
Abstract
Fungal secondary metabolites play multiple biological roles in development, virulence, defense, adaptation, and stress response. Many of these low-molecular-mass compounds are of intense interest to humankind due to their economic applications (antibiotics and drugs) and/or adverse effects (mycotoxins). Many filamentous fungal secondary metabolites are synthesized by enzymes and related regulator(s) are encoded by clustered genes. Expression of these clusters are governed by various transcription factors and signaling elements. Recent studies have further revealed that fungal secondary metabolite genes are subject to epigenetic regulation. This chapter presents a concise summary of the epigenetic modifications and remodeling of the genes associated with fungal secondary metabolism, focusing on the genetic elements involved in epigenetic regulation, including HepA, (LaeA), complex proteins associated with Set1 (COMPASS) complex, histone deacetylases (HDACs), Spt-Ada-Gcn5-acetyltransferase (SAGA/ADA) complex, and small ubiquitin-like modifier (SUMO). Finally, the current applications of these epigenetic regulators are discussed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Epigenetics
- Fungi
- Secondary metabolism
- Gene Cluster
- Regulation
- Chromatin
- Histone
- Methylation
- Acetylation
- Sumoylation
Fungal Secondary Metabolism and Epigenetics
Fungi produce a diverse array of low molecular weight , bioactive secondary metabolites are not essential for their survival. Secondary metabolism (SM) is defined as “the production of ancillary metabolites and ‘useful’ compounds, initiated after using preferred carbon and nitrogen sources” [1, 2]. Secondary metabolites are not necessary for normal growth, but are considered important for the producing fungus to flourishing in its niche [3–5], stress tolerance [6, 7], or defense against hostile and/or competing organisms [1, 8]. They are important for day-to-day human life as beneficial antibiotics, pharmaceuticals, and/or harmful mycotoxins [9]. However, the true biological functions of many fungal secondary metabolites in producing fungi are largely cryptic.
Fungal SM is a complex process, which is often tightly related with morphological development [10]. Due to the importance of fungal secondary metabolites, an increasing number of genes associated with SM have been identified and characterized. Furthermore, the availability of fungal genomes accelerates the identification of biosynthetic genes for secondary metabolites. However, the role and regulatory mechanisms of many of the newly defined genes remain to be investigated [11]. In fungi, secondary metabolite biosynthetic and regulatory genes are usually clustered and not evenly distributed across the genomes [12–14]. Many of the clusters are silent under the standard laboratory culture conditions, which makes it difficult to elucidate their functions and regulatory mechanisms. It is both time and resource consuming to find the appropriate conditions to express the gene clusters of interest. A promising strategy to investigate unknown SM cluster(s) is via modifying global epigenetic regulators to activate the silenced SM clusters [15, 16].
Epigenetic phenomena are defined as reversible and heritable changes in gene expression levels without altering the DNA sequences. Epigenetic phenomena can derive from DNA-, chromatin- , and RNA-based effects, and include DNA methylation, position effects, RNA silencing systems, centromere/telomere location, and chromatin structure changes. Many of the aforementioned phenomena occur in fungi throughout the life cycle [17] which makes fungi an excellent model system to understand the fundamental principles of epigenetics . During the life cycle, fungi regulate development by several epigenetic mechanisms. Most steps or cell types are known to be under control by DNA methylation, which is regulated by changes in the chromatin state. Methylation induced premeiotically (MIP) and repeat-induced point mutation (RIP) occur during dikaryon formation and conjugated nuclear division [18–25]. MIP is regulated by DNA methylation, and RIP may also be regulated by it. Moreover, filamentous fungi share conserved silencing systems with higher eukaryotes, such as RNA interference (RNAi) and DNA methylation [26–31]. However, it is uncertain whether RNAi, which regulates the parasexual cycle and germination, is related to DNA- or chromatin-mediated epigenetic phenomena [32]. In addition, meiotic silencing by unpaired DNA, also known as meiotic silencing (MSUD), is another RNA silencing mechanism, that occurs throughout meiosis [33, 34].
As mentioned, fungal secondary metabolite synthetic and regulatory genes tend to be clustered. Gene clusters may originate from the horizontal transfer of genes from bacteria to fungi [35–40]. However, some SM gene clusters—e.g., gibberellin (GB) gene cluster—are unlikely a result of horizontal transfer [41]. The clustered SM genes are likely subject to co-regulation by epigenetic changes. An emerging field, chemical epigenetics, has been evolving to stimulate expression of secondary metabolite gene clusters by altering epigenetic status such as DNA and/or histone modifications [1, 42].
Epigenetic Modifications that Affect Secondary Metabolism
The epigenetic regulation of fungal SM is mainly through histone modifications: methylation, acetylation , and sumoylation (Fig. 3.1) [43–47]. Histone proteins are the primary protein components of chromatin and function as a scaffold for the nucleosome formation. Histone octamer consisting of two each of H2A, H2B, H3, and H4 is wrapped by DNA and forms a nucleosome [48].

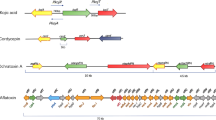
Examples of fungal secondary metabolites and their epigenetic regulators. Certain fungal secondary metabolites regulated by one or more epigenetic regulators are shown. PC Penicillin, LOV Lovastatin, GB Gibberellin, AT Asperthecin, GL Gliotoxin, OA Orsellinic Acid, TA Terrequinone A, ST Sterigmatocystin
Histone modifications can affect chromatin conformation and recruited proteins that cause epigenetic changes by interacting with histones [49]. Most histone modifications involve histones H3 and H4 [44, 45, 50]. The N-terminus of H3 and H4 are crucial to generate heterochromatin or euchromatin. In euchromatin, lysines in the H3 and H4 tails are hyperacetylated and H3K4 is trimethylated. In heterochromatin, in comparison, lysines in H3 and H4 are hypoacetylated and H3K9 is trimethylated [51]. By histone modifications, only a group of specific target genes inside of distinct regions of the chromosomes can be regulated, further supporting the advantage of SM genes being clustered [44, 45, 50].
