rev Microbiol 3:937–947
Kelly WL, Hillson NJ, Walsh CT (2005) Excision of the epothilone synthetase B cyclization domain and demonstration of in trans condensation/cyclodehydration activity. Biochem (Mosc) 44:13385–13393
Khaldi N, Wolfe KH (2011) Evolutionary origins of the fumonisin secondary metabolite gene cluster in Fusarium verticillioides and Aspergillus niger. Int J Evol Biol 2011 pp 1-7. doi:10.4061/2011/423821
Kihara J, Moriwaki A, Tanaka N, Tanaka C, Ueno M, Arase S (2008) Characterization of the BMR1 gene encoding a transcription factor for melanin biosynthesis genes in the phytopathogenic fungus Bipolaris oryzae. FEMS Microbiol Lett 281:221–227
Kim H, Woloshuk C (2008) Role of AREA, a regulator of nitrogen metabolism, during colonization of maize kernels and fumonisin biosynthesis in Fusarium verticillioides. Fungal Genet Biol 45:947–953
Kleinwaechter P, Schlegel B, Doerfelt H, Graefe U (2001) Spirobenzofuran, a new bioactive metabolite from Acremonium sp. HKI 0230. J Antibiot 54:526–527
Knox BP, Keller NP (2015) Key players in the regulation of fungal secondary metabolism. In: Zeilinger S, Martín J-F, García-Estrada C (eds) Biosynthesis and molecular genetics of fungal secondary metabolites, vol 2. Springer, New York, pp 13–28
Kopke K, Hoff B, Bloemendal S, Katschorowski A, Kamerewerd J, Kück U (2013) Members of the Penicillium chrysogenum velvet complex play functionally opposing roles in the regulation of penicillin biosynthesis and conidiation. Eukaryot Cell 12:299–310
Kornberg RD (1974) Chromatin structure: a repeating unit of histones and DNA. Science 184:868–871
Kudla B et al (1990) The regulatory gene areA mediating nitrogen metabolite repression in Aspergillus nidulans. Mutations affecting specificity of gene activation alter a loop residue of a putative zinc finger. EMBO J 9:1355–1364
Kumar A, Jaiswal V, Kumar V, Dey A, Kumar A (2018) Functional redundancy in Echinocandin B in-cluster transcription factor ecdB of Emericella rugulosa NRRL 11440. Biotechnol Rep 19:e00264. https://doi.org/10.1016/j.btre.2018.e00264
Lai L-ST, Hung C-S, Lo C-C (2007) Effects of lactose and glucose on production of itaconic acid and lovastatin by Aspergillus terreus ATCC 20542. J Biosci Bioeng 104:9–13. https://doi.org/10.1263/jbb.104.9
Lan H et al (2016) The Aspergillus flavus histone acetyltransferase AflGcnE regulates morphogenesis, aflatoxin biosynthesis, and pathogenicity. Front Microbiol 7:1324
Lim FY, Sanchez JF, Wang CC, Keller NP (2012) Toward awakening cryptic secondary metabolite gene clusters in filamentous fungi. Methods Enzymol 517:303
Lin H-C et al (2015) Elucidation of the concise biosynthetic pathway of the communesin indole alkaloids. Angew Chem Int Ed 54:3004–3007. https://doi.org/10.1002/anie.201411297
Linne U, Doekel S, Marahiel MA (2001) Portability of epimerization domain and role of peptidyl carrier protein on epimerization activity in nonribosomal peptide synthetases. Biochemistry (Mosc) 40:15824–15834. https://doi.org/10.1021/bi011595t
Liu L, Zhang Z, Shao C-L, Wang J-L, Bai H, Wang C-Y (2015) Bioinformatical analysis of the sequences, structures and functions of fungal polyketide synthase product template domains. Sci Rep 5:10463. https://doi.org/10.1038/srep10463
López-Berges MS et al (2013) The velvet complex governs mycotoxin production and virulence of Fusarium oxysporum on plant and mammalian hosts. Mol Microbiol 87:49–65
Luger K (2003) Structure and dynamic behavior of nucleosomes. Curr Opin Genet Dev 13:127–135. https://doi.org/10.1016/S0959-437X(03)00026-1
Macheleidt J et al (2016) Regulation and role of fungal secondary metabolites. Annu Rev Genet 50:371–392
MacPherson S, Larochelle M, Turcotte B (2006) A fungal family of transcriptional regulators: the zinc cluster proteins. Microbiol Mol Biol Rev 70:583–604
