
Abstract
The removal of arsenic from solutions has been investigated for decades and continues to be a topic of intense research studies. Many arsenic treatment techniques have been recommended, but paucity still exists where practically feasible and less expensive and eco-friendly technique for remediation of arsenic is urgently required. Redox behavior and chemical species of arsenic in acidic aqueous system, photochemical oxidation of trivalent arsenic and the molecular reaction mechanism, formation mechanism and characteristics of tooeleite are stated. Cascade sulfide precipitation and separation of copper from high-arsenic acid wastewater, and a new process to remove arsenic efficiently by Fe3O4 hierarchical particles via adsorption in aqueous solution are well detailed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
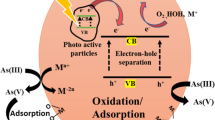

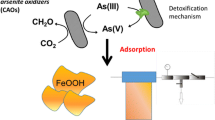
The removal of arsenic from solutions has been investigated for decades and continues to be a topic of intense research studies. The most commonly used arsenic removal methods are oxidation, precipitation, ion exchange, adsorption, lime treatment, biological removal processes, and reverse osmosis. A number of arsenic treatment techniques have been recommended or they are being used so far, but still paucity exists where practically feasible, less expensive, and eco-friendly technique for remediation of arsenic is urgently required.
3.1 Introduction
The over standard arsenic concentration has been detected in ground and surface waters, which arises from the discharge of arsenic-containing wastewaters [1, 2]. Arsenic is often found in association with nonferrous metal ore [3]. In nonferrous metal smelting process, acidic wastewater containing high arsenic is generated from the wet scrubber process of smelting fume [4] (acidic wastewater samples in Table 3.1 from a typical copper smelter and lead-zinc smelter).
Arsenic prevalently exists as arsenite (As(III)) and arsenate (As(V)) in aquatic environment. As(III) is predominant inorganic arsenic species in smelting and mining effluents [1, 5], and constitutes over 70% of dissolved arsenic in groundwater samples [6]. Including precipitation [7], coagulation [8, 9], ion exchange [10, 11], adsorption [12, 13], and so forth, many techniques are available for arsenic removal but inclined to remove As(V). Consequently, pre-oxidation is an essential anchor for As(III)-contained wastewater treatment. Various methods have been studied for As(III) oxidation (e.g., ozone [6], manganese and chlorine compounds [14, 15], ferrate [16], and hydrogen peroxide [17]). In comparison, the catalyzed oxidation systems are more attractive due to rapid reaction, such as, Fenton (Fe(II)/H2O2) [18], Fenton-like (Fe(III)/H2O2) [19], photo-Fenton (UV/H2O2) [20] TiO2 photocatalytic system (UV/TiO2) [21], etc. In these systems, the most important reactive species devoted to As(III) oxidation is determined as hydroxyl radical (HO·) and its reaction rate toward As(III) is very high (8.5 × 109 M−1 s−1) at acidic conditions [22]. Similar to HO·, sulfate radical (SO4·−) is also a strong oxidizing agent. It has been reported that SO4·− is more advantageous than HO· due to a longer half-life, and thus capable to escape from the solvent cage to oxidize As(III) [23, 24]. Moreover, SO4·− is a very strong electron acceptor enabling reactions which are impossible for HO· [25]. For example, perfluorinated carboxylic acids are inert toward HO· but can be degraded by SO4·− [26, 27]. A promising oxidation technique based on SO4·− has emerged over the past years to mineralize organic pollutants and oxidize As(III) [28,29,30,31]. The generation of SO4·− from peroxydisulfate (PDS, S2O82−) resembles that of HO· from H2O2. The peroxide bond in radical precursors PDS and H2O2 can be broken by heat, ultraviolet, ultrasound, radiolysis, etc. [28] Numerous activated methods (UV light [32], acoustic cavitation [31], and Fe(II) activation [29]) to generate SO4·− have thus been studied for As(III) oxidation. For comparison with the HO·-based oxidation, As(III) oxidation based on SO4·− should be discussed. The high intensive xenon (Xe) light can simulate natural solar light, which is effective to activate PDS and then stimulate As(III) oxidation at room temperature. In addition, we noticed that As(III) oxidation was rapid in the ferrous-activated PDS system, i.e., the initial few-minute reaction dominated in a 60-min oxidation process [29]. As for oxidation mechanism, both SO4·− and HO· were reported to play roles in As(III) oxidation via electron transfer reaction [29]. Tetravalent As(IV) as an intermediate has been postulated in the metal (Fe(II), Fe(III) and Cu(II)) catalyzed As(III)-PDS systems [33, 34]. In addition to the resultant As(OH)3·+ of electron transfer reaction, As(OH)4·, the product of HO·-based addition reaction, was also reported previously [35, 36]. Unfortunately, there is no suitable detection method for various arsenic intermediates and few studies on molecular mechanism of As(III) oxidation.
The behavior of arsenic in the environment strongly depends on its chemical forms. Most authors investigated adsorption and precipitation techniques for treatment of arsenic-containing wastewater and focused on the distribution of arsenic species such as H3AsO4, H2AsO4−, HAsO42−, AsO43−, H3AsO3, H2AsO3−, HAsO32−, and AsO33− [37]. Arsenic is removed from wastewaters mostly based on iron materials through precipitation and adsorption. The arsenic removal efficiency will be determined by the complexation between arsenic and iron to some extent. Arsenic and iron generally occur in water system as arsenite As(III), arsenate As(V), ferrous Fe(II), and ferric Fe(III). Many solid compounds of ferrous arsenite and ferrous arsenate have been reported [38] but no evidence for the existence of ferrous complexes with either arsenite or arsenate has been found. The arsenic disposal procedure currently favored by the industry involves the formation of an insoluble ferric arsenate compound [4]. Well-crystalline iron arsenate (e.g., scorodite) has advantages in lower ferric demand, higher density, and greater stability. Earlier synthesis of scorodite was conducted under autoclave conditions, i.e., under the high temperature and the high-pressure conditions [39]. Recently, atmospheric scorodite synthesis is of great concern due to lower capital investment [40]. Scorodite can be formed at temperatures as low as 40 °C [41], but amorphous precipitates such as ferric arsenate and its assemblages were more easily formed under lower temperatures [42]. Diverse precipitates in Fe–As-acid system are mainly depended on changed species in solutions. For example, we found the transformation of Fe(III)–As(V) complexes into colloid ferric arsenate previously [43]. Therefore, the species distribution and transformation in acidic Fe–As system is meaningful for developing novel phase with high potential in arsenic removal and stabilization by iron salts.
3.2 Redox Behavior and Chemical Species of Arsenic in Acidic Aqueous System
3.2.1 Redox Behavior of Arsenic
The Gibbs free energies of As(III) and As(V) species in HSC 7.0 database are in accordance with most reports [44,45,46,47,48,49]. An Eh-pH diagram for As (0.1 M)–H2O system was constructed by using HSC 7.0 software and depicted in Fig. 3.1[50]. The predominance fields for As(V) species under oxidizing conditions are similar to all published Eh-pH diagrams [51,52,53,54,55]. However, the field boundaries for As(III) species were different because of the inclusion of H3AsO3 and its conjugate base rather than HAsO2. The HAsO2 stoichiometry was supported by the mass spectrometry because of mass-to-charge-ratios (m/z) of 107 corresponding to AsO2− ion in the gas phase [56]. However, H3AsO3, a moiety comprising one arsenic atom coordinated by three OH ligands was supported by Raman and EXAFS spectroscopy [57,58,59]. The critical potentials of As(V)/As(III) couples against pH value indicate that the oxidation of As(III) to As(V) is more feasible in alkaline than in acidic solutions. Thus, the redox behavior of As(III) under strong acidic wastewater is challenging.

Reprinted from Ref. [50] Copyright 2017, with permission from Elsevier
The Eh (vs. SCE)-pH diagram in As (0.1 M)–H2O system at 25 °C and 1 bar.
The redox behavior of As(III) in sulfuric acid (pH = 1.0) was studied by cyclic voltammetry (CV) methods. Figure 3.2 showed the voltammogram of a solution containing 0.1 M As(III) and a blank solution. There are six peaks on the CV curve in 0.1 M As(III) solution (blue line). The peaks B and C correspond to the oxygen evolution and the reduction of the oxide layer on Au electrode. The location and current of peaks A, D, and F are relevant to the concentration of As(III). Peak A (Fig. 3.2) was identified as a multiple electron transfer oxidation of As(III) to As(V), which obviously splits into three small peaks when As(III) concentration is less than 5 mM (Fig. 3.3a). Moreover, the current of the first electron transfer reaction is linearly correlated with As(III) concentration (R2 = 0.9985, Fig. 3.3b) and the potential is ca. 0.9 V over the theoretical potential (ca. 0.3 V derived from Fig. 3.1). In addition, investigations at different scan rates showed a good linear relationship between the current of peak A and scan rate (v1/2) (Fig. 3.3d), therefore, As(III) oxidation is mainly diffusion-controlled.

Reprinted from Ref. [50] Copyright 2017, with permission from Elsevier
The voltammogram in 0.1 M As(III) solution (blue line) and blank solution (red line) (ν = 50 mV s−1).

Reprinted from Ref. [50] Copyright 2017, with permission from Elsevier
a The voltammogram in As(III) solutions, b and c linear calibration plots of peak current against As(III) concentration (As(III) = 0–5 mM, ν = 50 mV s−1), and d linear calibration plots of peak A current against v1/2 (As(III) = 0.1 M, v = 10, 20, 30, 40, 50 mV s−1).
Peak D corresponds to the reduction of As(III) and its peak current is linearly correlated to As(III) concentration. However, the different linear calibration plots of peak current against As(III) concentration were found in As(III) range of 0–1, 1–2, and 2–5 mM (Fig. 3.3c), which indicates that multilayer As(s) was electrodeposited from As(III) on Au electrode. The weak peak E can be explained as outmost As(s) electrodeposited from As(III) or from As(V). Although the reduction of As(V) to As(s) is thermodynamic sluggish, the electrogenerated H2 could chemically reduce As(V) to As(s) [60]. Following peak D and E, peak F is no doubt the oxidation of As(s) to As(III) and the linear correlation between peak current and As(III) concentration is in accordance with that of peak D. The consistent trend between peak D and F (Fig. 3.3c) confirmed that multilayer As(s) was electrodeposited on Au electrode.
3.2.2 Chemical Species of Arsenic
As(III) and As(V), respectively, exist as HnAsO3(n−3) and HnAsO4(n−3) (0 ≤ n ≤ 3) in As–H2O system. The speciation-pH diagram for As(III) and As(V) was calculated at ionic strength 0 based on thermodynamic formation constants reported by Marini et al. [61]. As depicted by thick line in Fig. 3.4a, As(V) predominantly existed as (1) H3AsO4 and H2AsO4− at pH 0–4.5, (2) H2AsO4− and HAsO42− at pH 4.5–9, (3) HAsO42− and AsO43− at pH 9–14. The major form of As(V) is neutral molecules H3AsO4 at pH < 2, thus it is hard to be removed by electrostatic adsorption. Figure 3.4b (thick line) showed that As(III) mainly existed as (1) H3AsO3 at pH 0–7.5, (2) H3AsO3 and H2AsO3− at pH 7.5–10.5, (3) H2AsO3− and HAsO32− at pH 10.5–12, and (4) H2AsO3−, HAsO32− and AsO33− at pH 12–14. Accordingly, H3AsO3 is the only As(III) species under acidic solutions. It was reported that undissociated H3AsO3 does not react with H2O2 [62]. However, rate constant k for the oxidation reaction between As(III) and H2O2 can be determined by pH, temperature, and IS in pH 7.5–10.3 [63]. This is mainly because the molar fraction of H2AsO3−, HAsO32−, and AsO33− species can be affected by pH, temperature, and IS. The speciation-pH diagram at IS = 0.2 M was determined based on the Davies equation. As depicted by thin line in Fig. 3.4, high IS facilitates deprotonation reaction. Accordingly, negative-charged species with higher fraction under high IS solutions can be absorbed on the positive-charged surfaces.

Reprinted from Ref. [50] Copyright 2017, with permission from Elsevier
Speciation-pH diagrams of a As(V) species and b As(III) species in As–H2O system at 25 °C. Thick line, IS = 0; thin line, IS = 0.2 M.
As(V) can complex with metal ion to form aqueous complexes [61]. Fe(III) is usually used as a precipitator for As(V) and Fe(III)–As(V) complexes are meaningful for understanding arsenic species and behaviors in Fe(III)–As(V)–H2O system. We investigated the complexation between As(V) and Fe(III) by UV-Vis spectroscopy in series of As–Fe–H2SO4–H2O acidic solutions (Table 3.2). A new peak at ca. 240–300 nm appeared as As(V) concentration increased and is attributed to Fe(III)–As(V) complexes (Fig. 3.5). The lgK of Fe(III)–As(V) complexes were reported by Marini et al. [64] and are in accordance with other reports excluding FeAsO4(a) (Table 3.3). The pH value, IS, and the fraction of various Fe(III) species were thus calculated by MINTEQ 3.1 with the consideration of Fe(III)–As(V) complexes or not. The calculated pH values are slightly higher than measured ones when Fe(III)–As(V) complexes were considered (Table 3.2). Without considering Fe(III)–As(V) complexes, the calculated Fe(III) species predominantly exists as FeSO4+ and Fe(OH)2+ when As(V) concentration is less than 45 mM and 45–50 mM, respectively (Fig. 3.6a). The band maxima of Fe(OH)2+ occur at the wavelengths of 300 nm [65], and thus the new peak at ca. 240–300 nm is not due to Fe(OH)2+. When Fe(III)–As(V) complexes were considered in species calculations (Fig. 3.6b), FeH2AsO42+ and FeHAsO4+ become major in 15–40 mM As(V) solutions and FeHAsO4+ is almost the only soluble Fe(III) species when As(V) is 40–50 mM. In addition, when the concentration of As(V) > 30 mM, the saturation index was higher than 0.577, and thus Hematite, Goethite, Lepidocrocite, FeAsO4·2H2O, etc., are oversaturated, which make experimental solutions turbid. Accordingly, Fe(III)–As(V) complexes are significant to be considered in Fe(III)–As(V)–H2O acidic solutions. A speciation-pH diagrams with inclusion of Fe(III)–As(V) complexes were calculated for Fe(III)–As(V)–H2SO4–H2O system, a representative system of As(V) removal by iron salts from acidic wastewater. As displayed in Fig. 3.7a, As(V) mainly exists as (1) FeH2AsO42+ and H3AsO4 at pH < 1, (2) FeH2AsO42+, FeHAsO4+, H3AsO4, and H2AsO4− at pH = 1–2, and (3) H2AsO4− at pH > 2. As shown in Fig. 3.7b, Fe(III) mainly exists as FeH2AsO42+ at pH < 2, and FeHAsO4+ at pH > 2. However, the total dissolved Fe(III) concentration decreased when pH is higher than 2. Therefore, As(V) mainly exists as FeH2AsO42+, FeHAsO4+, H3AsO4, and H2AsO4− in acidic wastewater treatment system.

Reprinted from Ref. [50] Copyright 2017, with permission from Elsevier
UV-Vis spectra of Fe2(SO4)3–Na3AsO4–H2SO4–H2O solution.

Reprinted from Ref. [50] Copyright 2017, with permission from Elsevier
Fe(III) species analyses in UV-Vis experiment solutions with a exclusion and b inclusion of Fe(III)–As(V) complexes.

Reprinted from Ref. [50] Copyright 2017, with permission from Elsevier
Speciation-pH diagrams of a As(V) and b Fe(III) with inclusion of Fe(III)–As(V) complexes. As(V) = Fe(III) = SO4 = 0.1 M.
3.2.3 Eh-pH Diagram in Fe–As–H2O System
Fe(III)–As(V) complexes are significant for the speciation of Fe(III) and As(V) and should also be included in the Eh-pH diagram for As–Fe–H2O system. Although Fe(III)–As(V), Fe(III)–As(III), Fe(II)–As(V), and Fe(II)–As(III) complexes were all predicted, Fe(III)–As(III) and Fe(II)–As(III) complexes are ruled out because of the possible redox reaction between Fe(III) and As(III) and the oxidation of As(III) in parallel to the dark oxidation of Fe(II) by dissolved O2 [69]. An Eh-pH diagram for Fe–As–H2O system with inclusion of Fe(III)–As(V) and Fe(II)–As(V) complexes were constructed and compared with the one with exclusion of these complexes. Figure 3.8a with exclusion of Fe–As complexes showed that FeAsO4(s) was predominant in the range of pH 1.2–3.4 under oxidizing conditions. It was reported that amorphous ferric arsenate could transform into scorodite [70]. The kinetics of scorodite formation and its transformation from ferric arsenic is strongly controlled by pH, for example, scorodite precipitated after ~384 h at pH 4.5 but ~13 h at pH 1 [71]. The pH of smelting acidic wastewater is usually less than 3, which is suitable for the production of ferric arsenate and scorodite. However, Fe(III)–As(V) complexes (FeH2AsO42+, FeHAsO4+, and FeH2AsO4+) were reported predominant under extremely acidic pH condition [72]. Thus, the Eh-pH diagram with inclusion of Fe–As complexes is more meaningful to understand the arsenic geochemistry in Fe–As–H2O system. Fe–As complexes were not considered in previous Eh-pH diagram mainly due to the unreliability of thermodynamic data. Accordingly, a new Eh-pH diagram for Fe–As–H2O system was constructed based on the estimates of equilibrium constants and Gibbs free energies. Figure 3.8b showed that Fe(III)–As(V) complexes shrank the stability field of H3AsO4 and FeAsO4(s). Moreover, Fe(II)–As(V) complexes occur at pH range of 0.5–3.0. The predominant field of Fe3(AsO4)2 was affected by Fe(II)–As(V) complexes. FeH2AsO42+, FeHAsO4+, and FeH2AsO4+ were all restricted at acidic pH conditions. Consequently, Fe–As(V) complexes are considerable in the arsenic removal from smelting acidic wastewaters by coprecipitation with iron salts.

Reprinted from Ref. [50] Copyright 2017, with permission from Elsevier
The Eh-pH diagram in 0.1 M As–0.1 M Fe–H2O system with the a exclusion and b inclusion of Fe–As complexes at 25 °C and 1 bar. Eh = E (vs. SCE) + 0.245.
3.2.4 Fe(III)–As(V) Complexes
In the sample solutions, Fe(III) concentration was 0.5 mmol L−1, and As(V) concentration systematically increased from 0 to 30 mmol L−1 in both HClO4 and H2SO4 systems. The pH value of all sample solutions was controlled at lower than 2.1, thus deprotonated As(V) species (H2AsO4−, HAsO42−, and AsO43−) and Fe(III) hydroxo complexes (Fe(OH)2+, Fe(OH)2+, Fe(OH)30, etc.) are ruled out [73, 74]. Fe(III)–As(V) complexes, if any, coexist with protonated As(V) (H3AsO4), free iron ion (Fe3+), and Fe(III)-sulfate complexes in sample solutions. The baseline-corrected UV-Vis spectra were shown in Fig. 3.9[75]. In HClO4 system, perchlorate (ClO4−) ion does not complex with Fe(III) [76], thus Fe3+ ion is the main Fe(III) species in As(V)-free solution. An absorption band on the spectrum of As(V)-free solution was located at ~240 nm (Fig. 3.9a), which is the feature band of Fe3+ [77]. When As(V) concentration increase, the spectra curves in the range of 250–340 nm gradually rise. An absorption band was located at around 280 nm, and its absorption strength is positively related to As(V) concentration. The absorption band of H3AsO4 is not close to 280 nm [43]. Therefore, only Fe(III)–As(V) complexes can explain the band at ~280 nm and the enhanced absorption in range of 250–340 nm. In H2SO4 system, sulfate can complex with Fe(III) to form Fe(III)-sulfate complexes (FeSO4+, Fe(SO4)2−, FeHSO42+, etc.) [19]. The absorption bands at ~305 and ~225 nm on the spectrum of As(V)-free solution are the feature bands of FeSO4+ [19, 78]. When As(V) concentration increases, spectra curves in the range of 240–300 nm gradually rise. A spectral band at ~300 nm is the feature band of Fe(SO4)2− [19]. Another band was located at about 280 nm and its absorption strength is also positively related to As(V) concentration. Accordingly, the ~280 nm band in both the HClO4 and H2SO4 system is probably owing to Fe(III)–As(V) complexes.