Genes Affecting Histone Methylation
HepA
HepA is the Aspergillus nidulans homolog of HP1 (the heterochromatin protein-1, SWI6 in Schizosaccharomyces pombe) [52–54]. Heterochromatin domains are silenced and have hypoacetylation of lysines in H3 and H4 [55] with different degrees of methylation of H3K9 (H3K9me) by a histone methyltransferase (Clr4 in S. pombe) [56, 57]. As a transcriptional repressor, HP1 recognizes H3K9me and directly binds to it, achieving both targeting and transcriptional repression by maintaining the heterochromatin structure [58–63]. Artificial recruitment of HP1 to a gene promoter region leads to gene repression, supporting that HP1 is essential in gene silencing [64, 65].
HepA acts as an epigenetic repressor in expression of secondary metabolite genes [52]. The deletion of HepA leads to derepression of secondary metabolite biosynthetic genes, including sterigmatocystin (ST), penicillin (PC), and terrequinone A (TA). Biochemical analysis shows that the silent ST gene cluster is marked by H3K9me3 and recruits high levels of HepA, leading to repression of ST production during growth phase. Upon growth arrest and activation of SM, HepA, and H3K9me3 levels decrease while the acetylated histone H3 increases [52]. HepA occupancy and H3K9me3 levels are counteracted by the global SM regulator (LaeA) (Fig. 3.2) .

Overview and the roles of the epigenetic regulators in fungal secondary metabolism. Many epigenetic regulators participate in fungal secondary metabolism. HepA, Clr4, COMPASS, and LaeA are involved in histone methylation (red box, red stars indicate histone methylation). Clr4 leads to H3K9 methylation, which enables HepA binding to histone. HepA binding stabilizes the heterochromatin structure and thus leads to silencing the secondary metabolic gene clusters. COMPASS methylates H3K4 and H3K9, and silences SM, while LaeA removes the histone methylation and HepA binding and induces SM. HDACs and SAGA/ADA complex play a role in controlling histone acetylation (blue box, blue stars indicate histone acetylation), which induces fungal SM. HDACs deacetylate the lysines of H3 and/or H4, while the SAGA/ADA complex acetylates them. SUMO (the scarlet decagon) conducts sumoylation of several epigenetic regulators, including Clr4, COMPASS, HDACs, and SAGA/ADA, and silences SM
LaeA
(loss of aflR expression-A) is a global regulator of SM and development in filamentous fungi. This nuclear protein was first reported in Aspergillus spp. [44] . The lack of laeA blocks expression of several metabolic gene clusters, including ST, PC, and lovastatin (LOV). The overexpression of laeA contrarily increases expression of ST and LOV gene clusters and subsequent ST and LOV production [44]. In Penicillium chrysogenum, the overexpression of laeA increases PC production (~ 125 %) and the lack of laeA dramatically reduces PC gene expression levels and PC production [50]. Similarly, LaeA serves as a positive regulator of GB production in Fusarium fujikuroi [66]. In addition, microarray analysis indicates that LaeA regulates up to 9.5 % of the Aspergillus fumigatus transcriptome and up to 13 of its 22 secondary metabolite gene clusters, containing NRPS, PKS, and P450 monooxygenase genes [67].
LaeA forms a key heterotrimeric complex with the two velvet proteins VelB and VeA. The VelB/VeA/LaeA trimeric complex coordinates light signals with fungal development and SM [68]. VeA physically interacts with VelB, and bridges it to LaeA. All three components in this complex are essential for sexual development and ST production in A. nidulans. Previous studies showed that LaeA and VeA interact in P. chrysogenum and F. fujikuroi, too [50, 66]. The successful cross-genus complementation between Fusarium, Aspergillus, and Penicillium indicates that the VelB/VeA/LaeA complex has undergone a divergence in specific functions mediating SM [66] .
LaeA-mediated SM regulation primarily depends on histone methylation. LaeA contains a predicted and functionally necessary S-adenosyl-methionine (SAM) binding domain [68–70], which is present in all members of the methylase superfamily [71], and has sequence similarity to histone and arginine methyltransferase [44, 72]. The laeA gene is negatively regulated by AflR, a Zn2/Cys6 transcription factor located in the aflatoxin and ST gene clusters, in a feedback loop [44]. In A. nidulans, the ST gene cluster expression analysis shows that LaeA-mediated regulation of the cluster is location specific. The placement of argB in the ST cluster results in argB silencing in the laeA deletion background, whereas the genes bordering the ST cluster are unaffected [69]. Similar location-specific effects on SM gene regulation have been reported in other Aspergillus species as well [73–75]. Notably, the location specific effect is only reported in Aspergillus and Neurospora [13, 76],
These findings indicate that LaeA may differentially affect histone protein methylation, which in turn allows the cluster region to be more accessible to gene transcription [69]. Biochemical analyses of laeA and heterochromatin mutants (e.g., histone deacetylase and histone methyltransferase mutants) in A. nidulans demonstrate that LaeA activates SM gene expression by being involved in the removal of heteromatin marks like H3K9 methylation and HepA binding [13, 52, 77]; i.e., the LaeA-involved machinery reverses the heterochromatic signature and activates the gene expression inside the SM cluster .