Magasanik B, Kaiser CA (2002) Nitrogen regulation in Saccharomyces cerevisiae. Gene 290:1–18
Mahmood ZA, Ahmed SW, Azhar I, Sualeh M, Baig MT, Zoha S (2010) Bioactive alkaloids produced by fungi I. Updates on alkaloids from the species of the genera boletus, Fusarium and psilocybe. Pak J Pharm Sci 23:349–357
Mann V et al (1994) Complex I, iron, and ferritin in Parkinson’s disease substantia nigra. Ann Neurol 36:876–881
Marshall CG, Burkart MD, Keating TA, Walsh CT (2001) Heterocycle formation in vibriobactin biosynthesis: alternative substrate utilization and identification of a condensed intermediate. Biochemistry (Mosc) 40:10655–10663
Martinez-Zamudio R, Ha HC (2012) Histone ADP-ribosylation facilitates gene transcription by directly remodeling nucleosomes. Mol Cell Biol. https://doi.org/10.1128/MCB.06667-11
Marzluf GA (1997) Genetic regulation of nitrogen metabolism in the fungi. Microbiol Mol Biol Rev 61:17–32
Mihlan M, Homann V, Liu TWD, Tudzynski B (2003) AREA directly mediates nitrogen regulation of gibberellin biosynthesis in Gibberella fujikuroi, but its activity is not affected by NMR. Mol Microbiol 47:975–991
Mooney JL, Yager LN (1990) Light is required for conidiation in Aspergillus nidulans. Genes Dev 4:1473–1482
Moss MO (2002) Mycotoxin review-1. Aspergillus and penicillium. Mycologist 16:116–119
Netzker T et al (2015) Microbial communication leading to the activation of silent fungal secondary metabolite gene clusters. Front Microbiol 6:299
Ni M, Yu J-H (2007) A novel regulator couples sporogenesis and trehalose biogenesis in Aspergillus nidulans. PLoS One 2:e970
Noh MJ et al (1999) Isolation of a novel microorganism, Pestalotia heterocornis, producing paclitaxel. Biotechnol Bioeng 64:620–623
Nützmann H-W et al (2011) Bacteria-induced natural product formation in the fungus Aspergillus nidulans requires Saga/Ada-mediated histone acetylation. Proc Natl Acad Sci 108:14282–14287
Nützmann H-W, Fischer J, Scherlach K, Hertweck C, Brakhage AA (2013) Distinct amino acids of histone H3 control secondary metabolism in Aspergillus nidulans. Appl Environ Microbiol 79:6102–6109
O’Callaghan J, Stapleton PC, Dobson ADW (2006) Ochratoxin A biosynthetic genes in Aspergillus ochraceus are differentially regulated by pH and nutritional stimuli. Fungal Genet Biol 43:213–221. https://doi.org/10.1016/j.fgb.2005.11.005
Omura S et al (1995) Arisugacin, a novel and selective inhibitor of acetylcholinesterase from Penicillium sp. FO-4259. J Antibiot 48:745–746
Ondeyka JG et al (1997) Nodulisporic acid A, a novel and potent insecticide from a Nodulisporium sp. isolation, structure determination, and chemical transformations. J Am Chem Soc 119:8809–8816
Pahl HL et al (1996) The immunosuppressive fungal metabolite gliotoxin specifically inhibits transcription factor NF-kappaB. J Exp Med 183:1829–1840
Palmer JM, Keller NP (2010) Secondary metabolism in fungi: does chromosomal location matter? Curr Opin Microbiol 13:431–436
Palmer JM et al (2010) Telomere position effect is regulated by heterochromatin-associated proteins and NkuA in Aspergillus nidulans. Microbiology 156:3522–3531
Park H-S, Nam T-Y, Han K-H, Kim SC, Yu J-H (2014) VelC positively controls sexual development in Aspergillus nidulans. PLoS One 9:e89883
Patananan AN, Palmer JM, Garvey GS, Keller NP, Clarke SG (2013) A novel automethylation reaction in the Aspergillus nidulans LaeA protein generates S-methylmethionine. J Biol Chem 288:14032–14045. https://doi.org/10.1074/jbc.M113.465765
Pedley KF, Walton JD (2001) Regulation of cyclic peptide biosynthesis in a plant pathogenic fungus by a novel transcription factor. Proc Natl Acad Sci 98:14174–14179
Perrin RM et al (2007) Transcriptional regulation of chemical diversity in Aspergillus fumigatus by LaeA. PLoS Pathog 3:e50