Reprinted from Ref. [50] Copyright 2017, with permission from Elsevier
UV-Vis spectra of sample solutions in a Fe(III)–As(V)–HClO4 system b Fe(III)–As(V)–H2SO4 system.
Quantitative analysis of UV-Vis data was performed and principal component analysis (PCA) was applied. PCA of the two series of spectra indicate that at least three and four absorbing species are believable to explain the UV-Vis data obtained in Fe(III)–As(V)–HClO4 system and Fe(III)–As(V)–H2SO4 system (Fig. 3.10). It is likely that Fe3+ and Fe(III)–As(V) complexes are responsible for the spectra obtained in the HClO4 system, while FeSO4+, Fe(SO4)2−, and Fe(III)–As(V) complexes are responsible in the H2SO4 system (Fig. 3.11). Aqueous Fe(III)–As(V) complexes are mononuclear structure with a formula of FeH2AsO42+, FeHAsO4+ or FeAsO40 rather than multinuclear structure [67, 72, 73, 79,80,81]. Gel-like FeAsO40 can make solution turbid and absorb visual light [43]. Hence, FeH2AsO42+ and FeHAsO4+ are proposed in limpid solutions. In addition, given the strong resemblance between H3AsO4 and H3PO4, FeH2AsO42+ and FeHAsO4+ are conceivable because FeH2PO42+ and FeHPO4+ are the main Fe(III)-phosphate complexes [82, 83]. Based on the “model-free” analysis, the molar absorbance spectrum curve of species A resembles the experimental spectrum of As(V)-free solution in HClO4 system, which implies that species A is Fe3+. The molar absorbance spectra of the other two species, B and C, have little discrepancy and have an absorption band at ~280 nm. Thus, they are postulated as Fe(III)–As(V) complexes FeH2AsO42+ and FeHAsO4+. Species D has two bands at about 305 and 225 nm, and species E has the feature band at ~300 nm. Thus, species D and E are FeSO4+ and Fe(SO4)2−, respectively. The spectra of species F and G are as same as B and C, which are identified as FeH2AsO42+ and FeHAsO4+ with the same absorption band at ~280 nm. Consequently, two Fe(III)–As(V) complexes, FeH2AsO42+, and FeHAsO4+, were reasonable in both Fe(III)–As(V)–HClO4 and Fe(III)–As(V)–H2SO4 systems, and they have a feature band at ~280 nm.

Reprinted from Ref. [50] Copyright 2017, with permission from Elsevier
Principal component analysis (PCA) on spectroscopic data in Fe(III)–As(V)–HClO4 and Fe(III)–As(V)–H2SO4 system. The grey line indicates the level of analytical uncertainty of 0.3%.

Reprinted from Ref. [50] Copyright 2017, with permission from Elsevier
Molar absorbance spectra from “model-free” analysis in a Fe(III)–As(V)–HClO4 system and b Fe(III)–As(V)–H2SO4 system.
X-ray absorption near edge structure (XANES) of Fe K-edge in Fe(III)–As(V)–HClO4 solutions dropped with increased As(V) concentration, but the primary feature was same (Fig. 3.12). The hexahydrate complex ([Fe(H2O)6]3+) with an octahedral Fe–O geometry [84, 85] is the only Fe(III) species in As(V)-free solution. The weak 1s → 3d transition (7110–7116 eV) was not enhanced as As(V) increased (see the inset in Fig. 3.12), which implies the Fe–O coordination of Fe(III)–As(V) complexes was still octahedral and same to [Fe(H2O)6]3+. In addition, the edge crest 1s → 4p becomes lower and broader as As(V) concentration increased, which shows that the Fe–O bond in Fe(III)–As(V) complexes will be inequivalent because of the common effects of complexation on distortion in octahedral geometry with shorter Fe–O distances to the anion(s) [86]. Moreover, the next higher energy feature after edge crest corresponds to single scattering of the photoelectron by the nearest neighbor O atom and shifts to higher energy with the increased As(V) concentration, implying that the nearest Fe–O distance became shorter due to the formation of Fe(III)–As(V) complexes in As(V)-contained solutions.

Reprinted from Ref. [50] Copyright 2017, with permission from Elsevier
XANES of Fe K-edge in Fe(III)–As(V)–HClO4 system (uncorrected for phase shift).
Since Fe–O coordination of Fe(III)–As(V) complexes is same as that of Fe(H2O)63+, it is feasible that HAsO42− and H2AsO4− substitute for one or two water molecules to form [FeH2AsO4(H2O)5]2+ and [FeHAsO4(H2O)5]+, or [FeH2AsO4(H2O)4]2+ and [FeHAsO4(H2O)4]+ (Fig. 3.13). Compared with the Fe–O bond in Fe(H2O)63+ (2.017), all Fe–O(H2) bonds of Fe(III)–As(V) complexes are longer than 2.017 Å. However, Fe–O(As) bonds are shorter than 2.017 Å. The average Fe–O distance of [FeH2AsO4(H2O)5]2+, [FeH2AsO4(H2O)4]2+, [FeHAsO4(H2O)5]+, and [FeHAsO4(H2O)4]+ are, respectively, 2.045 ± 0.102, 2.068 ± 0.055, 2.157 ± 0.07, and 2.065 ± 0.096 Å. Thus, the Fe–O bonds in Fe(III)–As(V) complexes are significantly inequivalent, which leads to irregular Fe sites and agrees with the XANES analysis. Additionally, the Fe–As distance of −2.7 Å in [FeH2AsO4(H2O)4]2+ and [FeHAsO4(H2O)4]+ is in accordance with that in bidentate edge-sharing (2E) surface complexes [87, 88]. The Fe–As distance in [FeHAsO4(H2O)5]+ is 3.38 Å and in [FeH2AsO4(H2O)5]2+ is 3.40 Å, which agrees with that in scorodite (FeAsO4·2H2O) [87, 89, 90]. The extended x-ray absorption fine structure (EXAFS) of Fe K-edge were analyzed based on the four optimized structures of Fe(III)–As(V) complexes. The k2-weighted x(k) functions fitted by monodentate structures ([FeHAsO4(H2O)5]+ and [FeH2AsO4(H2O)5]2+) are satisfied (Fig. 3.14a and Table 3.4). First Fe–O shell was satisfactorily fitted with a mean distance of 1.98 Å. ΔR = 0.10–0.18 Å implies a distorted FeO6 octahedra in Fe(III)–As(V) complexes. Similarly, FeO6 octahedron is also distorted in the scorodite structure with a Fe–O bond length spread of 0.167 Å [91]. Second Fe–As shell fitting gave a mean distance of 3.25–3.26 Å, which was a little shorter than that in crystalline scorodite (3.33 ± 0.01 Å) [92]. However, bidentate structures of Fe(III)–As(V) complexes ([FeHAsO4(H2O)4]+ and [FeH2AsO4(H2O)4]2+) with much shorter Fe–As distance (2.7 Å) are unlikely to be realistic due to unsatisfactory fitting. Therefore, Fe(III)–As(V) complexes are monodentate structures, i.e., [FeHAsO4(H2O)5]+ and [FeH2AsO4(H2O)5]2+. Meanwhile, the EXAFS spectra of As K-edge in sample solutions were all well fitted with AsO4 tetrahedra, for example, CN = 4.0 and R(As–O) = 1.68 (Fig. 3.14b). There was no As–Fe path because As(V) is great excessive and exists as H3AsO4 in sample solutions.

Reprinted from Ref. [50] Copyright 2017, with permission from Elsevier
The optimized structures of a [FeH2AsO4(H2O)5]2+, b [FeH2AsO4(H2O)4]2+, c [FeHAsO4(H2O)5]+, and d [FeHAsO4(H2O)4]+. Fe–O bond distances are shown.

Reprinted from Ref. [50] Copyright 2017, with permission from Elsevier
The Fourier transform of a Fe K-edge and b As K-edge in Fe(III)–As(V)–HClO4 system (uncorrected for phase shift).
TDDFT calculations were carried out at the CAM-B3LYP/6-311+G** level to illuminate the absorption spectroscopic nature of [FeHAsO4(H2O)5]+ and [FeH2AsO4(H2O)5]2+. The accuracy of spectra calculation was evaluated by Fe(H2O)63+ and the calculated feature band of Fe(H2O)63+ at around 240 nm is consistent with experimental spectrum. The absorptions of both [FeHAsO4(H2O)5]+ and [FeH2AsO4(H2O)5]2+ are in the range of 240–340 nm (Fig. 3.15), which is consistent with our experimental results, i.e., 250–340 nm in the HClO4 system and 240–300 nm in the H2SO4 system. Several strong electron excitations of [FeH2AsO4(H2O)5]2+ and [FeHAsO4(H2O)5]+ with significant oscillator strength (f ≥ 0.01) were considered to elucidate the absorption properties and listed in Table 3.5. There are three well-known types of excitations, including local excitation (LE), charge-transfer excitation (CT), and Rydberg excitation (R) [93]. The Δr is proposed as a quantitative indicator of electron excitation type. Δr ≤ 2 Å indicates LE, and Δr ≥ 2 Å identifies strong CT [93]. To a great extent, S6 of [FeH2AsO4(H2O)5]2+, S28–29 and S33 of [FeHAsO4(H2O)5]+ are in CT character, and other states are in LE character. Other parameters are auxiliary to identify the excitation types, which include the distance between centroid of hole and electron (D), and the integral of overlap of hole-electron (I) (Table 3.5). As depicted in Fig. 3.16, electron and hole are severally centered on O atoms and Fe atoms, which indicate that electron transfer proceeds from O atoms to Fe atoms. However, O atoms in Fe(III)–As(V) complexes are inequivalent, which can be defined as As-bonded O, Fe–As-bridged O and water O atoms in As–O(H), Fe–O−As and H–O−H structures, respectively.

Reprinted from Ref. [50] Copyright 2017, with permission from Elsevier
The calculated UV-Vis spectra of [FeH2AsO4(H2O)5]2+, and [FeHAsO4(H2O)5]+. The grey area indicates experimental absorption range of Fe(III)–As(V) complexes.

Reprinted from Ref. [50] Copyright 2017, with permission from Elsevier
Isosurface of hole and electron distribution. a and b respectively, represent [FeH2AsO4(H2O)5]2+ and [FeHAsO4(H2O)5]+ (e.g., a-S6 indicates S6 of [FeH2AsO4(H2O)5]2+). Blue and green isosurface represent electron and hole distributions, respectively. Brownish, red, and white spheres correspond to arsenic, oxygen, and hydrogen atoms, respectively. Iron atoms are wrapped by the isosurface of hole distribution.
According to the contribution of molecule orbitals (MO) to electron and hole, the significant electron transitions can be symbolized as corresponding frontier molecular orbital excitations. As depicted in Figs. 3.17 and 3.18, all related frontier singly occupied molecule orbitals (SOMO) are principally characterized as d-orbital of Fe atom. However, the frontier highest occupied molecule orbitals (HOMO) are totally different, which contributes to various electron excitations. The electron excitations S28 and S29 of [FeHAsO4(H2O)5]+ have absorptions at 330–340 nm, which are symbolized as HOMO-3 → SOMO and HOMO-2 → SOMO. HOMO-2 and HOMO-3 of [FeHAsO4(H2O)5]+ are localized mainly on As-bonded O atoms. It is accordingly an electron excitation from As-bonded O atoms to d-orbital of Fe, corresponding to a ligand-to-metal charge transfer (LMCT). In the case of S6 of [FeH2AsO4(H2O)5]2+ and S33 of [FeHAsO4(H2O)5]+, the involved HOMOs are mainly located on Fe–As-bridged O atoms, which yields an excitation around 300–310 nm. As shown in Fig. 3.16, a-S6 and b-S33, there is no significant overlap between the electron and hole, consequently LMCT type. Nearly equal excitation wavelengths for S36 and S38–39 of [FeHAsO4(H2O)5]+ is 280–290 nm. The involved HOMOs approximately spread over all O atoms, whereas contributions of Fe–As-bridged O are predominant. Accordingly, S36 and S38–39 of [FeHAsO4(H2O)5]+ are assigned to MC transitions. In a case of S20–21 of [FeH2AsO4(H2O)5]2+, their excitation wavelengths are around 250 nm, corresponding to MC transition with the major contributions of water O atoms. Since all SOMOs are mainly characterized as d-orbital of Fe, the excitation wavelength is blue-shifted from 340 to 250 nm along with the localization of HOMOs on As-bonded O, Fe–As-bridged O and water O atoms. Therefore, the experimental spectra in the range of 250–340 nm in Fe(III)–As(V)–HClO4 system and of 240–300 nm in Fe(III)–As(V)–H2SO4 system are attributed to different electron excitations of Fe(III)–As(V) complexes. Moreover, the experimental band at around 280 nm is attributed to the MC excitations chiefly from Fe–As-bridged O atoms to d-orbital of Fe, which can be considered a feature band of aqueous Fe(III)–As(V) complexes.

Reprinted from Ref. [50] Copyright 2017, with permission from Elsevier
Related frontier molecular orbitals of [FeHAsO4(H2O)5]+ calculated at the TDDFT/CAM-B3LYP/6-311+G** CPCM level of theory (isosurface value = 0.02 e/Å3).

Reprinted from Ref. [50] Copyright 2017, with permission from Elsevier
Related frontier molecular orbitals of [FeH2AsO4(H2O)5]2+ calculated at the TDDFT/CAM-B3LYP/6-311+G** CPCM level of theory (isosurface value = 0.02 e/Å3).
3.2.5 Species Transformation of Fe(III)–As(V) Complexes
The complexation of Fe(III) and As(V) was investigated spectrophotometrically in a series of Fe(III)–As(V)–HClO4–H2O solutions. The absorbance at 280 nm increased proportionally to the concentrations of As(V) due to the formation of Fe(III)–As(V) complexes shown in Fig. 3.19a. In order to verify that the absorbance at 280 nm was due to ferric–arsenate complex, same method was conducted to the Fe(III)–SO4 complexes and the absorbance at 305 nm corresponding to FeSO4+ [94, 95] was also shown in Fig. 3.19b. Here the correlation curve of absorbance at 305 nm versus sulfate was compared with that of absorbance at 280 nm versus arsenate in Fig. 3.20. Linear correlation with negative deviation at higher sulfate and arsenate concentrations was observed for the two correlation curves. The similar correlation trends of two curves gave indirect evidence that a peak at 280 nm corresponded to ferric–arsenate complex.

Reprinted from Ref. [43] by permission of The Royal Society of Chemistry 2015
The UV-Vis spectra of a ferric–arsenate sample solutions and b ferric–sulfate sample solutions measured after 24 h standing at 25 ± 0.1 °C.

Reprinted from Ref. [43] by permission of The Royal Society of Chemistry 2015
The correlation between absorbance of the feature band and sulfate/arsenate concentration.
To determine the equilibrium constant for Fe(III)–As(V) and Fe(III)–SO4 complexes, the interaction was analyzed according to Benesi–Hildebrand equations (Eq. (3.1)) based on Fig. 3.20 [96].
where [Fe(III)] represents the concentration of the ferric ions, l is the length of quartz cuvette, ΔA is the difference in absorbance between the complex and ferric ion, C is the concentration of ligand; ε and K are the molar absorptivity of the complex at λ and the equilibrium constant for complex formation, respectively. The highly linear relationship (R2 = 0.9969 and 0.9980) between 1/ΔA and 1/C indicated that the absorbance of the complex followed the Benesi–Hildebrand equation (Fig. 3.21). The molar absorptivity of Fe(III)–SO4 and Fe(III)–As(V) complexes is \( \varepsilon \)305 = 1.716 × 103 M−1 cm−1 and \( \varepsilon \)280 = 2.797 × 103 M−1 cm−1, respectively. Correspondingly, the equilibrium is Kf = 1.56 × 102 M−1 (log Kf = 2.2) and Kf = 1.28 × 102 M−1 (log Kf = 2.1).

Reprinted from Ref. [43] by permission of The Royal Society of Chemistry 2015
The Benesi–Hildebrand fitting of Fe(III)–As(V) complex (circle) and Fe(III)–SO4 complex (square).
In order to investigate what would happen in As(V)–SO4.−-rich system, UV-V is spectra of two series of solutions containing both As(V) and SO4.− were collected for comparison. In the first series of solutions, SO4.− concentration was 20 mM and As(V) increased from 0 to 20 mM. Specifically, the absorbance increased significantly in the range of 240–300 nm and decreased significantly in the range of 300–380 nm (Fig. 3.22a). Therefore, Fe(III)–As(V) complexes were formed even in the presence of SO4.−. In the second series of solutions, the concentration of As(V) was 20 mM and SO4.− increased from 0 to 20 mM, the absorbance of solutions showed no obvious change as SO4.− concentration increased (Fig. 3.22b). Accordingly, the formation of Fe(III)–SO4.− complexes were hindered by As(V), and thus the complexation of As(V) is stronger than SO4.− toward Fe(III).

Reprinted from Ref. [43] by permission of The Royal Society of Chemistry 2015
UV-Vis spectra of Fe(III)–As(V)–SO4–HClO4 solutions. a Fe: 0.5 mM; As(V): 20 mM; SO4: 0–20 mM. b Fe: 0.5 mM, As(V): 0–25 mM; SO4: 20 mM.
The pH value is one of the key parameters for the formation of ferric arsenate solid from Fe(III)–As(V)–H2O system. In a series of solutions containing 0.5 mM Fe(III) and 20 mM As(V), perchlorate acid was added to control pH values in the range of 0.35 to 6.81. Sample solutions were limpid at pH ≤ 1.57 and turned turbid at higher pH of 2.38 because gel-like material was formed. These findings by naked eye were in accordance with the results of UV-Vis spectra (Fig. 3.23). The turbid solutions absorbed both ultraviolet light and visual light at pH ≥ 2.38, however, only ultraviolet light was absorbed at pH ≤ 1.57. It is well-known that the hydrolysis of Fe(III) ion and deprotonation of H3AsO4 are closely related to pH values. As(V) existed as H3AsO4 in acidic, H2AsO4− in weak acidic and HAsO42− in nearly neutral solutions due to pK1 = 2.2 and pK2 = 6.96. The first hydrolysis constant of Fe3+ at 0.1 M NaClO4 was 2.54 [97] and thus FeOH2+ formed under higher pH conditions, which can react with negatively charged HAsO42− to form FeAsO4. Moreover, the deprotonation of FeH2AsO42+ and FeHAsO4+ with pH increase will also produce FeAsO4. Therefore, the formed gel-like material was speculated as FeAsO4 [98] under higher pH solutions.

Reprinted from Ref. [43] by permission of The Royal Society of Chemistry 2015
UV-Vis spectra at different pH values (Fe(III): 0.5 mM L−1; As(V): 20 mM L−1).
Three solutions containing 0.5 mM Fe(III) and 10, 20 and 30 mM As(V) were heated at 90 °C for 30 min and then became turbid. The ultraviolet light was absorbed whether the solution was heated or not, and only visual light was absorbed after being heated (Fig. 3.24a). Thus, it can be concluded that higher temperature (T ≥ 90 °C) benefits for the formation of gel-like material. A continuous heating process was conducted for the solution containing 20 mM As(V) and the absorbance at 280 nm increased slightly at the early stage and increased dramatically later (Fig. 3.24b). An obvious boundary of the time was about 3000 s and the temperature was about 90 °C. Therefore, enough heat is helpful to form the gel-like material.

Reprinted from Ref. [43] by permission of The Royal Society of Chemistry 2015
a UV-Vis spectra of sample solutions before and after heated (Fe(III): 0.5 mM L−1; As(V): 10, 20 and 30 mM L−1; Heating time: 20 min), b absorbance at 280 nm in a continuous heating process (Fe(III):0.5 mM L−1; As(V): 20 mM L−1).
Gel-like material formed in 0.1 M perchlorate acid solutions containing 45 mM As(V) and 0.5 mM Fe(III) was separated and dried at 60 °C. The molar ratio of As: Fe: O was 1: 1.11: 3.77 in this solid, which is close to the atomic ratio in FeAsO4. XRD indicated that the gel-like material was poorly crystalline ferric arsenate characterized by two broad peaks centered at 2θ values of 28° and 58° (Fig. 3.25) [99]. Poorly crystalline ferric arsenate was reported as scorodite precursor with a formula of FeAsO4·(2 x) H2O (0 < x < 1) [100]. The gel-like solid was further characterized using FTIR (Fig. 3.26). A strong band at 1630–1636 cm−1 and a broad band near 3445 cm−1 were attributed to the O–H bending and stretching vibration of water. The stretching vibration of As–O–Fe was located at 838 cm−1 and generally with a shoulder at ~750 cm−1, which is probably caused by the hydrogen bonding between H2O and AsO4. An important IR absorption at 835 cm−1 in Fig. 3.25 with a shoulder at ~750 cm−1 belonged to the ν3 mode of AsO4 (~833 cm−1), identified as arsenate bonding structures of poorly crystalline ferric arsenate [99]. Other IR absorption bands at about 2363, 1384, and 1088 cm−1 were, respectively, antisymmetric stretching mode of carbon dioxide (−OH···O=C=O) adsorbed on hydroxyl groups [101], characteristic IR absorption of NO3− (1384 cm−1) [102] and ClO4− (1088 cm−1) [103] anions. These impurities were introduced by iron nitrate and perchloric acid. The results of XRF, XRD, and FTIR demonstrated that gel-like material in our experiments was poorly crystallized ferric arsenate. Scanning electron microscopy (SEM) images showed that the sample was bulk and conchoidal (Fig. 3.27). It was different from other reported precipitates in Fe(III)–As(V)–H2O system which were aggregated particles [100]. This may be because the poorly crystalline ferric arsenate was formed from precursors of FeHAsO4+ and FeH2AsO42+, while other amorphous ferric arsenate was formed through transformation of the arsenate ions adsorption on the surface of the ferrihydrite.