COMPASS
COMPASS (complex proteins associated with Set1) is a multi-subunit complex consisting of Set1, Bre2, Sdc1, Spp1, Swd1, Swd2, and Swd3 [78–80]. COMPASS is involved in H3K4 mono-, di-, and tri-methylation [77, 79–82], which is necessary for RNA Pol II binding and transcriptional activity in development and differentiation [79, 80, 83] in Saccharomyces cerevesiae. Three core components, Set1, Swd1, and Swd3 are essential for COMPASS [78]. Swd2, Bre2, Sdc1, and Spp1 affect the degree of Set1 methylation [84–86]. Set1 has the SET domain, which possesses histone or lysine methyltransferase (HMTase or KMTase) activity [87].
CclA (Bre2 in S. cerevisiae) is one of the eight members of COMPASS in A. nidulans. The lack of CclA leads to reduced levels of H3K4 and H3K9 di- and tri-methylation, as well as reduced H3 acetylation [88]. H3K4 di- and tri-methylation is associated with actively expressed genes and are required for telomere silencing in S. cerevesiae [79, 80, 89–91], and activating A. nidulans SMs, e.g., monodictyphenone, emodins, and the polyketides F9775A and F9775B [52, 77]. In A. fumigatus, loss of CclA results in slow fungal growth and increased SM production like gliotoxin [92]. Based on 6-azauracil (6AU) sensitivity test result, CclA plays a role in transcription elongation [92] .
Genes Influencing Histone Acetylation
Histone Deacetylases (HDAC)
Histone deacetylases (HDACs) and histone acetyltransferases (HATs) play critical roles in fungal epigenetic regulatory mechanism. Histone acetylation is reversible and controlled by HDACs and HATs [51]. HDACs are classified into three main groups based on their homology to yeast proteins: Class I, HDACs have homology to yeast Rpd3; Class II, HDACs have homology to yeast Hda1; Class III, HDACs have homology to yeast Sir2. Both Classes I and II HDACs contain zinc in their catalytic site, and are known as epigenetic regulators in fungal SM. Class III HDACs do not have zinc in the catalytic site but require NAD+ instead [93].
A. nidulans RpdA is a Class I HDAC and the homolog of the global repressor Rpd3 in S. cerevisiae. RpdA is necessary for growth, conidiation, and gene regulation. The lack of Rpd3 leads to increased acetylation of H4K5, H4K12, and H3K18 in derepressed genes [94]. The absence of RpdA is lethal in A. nidulans and Neurospora crassa [95]. Silencing of RpdA in A. nidulans reveals that RpdA is involved in normal growth and H3 and H4 deacetylation [96].
Histone deacetylase A (HdaA) is a Class II HDAC playing a counter role to LaeA in SM regulation in A. nidulans. Loss of hdaA causes precocious and increased expression of ST and PC biosynthetic genes. The deletion of hdaA causes derepression of SM gene clusters that are located close to the telomeres in A. nidulans [97]. In A. fumigatus, HdaA plays a similar role in SM regulation [98]. Inhibition of most HDACs induces the production of unknown SMs in Penicillium expansum [97]. Treating the fungus with HDAC inhibitors leads to overproduction of several secondary metabolites, suggesting that HDAC-mediated repression of certain SM gene clusters is conserved in fungi [97].
SAGA/ADA Complex
The Spt-Ada-Gcn5-acetyltransferase (SAGA/ADA) coactivator complex regulates numerous cellular processes by posttranslational modifications of histones [99]. SAGA/ADA contains a HAT, Gcn5, and acetylates multiple lysine residues at the N-terminal tails of H3 and H2B. In A. nidulans, GcnE (Gcn5 homolog in A. nidulans) regulates PC biosynthesis gene cluster located on chromosome VI by histone acetylation [45, 100]. The Ada1–5 proteins (Alteration/deficiency in activation) are components of SAGA/ADA in S. cerevisiae [101]. Ada2/Ada3/Gcn5 complex is sufficient for robust histone and nucleosomal HAT activity in yeast [102] .
Both GcnE and AdaB are required for induction of the orsellinic acid gene cluster in A. nidulans. Similarly, SAGA/ADA plays a major role in specific induction of other SM gene clusters, such as ST, PC, and terrequinone [45]. Chromatin immunoprecipitation (ChIP) data shows that SAGA/ADA increases acetylation at H3K9 and H3K14 in A. nidulans. Interestingly, the increase of H3K14 acetylation is a global phenomenon of the whole genome, while the increase of H3K9 acetylation can be only observed within SM gene clusters [45].
Genes Impacting Sumoylation
SUMO
Small ubiquitin-like modifier (SUMO) is a small protein that has high structural similarity to ubiquitin, despite its low similarity at the level of the amino acid sequence [103–106]. SUMO covalently attaches to other proteins through the activities of an enzyme cascade (E1-E2-E3) similar to that of ubiquitination, and is known to play a role in histone modification like ubiquitin [105, 107–111]. Histone sumoylation mediates gene silencing through recruitment of HDAC and Hp1 both in vitro and in vivo in human cells [112, 113]. SUMO also modifies Gcn5, a member of the SAGA/ADA complex, and results in gene silencing in yeast [114].