Picot A, Barreau C, Pinson-Gadais L, Caron D, Lannou C, Richard-Forget F (2010) Factors of the Fusarium verticillioides-maize environment modulating fumonisin production. Crit Rev Microbiol 36:221–231
Proctor RH, Hohn TM, McCormick SP, Desjardins AE (1995) Tri6 encodes an unusual zinc finger protein involved in regulation of trichothecene biosynthesis in Fusarium sporotrichioides. Appl Environ Microbiol 61:1923–1930
Purschwitz J et al (2008) Functional and physical interaction of blue-and red-light sensors in Aspergillus nidulans. Curr Biol 18:255–259
Purschwitz J, Müller S, Fischer R (2009) Mapping the interaction sites of Aspergillus nidulans phytochrome FphA with the global regulator VeA and the White Collar protein LreB. Mol Gen Genomics 281:35–42
Ramawat KG, Mérillon J-M (2013) Natural products: phytochemistry, botany and metabolism of alkaloids, phenolics and terpenes. Springer, Berlin/Heidelberg
Raper KB (1946) The development of improved penicillin-producing molds. Ann N Y Acad Sci 48:41–56
Ravagnani A et al (1997) Subtle hydrophobic interactions between the seventh residue of the zinc finger loop and the first base of an HGATAR sequence determine promoter-specific recognition by the Aspergillus nidulans GATA factor AreA. EMBO J 16:3974–3986
Reyes-Dominguez Y et al (2008) Nucleosome positioning and histone H3 acetylation are independent processes in the Aspergillus nidulans prnD-prnB bidirectional promoter. Eukaryot Cell 7:656–663
Reyes-Dominguez Y et al (2010) Heterochromatic marks are associated with the repression of secondary metabolism clusters in Aspergillus nidulans. Mol Microbiol 76:1376–1386
Roberts MF (2013) Alkaloids: biochemistry, ecology, and medicinal applications. Springer, New York
Rosas-Hernández LL et al (2008) yKu70/yKu80 and Rif1 regulate silencing differentially at telomeres in Candida glabrata. Eukaryot Cell 7:2168–2178
Rossetto D, Avvakumov N, Côté J (2012) Histone phosphorylation: a chromatin modification involved in diverse nuclear events. Epigenetics 7:1098–1108
Ruijter GJ, Visser J (1997) Carbon repression in Aspergilli. FEMS Microbiol Lett 151:103–114
Sándor E et al (1998) Allosamidin inhibits the fragmentation of Acremonium chrysogenum but does not influence the cephalosporin-C production of the fungus. FEMS Microbiol Lett 164:231–236
Schmitt EK, Kück U (2000) The fungal CPCR1 protein, which binds specifically to β-lactam biosynthesis genes, is related to human regulatory factor X transcription factors. J Biol Chem 275:9348–9357
Schönig B, Brown DW, Oeser B, Tudzynski B (2008) Cross-species hybridization with Fusarium verticillioides microarrays reveals new insights into Fusarium fujikuroi nitrogen regulation and the role of AreA and NMR. Eukaryot Cell 7:1831–1846
Schrettl M et al (2010) HapX-mediated adaption to iron starvation is crucial for virulence of Aspergillus fumigatus. PLoS Pathog 6:e1001124
Schwerdtfeger C, Linden H (2000) Localization and light-dependent phosphorylation of white collar 1 and 2, the two central components of blue light signaling in Neurospora crassa. Eur J Biochem 267:414–422
Schwerdtfeger C, Linden H (2001) Blue light adaptation and desensitization of light signal transduction in Neurospora crassa. Mol Microbiol 39:1080–1087
Schwerdtfeger C, Linden H (2003) VIVID is a flavoprotein and serves as a fungal blue light photoreceptor for photoadaptation. EMBO J 22:4846–4855
Scott P, Wv W, Harwig J, Fennell D (1970) Occurrence of a mycotoxin, ochratoxin A, in wheat and isolation of ochratoxin A and citrinin producing strains of Penicillium viridicatum. Can J Plant Sci 50:583–585
Scott B et al (2013) Deletion and gene expression analyses define the paxilline biosynthetic gene cluster in Penicillium paxilli. Toxins 5:1422–1446