Reprinted from Ref. [43] by permission of The Royal Society of Chemistry 2015
XRD patterns of the gel-like material in solid phase.

Reprinted from Ref. [43] by permission of The Royal Society of Chemistry 2015
FTIR of the gel-like material in solid phase.

Reprinted from Ref. [43] by permission of The Royal Society of Chemistry 2015
SEM of the gel-like material in solid phase.
3.3 Photochemical Oxidation of As(III) and the Molecular Reaction Mechanism
3.3.1 The SO4·−-Based As(III) Oxidation
Peroxydisulfate (PDS) was more effective than H2O2 for oxidizing monomethylarsonic acid (MMA) and dimethylarsinic acid (DMA) under UV irradiation [104]. PDS is advantageous over H2O2 for As(III) oxidation because the oxidation yields in PDS system were all higher than in H2O2 system, especially under Xe lamp irradiation (Fig. 3.28) [105]. The oxidation yield is 56% in PDS/Xe combination, whereas only 12% and 17% in PDS/dark and PDS/daylight combination, respectively. Therefore, PDS/Xe is an effective combination for As(III) oxidation, where SO4·− may be generated due to the decomposition of PDS under Xe lamp irradiation [106]. Xe lamp solar simulator can emit the light of continuous band from UVA to visible range. As far as we know, UVA (320–400 nm) rays of solar radiation are natural resources on ground, which penetrate clouds more readily than UVB and UVC rays. Consequently, As(III) oxidation in PDS/Xe combination is meaningfully investigated to assess the feasibility of As(III) oxidation under solar radiation.

Reprinted from Ref. [105] Copyright 2017, with permission from Elsevier
The oxidation yield of As(III) by PDS or H2O2 after 30-min reaction under dark, daylight and Xe lamp. [As(III)]0 = 20 μmol L−1, [PDS]0 = [H2O2]0 = 10 μmol L−1.
When As(III) oxidation under PDS/Xe combination was compared with that under Xe lamp alone and PDS alone conditions, the reaction between As(III) and PDS is very slow in absence of Xe lamp (PDS alone), and tiny As(III) (−10%) was oxidized after 30-min reaction (Fig. 3.29). However, about 55% As(III) was oxidized after 30 min with Xe lamp alone (without PDS), which is coincidentally equal to the oxidation yield in PDS/Xe combination at Rox/As(III) = 0.5 in Fig. 3.28. When PDS was dosed in excess, i.e., Rox/As(III) = 2.5, the reaction process in PDS/Xe combination is totally different from that of Xe lamp alone. In PDS/Xe combination at Rox/As(III) = 2.5, a rapid oxidation was observed in the initial 2 min and the oxidation yield remained at –82% afterwards. Similarly, a pronounced decrease of As(III) concentration was observed within the initial 2 min in Fe(II)-persulfate system, where SO4·− and HO· were both determined to play key roles [106].

Reprinted from Ref. [105] Copyright 2017, with permission from Elsevier
Comparison results in different reaction system. [As(III)]0 = 20 μmol L−1.
SO4·− and HO· were involved either individually or simultaneously during the activation of PDS due to the reaction of Eq. (3.2) or (3.3) [107].
ESR was employed here to identify the radicals involved in As(III) oxidation process(Fig. 3.30). ESR signal of DMPO-HO· adduct was obviously detected in PDS/Xe system in the presence and absence of As(III). Additionally, no DMPO-SO4·− adduct was detected even DMPO was added in great excess. It can be explained by the conversion from SO4·− to HO· then followed by DMPO spin-trapping. In an acidic system, only SO4·− was expected to be the main reactive species [105]. However, only strong signal of DMPO-HO· adduct was detected even in the presence of 2.5 mmol H2SO4 (pH = −0). Therefore, a fast disappearance of DMPO-SO4·− adduct and simultaneous generation of DMPO-HO· adduct can be explained by the reaction of Eq. (3.4) [108, 109].

Reprinted from Ref. [105] Copyright 2017, with permission from Elsevier
ESR spectra obtained from different systems within 2-min reaction. As(III) = 0.5 μmol. a–e PDS = 10 μmol, DMPO = 80 μmol, H2SO4 = 2.5 mmol; f PDS = 50 μmol, DMPO = 560 μmol. Signal of DMPO-HO· marked in red rectangle and DMPO-SO4·− marked in blue foursquare.
Moreover, DMPO-HO· adduct signal was not obtained in the absence of PDS, which confirmed that DMPO-HO· adduct formation is concomitant with SO4·−. Accordingly, SO4·− is the main reactive species, which simultaneously contributes to the oxidation of As(III) and the generation of HO· in PDS/Xe system.
In quenching studies, methanol and isopropanol have been used as radical scavenger, which can readily react with both SO4·− and HO· to confirm the radical and nonradical reaction [110] (Fig. 3.31). Tert-butanol was used as a selective scavenger because it reacts 1000-fold faster with HO· (6 × 108 M−1 s−1) [111] compared to SO4·− (4–9.1 × 105 M−1 s−1) [112]. The oxidation of As(III) was almost completely stopped by 100 mmol L−1 methanol and isopropanol, individually. Accordingly, the rapid As(III) oxidation is a radical reaction. In the presence of 1, 10 and 100 mmol L−1 tert-butanol, the oxidation also occurred mainly in the initial few minutes, but the oxidation yield became lower in the presence of more tert-butanol. Compared with the complete quenching effect of methanol and isopropanol, the quenching effect of tert-butanol is much lower. For example, in comparison with the blank experiment (no scavenger), the oxidation yield decreased by −93% in the presence of 100 mmol L−1 methanol or isopropanol, whereas only −42% decrease was observed in the presence of the same amount of tert-butanol. Therefore, the contribution of SO4·− to As(III) oxidation is over 50% and the rest contribution (not more than 42%) was attributed to HO·. Consequently, both SO4·− and HO· was mainly responsible for As(III) oxidation.

Reprinted from Ref. [105] Copyright 2017, with permission from Elsevier
Quenching effect of methanol, isopropanol, and tert-butanol on the oxidation of As(III) in PDS/Xe combination system. [As(III)]0 = 20 μmol L−1, [PDS]0 = 50 μmol L−1.
3.3.2 Kinetics of Oxidation Reaction
SO4·− radical is a footstone for the rapid oxidation reaction occurred in the initial few minutes in PDS/Xe system and its production is controlled by PDS content but independent on light source because the power density of Xe lamp was maintained at 1.57 w cm−2. The As(III) oxidation reaction can be simply described as Eq. (3.5), where PDS rather than SO4·− is associated with As(III).
Although tetravalent As(IV) was reported as high reactive intermediate, it can be fast oxidized to As(V) and reduced to As(III) [113]. Therefore, the decrease of As(III) can represent the amount of generated As(V). Initial As(III) ([As(III)]0) and residual As(III) at reaction time t ([As(III)]t) were detected to study the reaction kinetics. In addition, oxidation yields of As(III) were calculated based on Eq. (3.6).
It can be seen that As(III) oxidation yields increased faster as [PDS]0 increased in the initial 2 min (Fig. 3.32a), which illustrated higher [PDS]0 facilitated the rapid As(III) oxidation reaction. Moreover, when [PDS]0 < 50 μmol L−1, oxidation yield increased to −80% as prolonged reaction time. However, oxidation yield maintained unchanged when [PDS]0 > 50 μmol L−1. The cleavage of S2O82− produces two SO4·− radicals, which are sufficient to oxidize As(III) to As(IV) and subsequently to As(V) [114], i.e., it is very likely that the stoichiometric ratio of As(III) and PDS is R = 1. Therefore, [PDS]0 > 50 μmol L−1 were overdosed for 20 μmol L−1 [As(III)]0 and the oxidation reaction finished quickly in the initial 2 min. However, the oxidation yields decreased as the increase of [As(III)]0 in the initial 2 min. Especially, when [As(III)]0 > 50 μmol L−1, the oxidation yields decreased dramatically as the increase of [As(III)]0. This is because [PDS]0 = 50 μmol L−1 is insufficient for [As(III)]0 > 50 μmol L−1. Fortunately, the lower oxidation yields can be elevated in the subsequent reaction time. For example, reaction proceeding from 2 to 30 min, the oxidation yield increased from 56 to 81% when [As(III)]0 = 70 μmol L−1 (Fig. 3.32b). This may be attributed to superoxide radical (HO2·/O2·−), H2O2 and O2, which are generated via reactions in Eqs. (3.7) and (3.8) [115].

Reprinted from Ref. [105] Copyright 2017, with permission from Elsevier
Oxidation yield under various initial concentrations of PDS and As(III). a [As(III)]0 = 20 μmol L−1 and [PDS]0 = 15–120 μmol L−1, b [As(III)]0 = 5–80 μmol L−1 and [PDS]0 = 50 μmol L−1.
In the rapid As(III) oxidation process, PDS concentration is unambiguously known and no subproducts can be detected. The initial reaction rate method [116, 117] is chosen to study reaction kinetics. As(III) concentrations were fitted against reaction time and the derivative at zero time was determined to get the initial reaction rate (r0). The reaction rate equation is constructed as Eq. (3.9).
where square brackets represent concentration, a and b are the apparent reaction order toward As(III) and PDS, respectively.
Because [As(III)]0 > 50 μmol L−1 was over-stoichiometrical for 50 μmol L−1 [PDS]0, the kinetics fitting was only conducted for [As(III)]0 < 50 μmol L−1. The apparent reaction order toward [As(III)]0 derived from the slope of fitting of lnr0 and [As(III)]0 is 0.51, i.e., a = 0.51 (Fig. 3.33a). In addition, the apparent reaction order toward [PDS]0 based on the fitting of lnr0 and ln[PDS]0 is 0.64, i.e., b = 0.64. The apparent reaction rate equation can thus be expressed as Eq. (3.10). The apparent reaction order with respect to [PDS]0 and [As(III)]0 is close and the apparent reaction stoichiometric ratio of 1.25, which is close to the theoretical value R = 1. Besides, the rate constant with respect to [As(III)]0 (ka1) and with respect to [PDS]0 (ka2) is 8.4 × 10−2 and 2.5 × 10−2 M−0.15 s−1, respectively.

Reprinted from Ref. [105] Copyright 2017, with permission from Elsevier
Linear fitting of a lnr0 versus ln[As(III)]0 and b lnr0 versus ln[PDS]0. a [As(III)]0 = 5–40 μmol L−1 and [PDS]0 = 50 μmol L−1, b [As(III)]0 = 20 μmol L−1 and [PDS]0 = 15–120 μmol L−1.
The rapid oxidation process is different under various temperatures (293–330 K) (Fig. 3.34a). The initial reaction rates r0 under different temperatures were figured out according to the apparent reaction rate equation (Eq. (3.10)). The reaction rate constant at 293 K is 6.25 × 10−2 M−0.15 s−1, which lies in our postulated range of 2.5–8.4 × 10−2 M−0.15 s−1. Following the Arrhenius equation shown in Eq. (3.11), lnk and 1/T was fitted linearly (Fig. 3.34b) and the apparent activation energy Ea is 46.09 kJ mol−1. It is much lower than the average activation energy of As(III) oxidation by H2O2 (96.6 kJ mol−1) and by dissolved O2 (73.2 kJ mol−1) [118, 119].

Reprinted from Ref. [105] Copyright 2017, with permission from Elsevier
a Oxidation of As(III) under different temperatures and b lnk versus T−1 plot. [As(III)]0 = 20 μmol L−1 and [PDS]0 = 50 μmol L−1.
The effect of initial pH value (pH0 < 3) showed that the rapid As(III) oxidation was partly retarded under lower pH0 (Fig. 3.35a). The rate constant increased from 0.7 × 10−2 to 5.4 × 10−2 M−0.15 s−1 with pH0 increasing from 0.68 to 2.81. The linear fitting of logk and pH0 (Fig. 3.35b) indicates that the effect of pH0 on the reaction rate constant can be described as Eq. (3.12). The slight inhibition effect of pH0 can be attributed to the decrease of HO· under acidic conditions. Therefore, the mixture of SO4·− and HO· is beneficial for the rapid As(III) oxidation under neutral and weak alkaline conditions. However, oxidation yields after 30-min reaction was not affected by pH0. We noticed a pH drop in the 30-min reaction process, e.g., pH value went down from 6.02 to 3.96 after 30-min reaction. It is previously reported that the dropped pH is due to H+ production in the reaction between SO4·− and solvent H2O molecule (Eq. (3.2)) [120]. It was also found that higher pH0 resulted in greater pH decrease. As listed in Table 3.8, when pH0 = 6.02, 2.81, 2.15 and 0.68–1.49, pH decrease was, respectively, 1.74, 0.22, 0.03 and −0 after 10-min reaction. Similarly, the greater pH decrease was reported in higher pH0 reaction system of Fe(II)/PDS [29]. Additionally, the pH decrease was not limited in the initial 2 min but proceeded in the subsequent time with a slower rate. The production of H+ in 2–30 min cannot be explained by the reaction of SO4·− and solvent H2O due to no available SO4·−. Therefore, we postulated that deprotonation reaction of resultant H3AsO4 may also contribute to the pH decrease. Based on this, (1) no pH decrease was found at pH0 < 2 because pKa1 of H3AsO4 is 2.2 [72], (2) more H+ can be generated under higher pH conditions due to the significant deprotonation of H3AsO4 under higher pH conditions, (3) the quick pH decrease in the initial 2 min is due to the rapid formation of resultant H3AsO4.

Reprinted from Ref. [105] Copyright 2017, with permission from Elsevier
a Oxidation of As(III) under different pH0 and b logk versus pH0 plot. [As(III)]0 = 20 μmol L−1 and [PDS]0 = 50 μmol L−1.
3.3.3 Molecular Reaction Mechanism
H3AsO3 is the main species of As(III) under acidic and weak alkaline conditions due to its pKa1 = 9.2 [59]. SO4·− is mainly responsible for As(III) oxidation in PDS/Xe combination system, however, HO· induced by SO4·− can also be involved in As(III) oxidation process, especially under neutral and weak alkaline conditions. The oxidation of H3AsO3 by SO4·− and HO· was both envisaged to occur via reactions displayed in Table 3.9. The calculations were conducted by Gaussian 09 software at IEFPCM (water)/M06-2X/6-311++G**//M06-2X/6-31G* level. Seven pathways were supposed for the oxidation from H3AsO3 to H3AsO4 under the single function of HO· (Table 3.10) and the second and forth pathway has least elementary reactions and the lowest Gibbs free energy in the controlling step. Although SO4·− can play a role in several elementary reactions, there is no available pathway for H3AsO3 oxidized by only SO4·− to H3AsO4. In mixed SO4·− and HO· system, six different pathways were proposed for the oxidation from H3AsO3 to H3AsO4 and the first pathway has least elementary reactions and the lowest Gibbs free energy in the controlling step (Fig. 3.36). Therefore, the most favorable pathways for H3AsO3 oxidation to H3AsO4 is H3AsO3 → H4AsO4· → H3AsO4 and H3AsO3 → H2AsO3· → H3AsO4 in HO·-based system, and H3AsO3 → H4AsO4· → H3AsO4 in mixed SO4·− and HO· system where SO4·− plays a role in the first step.

Reprinted from Ref. [105] Copyright 2017, with permission from Elsevier
Possible pathways of H3AsO3 oxidation to H3AsO4 by SO4·− and HO· and the Gibbs free energy (kcal mol−1).
3.4 Formation Mechanism and Characteristics of Tooeleite from High-Arsenic Acid Wastewater
The sequestering of arsenic in tooeleite is considered to be the best environmentally friendly possible waste form for stable outdoor storage.
3.4.1 Direct Removal of As(III) by the Formation of Tooeleite
The formation of tooeleite was investigated in pH range from 1.8 to 9. XRD patterns (Fig. 3.37b [121]) confirmed that the precipitate obtained in pH range of 1.8 to 4.5 was tooeleite, consistent with PDF44-1468. This pH range for tooeleite formation is wider than the previous report (2.0–3.5) [122]. However, tooeleite would transform to amorphous ferric arsenite when pH > 4.5. As(III) and Fe(III) removal efficiency increased with the increase of pH, for example, As(III) removal sharply increased from 30% at pH 1.8 to around 90% at pH 2.8 (Fig. 3.37a). This is because that the mixed solution was in the metastable state at pH 1.8, and primary nucleation of tooeleite may carry a portion of As(III) and Fe(III) after a period of inducing time [123]. Considering hydrolysis reaction, Fe(III) is supposed to start precipitating at pH 1.76 and complete at pH 3.17 and thus the majority of Fe(III) will be removed with the increase of pH from 1.8 to 2.8. The formation of iron hydroxides makes contributions to the removal of As(III) as well as the formation of tooeleite. However, As(III) removal decreased at pH 4–5 and then increase again with continuous pH increase. The decrease can be explained by As releasing from the partial transform of tooeleite to amorphous FeOOH(As) [122] and the formation of FeAsO3 at pH 5–9 lead to the reprecipitation of As(III) [124]. The removal of As(III) can reach above 99% at pH 4 by forming well-crystallized tooeleite. Therefore, pH 4 is suggested for the treatment of acid wastewater containing a high concentration of As(III).

Reprinted from Ref. [121] Copyright 2017, with permission from Elsevier
As(III) and Fe(III) removal efficiency (a) and XRD patterns of As-Fe precipitates(b) at pH 1.8–9.
The As(III) concentration from 0.75 to 7.5 g L−1 is suitable for forming tooeleite but higher As(III) facilitates the crystallization of tooeleite (Fig. 3.38b). The higher As(III) also facilitates As(III) removal efficiency but has no significant effect on iron removal. Since the solution pH is 4, precipitation of iron leads to persistent high iron removal but different As(III) removal due to different initial As(III) concentration (Fig. 3.38a). When As(III) concentration lowed to 0.5 g L−1, As(III) removal efficiency was 60% but no tooeleite was found. Adsorption of As(III) on iron hydroxides benefits As(III) removal [125], with the increase of As(III) initial concentration, As(III) was incorporated into the growing iron hydroxides phase and finally co-precipitated as tooeleite [126].

Reprinted from Ref. [121] Copyright 2017, with permission from Elsevier
As(III) and Fe(III) removal (a) and XRD patterns of As–Fe precipitates (b) as affected by initial As(III) concentration varied from 0.5 to 7.5 g L−1.
The formation of tooeleite can be realized at the Fe/As from 0.8 to 2 and the crystallinity degree increased with Fe/As (Fig. 3.39b). For all Fe/As, Fe(III) removal maintained over 99%, however, As(III) removal linearly increased from 46 to 97% against Fe/As from 0.8 to 2 (Fig. 3.39a). As(III) removal exhibited a good Fe-dependency and we hypothesized that iron compounds like ferrihydrite were formed due to the introduction of NaOH and increased As(III) promoted the transformation of ferrihydrite to tooeleite [127].

Reprinted from Ref. [121] Copyright 2017, with permission from Elsevier
As(III) and Fe(III) removal (a) and XRD patterns of As–Fe precipitates (b) as affected by Fe/As varied from 0.8 to 2.
3.4.2 Characterization of Tooeleite
SEM images of tooeleite obtained at pH 4 exhibited a flower structure of platy particles (1 μm) aggregating together (Fig. 3.40a). Quantitative chemical analysis shows the As content of tooeleite is 23.38%, which is much higher than normal arsenic-bearing stabilized slag (5–15%) [128]. The XPS As 3d spectrum confirmed that arsenic exists as As(III) in tooeleite precipitate (Fig. 3.40b). This provides strong evidence that As(III) can be directly removed and solidified in tooeleite from high-As(III) acid wastewater. IR spectrum (Fig. 3.40c) confirmed stretching modes and bending vibrations of adsorbed water at 3396 and 1632 cm−1, three unique SO42− absorption bands in tooeleite (sorbed v3(SO4) at 1112 cm−1, outer sphere bound v1(SO4) at 990 cm−1 and structural v4(SO4) at 618 cm−1) [129], band of AsO33− at 778 cm−1 and Fe–O–As band at 513 cm−1 [130].

Reprinted from Ref. [121] Copyright 2017, with permission from Elsevier
SEM image (a), XPS analysis (b) and FTIR analysis (c) of precipitate obtained at pH 4.0.
3.4.3 Safety Evaluation
The short-term stability experiments (Fig. 3.41a) showed that As leaching at pH 1.8, 2.8 and 4 was far less than at pH 6 and 9. This is because tooeleite was formed at pH below 4, while As–Fe compound with poor crystal quality at pH 6 and 9 (Fig. 3.41b). Therefore, tooeleite is much more stable than poor-crystallized precipitates obtained at higher pH. However, much lower pH is unfavorable for the stability of tooeleite due to the adsorption of some As on the surface of tooeleite. Tooeleite prepared at pH 1.8 showed the lowest arsenic leaching, however, it still exceeds the national criteria (5 mg L−1). This puts us heavy pressure to find some ways to improve the stability of tooeleite.