In A. nidulans, SUMO represses sexual development and is involved in accurate induction and light stimulation of asexual development [104, 115]. CclA and SetA, two members of COMPASS, connects the SUMO network to histone modification. The interplay of the fungal sumoylation network controls temporal and spatial steps in cell differentiation [104] .
SUMO is also essential for sexual fruiting body formation and SM in A. nidulans [47, 116]. Deleting sumo causes about 200-fold increase of asperthecin production but decreases production of austinol/dehydroaustinol and ST [47]. The effect of sumoylation on SM may occur at several levels, such as silencing the secondary metabolite gene clusters at the chromatin level or regulating TFs involved in the SM regulation [47]. Additional work needs to be done to elucidate how SUMO regulates specific secondary metabolite production.
Application of Epigenetic Regulators of Fungal Secondary Metabolites
Understanding the SM epigenetic regulators can accelerate fungal SM studies by activating certain SM gene clusters that are often silent and cryptic in lab culture conditions. Suberoylanilide hydroxamic acid (SAHA), an HDAC inhibitor, has been used to stimulate the production of new cladochromes and calphostin B, a known protein kinase C inhibitor [117], in Cladosporium cladosporioides [118]. Treatment with SAHA can boost nygerone A production in Aspergillus niger [119, 120] and orsellinic acid production in A. nidulans without coculturing with Streptomyces rapamycincus [45]. In addition, using a global SM regulator is a new approach to identify new secondary metabolic genes. For example, LaeA is an excellent genomic mining tool and has successfully been manipulated to uncover several novel secondary metabolites such as terrequinone A [15, 16]. Another way to alter expression of SM gene clusters is to manipulate histone modification, for example, by the deletion of hepA [52] or hdaA [97].
Conclusion
Fungi produce a wide range of secondary metabolites. These low molecular weight compounds are diverse in structure and perform important yet often cryptic biological functions. The scientific community shows great interest in fungal secondary metabolites due to their importance to humankind. However, sequencing data of the fungal genomes indicate that a large number of fungal secondary metabolites are yet to be uncovered and characterized. As most fungal secondary metabolic gene clusters are silent under standard laboratory conditions, the importance of global regulators and epigenetic regulatory mechanism has been increasingly recognized. Various proteins and their complexes play a role in the regulation of fungal SM gene clusters through histone modification. Some of these epigenetic regulators mediate modification at distinct sites, such as methylation, acetylation, and sumoylation, whereas others inhibit such alterations (Fig. 3.2).
In this chapter, we have reviewed several known epigenetic regulators that are involved in regulating fungal SM. Epigenetics is an emerging area for investigating fungal SM, and a better understanding of SM epigenetic regulation would lead to the discovery of new drugs.
References
Brakhage AA, Schroeckh V (2011) Fungal secondary metabolites—strategies to activate silent gene clusters. Fungal Genet Biol 48(1):15–22
Brakhage AA, Schuemann J, Bergmann S, Scherlach K, Schroeckh V, Hertweck C (2008) Activation of fungal silent gene clusters: a new avenue to drug discovery. Natural compounds as drugs. Springer, Basel p. 1–12
Howard RJ, Valent B (1996) Breaking and entering: host penetration by the fungal rice blast pathogen Magnaporthe grisea. Annu Rev Microbiol 50(1):491–512
Kimura N, Tsuge T (1993) Gene cluster involved in melanin biosynthesis of the filamentous fungus Alternaria alternata. J Bacteriol 175(14):4427–4435
Tsai H-F, Chang YC, Washburn RG, Wheeler MH, Kwon-Chung KJ (1998) The developmentally regulated alb1 gene of Aspergillus fumigatus: Its role in modulation of conidial morphology and virulence. J Bacteriol 180(12):3031–3038
Leonard KJ (1977) Virulence, temperature optima, and competitive abilities of isolines of races T and O of Bipolaris maydis. Phytopathology 67(11):1273–1279
Klittich CJR, Bronson CR (1986) Reduced fitness associated with tox1 of Cochliobolus heterostrophus. Phytopathology 76(12):1294–1298
Yim G, Wang HH (2007) Antibiotics as signalling molecules. Philos Trans R Soc B Biol Sci 362(1483):1195–1200
Yu J-H, Keller N (2005) Regulation of secondary metabolism in filamentous fungi. Annu Rev Phytopathol 43:437–458
Calvo AM, Wilson RA, Bok JW, Keller NP (2002 Sep) Relationship between secondary metabolism and fungal development. Microbiol Mol Biol Rev 66(3):447–459
Fox EM, Howlett BJ (2008) Secondary metabolism: regulation and role in fungal biology. Curr Opin Microbiol 11(6):481–487