Sekiguchi J, Gaucher GM (1977) Conidiogenesis and secondary metabolism in Penicillium urticae. Appl Environ Microbiol 33:147–158
Shen B (2003) Polyketide biosynthesis beyond the type I, II and III polyketide synthase paradigms. Curr Opin Chem Biol 7:285–295
Shimizu T, Kinoshita H, Nihira T (2007) Identification and in vivo functional analysis by gene disruption of ctnA, an activator gene involved in citrinin biosynthesis in Monascus purpureus. Appl Environ Microbiol 73:5097–5103
Smith KM et al (2008) The fungus Neurospora crassa displays telomeric silencing mediated by multiple sirtuins and by methylation of histone H3 lysine 9. Epigenetics Chromatin 1:1–20. https://doi.org/10.1186/1756-8935-1-5
Soukup AA et al (2012) Overexpression of the Aspergillus nidulans histone 4 acetyltransferase EsaA increases activation of secondary metabolite production. Mol Microbiol 86:314–330
Staunton J, Weissman KJ (2001) Polyketide biosynthesis: a millennium review. Nat Prod Rep 18:380–416
Stein T, Vater J, Kruft V, Otto A, Wittmann-Liebold B, Franke P, Panico M, McDowell R, Morris HR (1996) The multiple carrier model of nonribosomal peptide biosynthesis at modular multienzymatic templates. J Biol Chem 271:15428–15435
Sterner DE, Berger SL (2000) Acetylation of histones and transcription-related factors. Microbiol Mol Biol Rev 64:435–459
Stierle A, Strobel G, Stierle D (1993) Taxol and taxane production by Taxomyces andreanae, an endophytic fungus of Pacific yew. Science 260:214–216
Stinnett SM, Espeso EA, Cobeño L, Araújo-Bazán L, Calvo AM (2007) Aspergillus nidulans VeA subcellular localization is dependent on the importin α carrier and on light. Mol Microbiol 63:242–255
Strauss J, Reyes-Dominguez Y (2011) Regulation of secondary metabolism by chromatin structure and epigenetic codes. Fungal Genet Biol 48:62–69. https://doi.org/10.1016/j.fgb.2010.07.009
Strieker M, Tanović A, Marahiel MA (2010) Nonribosomal peptide synthetases: structures and dynamics. Curr Opin Struct Biol 20:234–240. https://doi.org/10.1016/j.sbi.2010.01.009
Strobel GA, Miller RV, Martinez-Miller C, Condron MM, Teplow DB, Hess WM (1999) Cryptocandin, a potent antimycotic from the endophytic fungus Cryptosporiopsis cf. quercina. Microbiology 145:1919–1926. https://doi.org/10.1099/13500872-145-8-1919
Talora C, Franchi L, Linden H, Ballario P, Macino G (1999) Role of a white collar-1-white collar-2 complex in blue-light signal transduction. EMBO J 18:4961–4968
Tamayo EN, Villanueva A, Hasper AA, de Graaff LH, Ramón D, Orejas M (2008) CreA mediates repression of the regulatory gene xlnR which controls the production of xylanolytic enzymes in Aspergillus nidulans. Fungal Genet Biol 45:984–993
Thomas JO, Kornberg RD (1975) An octamer of histones in chromatin and free in solution. Proc Natl Acad Sci 72:2626–2630
Trapp S, Hohn T, McCormick S, Jarvis B (1998) Characterization of the gene cluster for biosynthesis of macrocyclic trichothecenes in Myrothecium roridum. Mol Gen Genet MGG 257:421–432
Trushina N, Levin M, Mukherjee PK, Horwitz BA (2013) PacC and pH–dependent transcriptome of the mycotrophic fungus Trichoderma virens. BMC Genomics 14:138
Tsuji G et al (2000) Novel fungal transcriptional activators, Cmr1p of Colletotrichum lagenarium and Pig1p of Magnaporthe grisea, contain Cys2His2 zinc finger and Zn (II) 2Cys6 binuclear cluster DNA-binding motifs and regulate transcription of melanin biosynthesis genes in a developmentally specific manner. Mol Microbiol 38:940–954
Tudzynski B (1999) Biosynthesis of gibberellins in Gibberella fujikuroi: biomolecular aspects. Appl Microbiol Biotechnol 52:298–310
Tudzynski B (2014) Nitrogen regulation of fungal secondary metabolism in fungi. Front Microbiol 5:656