Leaching concentration of arsenic (a) and XRD patterns after leaching experiments (b) at pH 1.85, 2.80. 4.00, 6.00 and 9.00
3.4.4 Tooeleite Formation Mechanism
Samples were collected during the formation process of tooeleite and their color changed from reddish brown to lemon yellow as time goes by (Fig. 3.42). XRD (Fig. 3.43b) of the sample at 1 h exhibited two broad bands at 2θ of 31° (d ≈ 2.86 Å) and 60° (d ≈ 1.55 Å). Comparing with ferrihydrite [131], a little shift toward higher d-spacing was ascribed to the adsorption of As(III) [129]. Samples obtained after 17 h showed the enhanced diffraction peaks at 9.06, 27.78, and 29.28°, respectively, marked by the indices (020), (200) and (061) of tooeleite. In addition, FTIR spectra (Fig. 3.44) of samples obtained at 1, 6, 17 and 24 h showed weakened characteristic peaks of ferrihydrite at 1193 and 1046 cm−1 and enhanced Fe–O–As bond at 510 cm−1 [130]. During the transformation process, Fe(III) was almost removed and As(III) removal increased from 90% at beginning to −100% at 5 day (Fig. 3.43a).

Reprinted from Ref. [121] Copyright 2017, with permission from Elsevier
Color change of obtained samples as affected by reaction time.

Reprinted from Ref. [121] Copyright 2017, with permission from Elsevier
As(III) and Fe(III) removal efficiency (a) and XRD patterns of As–Fe precipitates (b) as affected by reaction time varied from 1 to 96 h.

Reprinted from Ref. [121] Copyright 2017, with permission from Elsevier
FTIR of As–Fe precipitates (1, 6, 17, 24 h).
Samples were obtained after 24-h reaction in solutions with Fe/As = 0.8 and after another 24-h reaction when Fe/As was adjusted by iron sulfate to 1.5. The two group samples were all confirmed as tooeleite (Fig. 3.45b). However, the removal of As(III) and Fe in the latter group are a little higher than in the former group (Fig. 3.45a). The formation mechanism of tooeleite can be described as that Fe(III) precipitates as ferrihydrite at pH up to 4.0 and adsorbs As and sulfate, and then transforms to tooeleite (Fig. 3.46).

Reprinted from Ref. [121] Copyright 2017, with permission from Elsevier
As(III) and Fe(III) removal efficiency (a) and XRD patterns (b) on the two Groups.

Reprinted from Ref. [121] Copyright 2017, with permission from Elsevier
Schematic diagram of tooeleite formation process.
3.5 Cascade Sulfide Precipitation and Separation of Copper and Arsenic from Acidic Wastewater
Many industrial processes, especially in the mining and metallurgical processing industry, discharge acidic effluents containing significant amounts of metals such as copper, nickel, zinc, lead, and arsenic [132, 133]. Copper is often associated with arsenic in the mixed sulfide minerals such as enargite (Cu3AsS4) and tennantite (Cu12As4S13) [134], therefore, among these heavy metals, copper and arsenic are found to behave similarly and exist simultaneously in the wastewater [135].
Arsenic contamination has greatly threatened water safety due to its high toxicity and carcinogenicity [136, 137]. In the case of liquid effluents, there are many methods available for the removal of heavy metals, such as chemical precipitation, adsorption, coagulation, ion exchange, microbial reduction, and so on [138,139,140]. On the other hand, dissolved air flotation was used to separate and recover heavy metals. Stalidis et al. [141] separated copper, zinc and arsenic ions from dilute aqueous solutions by the dissolved air technique via the production of fine gas bubbles. It seems to be complex and expensive for the treatment of acidic wastewater. Sulfide precipitation is indeed an effective process for the treatment of toxic heavy metals ions [142, 143]. One of the primary advantages of sulfide precipitation is that the process can achieve a high degree of metal removal over a broad pH range. Metal sulfide sludge also exhibits better thickening and dewatering characteristics than the corresponding metal hydroxide sludge [144]. Bhattacharyya et al. [145] separated arsenic and other heavy metals using sodium sulfide. The removals of cadmium, zinc, and copper from the actual wastewaters are greater than 99%, and those of arsenic and selenium are 98% and > 92%, respectively. However, it is H2S that reacts with heavy metals by gas–liquid reaction as sulfur exists almost in the form of H2S rather than S2− and HS− in acid conditions. It takes 5–10 times excess of theoretical amount of sodium sulfide, moreover, a lot of hydrogen sulfide gas escapes, which leads to serious secondary pollution [146].
A novel process of treating acidic wastewater containing copper and arsenic is proposed, involving primary separation of copper and arsenic by sulfide precipitation and further separation by the replacement of arsenic in the precipitates by copper in the solution [147].
3.5.1 Thermodynamics for Separation of Copper and Arsenic
With the introduction of S2−, Cu2+, and As(III) precipitated in the form of CuS and As2S3, respectively. The solubility equilibrium reaction of CuS and As2S3 can be expressed as follows:
The sulfur-containing aqueous solution consists of [H+], [OH−], [H2S], [HS−], [S2−], and the total sulfur ion of the system can be expressed as follows:
The stepwise stability constants for each ion at 298.15 K are used as follows:
It can be deduced from Eqs. (3.17) and (3.19) to Eq. (3.21):
The total copper of the solution system can be expressed as follows:
Similarly, arsenic in the water has the following chemical equilibrium:
It can be deduced from Eq. (3.25) to Eq. (3.27):
Combined Eqs. (3.17) and (3.23) with Eq. (3.28), the total arsenic can be expressed as follows:
For \( [{\text{H}}^{ + } ]= 1 0^{-\text{pH}} \), it can be deduced from Eqs. (3.24) and (3.29):
Hypothesizing that CuS and As2S3 stoichiometrically achieved dissolution equilibrium, then [Cu]T = [S]T, 3[As]T = 2[S]T, log[Cu]T and log[As]T as a function of pH value are shown in Fig. 3.47. With the increase of pH value, the solubility of CuS decreases first and then increases. The minimum solubility of CuS is 1.733 × 10−14 mol L−1 when the pH value is 7.75. The solubility of As2S3 is greater than that of CuS and increases with the increasing pH value, which favors the possibility of cascade sulfidation of copper and arsenic in the solution.

Reprinted from Ref. [147] Copyright 2017, with permission from Elsevier
Log[M]T – pH curves of CuS and As2S3.
3.5.2 Cascade Sulfide Precipitation and Separation Technique
The experimental program was divided into two stages. In the first stage, copper and arsenic were primarily separated by sulfide precipitation. The schematic diagram of the experimental apparatus is shown in Fig. 3.48. In the second stage, precipitates obtained in the first stage were recycled to the initial solution to improve the grade of copper in the precipitates through the replacement of arsenic by copper.

Reprinted from Ref. [147] Copyright 2017, with permission from Elsevier
Schematic diagram of the experimental apparatus.
Gaseous sulfide source (H2S) generated was collected by displacement of water. Two-way valve was turned off at first. The gas tank was filled with the saturated H2S solution and sealed with paraffin. Gaseous sulfide generated in the three-necked flask was then introduced into the tank. Solution in the gasholder was pushed into the fluid reservoir when the two-way valve was opened. A certain volume fraction of H2S gas was obtained by introducing nitrogen into the gasholder when the pressure gauge of cylinder was adjusted to the specified pressure with nitrogen cylinder open. The operating temperature and pH were controlled at a desired level. H2S gas was then delivered to the reactor and the simulated copper sulfate was pumped to the reactor by peristaltic pump.
3.5.3 Removal Efficiencies of Copper and Arsenic
(1) Reaction time
Arsenic and copper removal efficiency, barely influenced by reaction time (Fig. 3.49), are constant at 10% and 30%, respectively, as the reaction time increases from 0.5 to 4 min with addition of 10 mmol sulfide. However, it becomes 100% for both arsenic and copper when 70 mmol sulfide was added into the solution, which implies that sulfidation of arsenic and copper has finished within 0.5 min.

Reprinted from Ref. [147] Copyright 2017, with permission from Elsevier
Arsenic and copper removal efficiency at different reaction times (50 °C).
(2) Temperature
The copper removal efficiency increases from 60 to 80% when temperature increases from 24 to 60 °C, whilst arsenic removal efficiency decreases from 25 to 15%, indicating significant effect of temperature on arsenic and copper removal efficiency (Fig. 3.50). The improvement of separation efficiency of copper and arsenic can be achieved via raising the temperature. Besides, the temperature of acidic wastewater discharged from the industry process is as high as 55 °C, so the separation of arsenic and copper under the high temperature could be realized practically.

Reprinted from Ref. [147] Copyright 2017, with permission from Elsevier
Arsenic and copper removal efficiency at different temperatures.
(3) Sulfide dosage
Arsenic and copper removal efficiencies increase as the amount of sulfide increases (Fig. 3.51), which are consistent with results previously reported by van Hille and coworkers [148] for copper sulfide precipitation in a fluidized bed reactor. In all cases, the copper removal efficiency increases more significantly than the arsenic removal efficiency does. Both arsenic and copper removal efficiency are as high as nearly 100% when the addition of sulfide is more than 70 mmol. Compared the results obtained at 30, 40, and 50 °C, the difference between copper and arsenic removal efficiency increases with the temperature increasing. The copper removal efficiency is higher than 80% and arsenic removal efficiency is only 20% when a certain amount of sulfide is added into the solution at 50 °C. The results indicate that separation efficiency of copper and arsenic become higher with the temperature increases which consists of the results mentioned above.

Reprinted from Ref. [147] Copyright 2017, with permission from Elsevier
Arsenic and copper removal efficiency and concentration as a function of sulfide dosage at different temperatures (arsenic concentration of 600 mg L−1, copper concentration of 2000 mg L−1).
In the first stage of primary separation, more than 80% copper and nearly 20% arsenic are precipitated with 20 mmol L−1 H2S at 50 °C within 0.5 min.
3.5.4 Replacement of Arsenic by Copper
(1) Solid to liquid ratio
Arsenic and copper concentration in acidic wastewater when precipitates generated in the first stage are recycled to the initial solution with different solid to liquid ratios (Fig. 3.52). The results show that arsenic concentration in the initial solution increases as solid to liquid ratio increases, while copper concentration becomes lower at the similar conditions. It attributes to the replacement reaction between As2S3 and copper ions; it could be described as follows:

Reprinted from Ref. [147] Copyright 2017, with permission from Elsevier
Arsenic and copper concentration in acidic wastewater at different solid/liquid ratios.
Most of the copper in the initial acidic wastewater is distributed into precipitates and the copper concentration in the initial acidic wastewater is lower than 0.1 mg L−1 when the solid to liquid ratio is more than 10%. Arsenic was precipitated into the residue before dissolves into the solution. Therefore, the addition of precipitates generated in the first separation stage into the initial acidic wastewater with certain solid to liquid ratio can promote the separation of arsenic and copper effectively.
(2) Reaction time
The data of arsenic and copper concentration in acidic wastewater as a function of reaction time (Fig. 3.53) show that arsenic concentration increases within the first 10 min and then becomes constant at about 3200 mg L−1 as time increasing, while copper concentration decreases rapidly from 110 to 0.5 mg L−1 within the first 5 min and further decreases to lower than 0.1 mg L−1 when time is longer than 10 min. The separation efficiency of arsenic and copper is more than 99%. It indicated that separation of arsenic and copper is finished within 10 min which is favorable to the application in the industry.

Reprinted from Ref. [147] Copyright 2017, with permission from Elsevier
Arsenic and copper concentration in acidic wastewater with different reaction time.
(3) Temperature
Second separation of arsenic and copper is temperature-independent represented by the results that copper concentration in the solution scarcely changes when temperature increases (Fig. 3.54). However, arsenic concentration increases slightly as temperature increasing, which attributes to the redissolution of arsenic sulfide in the precipitates at high temperature.

Reprinted from Ref. [147] Copyright 2017, with permission from Elsevier
Arsenic and copper concentration in acidic wastewater at different temperatures.
In the second stage of separation, the separation efficiencies of copper and arsenic are more than 99% with precipitates generated in the first stage recycled to the initial solution when solid to liquid ratio is not less than 10% at 20 °C within 10 min.
Generally, the morphology of the precipitate (Fig. 3.55) exhibits agglomeration of dense, amorphous particles in the micron range. The mole ratio of Cu to S is 1.15, which is approximate to 1, indicating that the precipitate mainly consists of CuS. The copper content of precipitate is as high as 63.38% in mass fraction.

Reprinted from Ref. [147] Copyright 2017, with permission from Elsevier
SEM-EDS image of the precipitate.
3.6 Arsenic Removal by Fe3O4-Based Nanomaterials via Adsorption
3.6.1 Controllable Synthesis of Hierarchical Porous Fe3O4 Particles for Arsenic Removal
In terms of the application of Fe3O4 as an adsorbent, decreasing the Fe3O4 particle size from micrometers to nanometers would increase the available adsorptive areas by 100–1000 times [149,150,151]. However, as the Fe3O4 particle size decreases to nanometers, its response to an external magnetic field undesirably decreases, which will not be large enough to overcome Brownian motion and no efficient magnetic separation will occur [149, 152, 153]. To tackle this problem, one practical strategy is to prepare magnetic hierarchical structures, which are constructed with building blocks of nano units. The hierarchical nanostructures not only exhibit high specific surface area because of the abundant interparticle spaces or intraparticle pores, but also possess satisfactory magnetic response because of their larger size and weaker Brownian motion, which therefore show great superiority to individual nanometer- and micrometer-sized materials [154,155,156,157,158,159]. Hence, we controllably prepare Fe3O4 hierarchical particles via modulating the grain property for arsenic adsorption [160].
(1) Morphology, structure, and properties of Fe 3 O 4
The SEM images and size distribution of the Fe3O4-i (i = 1–6, representing the dosage of PDDA from 1 to 6 g) particles are presented in Fig. 3.56. The size of monodispersed hierarchical particles monotonously decreases from (A) 420 nm to (F) 100 nm, as increasing the poly (diallyl dimethyl ammonium chloride) (PDDA) dosage from 1 to 6 g. Correspondingly, the morphology of hierarchical particles gradually becomes coarse and porous, with the increase of PDDA dosage. HRTEM images were also conducted to give further insight into the grain assembly (Fig. 3.57). Taking Fe3O4-4 as an example, the particle shows pineal-like morphology with fringe spacing of 0.48 nm, corresponding to the (111) lattice planes of Fe3O4. The result indicates the possible oriented assembly of grain along (111) plane, which is the crystallographic plane with the highest energy and preferential for oriented attachment [161]. The structures and grain size of Fe3O4 were further measured by XRD, as shown in Fig. 3.58. All the diffraction peaks at 18.32 ± 0.03, 30.10 ± 0.05, 35.48 ± 0.03, 43.10 ± 0.02, 53.40 ± 0.04, 57.02 ± 0.05, and 62.58 ± 0.08° can be indexed to the indices (111), (220), (311), (400), (422), (511), and (440) of Fe3O4. Briefly speaking, PDDA-modulated solvothermal method successfully modulate products morphology, particle size, grain size, and facilitate the oriented grain assembly.

Reprinted with the permission from Ref. [160] Copyright 2013, American Chemical Society
SEM images and the corresponding hierarchical particle size distribution of a Fe3O4-1, b Fe3O4-2, c Fe3O4-3, d Fe3O4-4, e Fe3O4-5, and f Fe3O4-6 at initial PDDA dosage varying from 1 to 6 g.

Reprinted with the permission from Ref. [160] Copyright 2013, American Chemical Society
HRTEM images (a–c) of Fe3O4-4; b and c represent the magnification of the dotted area in (a).

Reprinted with the permission from Ref. [160] Copyright 2013, American Chemical Society
XRD patterns of Fe3O4 particles obtained at different PDDA dosage: (a) 2 g, (b) 3 g, (c) 4 g, (d) 5 g, (e) 6 g.
On the other hand, the surface area and pore size distribution of as-synthesized Fe3O4-i (i = 2, 4, 5, 6) were determined by nitrogen adsorption–desorption measurements (Fig. 3.59). Fe3O4-6 (Fig. 3.59d) synthesized with the highest PDDA dosage possesses surface area and pore volume of 32.75 m2 g−1 and 0.12 cm3 g−1, respectively, both of which are higher than that of the Fe3O4-5 (31.16 cm2 g−1 and 0.117 cm3 g−1, Fig. 3.59c), Fe3O4-4 (19.13 m2 g−1 and 0.07 cm3 g−1, Fig. 3.59b) and Fe3O4-2 (7.05 m2 g−1 and 0.015 cm3 g−1, Fig. 3.59a). All the samples pose pore sizes in the range of 7–12 nm. The results above can be ascribed to the fact that smaller grain assembly possesses more channels, leading to the increased surface area and pore amount. Hence, increasing the PDDA dosage yields Fe3O4 hierarchical particles composing of smaller grain, which exhibit higher surface area and porosity.

Reprinted with the permission from Ref. [160] Copyright 2013, American Chemical Society
Nitrogen adsorption–desorption isotherms and pore size distribution curves (the corresponding insert) of a as-obtained Fe3O4-2, b Fe3O4-4, c Fe3O4-5, and d Fe3O4-6, respectively.
The magnetic property of Fe3O4 hierarchical particles was evaluated, as shown in Fig. 3.60. The Ms for the Fe3O4-i (i = 2–6) is in the range of 50–80 emu g−1, which is comparable with many other magnetic particles [154].

Reprinted with the permission from Ref. [160] Copyright 2013, American Chemical Society
Room temperature hysteresis loops for the Fe3O4-i (i = 2–6) with initial PDDA dosage increasing from 2 to 6 g.
Briefly speaking, hierarchical porous Fe3O4 particles with high magnetism were synthesized by facile PDDA-modulated solvothermal method, which is achieved in one-pot solution reaction and avoids the time/energy consuming precursor calcination process. Furthermore, PDDA-induced grain size tunable strategy has been proved to be an efficient way to enhance the surface area and porosity of particles.
(2) Mechanism for the formation of Fe 3 O 4 hierarchical particles mediated by PDDA
The morphology and structure of the products with initial PDDA dosage of 4 g at various reaction time were examined by TEM, FTIR and XRD to preliminarily understand the morphology and structure evolution of Fe3O4 hierarchical particles (Fig. 3.61).

Reprinted with the permission from Ref. [160] Copyright 2013, American Chemical Society
a–f TEM, g XRD, and f FT-IR spectra of Fe3O4-4-xh samples (x = 1.5, 2.5, 4, 5, 6, 8).
TEM results give insight into the morphology evolution of mesoporous Fe3O4 (Fig. 3.61a–f), three typical stages were observed for the formation of Fe3O4, namely, the formation of spindle precursor with length of 5–10 nm (0–1.5 h), the formation and assembly of grain to sphere particles (1.5–4 h), and the oriented assembly/Ostwald ripening process of preformed sphere into porous particles (4–8 h).
The XRD patterns (Fig. 3.61g) of Fe3O4-4 at 1.5 h depict a strong peak at 7.55° along with a broad weak one at 25° probably originated from (001) and (013) planes of an iron oxide acetate hydroxide hydrate with a formula of Fe2O(CH3COO) (OH)3· H2O according to JCPDS. Then, XRD patterns of the samples obtained at the time from 2.5 to 4 h show gradually enhanced peaks at 30.00°, 35.48°, 43.14°, 53.44°, 57.04°, and 62.58°, marked by the indices (220), (311), (400), (422), (511), and (440) of Fe3O4 phases. When the reaction time was 6–8 h, the produced aggregates were pure Fe3O4.
The FT-IR spectra (Fig. 3.61h) of Fe3O4-1.5 h and Fe3O4-2.5 h show reduced absorption peaks at 1578 and 1445 cm−1 due to the asymmetric and symmetric stretching of COO– group, band at 1090 cm−1 owing to C–O stretching of the COO– group, band at 887 cm−1 due to the OH bending [162]. The spectra of the samples within 4–8 h show broad strong band at 591 cm−1 due to the Fe–O lattice mode of Fe3O4 [157]. Except for the anticipated typical peak for iron composite (Fe2O(CH3COO) (OH)3·H2O or Fe3O4), the peak at 1125 cm−1 was ascribed to the C–N symmetric stretching vibration of PDDA. The PDDA also exhibits weak CH2 bending vibrations (around 1474, 1326, and 960 cm−1), C-H asymmetric, and C-H symmetric stretching frequencies (2918 and 2867 cm−1) [163, 164]. Thus, both the XRD and FT-IR indicate the gradual formation of Fe3O4 phase at the expense of preformed Fe2O(CH3COO) (OH)3·H2O phase.
Thus, a possible mechanism was proposed to elucidate the PDDA-induced grain size tunable strategy for the controllable synthesis of porous Fe3O4 hierarchical particles. A mixture composed of FeCl3, EG, NaAc, and PDDA was first obtained and the viscosity of mixture was greatly enhanced by PDDA (Fig. 3.62). Spindle particles were then obtained with PDDA as capping agents, which improved the particle dispersibility. As time goes on, hierarchical Fe3O4 particles were eventually produced, and meanwhile PDDA function on capping effect and increasing viscosity declines particle and grain size, facilitates oriented assembly, thus synchronously enhancing surface area and porosity. Briefly speaking, a PDDA-modulated solvothermal method can controllably prepare porous Fe3O4 hierarchical particles.