Ma L-J, Van Der Does HC, Borkovich KA, Coleman JJ, Daboussi M-J, Di Pietro A et al (2010) Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature 464(7287):367–373
Palmer JM, Keller NP (2010) Secondary metabolism in fungi: does chromosomal location matter? Curr Opin Microbiol 13(4):431–436
Fedorova ND, Khaldi N, Joardar VS, Maiti R, Amedeo P, Anderson MJ et al (2008) Genomic islands in the pathogenic filamentous fungus Aspergillus fumigatus. PLoS Genet 4(4):e1000046
Bouhired S, Weber M, Kempf-Sontag A, Keller NP, Hoffmeister D (2007) Accurate prediction of the Aspergillus nidulans terrequinone gene cluster boundaries using the transcriptional regulator LaeA. Fungal Genet Biol 44(11):1134–1145
Bok JW, Hoffmeister D, Maggio-Hall LA, Murillo R, Glasner JD, Keller NP (2006) Genomic mining for Aspergillus natural products. Chem Biol 13(1):31–37
Freitag M, Selker EU (2005) Controlling DNA methylation: many roads to one modification. Curr Opin Genet Dev 15(2):191–199
Selker EU, Jensen BC, Richardson GA (1987) A portable signal causing faithful DNA methylation de novo in Neurospora crassa. Science 238(4823):48–53
Singer MJ, Marcotte BA, Selker EU (1995) DNA methylation associated with repeat-induced point mutation in Neurospora crassa. Mol Cell Biol 15(10):5586–5597
Miao VPW, Freitag M, Selker EU (2000) Short tpa-rich segments of the ζ-η region induce DNA methylation in Neurospora crassa. J Mol Biol 300(2):249–273
Tamaru H, Selker EU (2003) Synthesis of signals for de novo DNA methylation in Neurospora crassa. Mol Cell Biol 23(7):2379–2394
Rossignol JL, Faugeron G (1995) Mip: An epigenetic gene silencing process in Ascobolus immersus. Gene silencing in higher plants and related phenomena in other eukaryotes: Springer, Verlag p. 179–191
Barry C, Faugeron G, Rossignol J-L (1993) Methylation induced premeiotically in Ascobolus: coextension with DNA repeat lengths and effect on transcript elongation. Proc Natl Acad Sci 90(10):4557–4561
Rhounim L, Rossignol J-L, Faugeron G (1992) Epimutation of repeated genes in Ascobolus immersus. EMBO J 11(12):4451
Faugeron G, Rhounim L, Rossignol J-L (1990) How does the cell count the number of ectopic copies of a gene in the premeiotic inactivation process acting in Ascoborus immersus? Genetics 124(3):585–591
Jorgensen RA, Que Q, Stam M (1999) Do unintended antisense transcripts contribute to sense cosuppression in plants? Trends Genet 15(1):11–12
Catalanotto C, Azzalin G, Macino G, Cogoni C (2000) Transcription: gene silencing in worms and fungi. Nature 404(6775):245–245
Romano N, Macino G Quelling: transient inactivation of gene expression in Neurospora crassa by transformation with homologous sequences. Mol Microbiol 6(22):3343–3353
Chen B, Choi GH, Nuss DL (1994) Attenuation of fungal virulence by synthetic infectious hypovirus transcripts. Science 264(5166):1762–1764
Choi GH, Nuss DL (1992) Hypovirulence of chestnut blight fungus conferred by an infectious viral cDNA. Science 257(5071):800–803
Nuss DL (2011) Mycoviruses, RNA silencing, and viral RNA recombination. Adv Virus Res 80:25
Hammond TM, Keller NP (2005) RNA silencing in Aspergillus nidulans is independent of RNA-dependent rna polymerases. Genetics 169(2):607–617
Aramayo R, Metzenberg RL (1996) Meiotic transvection in fungi. Cell 86(1):103–113
Shiu PKT, Raju NB, Zickler D, Metzenberg RL (2001) Meiotic silencing by unpaired DNA. Cell 107(7):905–916
Keller NP, Hohn TM (1997) Metabolic pathway gene clusters in filamentous fungi. Fungal Genet Biol 21(1):17–29
Rosewich UL, Kistler HC (2000) Role of horizontal gene transfer in the evolution of fungi. Annu Rev Phytopathol 38(1):325–363
Khaldi N, Collemare J, Lebrun M-H, Wolfe KH (2008) Evidence for horizontal transfer of a secondary metabolite gene cluster between fungi. Genome Biol 9(1):R18
Lawrence JG, Roth JR (1996) Selfish operons: horizontal transfer may drive the evolution of gene clusters. Genetics 143(4):1843–1860
Lawrence JG (1999) Gene transfer, speciation, and the evolution of bacterial genomes. Curr Opin Microbiol 2(5):519–523
Smith MW, Feng D-F, Doolittle RF (1992) Evolution by acquisition: the case for horizontal gene transfers. Trends Biochem Sci 17(12):489–493
Tudzynski B, Hedden P, Carrera E, Gaskin P (2001) The p450–4 gene of Gibberella fujikuroi encodes ent-kaurene oxidase in the gibberellin biosynthesis pathway. Appl Environ Microbiol 67(8):3514–3522
Wang X, Sena Filho JG, Hoover AR, King JB, Ellis TK, Powell DR et al (2010) Chemical epigenetics alters the secondary metabolite composition of guttate excreted by an atlantic-forest-soil-derived Penicillium citreonigrum. J Nat Prod 73(5):942–948
Shilatifard A (2006) Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Annu Rev Biochem 75:243–269
Bok JW, Keller NP (2004) LaeA, a regulator of secondary metabolism in Aspergillus spp. Eukaryotic cell 3(2):527–535