Tudzynski B, Liu S, Kelly JM (2000) Carbon catabolite repression in plant pathogenic fungi: isolation and characterization of the Gibberella fujikuroi and Botrytis cinerea creA genes. FEMS Microbiol Lett 184:9–15
Umezawa H, Maeda K, Takeuchi T, Okami Y (1966) New antibiotics, bleomycin A and B. J Antibiot 19:200
Van Den Berg MA, Hans M (2011) Improved statin production. In DSM IP Assets BV, United States
Verdone L, Agricola E, Caserta M, Di Mauro E (2006) Histone acetylation in gene regulation. Brief Funct Genomics 5:209–221
Vertesy L, Kurz M, Schiell M, Hofmann J (2003) Cephaibols: novel antiparasitics from Acremonium tubakii process for their production, and use thereof. In Google Patents
Wakimoto T, Mori T, Morita H, Abe I (2011) Cytotoxic tetramic acid derivative produced by a plant type-III polyketide synthase. J Am Chem Soc 133:4746–4749
Waller GR, Burström H (1969) Diterpenoid alkaloids as plant growth inhibitors. Nature 222:576. https://doi.org/10.1038/222576a0
Walsh CT et al (2001) Tailoring enzymes that modify nonribosomal peptides during and after chain elongation on NRPS assembly lines. Curr Opin Chem Biol 5:525–534
Wansi JD, Devkota KP, Tshikalange E, Kuete V (2013) 14 – Alkaloids from the medicinal plants of Africa. In: Kuete V (ed) Medicinal plant research in Africa. Elsevier, Oxford, pp 557–605
Wee J (2015) Regulation and subcellular localization of aflatoxin biosynthesis in Aspergillus parasiticus. Michigan State University, Michigan
Wei S, Zhao F, Jiang Z, Zhou D (2013) Microbial conversion of waste cooking oil into riboflavin by Ashbya gossypii [Conversão microbiana de óleo de cozinha recolhido em riboflavina por Ashbya gossypii]. Biosci J 29:1000–1006
Weissman KJ (2009) Introduction to polyketide biosynthesis. Methods Enzymol 459:3–16
William CN et al (2005) Secondary metabolite biosynthetic gene clusters in filamentous fungi. In: Fungal genetics conference 23rd. Fungal Genetics Newsletter, California, p 190
Wilson NR, Hochstrasser M (2016) The regulation of chromatin by dynamic SUMO modifications. In: SUMO. Springer, New York, pp 23–38
Wiseman DW, Buchanan RL (1987) Determination of glucose level needed to induce aflatoxin production in Aspergillus parasiticus. Can J Microbiol 33:828–830
Wolf J, Mirocha C (1973) Regulation of sexual reproduction in Gibberella zeae (Fusarium roseum ‘Graminearum’) by F-2 (zearalenone). Can J Microbiol 19:725–734
Wong KH, Hynes MJ, Todd RB, Davis MA (2007) Transcriptional control of nmrA by the bZIP transcription factor MeaB reveals a new level of nitrogen regulation in Aspergillus nidulans. Mol Microbiol 66:534–551
Wong KH, Hynes MJ, Todd RB, Davis MA (2009) Deletion and overexpression of the Aspergillus nidulans GATA factor AreB reveals unexpected pleiotropy. Microbiology 155:3868–3880
Xu W, Gavia DJ, Tang Y (2014) Biosynthesis of fungal indole alkaloids. Nat Prod Rep 31:1474–1487
Yin W, Keller NP (2011) Transcriptional regulatory elements in fungal secondary metabolism. J Microbiol 49:329–339
Ziegler J, Facchini PJ (2008) Alkaloid biosynthesis: metabolism and trafficking. Annu Rev Plant Biol 59:735–769
Zutz C, Gacek A, Sulyok M, Wagner M, Strauss J, Rychli K (2013) Small chemical chromatin effectors alter secondary metabolite production in Aspergillus clavatus. Toxins 5:1723–1741
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Kumar, A., Kumar, A. (2019). Synthesis and Regulation of Fungal Secondary Metabolites. In: Arora, P. (eds) Microbial Technology for the Welfare of Society. Microorganisms for Sustainability, vol 17. Springer, Singapore. https://doi.org/10.1007/978-981-13-8844-6_2
Download citation
DOI: https://doi.org/10.1007/978-981-13-8844-6_2
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-8843-9
Online ISBN: 978-981-13-8844-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)