Reprinted with the permission from Ref. [160] Copyright 2013, American Chemical Society
Scheme of the formation of hierarchical Fe3O4 particles mediated by PDDA.
(3) Arsenic adsorption performance of Fe 3 O 4
The adsorption capacity of Fe3O4-i (i = 2–6) for As(V) and As(III) was evaluated using the equilibrium adsorption isotherm by varying the initial As(V) and As(III) concentrations. The adsorption capacity of As(V) and As(III) monotonously increased from 1.93 and 1.57 mg g−1 for Fe3O4-2 to 7.23 and 6.77 mg g−1 for Fe3O4-6 (Fig. 3.63A, C), indicating that the morphology mediated by PDDA greatly facilitates the absorption performance of particles. Fe3O4-5 and Fe3O4-6 exhibit higher adsorption capacity than commercial Fe3O4 particles (1.35 mg g−1 for As(V) and 0.76 mg g−1 for As(III)). The adsorption of As(V) onto Fe3O4 fits the Langmuir isotherm model (Fig. 3.63B), which interprets the adsorption process as a monolayer adsorption on a homogeneous surface. In contrast, the adsorption of As(III) onto Fe3O4 fits well with the Freundlich isotherm model (Fig. 3.63D), indicating that the adsorption process is a multilayer adsorption on a homogeneous surface.

Reprinted with the permission from Ref. [160] Copyright 2013, American Chemical Society
Adsorption isotherms of A As(V) and C As(III) onto (a) Fe3O4-2, (b) Fe3O4-3, (c) Fe3O4-4, (d) Fe3O4-5, and (e) Fe3O4-6 samples with the initial PDDA dosage of 2 g, 3 g, 4 g, 5 g, and 6 g, respectively. B Linearized Langmuir isotherm for As(V) adsorption and D linearized Freundlich isotherm for As(III) adsorption by (a) Fe3O4-2, (b) Fe3O4-3, (c) Fe3O4-4, (d) Fe3O4-5, and (e) Fe3O4-6 samples (T = 25 °C; adsorbent doses = 0.5 g L−1; pH = 5 ± 0.2).
The two different adsorption isotherm models may be attributed to the different surface charge effects of As(V) and As(III) species under the environment of pH 5 [165]. For As(V), there exists an electrostatic attraction between positively charged Fe3O4 samples and negatively charged As(V) species. However, As(III) exists predominantly as noncharged H3AsO3 [166]. The interaction between Fe3O4 samples and noncharged As(III) species is little so that the adsorption of As(III) should continue to increase with the increase of As(III) concentration [165].
The adsorption of As(V) or As(III) is rapid at first and then slows considerably (Fig. 3.64). The rapid adsorption at first is ascribed to the process of arsenic adsorption on the exterior surface of the Fe3O4 particles. The slower adsorption rate followed might be partially due to the higher diffusion resistance as the arsenic begins to enter and move into the interior of the Fe3O4 particles via the nanopores [165]. All the above adsorption kinetic experimental data can be best fitted into a pseudo-second-order rate kinetic model.

Reprinted with the permission from Ref. [160] Copyright 2013, American Chemical Society
(a, c) Adsorption rate of As(V) and As(III) by Fe3O4-3 (1), Fe3O4-4 (2), and Fe3O4-5 (3) samples. (b, d) Pseudo-second-order kinetic plots for the adsorption of As(V) and As(III). (T = 25 °C; adsorbent doses = 0.5 g L−1; pH = 5 ± 0.2).
The full-range XPS spectra (Fig. 3.65a) show the appearance of arsenic species and the increase of O intensity after arsenic adsorption, validating the arsenic adsorption. XPS of Fe2p of all samples (Fig. 3.65b) exhibit the binding energies of Fe 2p1/2 at 724.4 eV and Fe 2p3/2 at 710.5 eV [167, 168]. XPS of As3d (Fig. 3.65c) in Fe3O4 adsorbed As(V) shows a peak located at 45.1 eV, attributing to As(V)–O bonding, and that of As3d in Fe3O4 adsorbed As(III) indicates fitted peak located at 43.9 eV, corresponding to As(III)–O, respectively [169,170,171,172,173]. The results confirmed no major differences in the valence state of the Fe and As species in arsenic adsorption. O1s XPS spectrum (Fig. 3.65d) can be deconvoluted into peaks located at 530.0, 531.5, and 533.0 eV, which are attributed to oxygen in the lattice (e.g., Fe–O or As–O), oxygen atoms in the surface hydroxyl groups (H–O), and oxygen in the outermost layer of H2O or CO2 adsorbed [169, 171, 174,175,176]. The high peak intensity of H–O species of Fe3O4 confirms the existence of many hydroxyl groups on the surface of Fe3O4 spheres, which plays a vitally important role in the arsenic removal [169]. Moreover, after arsenic adsorption, the shift of the O 1s binding energy to low energy, the proportion decrease of the H–O (531.5 eV) and the proportion increase of O in the lattice (530.0 eV) suggest that the adsorption mechanism was mainly ascribed to the substitution of Fe–OH groups by arsenic species.

Reprinted with the permission from Ref. [160] Copyright 2013, American Chemical Society
a Full-range, b Fe 2p, c As 3d, and d O 1s XPS spectra of several samples of interests including the Fe3O4, Fe3O4 adsorbed As(V), and Fe3O4 adsorbed As(III) particles.
3.6.2 Arsenic Adsorption by Cu Doped Fe3O4 Magnetic Adsorbent
The transformation of As(III) to As(V) is necessary for arsenic removal [177,178,179,180]. Doping is an effective approach to modify particle structure, such as increased amount of surface defects [181, 182], increased hydroxyl amount [183, 184], tunable surface charge [185] and high electronic conduction properties [186], which could enhance adsorption performance and broaden applications [187]. Adding oxidant to convert As(III) to As(V) is normal and efficient route. Most recently, Cu doping has attracted great attention, copper ions were able to act as electron mediation centers to accelerate the oxidation reaction based on Cu(II)/Cu(I) couple [188, 189]. It is rational to dope copper ions in magnetic adsorbents, which are expected to simultaneously possess good adsorption performance, catalytic oxidation and readily magnetic separation property.
(1) Morphology and structure of Fe 3 O 4 :Cu
As the initial amount of Cu2+ increased from 0 to 2.5 mM, the sphere size decreased from −560 to −120 nm (Fig. 3.66). The particle structure transformed from solid spheres to porous spheres with decreased size accordingly. EDS-mapping images of Fe3O4:Cu-2.5 in Fig. 3.75g–i revealed that Cu, Fe, and O elements distributed uniformly on the Fe3O4:Cu-2.5 particles. Microscopic images indicated that indicated that porous Fe3O4:Cu particles with Cu, Fe, O uniformly distributed shows particle size and structure dependent on initial Cu2+ dosage.

Reprinted from Ref. [136] by permission of The Royal Society of Chemistry 2015
SEM of Fe3O4:Cu-0 (a), Fe3O4:Cu-0.05 (b), Fe3O4:Cu-0.25 (c), Fe3O4:Cu-0.5 (d), Fe3O4:Cu-1.0 (e), Fe3O4:Cu-2.5 (f) particles, EDS-mapping images Cu (g), Fe (h) and O (i) of Fe3O4:Cu-2.5 particles. The inset shows the corresponding TEM image.
The XRD spectra of all Fe3O4:Cu particles in Fig. 3.67 show well–defined diffraction lines suggesting the well-crystallized structure. As expected, the typical crystal planes (111), (220), (311), (400), (511), and (440) of Fe3O4 (JCPDS no. 86-1344) were observed for all samples. With the increase of initial Cu2+ dosage, the intensity of diffraction peaks at 43.31, 50.44, and 74.10 correspond to the crystal planes (111), (200), and (220) of Cu increase, indicating the augment in the amount of metal Cu (JCPDS no. 85-1326). The metal Cu might be produced due to the reduction of Cu2+ by ethylene glycol [190]. No peaks attributed to CuO or Cu2O were detected [191, 192].

Reprinted from Ref. [136] by permission of The Royal Society of Chemistry 2015
XRD spectra (a) and the magnified peak (311) (b) of Fe3O4:Cu-0 (1), Fe3O4:Cu-0.05 (2), Fe3O4:Cu-0.25 (3), Fe3O4:Cu-1.0 (4), and Fe3O4:Cu-2.5 (5) particles. Rhombic and stellate icons represent the crystal place of Fe3O4 and Cu, respectively.
The combined microscopic and XRD results indicated the concurrent presence of doped Cu2+ and the Cu nanocrystal on Fe3O4:Cu particles. The mixture of Cu0 and Cu2+ doped Fe3O4 nanograin constituted Fe3O4:Cu particles with Fe, Cu, and O uniformly distributed. The Cu loading decreased the particle size, which should be ascribed to the nucleation effect of the doped Cu2+ [193, 194].
The pH point of zero charged (pHPZC) of Fe3O4:Cu increased from 6.97 to 8.86 as increasing initial dosage of copper ions from 0 to 2.5 mM (Fig. 3.68). The function of Cu incorporation in enhancing pHPZC should be that the doping of Cu2+ caused more defects in Fe3O4 crystals, which was favorable for the adsorption of hydroxyl on particles and then increased the affinity to proton, eventually leading to the increase in pHPZC [195]. The increased pHPZC indicates that the adsorbent has a high tendency to be positive and thus has the potential of high affinity to negative arsenic species [196, 197].

Reprinted from Ref. [136] by permission of The Royal Society of Chemistry 2015
Zeta potential of Fe3O4:Cu particles as a function of pH. The dashed line represents zero zeta potential.
(2) Arsenate adsorption performance
As the initial dosage of copper ions increase from 0 to 2.5 mM, the adsorption capacity for As(V) and As(III) increases from 7.32 to 42.90 mg g−1 and from 8.12 to 32.97 mg g−1 (Fig. 3.69a and c), respectively. The adsorption capacity of Fe3O4:Cu-2.5 particle is much higher than many reported related adsorbents. Adsorbents with higher zeta potential possessed positive-charged surface and thus high affinity toward negative As(V) species. However, the modified surface charged state could not explain increased affinity for As(III). The speciation of adsorbed As(III) was then characterized in detail to explain the phenomenon.

Reprinted from Ref. [136] by permission of The Royal Society of Chemistry 2015
Adsorption isotherms of As(V) (a and b) and As(III) (c and d) on Fe3O4:Cu-0 (1), Fe3O4:Cu-0.05 (2), Fe3O4:Cu-0.25 (3), Fe3O4:Cu-0.5 (4), Fe3O4:Cu-1 (5), and Fe3O4:Cu-2.5 (6) particles at 298 K.
The Langmuir models can well describe the adsorption behavior of As(V) on Fe3O4:Cu (Fig. 3.69b), while Freundlich models well describe the adsorption behavior of As(III) on Fe3O4:Cu (Fig. 3.69d). The two different adsorption isotherm models may be attributed to the different surface charges of arsenic species, namely negative-charged As(V) and noncharged As(III) [197, 198]. With the increase of Cu amount, the calculated Langmuir maximum adsorption capacities for As(V) increased from 7.90 to 43.37 mg g−1.
The regeneration and reuse of the adsorbent are important in considering the practical applicability. The Fe3O4:Cu adsorbent was magnetically separated within 10 s and readily re-dispersed by slightly stirring. The regeneration of Fe3O4:Cu was conducted using 0.1 M NaOH solution as eluent. It was found that the removal efficiency remained 80% after six cycles (82.4% for As(V) and 81.8% for As(III)), which indicated the feasibility of regenerating the Fe3O4:Cu adsorbent (Fig. 3.70).

Reprinted from Ref. [136] by permission of The Royal Society of Chemistry 2015
Arsenic removal efficiency of Fe3O4:Cu-2.5 particles for As(V) (1) and As(III) (2) in different cycle numbers. The inset: separation/redispersion property of Fe3O4:Cu-2.5 under external magnet (M).
(3) Arsenate adsorption mechanism
Full-range XPS spectra of Fe3O4:Cu-2.5 exhibited the information of Fe, Cu and O element (Fig. 3.71). As 3d peaks appeared after As(V) and As(III) adsorption. High-resolution XPS spectra of the As 3d peak are shown in Fig. 3.71a. Fe3O4:Cu-2.5 with As(V) adsorbed shows the peak at 45.3 eV ascribed to As(V)–O, indicating no valence change for As(V) [169, 199]. The adsorption of As(III) on Fe3O4:Cu was conducted under atmosphere, N2 protection, and O2 bubbling condition, respectively. The As(V)–O were estimated as 9.31%, 86.71%, and 94.29% for N2 protection, atmosphere, and O2 bubbling condition, respectively. The results suggested that As(III) was efficiently oxidized to As(V) by O2 on Fe3O4:Cu particles. A few oxidation efficiencies for N2 protection were attributed to trace rudimental oxygen in solution.

Reprinted from Ref. [136] by permission of The Royal Society of Chemistry 2015
As XPS 3d spectra (a) of Fe3O4:Cu-2.5 with arsenic adsorbed and O XPS 1s spectra (b) of Fe3O4:Cu-2.5 before and after arsenic adsorption.
The O 1s spectra of Fe3O4:Cu-2.5 (Fig. 3.71b) can be deconvoluted into three overlapped peaks corresponding to X–O (Fe–O, As–O), hydroxyl groups (–OH) and adsorbed water (H2O), respectively. After arsenic adsorption, the peak of the X–O has shifted from 530.11 to 530.50 eV and the area ratio attributed to X–O increased from 50.43% to 79.86% and 77.65% after As(V) and As(III) adsorption, respectively. The variation for X–O may be due to: (i) the formation of Fe-O on the surface after the reaction between adsorbents and adsorbates; (ii) the As–O in adsorbed arsenic species on the surface. The intensity of the –OH peak slightly decreased from 38.10 to 19.16% and 21.08% after arsenic adsorption, implying that the hydroxyl groups have exchanged with arsenic species.
XANES spectra of As(V) adsorbed and As(III) adsorbed Fe3O4:Cu particles were recorded in Fig. 3.72. For comparison, the XANES spectrum of Na3AsO·12H2O and NaAsO2 were shown, which exhibited peaks at 11874.1 eV for As(V)–O and peaks at 11870.3 eV for As(III)–O, respectively [200]. The XANES spectrum of all Cu–Fe3O4 samples after As(V) adsorption exhibited a typical peak for As(V)–O at 11874.1 eV (Fig. 3.72a). After As(III) adsorption (Fig. 3.72b), two peaks were observed, a peak at 11870.3 eV ascribed to As(III)–O and a peak at 11874.1 eV ascribed to As(V)–O. The intensity of As(V)–O peak increased with the increase of Cu amount, higher than the negligible peak for As(V)–O on Fe3O4:Cu-0. The results indicate that the oxidation efficiency of As(III) was highly dependent on the amount of Cu on adsorbent. The mechanism is Cu2+ acting as electron transfer center between reaction agents. Based on Cu(II)/(I), the proposed pathway is that Cu2+ first acted as electron receptor, producing oxidation product and Cu+ as transient species. The active Cu+ then serves as electron donor, which readily transfers electron to oxidants, yielding reduction product and Cu2+ [201, 202].

Reprinted from Ref. [136] by permission of The Royal Society of Chemistry 2015
Normalized absorbance of As K-edge XANES spectra for As(V) absorbed (a) and As(III) absorbed (b) Fe3O4:Cu-0 (1), Fe3O4:Cu-0.05 (2), Fe3O4:Cu-0.25 (3), Fe3O4:Cu-0.5 (4), Fe3O4:Cu-1 (5), and Fe3O4:Cu-2.5 (6) under atmosphere condition.
The proposed mechanism of arsenic adsorption on Fe3O4:Cu particle was given in Fig. 3.73. The mixture of Cu0 and Cu2+ doped Fe3O4 nanograin constituted Fe3O4:Cu particles. Due to the Cu incorporation, the Fe3O4:Cu particles have high surface area, high porosity, abundant hydroxyl groups, and positive-charged surface, which are beneficial for As(V) adsorption. When Fe3O4:Cu adsorbent was applied for As(III) adsorption, As(III) was efficiently oxidized to As(V) by O2 based on Cu2+ as electron mediation agent between As(III) and O2. The produced negative As(V) species was then in situ adsorbed on Fe3O4:Cu particles based on the electrostatic interaction and hydroxyl exchange. Thus, the Fe3O4:Cu particles were efficient adsorbents for both As(III) and As(V).

Reprinted from Ref. [136] by permission of The Royal Society of Chemistry 2015
The As(III) redox and adsorption on Fe3O4:Cu particle.
3.6.3 Arsenic Adsorption by Fe3O4@Cu(OH)2 Composites
Magnetic nanoparticles (MNPs) have aroused great attention because of the unique merits of facile magnetic separation property [203, 204], such as porous Fe3O4 [197], flower-like Fe3O4 adsorbent [203], superparamagnetic high-surface-area Fe3O4 nanoparticles [205]. Nevertheless, these materials always suffer from undesirable adsorption performances because of high tendency of self-aggregation, less quantity of functional groups, and weak affinity to contaminants [206]. To address the problems, surface functional coating provides a prospective solution for MNPs performance improvement by combining multiple functionalities of introduced chemicals or special structures [207], which attracted increasing interests in specific drug targeting, magnetic cell separation, magnetic sealing, etc. Copper-based nanomaterials have received great attention because of their potential application in optoelectronic devices, catalysis, and superconductors [208, 209]. Thus, Fe3O4@Cu(OH)2 composites were synthesized and used for arsenic adsorption.
(1) Characterization of Fe 3 O 4 @Cu(OH) 2 composites
From the TEM images, bare Fe3O4 particles apparently showed regular microspheres of size around 250 nm without Cu addition (Fig. 3.74a) [210]. When Cu was added initially, some tiny flocculent like structures can be observed obviously along the surface of Fe3O4 (Fig. 3.74b), implying obtained copper hydroxide was preferably assembled on the particles. It is more interesting that as the amount of CuCl2 increased successively, the initial dendrites gradually grew up to constitute final leaflike nanostructures around the Fe3O4 particles with ca. 300–500 nm in length and at most 200 nm in width (Fig. 3.74c–e).

Reprinted from Ref. [210] Copyright 2016, with permission from Elsevier
TEM images of Fe3O4 (a), Fe3O4@Cu(OH)2-1 (b), Fe3O4@Cu(OH)2-2 (c), Fe3O4@Cu(OH)2-3 (d) and Fe3O4@Cu(OH)2-4 (e).
The phase and composition of the Fe3O4 and Fe3O4@Cu(OH)2 composites were characterized by XRD (Fig. 3.75). All the diffraction peaks located at about 30.24°, 35.52°, 43.12°, 57.04°, and 62.66° can be assigned to the diffraction of Fe3O4 crystal with inverse spinel structure from the (220), (311), (400), (511), and (440) planes, respectively (PDF#75-1610). Compared with curves 1, 2, 3, 4, and 5, Fe3O4@Cu(OH)2 exhibited the typical magnetic Fe3O4 peak patterns indicating that the prepared Fe3O4@Cu(OH)2 retained the magnetic property of Fe3O4.

Reprinted from Ref. [210] Copyright 2016, with permission from Elsevier
X-ray diffraction pattern of Fe3O4 (1), Fe3O4@Cu(OH)2-1 (2), Fe3O4@Cu(OH)2-2 (3), Fe3O4@Cu(OH)2-3 (4) and Fe3O4@Cu(OH)2-4 (5).
Figure 3.76 presents the FTIR spectra of the Fe3O4@Cu(OH)2 composites with various Cu addition. The broad adsorption between 3500 and 3000 cm−1 represents the stretching/bending vibration of –OH belonging to free surface water and metal oxides (M–OH, M = Fe, Cu). The strong –OH absorption bands at 1618 cm−1 arise from physically adsorbed water molecules [211]. The characteristic peaks at 1060 cm−1 can be assigned to the bending vibration of hydroxyl groups on metal oxides [212, 213]. Profoundly, as Cu/Fe ratios rose, the bands intensity of hydroxyl groups in the spectrum all showed increasing tendency, strongly demonstrating that Cu(OH)2 coating could significantly enhance the –OH group concentration in surface and lattices of obtained composites.

Reprinted from Ref. [210] Copyright 2016, with permission from Elsevier
FTIR spectra of Fe3O4 (1), Fe3O4@Cu(OH)2-1 (2), Fe3O4@Cu(OH)2-2 (3), Fe3O4@Cu(OH)2-3 (4) and Fe3O4@Cu(OH)2-4 (5).
(2) Magnetic property of Fe 3 O 4 @Cu(OH) 2
The saturation magnetization (Ms) of Fe3O4 (1), Fe3O4@Cu(OH)2-1 (2), Fe3O4@Cu(OH)2-2 (3), Fe3O4@Cu(OH)2-3 (4), and Fe3O4@Cu(OH)2-4 (5) was 98.84, 97.74, 91.42, 67.38 and 56.53 emu g−1, respectively (Fig. 3.77). It is evident that Cu coating has a negative effect on the Fe3O4 magnetic characteristic. The decreased Ms. can be attributed to the coating of Cu(OH)2 shell on the surface of Fe3O4 microsphere cores, which reduced the magnetite fraction in each microsphere [214,215,216,217]. However, the saturation magnetization value achieved with Fe3O4 and Fe3O4@Cu(OH)2 composites was high enough for magnetic separation [218]. The superior magnetic property guaranteed the convenient separation in adsorption application.