Nützmann H-W, Reyes-Dominguez Y, Scherlach K, Schroeckh V, Horn F, Gacek A et al (2011) Bacteria-induced natural product formation in the fungus Aspergillus nidulans requires saga/ada-mediated histone acetylation. Proc Natl Acad Sci 108(34):14282–14287
Strauss J, Reyes-Dominguez Y (2011) Regulation of secondary metabolism by chromatin structure and epigenetic codes. Fungal Genet Biol 48(1):62–69
Szewczyk E, Chiang Y-M, Oakley CE, Davidson AD, Wang CC, Oakley BR (2008) Identification and characterization of the asperthecin gene cluster of Aspergillus nidulans. Appl Environ Microbiol 74(24):7607–7612
Kornberg RD (1974) Chromatin structure: a repeating unit of histones and DNA. Science 184(4139):868–871
de la Cruz X, Lois S, Sánchez‐Molina S, Martínez‐Balbás MA (2005) Do protein motifs read the histone code? Bioessays 27(2):164–175
Kosalková K, García-Estrada C, Ullán RV, Godio RP, Feltrer R, Teijeira F et al (2009) The global regulator LaeA controls penicillin biosynthesis, pigmentation and sporulation, but not roquefortine C synthesis in Penicillium chrysogenum. Biochimie 91(2):214–225
Smith KM, Phatale PA, Bredeweg EL, Pomraning KR, Freitag M (2012) Epigenetics of filamentous fungi. In: Meyers RA (ed) Epigenetic regulation and epigenomics (Current Topics from the Encyclopedia of Molecular Cell Biolo). Wiley-VCH Verlag GmbH & Co., pp 1063–1107
Reyes‐Dominguez Y, Bok JW, Berger H, Shwab EK, Basheer A, Gallmetzer A et al (2010) Heterochromatic marks are associated with the repression of secondary metabolism clusters in Aspergillus nidulans. Mol Microbiol 76(6):1376–1386
Wang G, Ma A, Chow C-m, Horsley D, Brown NR, Cowell IG et al. (2000) Conservation of heterochromatin protein 1 function. Mol Cell Biol 20(18):6970–6983
Cryderman DE, Cuaycong MH, Elgin SC, Wallrath LL (1998) Characterization of sequences associated with position-effect variegation at pericentric sites in Drosophila heterochromatin. Chromosoma 107(5):277–285
Holbert MA, Marmorstein R (2005) Structure and activity of enzymes that remove histone modifications. Curr Opin Struct Biol 15(6):673–680
Rea S, Eisenhaber F, O’Carroll D, Strahl BD, Sun Z-W, Schmid M et al (2000) Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406(6796):593–599
Noma K-i, Allis CD, Grewal SI (2001) Transitions in distinct histone H3 methylation patterns at the heterochromatin domain boundaries. Science 293(5532):1150–1155
Fanti L, Pimpinelli S (2008) HP1: a functionally multifaceted protein. Curr Opin Genet Dev 18(2):169–174
Freitag M, Hickey PC, Khlafallah TK, Read ND, Selker EU (2004) HP1 is essential for DNA methylation in Neurospora. Mol Cell 13(3):427–434
Lewis ZA, Honda S, Khlafallah TK, Jeffress JK, Freitag M, Mohn F et al (2009) Relics of repeat-induced point mutation direct heterochromatin formation in Neurospora crassa. Genome Res 19(3):427–437
Sims RJ III, Nishioka K, Reinberg D (2003) Histone lysine methylation: a signature for chromatin function. TRENDS Genet 19(11):629–639
Bannister AJ, Zegerman P, Partridge JF, Miska EA, Thomas JO, Allshire RC et al (2001) Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature 410(6824):120–124
Lachner M, O’Carroll D, Rea S, Mechtler K, Jenuwein T (2001) Methylation of histone H3 lysine 9 creates a binding site for HP1 proteins. Nature 410(6824):116–120
Ayyanathan K, Lechner MS, Bell P, Maul GG, Schultz DC, Yamada Y et al (2003) Regulated recruitment of hp1 to a euchromatic gene induces mitotically heritable, epigenetic gene silencing: a mammalian cell culture model of gene variegation. Genes Dev 17(15):1855–1869
Li Y, Danzer JR, Alvarez P, Belmont AS, Wallrath LL (2003) Effects of tethering HP1 to euchromatic regions of the Drosophila genome. Development 130(9):1817–1824
Wiemann P, Brown DW, Kleigrewe K, Bok JW, Keller NP, Humpf HU et al (2010) FfVel1 and FfLae1, components of a velvet‐like complex in Fusarium fujikuroi, affect differentiation, secondary metabolism and virulence. Mol Microbiol 77(4):972–994
Perrin RM, Fedorova ND, Bok JW, Cramer RA Jr, Wortman JR, Kim HS et al (2007) Transcriptional regulation of chemical diversity in Aspergillus fumigatus by LaeA. PLoS Pathog 3(4):e50
Bayram Ö, Krappmann S, Ni M, Bok JW, Helmstaedt K, Valerius O et al (2008) VelB/VeA/LaeA complex coordinates light signal with fungal development and secondary metabolism. Science 320(5882):1504–1506
Bok JW, Noordermeer D, Kale SP, Keller NP (2006) Secondary metabolic gene cluster silencing in Aspergillus nidulans. Mol Microbiol 61(6):1636–1645
Hoffmeister D, Keller NP. (2007) Natural products of filamentous fungi: enzymes, genes, and their regulation. Natural Product Reports 24(2):393–416
Kozbial PZ, Mushegian AR (2005) Natural history of s-adenosylmethionine-binding proteins. BMC Struct Biol 5(1):19
Yin W, Keller NP (2011) Transcriptional regulatory elements in fungal secondary metabolism. J Microbiol 49(3):329–339
Chiou C-H, Miller M, Wilson DL, Trail F, Linz JE (2002) Chromosomal location plays a role in regulation of aflatoxin gene expression in Aspergillus parasiticus. Appl Environ Microbiol 68(1):306–315