Reprinted from Ref. [210] Copyright 2016, with permission from Elsevier
Magnetic hysteresis loop of Fe3O4 (1), Fe3O4@Cu(OH)2-1 (2), Fe3O4@Cu(OH)2-2 (3), Fe3O4@Cu(OH)2-3 (4), and Fe3O4@Cu(OH)2-4 (5).
(3) Arsenate adsorption isotherms
The arsenate adsorption capacity of Fe3O4@Cu(OH)2-1 to Fe3O4@Cu(OH)2-4 increased from 11.11 to 35.71 mg g−1 with the increase of Cu(OH)2 loading ratio (Fig. 3.78), indicating that Cu coating on bare magnetic particles played a significant role in enhancing adsorption performance of composites. Obviously, Fe3O4@Cu(OH)2 composites with high magnetic have a powerful arsenic removal capacity, thus making them promising as adsorbents for arsenic removal.

Reprinted from Ref. [210] Copyright 2016, with permission from Elsevier
Adsorption performance of Fe3O4@Cu(OH)2 composites toward As(V).
(4) Arsenate adsorption kinetics
The arsenate adsorption on the Fe3O4@Cu(OH)2 composites was rapid at first due to the large number of available adsorption sites at the initial stage (Fig. 3.79). With the increase of adsorption time, it had a slower removal rate and then reached equilibrium which was ascribed to the gradual occupation of available adsorption sites on Fe3O4@Cu(OH)2 composites by arsenate at the later time. The results further revealed that the Fe3O4@Cu(OH)2 composites had an excellent percentage of the arsenic adsorption.

Reprinted from Ref. [210] Copyright 2016, with permission from Elsevier
Adsorption rate of As(V) by Fe3O4@Cu(OH)2-2.
(5) Arsenate adsorption mechanism
XPS spectra of the adsorbent before and after As(V) adsorption at pH 5.0 were analyzed in Fig. 3.80. Figure 3.80A depicts two intense bands with binding energies of 711.7 eV and 724.0 eV that are assigned to Fe 2p3/2 and Fe 2p1/2, respectively, demonstrating the existence of the typical Fe3O4 structures [205, 206]. The Cu 2p XPS spectrum is shown in Fig. 3.80B. The peaks located at 934.5 and 954.2 eV are assigned to Cu 2p3/2 and Cu 2p1/2 [219, 220]. After arsenate adsorption, the binding energy of Fe and Cu has no changed, indicating that Fe3O4@Cu(OH)2 composites are stable in arsenate adsorption. XPS of As 3d peak appears after adsorption (Fig. 3.80D). The As 3d binding energies is 45.5 eV which is attributing to As(V)–O bonding [168, 207, 221,222,223], meaning that arsenate species have been loaded on the surface of the Fe3O4@Cu(OH)2 composites after the adsorption.

Reprinted from Ref. [210] Copyright 2016, with permission from Elsevier
Fe 2p (A), Cu 2p (B), O 1s (C) and As 3d (D) XPS spectra of Fe3O4@Cu(OH)2-2 (a), Fe3O4@Cu(OH)2-2 with arsenic adsorbed (b).
As for O 1s spectra in Fig. 3.80C, after arsenate adsorption, the O 1s peak locates at 531.5 eV decreased from 56.18 to 26.17% suggesting that the surface hydroxyl groups have surely important effects on arsenate adsorption, while the increased proportions of oxygen in the lattice at 530.0 eV from 39.33 to 67.12% maybe originate from the formation of M–O on the surface and As–O groups in the adsorbed arsenate species after arsenate adsorption [224]. That is to say, the M–OH groups on the Fe3O4@Cu(OH)2 composites surface are responsible for arsenic adsorption.
The possible mechanism of arsenate adsorption on Fe3O4@Cu(OH)2 composites was shown in Fig. 3.81. The M–OH groups on the Fe3O4@Cu(OH)2 composites surface played a key role in As(V) adsorption through the formation of coordination compounds between As–OH and M–OH and the corresponding M–O groups formed. Meanwhile, Fe3O4@Cu(OH)2 composites can be protonated in acidic solution to form the positively charged surface, the negative arsenate species can be directly adsorbed on the positive sites of the adsorbent via electrostatic attraction.

Reprinted from Ref. [210] Copyright 2016, with permission from Elsevier
As(V) adsorption mechanism on Fe3O4@Cu(OH)2 composites.
In summary, there were two different mechanisms contributing to adsorption: the formation of coordination compounds and electrostatic attraction between As(V) species and Fe3O4@Cu(OH)2 composites.
3.6.4 Arsenic Removal by Hollow Cu-Loaded Poly(m-Phenylenediamine) Particles
As a diamine derivative of polyaniline, poly (phenylenediamine) has many important applications in sensors, catalysis, electrodes, actuators, etc. [225,226,227]. In particular, widespread attention has been attracted to investigate the adsorption performance of poly (phenylenediamine) over recent decades due to its superior redox reversibility and chelation ability [228,229,230,231], and it can be used for removing various metals ions from water, such as As(V). A sustainable synthesis of poly (m-phenylenediamine) microparticles with hollow structures was developed via Cu-catalyzed air oxidation. The conversion efficiency of monomer in this research can reach close to 100%, which is higher than the highest yield reported (93.1%) [232]. Moreover, the nanoscaled Cu-loaded PmPD particle is a hopeful material in arsenic removal in aqueous solution.
(1) Morphology of PmPD particles
The PmPD particles were named as PmPD-Cux (x: 1:1, 1:0.5, 1:0.25, 1:0.125 and 1:0.1), where x corresponds to the mPD:Cu2+ molar ratio. Obviously, amount of Cu addition has a significant influence on the micro-morphology of PmPD particles. As shown in Fig. 3.82g, h, the product synthesized in nmPD:nCu = 1:0.125 just consisted of abundant irregular solid particles. However, great variation emerged by increasing Cu content. When mPD/Cu ratio became 1:0.5, a lot of distinguishable solid microspheres appeared actually in the product, though their surfaces are very rough (Fig. 3.82c, d). It is noted that some hollow nanostructures could be identified apparently in Fig. 3.82d. Especially, in a further experiment, the obtained PmPD-Cu1:1 particles were mainly composed of nanosized smooth spheres with the diameters of 300–500 nm (Fig. 3.82a, b). More importantly, the microspheres are hollow with a wall thickness of about 60–80 nm, as confirmed by the TEM image. Up to our knowledge, that is a new sustainable alternate to constitute hollow structure of PmPD particles.

SEM and TEM images: a and b PmPD-Cu1:1; c and d PmPD-Cu1:0.5; e and f PmPD-Cu1:0.25 and g and h PmPD-Cu1:0.125 (reaction time was 24 h)
The self-formation mechanism of the hollow structures can be described as follows. When in contact with Cu2+, mPD instantly reacted to generate the positively charged complex, which would increase the molecules amphiphilicity to constitute micelles in water. As higher amount of Cu(II) salt was added into the system, the counter ion concentration also increased, which can compress the electrical double layer and reduce charge repulsion, allowing the micelles to come closer to each other [233,234,235]. At the same time, the increased ionic strength can cause the molecules’ polar groups undergo more “dehydration” to enhance their hydrophobic character, which will greatly reduce monomer molecules exchange between the micelles and the bulk phase [236]. Hence, the effects worked together to drive micellar molecules to be rearranged to form enlarged stable micellar cluster or bilayer aggregates. When air was introduced into the bulk solution, the oxidation chain-propagation will principally take place around the surface of the aggregates to make them growing up [234]. However, when the reaction reached to a certain extent, the dynamic equilibrium cannot be maintained between the grown aggregates and polymer molecules. Particularly, if the total Cu addition is not enough, low ionic strength will go against keeping hollow aggregates stable and finally formed irregular solid particles.
(2) Structure of PmPD particles
The FTIR spectra of PmPD-Cux (x: 1:1, 1:0.5, 1:0.25, 1:0.125, 1:0.1) were similar (Fig. 3.83). The broad absorption centered between 3500 and 3000 cm−1 should be due to the stretching mode of –NH– [237,238,239]. The peak at ~1620 cm−1 was associated with phenazine and quinoid imine. And the peak at ~1500 cm−1 was attributed to benzenoid amine structures [240, 241]. Meanwhile, the peak at ~1250 cm−1 corresponded to the C–N stretching mode in the PmPD [242]. Moreover, it was found that the relative content of the two peaks changed obviously with the variation of the Cu/monomer molar ratio. With the increase of Cu2+, the peak at ~1620 cm−1 turned to stronger which indicated that content of phenazine and quinoid structure were increased.

FTIR spectra of PmPD-Cu1:1, PmPD-Cu1:0.5, PmPD-Cu1:0.25 and PmPD-Cu1:0.125
Figure 3.84 clearly indicates that the PmPD particles with Cu loaded are basically made up of carbon, oxygen, nitrogen, copper, and a small amount of chlorine. With the decrease of the initial dosage of Cu2+, the peak of Cu became weaker.

Wide energy range surface spectra of PmPD-Cu1:1; PmPD-Cu1:0.5; PmPD-Cu1:0.25 and PmPD-Cu1:0.125
The XPS spectra of N element of various PmPD particles (Fig. 3.85a–d) were investigated to study the relative molar contents of N-containing segments. The peak at 399.2 eV was associated with the neutral –N= in both quinoid imine and phenazine while the one at 400.2 eV was due to the –NH– in the benzenoid amine units. Besides these, there was a weak peak at 401.2 eV assigned to –N+= [243,244,245]. With the increase of Cu2+, the area of –N= and –N+= increased, indicating the enhancement of oxidation state. This is in agreement with the analysis of FTIR. Based on XPS analysis, it was interesting to see that the oxidation state of PmPD increased with the increase of Cu/monomer molar ratio.

Deconvolution results of N 1s (a–d) and Cu 2p (e–h) XPS spectra of PmPD-Cu1:1 (a–e), PmPD-Cu1:0.5 (b–f), PmPD-Cu1:0.25 (c–g) and PmPD-Cu1:0.125
The XPS spectra of Cu element of PmPD particles (Fig. 3.85e–h) indicates that copper existed in two oxidation states on the PmPD particles, Cu2+ and Cu+ [246, 247]. Cu+ contents tended to rise with the increase of Cu/mPD ratio. The variation of Cu+ contents proved that Cu2+ was involved in the mPD oxidation polymerization. It was the reduction of Cu2+ to Cu+ achieving the oxidation and the increased amount of Cu+ was possibly a direct reason for the increased oxidation state of PmPD. Therefore, it could be further correlated to the increase of conversion rate.
(3) Arsenate adsorption properties
When initial As(V) concentration is 70 mg L−1, PmPD-Cu0 exhibited a certain arsenic absorbance as low as about 8 mg g−1 (Fig. 3.86). As the Cu/mPD ratios rose, the absorbance of PmPD particles loaded with Cu significantly increased. The maximal absorbance (about 27.4 mg g−1) occurred at Cu/mPD ratios 0.25:1, 4 times as much as that of PmPD-Cu0. Subsequently, further enhancing Cu/mPD ratio had no great impacts upon the As(V) adsorption of PmPD particles with just a slight decrease to about 23–24 mg g−1. It is indicative that Cu played a crucial role in the arsenic removal of PmPD particles. Consequently, PmPD-Cu1:0.25 is more preferable adsorbent for As(V) removal. The Freundlich can better describe the adsorption since the correlation efficiency of Freundlich is all much higher than that of Langmuir. This suggests that PmPD particles adsorption of arsenic is a multicomponent adsorption isotherm.

Effects of initial concentration of PmPD particles (T = 35 °C; adsorbent dosage = 0.5 g L−1; pH = 5)
The adsorption kinetics of PmPD-Cu1:0.25 (Fig. 3.87) indicate the adsorption process can be roughly divided into fast and slow steps. The fast step lasted for about 10 min; the residue percentage of arsenic in the filtrate was sharply decreased to 18.5% for. This rapid process was caused by the adsorption of arsenic to the functional groups on the surface of PmPD particles. As prolonging the time, the adsorption became slow apparently, owing to the gradual diffusion of arsenic molecules from the surface to the inner structures of the PmPD particles. This rapid attainment of adsorption equilibrium is of great importance to the practical process of the obtained PmPD particles.

Effects of time on the adsorption performance of PmPD-Cu1:0.25 particles (T = 35 °C; adsorbent doses = 0.5 g L−1; pH = 5)
Arsenic removal property of PmPD-Cu1:0.25 was measured with initial pH value from 3 to 11 (Fig. 3.88). The removal rate was calculated according to the equations described in the literature [229]. When decreasing the pH from 5.0 to 3.0, the removal rate declined obviously from only 53.2 to 16.1%. This means that the acidic condition with pH lower than 5 is not beneficial for arsenic removal. While at pH 5.0–7.0, the removal rate varies little (<2%), implying that the weak acidic condition influences slightly on arsenic adsorption. As pH decreased from 5 decrease to 3, negative-charged H2AsO4− began to turn to neutral molecular state H3AsO4, which is difficult to be adsorbed, thus leading to the decrease of removal rate. As pH increased to 11, the removal rate of arsenic rapidly dropped to 0%, which is mainly due to OH− competition in alkaline condition. Consequently, the solution pH at 5.0–7.0 is suitable for arsenic adsorption of PmPD particles (Fig. 3.88).

Effect of pH on the adsorption performance of PmPD-Cu1:0.25 particles (T = 35 °C; adsorbent doses = 20 mg L−1)
(4) Adsorption mechanism
The As 3d and N 1s data (XPS) of PmPD particles and relevant calculation are given in Fig. 3.89 [248]. Based on the arsenic 3d data, the adsorption of arsenic on PmPD particles existed in two forms, 58.14% mol As(V) and 41.86% mol As(III). That means an oxidation process took place to make As(V) reduced to As(III) during the adsorption. As compared the data of N 1s before and after adsorption (in Fig. 3.88), it can be found that the content of –NH– declined by 5.7% after treating As(V), in turn the content of =N– and =N+– increased correspondingly.

Deconvolution results of N 1s and As 3d XPS spectra of PmPDCu1:0.25 before and after adsorption
As is known to all, –NH– of conjugated polymers can be oxidized readily and then hydrogen left, which was finally transformed to =N– [244]. 0.92% of –NH– was oxidized by As(V) form to =N–. On the other hand, the electron density of =N– is much higher than that of –NH– due to its different molecular configuration [249, 250], which allows it to interact with As(III) cations through coordination. The increased amount of =N+– after adsorption also strongly verifies this chelation between As(III) and =N–. It should be noted that part of the =N– in PmPD before adsorption has been already chelated by Cu ions to generate =N+– which can interact with negative ionic As(V) through static manner. Based on the discussion of XPS, the corresponding interaction was illustrated in Fig. 3.90 [244, 251, 252]. As soon as the addition of PmPD-Cu nanoparticles into the As(V) solution, abundant As(V) was adsorbed by =N+– components via electrostatic attraction. Meanwhile, the redox reaction between part of As(V) and nearby imine group (–NH–) occurred which produced the =N– and As(III). Therein, As(III) can readily tend to interact with =N– through coordination.