Roze LV, Arthur AE, Hong SY, Chanda A, Linz JE. (2007) The initiation and pattern of spread of histone H4 acetylation parallel the order of transcriptional activation of genes in the aflatoxin cluster. Molecular Microbiol 66(3):713–726
Smith CA, Woloshuk CP, Robertson D, Payne GA (2007) Silencing of the aflatoxin gene cluster in a diploid strain of Aspergillus flavus is suppressed by ectopic aflR expression. Genetics 176(4):2077–2086
Smith KM, Kothe GO, Matsen CB, Khlafallah TK, Adhvaryu KK, Hemphill M et al (2008) The fungus Neurospora crassa displays telomeric silencing mediated by multiple sirtuins and by methylation of histone H3 lysine 9. Epigenetics Chromatin 1(1):5
Bok JW, Chiang Y-M, Szewczyk E, Reyes-Dominguez Y, Davidson AD, Sanchez JF et al (2009) Chromatin-level regulation of biosynthetic gene clusters. Nat Chem Biol 5(7):462–464
Roguev A, Schaft D, Shevchenko A, Pijnappel WWM, Wilm M, Aasland R et al (2001) The Saccharomyces cerevisiae Set1 complex includes an Ash2 homologue and methylates histone 3 lysine 4. EMBO J 20(24):7137–7148
Krogan NJ, Dover J, Khorrami S, Greenblatt JF, Schneider J, Johnston M et al (2002) Compass, a histone H3 (lysine 4) methyltransferase required for telomeric silencing of gene expression. J Biol Chem 277(13):10753–10755
Nagy PL, Griesenbeck J, Kornberg RD, Cleary ML (2002) A trithorax-group complex purified from Saccharomyces cerevisiae is required for methylation of histone H3. Proc Natl Acad Sci 99(1):90–94
Briggs SD, Bryk M, Strahl BD, Cheung WL, Davie JK, Dent SYR et al (2001) Histone H3 lysine 4 methylation is mediated by Set1 and required for cell growth and rdna silencing in Saccharomyces cerevisiae. Genes Dev 15(24):3286–3295
Santos-Rosa H, Bannister AJ, Dehe PM, Géli V, Kouzarides T (2004) Methylation of H3 lysine 4 at euchromatin promotes Sir3p association with heterochromatin. J Biol Chem 279(46):47506–47512
Eissenberg JC, Shilatifard A (2010) Histone H3 lysine 4 (H3K4) methylation in development and differentiation. Dev Biol 339(2):240–249
Nedea E, Nalbant D, Xia D, Theoharis NT, Suter B, Richardson CJ et al (2008) The Glc7 phosphatase subunit of the cleavage and polyadenylation factor is essential for transcription termination on snorna genes. Mol Cell 29(5):577–587
Dichtl B, Aasland R, Keller W (2004) Functions for S. cerevisiae Swd2p in 3’ end formation of specific mrnas and snornas and global histone 3 lysine 4 methylation. RNA 10(6):965–977
Cheng H, He X, Moore C (2004) The essential WD repeat protein Swd2 has dual functions in RNA polymerase ii transcription termination and lysine 4 methylation of histone H3. Mol Cell Biol 24(7):2932–2943
Shilatifard A (2012) The compass family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem 81:65.
Meyers RA (2012) Epigenetic regulation and epigenomics: advances in molecular biology and medicine. Wiley-Blackwell, Chichester
Mueller JE, Canze M, Bryk M (2006) The requirements for COMPASS and Paf1 transcriptional silencing and methylation of histone H3 in Saccharomyces cerevisiae. Genetics 173(2):557–567
Nislow C, Ray E, Pillus L (1997) Set1, a yeast member of thetrithorax family, functions in transcriptional silencing and diverse cellular processes. Mol Biol Cell 8(12):2421–2436
Schneider J, Wood A, Lee J-S, Schuster R, Dueker J, Maguire C et al (2005) Molecular regulation of histone H3 trimethylation by compass and the regulation of gene expression. Mol Cell 19(6):849–856
Palmer JM, Bok JW, Lee S, Dagenais TRT, Andes DR, Kontoyiannis DP et al (2013) Loss of CclA, required for histone 3 lysine 4 methylation, decreases growth but increases secondary metabolite production in Aspergillus fumigatus. PeerJ 1:e4
Dokmanovic M, Clarke C, Marks PA (2007) Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res 5(10):981–989
Robyr D, Suka Y, Xenarios I, Kurdistani SK, Wang A, Suka N et al (2002) Microarray deacetylation maps determine genome-wide functions for yeast histone deacetylases. Cell 109(4):437–446
Smith KM, Dobosy JR, Reifsnyder JE, Rountree MR, Anderson DC, Green GR et al (2010) H2B-and H3-specific histone deacetylases are required for DNA methylation in Neurospora crassa Genetics 186(4):1207–1216
Tribus M, Bauer I, Galehr J, Rieser G, Trojer P, Brosch G et al (2010) A novel motif in fungal class 1 histone deacetylases is essential for growth and development of Aspergillus. Mol Biol Cell 21(2):345–353
Shwab EK, Bok JW, Tribus M, Galehr J, Graessle S, Keller NP (2007) Histone deacetylase activity regulates chemical diversity in Aspergillus. Eukaryotic Cell 6(9):1656–1664
Lee I, Oh J-H, Keats Shwab E, Dagenais TR, Andes D, Keller NP (2009) Hdaa, a class 2 histone deacetylase of Aspergillus fumigatus, affects germination and secondary metabolite production. Fungal Genet Biol 46(10):782–790
Baker S, Grant P (2007) The saga continues: Expanding the cellular role of a transcriptional co-activator complex. Oncogene 26(37):5329–5340