Possible mechanism for arsenic adsorption with PmPD particles
References
Luo, T., Cui, J., Hu, S., et al.: Arsenic removal and recovery from copper smelting wastewater using TiO2. Environ. Sci. Technol. 44(23), 9094–9098 (2010)
Baig, J.A., Kazi, T.G., Shah, A.Q., et al.: Speciation and evaluation of arsenic in surface water and groundwater samples: a multivariate case study. Ecotoxicol. Environ. Saf. 73(5), 914–923 (2010)
Mandal, B.K., Suzuki, K.T.: Arsenic round the world: a review. Talanta 58(1), 201–235 (2002)
Riveros, P.A., Dutrizac, J.E., Spencer, P.: Arsenic disposal practices in the metallurgical industry. Can. Metall. Q. 40(4), 395–420 (2001)
Emett, M.T., Khoe, G.H.: Photochemical oxidation of arsenic by oxygen and iron in acidic solutions. Water Res. 35(3), 649–656 (2001)
Kim, M.J., Nriagu, J.: Oxidation of arsenite in groundwater using ozone and oxygen. Sci. Total Environ. 247(1), 71–79 (2000)
Jia, Y.F., Zhang, D.N., Pan, R.R., et al.: A novel two-step coprecipitation process using Fe (III) and Al (III) for the removal and immobilization of arsenate from acidic aqueous solution. Water Res. 46(2), 500–508 (2012)
Cui, J.L., Jing, C.Y., Che, D.S., et al.: Groundwater arsenic removal by coagulation using ferric(III) sulfate and polyferric sulfate: a comparative and mechanistic study. J. Environ. Sci. 32, 42–53 (2015)
Mertens, J., Casentini, B., Masiond, A., et al.: Polyaluminum chloride with high Al30 content as removal agent for arsenic-contaminated well water. Water Res. 46(1), 53–62 (2012)
An, B., Liang, Q.Q., Zhao, D.Y.: Removal of arsenic(V) from spent ion exchange brine using a new class of starch-bridged magnetite nanoparticles. Water Res. 45(5), 1961–1972 (2011)
Pakzadeh, B., Batista, J.R.: Surface complexation modeling of the removal of arsenic from ion-exchange waste brines with ferric chloride. J. Hazard. Mater. 188(1–3), 399–407 (2011)
Zhang, G.S., Liu, F.D., Liu, H.J., et al.: Respective role of Fe and Mn oxide contents for arsenic sorption in iron and manganese binary oxide: an X-ray absorption spectroscopy investigation. Environ. Sci. Technol. 48(17), 10316–10322 (2014)
Dou, X.M., Mohan, D., Pittman Jr., C.U.: Arsenate adsorption on three types of granular schwertmannite. Water Res. 47(9), 2938–2948 (2013)
Na, L., Maohong, F., Johannes, V.L., et al.: Oxidation of As (III) by potassium permanganate. J. Environ. Sci. 19(7), 783–786 (2007)
Sorlini, S., Gialdini, F.: Conventional oxidation treatments for the removal of arsenic with chlorine dioxide, hypochlorite, potassium permanganate and monochloramine. Water Res. 44(19), 5653–5659 (2010)
Lee, Y.H., Um, I., Yoon, J.: Arsenic(III) oxidation by iron(VI) (ferrate) and subsequent removal of arsenic(V) by iron(III) coagulation. Environ. Sci. Technol. 37(24), 5750–5756 (2003)
Molnár, L., Virčíkova, E., Lech, P.: Experimental study of As (III) oxidation by hydrogen peroxide. Hydrometallurgy 35(1), 1–9 (1994)
Hug, S.J., Leupin, O.: Iron-catalyzed oxidation of arsenic(III) by oxygen and by hydrogen peroxide: pH-dependent formation of oxidants in the fenton reaction. Environ. Sci. Technol. 37(12), 2734–2742 (2003)
Laat, J.D., Le, T.G.: Kinetics and modeling of the Fe(III)/H2O2 system in the presence of sulfate in acidic aqueous solutions. Environ. Sci. Technol. 39(6), 1811–1818 (2005)
Sorlini, S., Gialdini, F., Stefan, M.: Arsenic oxidation by UV radiation combined with hydrogen peroxide. Water Sci. Technol. 61(2), 339–344 (2010)
Bissen, M., Vieillard-Baron, M.-M., Schindelin, A.J., et al.: TiO2-catalyzed photooxidation of arsenite to arsenate in aqueous samples. Chemosphere 44(4), 751–757 (2001)
Klaning, U.K., Bielski, B.H.J., Sehested, K.: Arsenic(IV). A pulse-radiolysis study. Inorg. Chem. 28(14), 2717–2724 (1989)
Rastogi, A., Al-Abed, S.R., Dionysiou, D.D.: Sulfate radical-based ferrous-peroxymonosulfate oxidative system for PCBs degradation in aqueous and sediment systems. Appl. Catal. B 85(3–4), 171–179 (2009)
Gomathi Devi, L., Girish Kumar, S., Mohan Reddy, K., et al.: Photo degradation of methyl orange an azo dye by advanced fenton process using zero valent metallic iron: Influence of various reaction parameters and its degradation mechanism. J. Hazard. Mater. 164(2–3), 459–467 (2009)
Yang, Y., Pignatello, J.J., Ma, J., Mitch, W.A.: Comparison of halide impacts on the efficiency of contaminant degradation by sulfate and hydroxyl radical-based advanced oxidation processes (AOPs). Environ. Sci. Technol. 48(4), 2344–2351 (2014)
Hori, H., Hayakawa, E., Einaga, H., et al.: Decomposition of environmentally persistent perfluorooctanoic acid in water by photochemical approaches. Environ. Sci. Technol. 38(22), 6118–6124 (2004)
Schröder, H.F., Meesters, R.J.W.: Stability of fluorinated surfactants in advanced oxidation processes–a follow up of degradation products using flow injection-mass spectrometry, liquid chromatography-mass spectrometry and liquid chromatography-multiple stage mass spectrometry. J. Chromatogr. A 1082(1), 110–119 (2005)
Zhang, B.T., Zhang, Y., Teng, Y.G., et al.: Sulfate radical and its application in decontamination technologies. Crit. Rev. Environ. Sci. Technol. 45(16), 1756–1800 (2015)
Lei, Z., Wei, Z., Jinfeng, Z., et al.: Ferrous-activated persulfate oxidation of arsenic (III) and diuron in aquatic system. J. Hazard. Mater. 263(Part 2), 422–430 (2013)
Jing, X., Wei, D., Feng, W., et al.: Rapid catalytic oxidation of arsenite to arsenate in an iron (III)/sulfite system under visible light. Appl. Catal. B Environ. 186, 56–61 (2016)
Neppolian, B., Doronila, A., Ashokkumar, M.: Sonochemical oxidation of arsenic (III) to arsenic (V) using potassium peroxydisulfate as an oxidizing agent. Water Res. 44(12), 3687–3695 (2010)
Neppolian, B., Celik, E., Choi, H.: Photochemical oxidation of arsenic(III) to arsenic(V) using peroxydisulfate ions as an oxidizing agent. Environ. Sci. Technol. 42(16), 6179–6184 (2008)
Woods, R., Kolthoff, I.M., Meehan, E.J.: Arsenic(IV) as an intermediate in the induced oxidation of arsenic(III) by the iron(II)- persulfate reaction and the photoreduction of iron(III). I. Absence of oxygen. J. Am. Chem. Soc. 85(16), 2385–2390 (1963)
Woods, R., Kolthoff, I.M., Meehan, E.J.: Arsenic(IV) as an intermediate in the iron(III) and copper(II) catalyzed arsenic(III)-per sulfate reaction. Inorg. Chem. 4(5), 697–704 (1965)
Takahashi, M., Chiba, K., Li, P.: Formation of hydroxyl radicals by collapsing ozone microbubbles under strongly acidic conditions. J. Phys. Chem. B 111(39), 11443–11446 (2007)
Li, Y., Cai, X.J., Guo, J.W., et al.: UV-induced photoactive adsorption mechanism of arsenite by anatase TiO2 with high surface hydroxyl group density. Colloids Surf. A 462, 202–210 (2014)
Robins, R.G.: The solubility of metal arsenates. Metall. Trans. B 12(1), 103–109 (1981)
Trigub, A.L., Tagirov, B.R., Kvashnina, K.O., et al.: X-ray spectroscopy study of the chemical state in “invisible” Au in synthetic minerals in the Fe-As-S system. Am. Miner. 102(5), 1057–1065 (2017)
Gonzalez-Contreras, P., Weijma, J., Buisman, C.J.N.: Bioscorodite crystallization in an airlift reactor for arsenic removal. Cryst. Growth Des. 12(5), 2699–2706 (2012)
Fujita, T., Taguchi, R., Abumiya, M., et al.: Novel atmospheric scorodite synthesis by oxidation of ferrous sulfate solution. Hydrometallurgy 90(2–4), 92–102 (2008)
Paktunc, D., Dutrizac, J., Gertsman, V.: Synthesis and phase transformations involving scorodite, ferric arsenate and arsenical ferrihydrite: implications for arsenic mobility. Geochim. Cosmochim. Acta 72(11), 2649–2672 (2008)
Dutrizac, J.E., Jambor, J.L.: The synthesis of crystalline scorodite, FeAsO4·2H2O. Hydrometallurgy 19(3), 377–384 (1988)
Yang, J.Q., Chai, L.Y., Yue, M.Q., et al.: Complexation of arsenate with ferric ion in aqueous solutions. RSC Adv. 5(126), 103936–103942 (2015)
Sergeyeva, E.I., Khodakovskiy, I.L.: Physicochemical conditions of formation of native arsenic in hydrothermal deposits. Geochem. Int. 846–859 (1969)
Technical Note 270-7. Schumm, R.H., Wagman, D.D., Evans, W.H., et al.: Selected Values of Chemical Thermodynamic Properties. National Bureau of Standards (1973)
Wagman, D.D., Evans, W.H., Parker, V.B., et al.: Erratum: The NBS tables of chemical thermodynamic properties. J. Phys. Chem. Ref. Data 18(4), 1807–1812 (1989)
Bard, A.J., Parsons, R., Jordan, J.: Standard Potentials in Aqueous Solution. CRC Press, New York (1985)
Shock, E.L., Helgeson, H.C.: Calculation of the thermodynamic and transport properties of aqueous species at high pressures and temperatures: correlation algorithms for ionic species and equation of state predictions to 5 kb and 1000 °C. Geochim. Cosmochim. Acta 52(8), 2009–2036 (1988)
Nordstrom, D.K., Archer, D.G.: Arsenic Thermodynamic Data and Environmental Geochemistry. Arsenic in Ground Water, pp. 1–25. Springer, (2003)
Jin-qin, Y.A.N.G., Li-yuan, C.H.A.I., Qing-zhu, L.I., et al.: Redox behavior and chemical species of arsenic in acidic aqueous system. Trans. Nonferrous Species Arsen. Acidic Aqueous 27(9), 2063–2072 (2017)
Long, H.: A Fundamental Study of the Acidic Pressure Oxidation of Orpiment and Pyrite at High Temperature. University of British Columbia, British Columbia (2000)
Masscheleyn, P.H., Delaune, R.D., Patrick, J.R.W.H.: Effect of redox potential and pH on arsenic speciation and solubility in a contaminated soil. Environ. Sci. Technol. 25(8), 1414–1419 (1991)
Pieczaba, E., Sanak-rydlewska, S., Zieba, D.: Removal of arsenic from aqueous solutions by the method of precipitate flotation. Arch. Min. Sci. 50(1), 131–142 (2005)
Yazdi, M.R.S., Darban, A.K.: Effect of arsenic speciation on remediation of arsenic-contaminated soils and waters. In: 15th International Conference on Heavy Metals in the Environment (ICHMET), Gdansh, pp. 492–495 (2010)
Yi, X.W.: An empirical estimation of standard entropy for some complex cationa and the E-pH diagram of As-H2O system at elevated temperature. J. Kunming Univ. Sci. Technol. 3, 58–73 (1982)
Debusschere, L., Demesmay, C., Rocca, J.L.: Arsenic speciation by coupling capillary zone electrophoresis with mass spectrometry. Chromatographia 51(5–6), 262–268 (2000)
Gout, R., Pokrovski, G., Schott, J., et al.: Raman spectroscopic study of arsenic speciation in aqueous solutions up to 275 °C. J. Raman Spectrosc. 28(9), 725–730 (1997)
Loehr, T.M., Plane, R.A.: Raman spectra and structures of arsenious acid and arsenites in aqueous solution. Inorg. Chem. 7(9), 1708–1714 (1968)
Mähler, J., Persson, I., Herbert, R.B.: Hydration of arsenic oxyacid species. Dalton Trans. 42(5), 1364–1377 (2013)
Bu, L.J., Gu, T., Ma, Y.X., et al.: Enhanced cathodic preconcentration of As(0) at Au and Pt electrodes for anodic stripping voltammetry analysis of As(III) and As(V). J. Phys. Chem. C 119(21), 11400–11409 (2015)
Marini, L., Accornero, M.: Prediction of the thermodynamic properties of metal–arsenate and metal–arsenite aqueous complexes to high temperatures and pressures and some geological consequences. Environ. Geol. 52(7), 1343–1363 (2007)
Pettine, M., Campanella, L., Millero, F.J.: Arsenite oxidation by H2O2 in aqueous solutions. Geochim. Cosmochim. Acta 63(18), 2727–2735 (1999)
Vink, B.W.: Stability relations of antimony and arsenic compounds in the light of revised and extended Eh-pH diagrams. Chem. Geol. 130(1–2), 21–30 (1996)
Marini, L., Accornero, M.: Erratum to: Prediction of the thermodynamic properties of metal–arsenate and metal–arsenite aqueous complexes to high temperatures and pressures and some geological consequences. Environ. Earth Sci. 59(7), 1601–1606 (2010)
Knight, R.J., Sylva, R.N.: Spectrophotometric investigation of iron (III) hydrolysis in light and heavy water at 25 °C. J. Inorg. Nucl. Chem. 37(3), 779–783 (1975)
Langmuir, D., Mahoney, J., Rowson, J.: Solubility products of amorphous ferric arsenate and crystalline scorodite (FeAsO4·2H2O) and their application to arsenic behavior in buried mine tailings. Geochim. Cosmochim. Acta 70(12), 2942–2956 (2006)
Robins, R.G.: The stability and solubility of ferric arsenate: an update. In: EPD Congress’90, pp. 93–104 (1990)
Whiting, K.S.: The Thermodynamics and Geochemistry of Arsenic with Application to Subsurface Waters at the Sharon Steel Superfund Site at Midvale, Utah. Colorado School of Mines, Midvale (1992)
Hug, S.J., Canonica, L., Wegelin, M., et al.: Solar oxidation and removal of arsenic at circumneutral pH in iron containing waters. Environ. Sci. Technol. 35(10), 2114–2121 (2001)
Wang, K.L., Jia, Y.F.: Effects of temperature and pH on the transformation of ferric arsenate to scorodite in acidic solution. Adv. Mater. Res. 726–731, 2165–2168 (2013)
Paktunc, D., Dutrizac, J., Gertsman, V.: Synthesis and phase transformations involving scorodite, ferric arsenate and arsenical ferrihydrite: implications for arsenic mobility. Geochim. Cosmochim. Acta 72(11), 2649–2672 (2008)
Welham, N.J., Malatt, K.A., Vukcevic, S.: The effect of solution speciation on iron–sulfur–arsenic–chloride systems at 298 K. Hydrometallurgy 57(3), 209–223 (2000)
Raposo, J.C., Olazabal, M.A., Madariaga, J.M.: Complexation and precipitation of arsenate and iron species in sodium perchlorate solutions at 25 °C. J. Solut. Chem. 35(1), 79–94 (2006)
Khoe, G.H., Robins, R.G.: ChemInform abstract: the complexation of iron(III) with sulfate, phosphate, or arsenate ion in sodium nitrate medium at 25 °C. J. Chem. Soc. Dalton Trans. 8, 2015–2021 (1988)
Chai, L., Yang, J., Zhang, N., et al.: Structure and spectroscopic study of aqueous Fe(III)-As(V) complexes using UV-Vis, XAS and DFT-TDDFT. Chemosphere 182, 595–604 (2017)
Ikeda-Ohno, A., Hennig, C., Tsushima, S., et al.: Speciation and structural study of U(IV) and (VI) in perchloric and nitric acid solutions. Inorg. Chem. 48(15), 7201–7210 (2009)
Stefansson, A., Lemke, K.H., Seward, T.M.: Iron(III) complexation in hydrothermal solutionse an experimental and theoretical study. In: 15th International Conference on the Properties of Water and Steam, Berlin (2008)
Chen, Z., Zhu, Y.G., Liu, W.J., et al.: Direct evidence showing the effect of root surface iron plaque on arsenite and arsenate uptake into rice (Oryza sativa) roots. New Phytol. 165(1), 91–97 (2005)
Glastras, M.: The Precipitation of Arsenic from Aqueous Solutions. University of New South Wales, Sydney (1988)
Lee, J.S., Nriagu, J.O.: Stability constants for metal arsenates. Environ. Chem. 4(2), 123–133 (2007)
Nordstrom, D.K., Parks, G.A.: Solubility and stability of scorodite, FeAsO4•2H2O: discussion. Am. Miner. 72(7–8), 849–851 (1987)
Galal-Gorchev, H., Stumm, W.: The reaction of ferric iron with ortho-phos-phate. J. Inorg. Nucl. Chem. 25(5), 567–574 (1963)
Wilhelmy, R.B., Patel, R.C., Matijevic, E.: Thermodynamics and kinetics of aqueous ferric phosphate complex formation. Inorg. Chem. 24(20), 3290–3297 (1985)
Harris, D., Loew, G.H., Komornicki, A.: Structure and relative spin-state ener-getics of [Fe(H2O)6]3+: a comparison of UHF, møller-plesset, nonlocal DFT, and semiempircal INDO/S calculations. J. Phys. Chem. A 101(21), 3959–3965 (1997)
Jarzecki, A.A., Anbar, A.D., Spiro, T.G.: DFT analysis of [Fe(H2O)6]3+ and [Fe(H2O)6]2+ structure and vibrations; implications for isotope fractionation. J. Phys. Chem. A 108(14), 2726–2732 (2004)
Collins, R.N., Rosso, K.M., Rose, A.L., et al.: An in situ XAS study of ferric iron hydrolysis and precipitation in the presence of perchlorate, nitrate, chloride and sulfate. Geochim. Cosmochim. Acta 177, 150–169 (2016)
Sherman, D.M., Randall, S.R.: Surface complexation of arsenic (V) to iron(III) (hydr)oxides: structural mechanism from ab initio molecular geometries and EXAFS spectroscopy. Geochim. Cosmochim. Acta 67(22), 4223–4230 (2003)
Waychunas, G.A., Rea, B.A., Fuller, C.C.: Surface chemistry of ferri-hydrite: Part 1. EXAFS studies of the geometry of coprecipitated and adsorbed arsenate. Geochim. Cosmochim. Acta 57(10), 2251–2269 (1993)
Kitahama, K., Kiriyama, R., Baba, Y.: Refinement of the crystal structure of scorodite. Acta Crystallogr. Sect. B 31(1), 322–324 (1975)
Mikutta, C., Mandaliev, P.N., Kretzschmar, R.: New clues to the local atomic structure of short-range ordered ferric arsenate from extended X-ray absorption fine structure spectroscopy. Environ. Sci. Technol. 47(22), 13201–13202 (2013)
Chen, N., Jiang, D.T., Cutler, J., et al.: Structural characterization of poorly-crystalline scorodite, iron(III) earsenate co-precipitates and uranium mill neutralized raffinate solids using X-ray absorption fine structure spectroscopy. Geochim. Cosmochim. Acta 73(11), 3260–3276 (2009)
Mikutta, C., Michel, F.M., Mandaliev, P., et al.: Structure of Amorphous Ferric Arsenate from EXAFS Spectroscopy and Total X-ray Scattering. EGU General Assembly, Vienna (2013)
Guido, C.A., Cortona, P., Mennucci, B., et al.: On the metric of charge transfer molecular excitations: a simple chemical descriptor. J. Chem. Theory Comput. 9(7), 3118–3126 (2013)
Goguel, R.: Direct spectrophotometric determination of sulfate in natural waters by formation of the ferric sulfate complex. Anal. Chem. 41(8), 1034–1038 (1969)
Harharan, A., Sudhakar, C.H., Rao, B.V.: Studies on the solvent extraction of iron (III) with tri-iso-octylamine from aqueous mineral acid solutions. Orient. J. Chem. 28(4), 1785–1790 (2012)
Sen, B., Mukherjee, M., Pal, S., et al.: A water soluble copper(II) complex as a HSO4− ion selective turn-on fluorescent sensor applicable in living cell imaging. RSC Adv. 5(62), 50532–50539 (2015)
Drahota, P., Filippi, M.: Secondary arsenic minerals in the environment: a review. Environ. Int. 35(8), 1243–1255 (2009)
Parker, V.B., Khodakovskii, I.L.: Thermodynamic properties of the aqueous ions (2+ and 3+) of iron and the key compounds of iron. J. Phys. Chem. Ref. Data 24(5), 1699 (1995)
Jia, Y.F., Xu, L.Y., Wang, X., et al.: Infrared spectroscopic and X-ray diffraction characterization of the nature of adsorbed arsenate on ferrihydrite. Geochim. Cosmochim. Acta 71(7), 1643–1654 (2007)
Le Berre, J.F., Gauvin, R., Demopoulos, G.P.: Characterization of poorly-crystalline ferric arsenate precipitated from equimolar Fe(III)-As(V) solutions in the pH range 2 to 8. Metall. Mater. Trans. B 38(5), 751–762 (2007)
Roque-Malherbe, R., Polanco-Estrella, R., Marquez-Linares, F.: Study of the interaction between silica surfaces and the carbon dioxide molecule. J. Phys. Chem. C 114(41), 17773–17787 (2010)
Zhu, H.F., Tang, P.G., Feng, Y.J., et al.: Intercalation of IR absorber into layered double hydroxides: preparation, thermal stability and selective IR absorption. Mater. Res. Bull. 47(3), 532–536 (2012)
Kim, C.R., Noh, T.H., Yoo, K.H., et al.: Anionic indicators on the surface of submicrospheres consisting of ionic palladium(II) complex. Bull. Korean Chem. Soc. 30(12), 3057–3060 (2009)
Yoon, S.H., Lee, S., Kim, T.H., et al.: Oxidation of methylated arsenic species by UV/S2O82−. Chem. Eng. J. 173(2), 290–295 (2011)
Chai, L., Yang, J., Liao, F., et al.: Kinetics and molecular mechanism of arsenite photochemical oxidation based on sulfate radical. Mol. Catal. 438, 113–130 (2017)
Neppolian, B., Celik, E., Choi, H.: Photochemical oxidation of arsenic(III) to arsenic(V) using peroxydisulfate ions as an oxidizing agent. Environ. Sci. Technol. 42(16), 6179–6184 (2008)
Zhou, L., Zheng, W., Ji, Y.F., et al.: Ferrous-activated persulfate oxidation of arsenic(III) and diuron in aquatic system. J. Hazard. Mater. 263(Part 2), 422–430 (2013)
Davies, M.J., Gilbert, B.C., Stell, J.K., et al.: Nucleophilic substitution reactions of spin adducts. Implications for the correct identification of reaction intermediates by EPR/spin trapping. J. Chem. Soc. Perkin Trans. 2(3), 333–335 (1992)
Mottley, C., Mason, R.P.: Sulfate anion free radical formation by the peroxidation of (Bi) sulfite and its reaction with hydroxyl radical scavengers. Arch. Biochem. Biophys. 267(2), 681–689 (1988)
Wang, Z., Bush, R.T., Sullivan, L.A., et al.: Selective oxidation of arsenite by peroxymonosulfate with high utilization efficiency of oxidant. Environ. Sci. Technol. 48(7), 3978–3985 (2014)