Spröte P, Hynes MJ, Hortschansky P, Shelest E, Scharf DH, Wolke SM et al (2008) Identification of the novel penicillin biosynthesis gene aatB of Aspergillus nidulans and its putative evolutionary relationship to this fungal secondary metabolism gene cluster. Mol Microbiol 70(2):445–461
Barrios A, Selleck W, Hnatkovich B, Kramer R, Sermwittayawong D, Tan S (2007) Expression and purification of recombinant yeast Ada2/Ada3/Gcn5 and Piccolo NuA4 histone acetyltransferase complexes. Methods 41(3):271–277
Marcus GA, Silverman N, Berger SL, Horiuchi J, Guarente L (1994) Functional similarity and physical association between Gcn5 and Ada2: putative transcriptional adaptors. EMBO J 13(20):4807
Bayer P, Arndt A, Metzger S, Mahajan R, Melchior F, Jaenicke R et al (1998) Structure determination of the small ubiquitin-related modifier sumo-1. J Mol Biol 280(2):275–286
Harting R, Bayram Ö, Laubinger K, Valerius O, Braus GH (2013) Interplay of the fungal sumoylation network for control of multicellular development. Mol Microbiol 90(5):1125–1145
Pickart CM, Eddins MJ (2004) Ubiquitin: structures, functions, mechanisms. Biochim Biophys Acta 1695(1):55–72 ((BBA)-Molecular Cell Research)
Schwartz DC, Hochstrasser M (2003) A superfamily of protein tags: ubiquitin, SUMO and related modifiers. Trends Biochem Sci 28(6):321–328
Sampson DA, Wang M, Matunis MJ (2001) The small ubiquitin-like modifier-1 (SUMO-1) consensus sequence mediates Ubc9 binding and is essential for sumo-1 modification. J Biol Chem 276(24):21664–21669
Bernier-Villamor V, Sampson DA, Matunis MJ, Lima CD (2002) Structural basis for E2-mediated sumo conjugation revealed by a complex between ubiquitin-conjugating enzyme Ubc9 and RanGAP1. Cell 108(3):345–356
Ohi MD, Vander Kooi CW, Rosenberg JA, Chazin WJ, Gould KL (2003) Structural insights into the U-box, a domain associated with multi-ubiquitination. Nat Struct Mol Biol 10(4):250–255
Swanson R, Locher M, Hochstrasser M (2001) A conserved ubiquitin ligase of the nuclear envelope/endoplasmic reticulum that functions in both ER-associated and Matα2 repressor degradation. Genes Dev 15(20):2660–2674
Pichler A, Gast A, Seeler JS, Dejean A, Melchior F (2002) The nucleoporin RanBP2 has SUMO1 E3 ligase activity. Cell 108(1):109–120
Shiio Y, Eisenman RN (2003) Histone sumoylation is associated with transcriptional repression. Proceedings of the National Academy of Sciences 100(23):13225–13230
Shin JA, Choi ES, Kim HS, Ho JCY, Watts FZ, Park SD et al (2005) SUMO modification is involved in the maintenance of heterochromatin stability in fission yeast. Mol Cell 19(6):817–828
Sterner DE, Nathan D, Reindle A, Johnson ES, Berger SL (2006) Sumoylation of the yeast Gcn5 protein. BioChemistry 45(3):1035–1042
Harting R (2013) The sumoylation and neddylation networks in Aspergillus nidulans development. Dissertation, der Georg-August University
Wong KH, Todd RB, Oakley BR, Oakley CE, Hynes MJ, Davis MA (2008) Sumoylation in Aspergillus nidulans: sumO inactivation, overexpression and live-cell imaging. Fungal Genet Biol 45(5):728–737
Kobayashi E, Ando K, Nakano H, Iida T, Ohno H, Morimoto M et al (1989) Calphostins (UCN-1028), novel and specific inhibitors of protein kinase CI fermentation, isolation, physico-chemical properties and biological activities. J Antibiot (Tokyo) 42(10):1470–1474
Williams RB, Henrikson JC, Hoover AR, Lee AE, Cichewicz RH (2008) Epigenetic remodeling of the fungal secondary metabolome. Organic Biomol Chem 6(11):1895–1897
Fisch K, Gillaspy A, Gipson M, Henrikson J, Hoover A, Jackson L et al (2009) Chemical induction of silent biosynthetic pathway transcription in Aspergillus niger. J Ind Microbiol Biotechnol 36(9):1199–1213
Henrikson JC, Hoover AR, Joyner PM, Cichewicz RH (2009) A chemical epigenetics approach for engineering the in situ biosynthesis of a cryptic natural product from Aspergillus niger. Organic Biomol Chem 7(3):435–438
Acknowledgements
We thank Dr. Ellin Doyle for critically reviewing the manuscript. This work was supported by USDA Hatch (WIS01665) and the Intelligent Synthetic Biology Center of Global Frontier Project (2011-0031955) funded by the Ministry of Education, Science and Technology grants to JHY.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer Science+Business Media New York
About this chapter
Cite this chapter
Wu, MY., Yu, JH. (2015). Epigenetics of Fungal Secondary Metabolism Related Genes. In: Zeilinger, S., Martín, JF., García-Estrada, C. (eds) Biosynthesis and Molecular Genetics of Fungal Secondary Metabolites, Volume 2. Fungal Biology. Springer, New York, NY. https://doi.org/10.1007/978-1-4939-2531-5_3
Download citation
DOI: https://doi.org/10.1007/978-1-4939-2531-5_3
Published:
Publisher Name: Springer, New York, NY
Print ISBN: 978-1-4939-2530-8
Online ISBN: 978-1-4939-2531-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)