Buxton, G.V., Greenstock, C.L., Helman, W.P., et al.: Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (OH/·O− in aqueous solution. J. Phys. Chem. Ref. Data 17(2), 513–886 (1988)
Neta, P., Grodkowski, J., Ross, A.B.: Rate constants for reactions of aliphatic carbon-centered radicals in aqueous solution. J. Phys. Chem. Ref. Data 25(3), 709–1050 (1996)
Klaening, U.K., Bielski, B.H.J., Sehested, K.: Arsenic(IV). A pulse-radiolysis study. Inorg. Chem. 28(14), 2717–2724 (1989)
Yamazaki-Nishida, S., Kimura, M.: Kinetics of the oxidation reaction of arsenious acid by peroxodisulfate ion, induced by irradiation with visible light of aqueous solutions containing tris(2,2′-bipyridine) ruthenium(II) ion. Inorg. Chim. Acta 174(2), 231–235 (1990)
Yoon, S.H., Lee, J.H., Oh, S.E., et al.: Photochemical oxidation of As(III) by vacuum-UV lamp irradiation. Water Res. 42(13), 3455–3463 (2008)
Fan, Z.Y., Huang, J.L., Wang, P., et al.: Kinetics of aniline oxidation with chlorine dioxide. J. Environ. Sci. 16(2), 238–241 (2004)
Gomes, A.C., Nunes, J.C., Simões, R.M.S.: Determination of fast ozone oxidation rate for textile dyes by using a continuous quench-flow system. J. Hazard. Mater. 178(1–3), 57–65 (2010)
Iwai, M., Majima, H., Awakura, Y.: Oxidation of As(III) with dissolved molecular oxygen in alkaline solutions. Trans. Jpn. Inst. Met. 26(7), 492–498 (1985)
Pettine, M., Campanella, L.G., Millero, F.J.: Arsenite oxidation by H2O2 in aqueous solutions. Geochim. Cosmochim. Acta 63(18), 2727–2735 (1999)
Lau, T.K., Chu, W., Graham, N.J.D.: The aqueous degradation of butylated hydroxyanisole by UV/S2O82-: study of reaction mechanisms via dimerization and mineralization. Environ. Sci. Technol. 41(2), 613–619 (2007)
Chai, L., Yue, M., Yang, J., et al.: Formation of tooeleite and the role of direct removal of As(III) from high-arsenic acid wastewater. J. Hazard. Mater. 320, 620–627 (2016)
Nishimura, T., Robins, R.G.: Confirmation that tooeleite is a ferric arsenite sulfate hydrate, and is relevant to arsenic stabilization. Miner. Eng. 21(4), 246–251 (2008)
McLeod, J., Paterson, A.H.J., Jones, J.R., et al.: Primary nucleation of alpha-lactose monohydrate: the effect of supersaturation and temperature. Int. Dairy J. 21(7), 455–461 (2011)
Dang, S.V., Kawasaki, J., Abella, L.C., et al.: Removal of arsenic from simulated groundwater by adsorption using iron-modified rice husk carbon. J. Water Environ. Technol. 7(2), 43–56 (2009)
Mercer, K.L., Tobiason, J.E.: Removal of arsenic from high ionic strength solutions: effects of ionic strength, pH, and preformed versus in situ formed HFO. Environ. Sci. Technol. 42(10), 3797–3802 (2008)
Wang, Y.X., Duan, J.M., Liu, S.X., et al.: Removal of As(III) and As(V) by ferric salts coagulation—implications of particle size and zeta potential of precipitates. Sep. Purif. Technol. 135, 64–71 (2014)
Li, X.F., Zhao, F.H., Deng, S.M.: The removal of arsenic(III) from acid mine drainage by mineral trap of tooeleite (Fe6(AsO3)4SO4(OH)4·4H2O). In: An Interdisciplinary Response to Mine Water Challenges, pp. 671–674 (2014)
Swash, P.M., Monhemius, A.J.: Comparison of the solubilities of arsenic-bearing wastes from hydrometallurgical and pyrometallurgical processes. GDMB 83, 141–152 (2000)
Paikaray, S., Göttlicher, J., Peiffer, S.: As(III) retention kinetics, equilibrium and redox stability on biosynthesized schwertmannite and its fate and control on schwertmannite stability on acidic (pH 3.0) aqueous exposure. Chemosphere 86, 557–564 (2012)
Rahman, N., Haseen, U.: Development of polyacrylamide chromium oxide as a new sorbent for solid phase extraction of As(III) from food and environmental water samples. RSC Adv. 5, 7311–7323 (2015)
Tokoro, C., Yatsugi, Y., Koga, H., et al.: Sorption mechanisms of arsenate during coprecipitation with ferrihydrite in aqueous solution. Environ. Sci. Technol. 44, 638–643 (2010)
Kaksonen, A.H., Riekkola-Vanhanen, M.L., Puhakka, J.A.: Optimization of metal sulfide precipitation in fluidized-bed treatment of acidic wastewater. Water Res. 37(2), 255–266 (2003)
Mokone,Cas T.P., van Hille, R.P., Lewis, A.E.: Effect of solution chemistry on particle characteristics during metal sulfide precipitation. J. Colloid Interface Sci. 351(1), 10–18 (2010)
Long, G., Peng, Y.J., Bradshaw, D.: Flotation separation of copper sulfides from arsenic minerals at Rosebery copper concentrator. Miner. Eng. 66–68, 207–214 (2014)
Padilla, R., Rodriguez, G., Ruiz, M.C.: Copper and arsenic dissolution from chalcopyrite–enargite concentrate by sulfidation and pressure leaching in H2SO4–O2. Hydrometallurgy 100(3–4), 152–156 (2010)
Wang, T., Yang, W.C., Song, T.T., et al.: Cu doped Fe3O4 magnetic adsorbent for arsenic: synthesis, property, and sorption application. RSC Adv. 5(62), 50011–50018 (2015)
Wang, Z.F., Cui, Z.J., Liu, L., et al.: Toxicological and biochemical responses of the earthworm eisenia fetida exposed to contaminated soil: effects of arsenic species. Chemosphere 154, 161–170 (2016)
Yan, X., Li, Q.Z., Chai, L.Y., et al.: Formation of abiological granular sludge-A facile and bioinspired proposal for improving sludge settling performance during heavy metal wastewater treatment. Chemosphere 113, 36–41 (2014)
Wang, T., Zhang, L.Y., Li, C.F., et al.: Synthesis of core-shell magnetic Fe3O4@poly(m-phenylenediamine) particles for chromium reduction and adsorption. Environ. Sci. Technol. 49(9), 5654–5662 (2015)
Chai, L.Y., Wang, Q.W., Li, Q.Z., et al.: Enhanced removal of Hg(II) from acidic aqueous solution using thiol-functionalized biomass. Water Sci. Technol. 62(9), 2157–2165 (2010)
Stalidis, G.A., Matis, K.A., Lazaridis, N.K.: Selective separation of Cu, Zn, and As from solution by flotation techniques. Sep. Sci. Technol. 24(1–2), 97–109 (1989)
Alison Emslie Lewis: Review of metal sulfide precipitation. Hydrometallurgy 104(2), 222–234 (2010)
Lian-hua, Z., Yu-lan, X.: Sulfide precipitation flotation for treatment of acidic mine waste water. Trans. Nonferrous Met. Soc. China 10, 106–109 (2000)
Huisman, J.L., Schouten, G., Schultz, C.: Biologically produced sulfide for purification of process streams, effluent treatment and recovery of metals in the metal and mining industry. Hydrometallurgy 83(1), 106–113 (2006)
Bhattacharyya, D., Jumawan Jr., A.B., Grieves, R.B.: Separation of toxic heavy metals by sulfide precipitation. Sep. Sci. Technol. 14(5), 441–452 (1979)
Veeken, A.H.M., de Vries, S., van Der Mark, A., et al.: Selective precipitation of heavy metals as controlled by a sulfide-selective electrode. Sep. Sci. Technol. 38(1), 1–19 (2003)
Jiang, G.M., Peng, B., Chai, L.Y., et al.: Cascade sulfidation and separation of copper and arsenic from acidic wastewater via gas–liquid reaction. Trans. Nonferrous Met. Soc. China 27(4), 925–931 (2017)
Zheng, J.X., Ye, H.Q., Huang, N.D., et al.: Selective separation of Hg(II) and Cd(II) from aqueous solutions by complexation-ultrafiltration process. Chemosphere 76(5), 706–710 (2009)
Yavuz, C.T., Mayo, J., Yu, W.W., et al.: Low-field magnetic separation of monodisperse Fe3O4 nanocrystals. Science 314(5801), 964–967 (2006)
Zeng, H., Singh, A., Basak, S., et al.: Nanoscale size effects on uranium(VI) adsorption to hematite. Environ. Sci. Technol. 43(5), 1373–1378 (2009)
Yean, S., Cong, L., Yavuz, C.T., et al.: Effect of magnetite particle size on adsorption and desorption of arsenite and arsenate. J. Mater. Res. 20(12), 3255–3264 (2005)
Cotten, G.B., Eldredge, H.B.: Nanolevel magnetic separation model considering flow limitations. Sep. Sci. Technol. 37(16), 3755–3779 (2002)
Kelland, D.R.: Magnetic separation of nanoparticles. IEEE Trans. Magn. 34(4), 2123–2125 (1998)
Mou, F.Z., Guan, J.G., Ma, H., et al.: Magnetic iron oxide chestnutlike hierarchical nanostructures: preparation and their excellent arsenic removal capabilities. ACS Appl. Mater. Interfaces 4(8), 3987–3993 (2012)
Ge, J.P., Huynh, T., Hu, Y.X., et al.: Hierarchical magnetite/silica nanoassemblies as magnetically recoverable catalyst–supports. Nano Lett. 8(3), 931–934 (2008)
Wei, Z.H., Xing, R., Zhang, X., et al.: Facile template-free fabrication of hollow nestlike α-Fe2O3 nanostructures for water treatment. ACS Appl. Mater. Interfaces 5(3), 598–604 (2013)
Wang, T., Zhang, L., Li, C., et al.: Synthesis of core–shell magnetic Fe3O4@ poly (m-phenylenediamine) particles for chromium reduction and adsorption. Environ. Sci. Technol. 49(9), 5654–5662 (2012)
Mou, F.Z., Guan, J.G., Xiao, Z.D., et al.: Solvent-mediated synthesis of magnetic Fe2O3 chestnut-like amorphous-core/γ-phase-shell hierarchical nanostructures with strong As(V) removal capability. J. Mater. Chem. 21(14), 5414–5421 (2011)
Wang, P., Lo, I.M.C.: Synthesis of mesoporous magnetic γ-Fe2O3 and its application to Cr(VI) removal from contaminated water. Water Res. 43(15), 3727–3734 (2009)
Wang, T., Zhang, L.Y., Wang, H.Y., et al.: Controllable synthesis of hierarchical porous Fe3O4 particles mediated by Poly(diallyldimethylammonium chloride) and their application in arsenic removal. ACS Appl. Mater. Interfaces 5(23), 12449–12459 (2013)
Jia, B.P., Gao, L.: Morphological transformation of Fe3O4 spherical aggregates from solid to hollow and their self-assembly under an external magnetic field. J. Phys. Chem. C 112(3), 666–671 (2008)
Fan, T., Pan, D., Zhang, H., et al.: Study on formation mechanism by monitoring the morphology and structure evolution of nearly monodispersed Fe3O4 submicroparticles with controlled particle sizes. Ind. Eng. Chem. Res. 50(15), 9009–9018 (2011)
Liu, Z.H., Yang, X.J., Makita, Y., et al.: Preparation of a polycation-intercalated layered manganese oxide nanocomposite by a delamination/reassembling process. Chem. Mater. 14(11), 4800–4806 (2002)
Liu, K.P., Zhang, J.J., Yang, G.H., et al.: Direct electrochemistry and electrocatalysis of hemoglobin based on poly(diallyldimethylammonium chloride) functionalized graphene sheets/room temperature ionic liquid composite film. Electrochem. Commun. 12(3), 402–405 (2010)
Yu, X.Y., Luo, T., Jia, Y., et al.: Porous hierarchically micro-/nanostructured MgO: morphology control and their excellent performance in As(III) and As(V) removal. J. Phys. Chem. C 115(45), 22242–22250 (2011)
Hang, C., Li, Q., Gao, S.A., et al.: As(III) and As(V) adsorption by hydrous zirconium oxide nanoparticles synthesized by a hydrothermal process followed with heat treatment. Ind. Eng. Chem. Res. 51(1), 353–361 (2012)
Zhong, L.S., Hu, J.S., Liang, H.P., et al.: Self-assembled 3D flowerlike iron oxide nanostructures and their application in water treatment. Adv. Mater. 18(18), 2426–2431 (2006)
Zhu, H., Hou, C., Li, Y.J., et al.: One-pot solvothermal synthesis of highly water-dispersible size-tunable functionalized magnetite nanocrystal clusters for lipase immobilization. Chem. Asian J. 8(7), 1447–1454 (2013)
Cao, C.Y., Qu, J., Yan, W.S., et al.: Low-cost synthesis of flowerlike α-Fe2O3 nanostructures for heavy metal ion removal: adsorption property and mechanism. Langmuir 28(9), 4573–4579 (2012)
Kanel, S.R., Greneche, J.M., Choi, H.: Arsenic(V) removal from groundwater using nano scale zero-valent iron as a colloidal reactive barrier material. Environ. Sci. Technol. 40(6), 2045–2050 (2006)
Nesbitt, H.W., Muir, I.J.: Oxidation states and speciation of secondary products on pyrite and arsenopyrite reacted with mine waste waters and air. Mineral. Petrol. 62(1–2), 123–144 (1998)
Gomes, J.A.G., Daida, P., Kesmez, M., et al.: Arsenic removal by electrocoagulation using combined Al–Fe electrode system and characterization of products. J. Hazard. Mater. 139(2), 220–231 (2007)
Chen, B., Zhu, Z.L., Ma, J., et al.: Surfactant assisted Ce–Fe mixed oxide decorated multiwalled carbon nanotubes and their arsenic adsorption performance. J. Mater. Chem. A 1(37), 11355–11367 (2013)
Wielant, J., Hauffman, T., Blajiev, O., et al.: Influence of the iron oxide acid-base properties on the chemisorption of model epoxy compounds studied by XPS. J. Phys. Chem. C 111(35), 13177–13184 (2007)
Ramos, M.A.V., Yan, W.L., Li, X.Q., et al.: Simultaneous oxidation and reduction of arsenic by zero-valent iron nanoparticles: understanding the significance of the core-shell structure. J. Phys. Chem. C 113(33), 14591–14594 (2009)
Lim, S.F., Zheng, Y.M., Chen, J.P. Organic arsenic adsorption onto a magnetic sorbent. Langmuir 25(9), 4973–4978 (2009)
Manning, B.A., Hunt, M.L., Amrhein, C., et al.: Arsenic (III) and arsenic (V) reactions with zerovalent iron corrosion products. Environ. Sci. Technol. 36(24), 5455–5461 (2002)
Pena, M., Meng, X., Koratis, G.P., et al.: Adsorption mechanism of arsenic on nanocrystalline titanium dioxide. Environ. Sci. Technol. 40(4), 1257–1262 (2006)
Sandoval, R., Cooper, A.M., Aymar, K., et al.: Removal of arsenic and methylene blue from water by granular activated carbon media impregnated with zirconium dioxide nanoparticles. J. Hazard. Mater. 193, 296–303 (2011)
Xu, W.H., Wang, J., Wang, L., et al.: Enhanced arsenic removal from water by hierarchically porous CeO2–ZrO2 nanospheres: role of surface-and structure-dependent properties. J. Hazard. Mater. 260, 498–507 (2013)
Lv, X.J., Yang, W.G., Quan, Z.W., et al.: Enhanced electron transport in Nb-Doped TiO2 nanoparticles via pressure-induced phase transitions. J. Am. Chem. Soc. 136(1), 419–426 (2014)
Li, H., Zhang, L.Z.: Oxygen vacancy induced selective silver deposition on the 001 facets of BiOCl single-crystalline nanosheets for enhanced Cr(VI) and sodium pentachlorophenate removal under visible light. Nanoscale 6(14), 7805–7810 (2014)
Zhang, Y., Yang, M., Dou, X.M., et al.: Arsenate adsorption on an Fe-Ce bimetal oxide adsorbent: Role of surface properties. Environ. Sci. Technol. 39(18), 7246–7253 (2005)
Warner, C.L., Chouyyok, W., Mackie, K.E., et al.: Manganese doping of magnetic iron oxide nanoparticles: tailoring surface reactivity for a regenerable heavy metal sorbent. Langmuir 28(8), 3931–3937 (2012)
Wang, Y.J., Chen, D.G., Wang, Y.D., et al.: Tunable surface charge of ZnS: Cu nano-adsorbent induced the selective preconcentration of cationic dyes from wastewater. Nanoscale 4(12), 3665–3668 (2012)
Neagu, D., Irvine, J.T.: Enhancing electronic conductivity in strontium titanates through correlated A and B-site doping. Chem. Mater. 23(6), 1607–1617 (2011)
Norris, D.J., Efros, A.L., Erwin, S.C.: Doped nanocrystals. Science 319(5871), 1776–1779 (2008)
Chen, Z., Pina, C.D., Falletta, E., et al.: A green route to conducting polyaniline by copper catalysis. J. Catal. 267(2), 93–96 (2009)
Chai, L.Y., Wang, T., Zhang, L.Y., et al.: A Cu–m-phenylenediamine complex induced route to fabricate poly(m-phenylenediamine)/reduced graphene oxide hydrogel and its adsorption application. Carbon 81, 748–757 (2015)
Deng, H., Li, X.L., Peng, Q., et al.: Monodisperse magnetic single-crystal ferrite microspheres. Angew. Chem. 117(18), 2842–2845 (2005)
Yin, A.Y., Guo, X.Y., Dai, W.L., et al.: The nature of active copper species in Cu-HMS catalyst for hydrogenation of dimethyl oxalate to ethylene glycol: new insights on the synergetic effect between Cu0 and Cu+. J. Phys. Chem. C 113(25), 11003–11013 (2009)
Derrouiche, S., Lauron-Pernot, H., Louis, C.: Synthesis and Treatment Parameters For Controlling Metal Particle Size And Composition in Cu/ZnO materials first evidence of Cu3Zn alloy formation. Chem. Mater. 24(12), 2282–2291 (2012)
McFarland, E.W., Metiu, H.: Catalysis by doped oxides. Chem. Rev. 113(6), 4391–4427 (2013)
Aldon, L., Kubiak, P., Picard, A., et al.: Size particle effects on lithium insertion into Sn-doped TiO2 anatase. Chem. Mater. 18(6), 1401–1406 (2006)
Deiana, C., Fois, E., Coluccia, S., et al.: Surface structure of TiO2 P25 nanoparticles: infrared study of hydroxy groups on coordinative defect sites. J. Phys. Chem. C 114(49), 21531–21538 (2010)
Chai, L.Y., Wang, Y.Y., Zhao, N., et al.: Sulfate-doped Fe3O4/Al2O3 nanoparticles as a novel adsorbent for fluoride removal from drinking water. Water Res. 47(12), 4040–4049 (2013)
Mohapatra, M., Sahoo, S.K., Anand, S., et al.: Removal of As(V) by Cu(II)-, Ni(II)-, or Co(II)-doped goethite samples. J. Colloid Interface Sci. 298(1), 6–12 (2006)
Yu, X.Y., Luo, T., Jia, Y., et al.: Porous hierarchically micro-/nanostructured MgO: morphology control and their excellent performance in As (III) and As (V) removal. J. Phys. Chem. C 115(45), 22242–22250 (2011)
Xu, W.H., Wang, L., Wang, J., et al.: Superparamagnetic mesoporous ferrite nanocrystal clusters for efficient removal of arsenite from water. CrystEngComm 15(39), 7895–7903 (2013)
Couture, R.M., Rose, J., Kumar, N., et al.: Sorption of arsenite, arsenate, and thioarsenates to iron oxides and iron sulfides: a kinetic and spectroscopic investigation. Environ. Sci. Technol. 47(11), 5652–5659 (2013)
Can, M., Uzun, S.: Oxidizing effect of the Cu(ClO4)2 on chemical polymerization of aniline in anhydrous media. Asian J. Chem. 22, 867–872 (2010)
Izumi, C.M.S., Constantino, V.R.L., Temperini, M.L.A.: Spectroscopic characterization of polyaniline formed by using copper (II) in homogeneous and MCM-41 molecular sieve media. J. Phys. Chem. B 109(47), 22131–22140 (2005)
Mou, F.Z., Guan, J.G., Ma, H.R., et al.: Magnetic iron oxide chestnutlike hierarchical nanostructures: preparation and their excellent arsenic removal capabilities. ACS Appl. Mater. Interfaces 4(8), 3987–3993 (2012)
Toulemon, D., Pichon, B.P., Cattoen, X., et al.: 2D assembly of non-interacting magnetic iron oxide nanoparticles via “click” chemistry. Chem. Commun. 47(43), 11954–11956 (2011)
Feng, L.Y., Cao, M.H., Ma, X.Y., et al.: Superparamagnetic high-surface-area Fe3O4 nanoparticles as adsorbents for arsenic removal. J. Hazard. Mater. 217–218, 439–446 (2012)
Saiz, J., Bringas, E., Ortiz, I.: Functionalized magnetic nanoparticles as new adsorption materials for arsenic removal from polluted waters. J. Chem. Technol. Biotechnol. 89(6), 909–918 (2014)
Chen, B., Zhu, Z.L., Ma, J., et al.: One-pot, solid-phase synthesis of magnetic multiwalled carbon nanotube/iron oxide composites and their application in arsenic removal. J. Colloid Interface Sci. 434, 9–17 (2014)
Li, H., Yu, S., Han, X.X.: Fabrication of CuO hierarchical flower-like structures with biomimetic superamphiphobic, self-cleaning and corrosion resistance properties. Chem. Eng. J. 283, 1443–1454 (2016)
Meshram, S.P., Adhyapak, P.V., Mulik, U.P., et al.: Facile synthesis of CuO nanomorphs and their morphology dependent sunlight driven photocatalytic properties. Chem. Eng. J. 204–206, 158–168 (2012)
Peng, B., Song, T., Wang, T., et al.: Facile synthesis of Fe3O4@Cu(OH)2 composites and their arsenic adsorption application. Chem. Eng. J. 299, 15–22 (2016)
Escudero, C., Fiol, N., Villaescusa, I., et al.: Arsenic removal by awaste metal (hydr)oxide entrapped into calcium alginate beads. J. Hazard. Mater. 164(2–3), 533–541 (2009)
Li, Z.J., Deng, S.B., Yu, G., et al.: As(V) and As(III) removal from water by a Ce–Ti oxide adsorbent: behavior and mechanism. Chem. Eng. J. 161(1–2), 106–113 (2010)
Yang, Y., Yang, M., Dou, X.M., et al.: Arsenate adsorption on an Fe-Ce bimetal oxide adsorbent: role of surface properties. Environ. Sci. Technol. 39(18), 7246–7253 (2005)
Zhang, S.W., Li, J.X., Wen, T., et al.: Magnetic Fe3O4@NiO hierarchical structures: preparation and their excellent As(V) and Cr(VI) removal capabilities. RSC Adv. 3(8), 2754–2764 (2013)
Wang, T., Zhang, L.Y., Li, C.F., et al.: Synthesis of core-shell magnetic Fe3O4 @poly(m-phenylenediamine) particles for chromium reduction and adsorption. Environ. Sci. Technol. 49(9), 5654–5662 (2015)
Xu, L.J., Wang, J.L.: Magnetic nanoscaled Fe3O4/CeO2 composite as an efficient fenton-like heterogeneous catalyst for degradation of 4-chlorophenol. Environ. Sci. Technol. 46(18), 10145–10153 (2012)
Gai, L.G., Li, Z.L., Hou, Y.H., et al.: Preparation of core–shell Fe3O4/SiO2 microspheres as adsorbents for purification of DNA. J. Phys. D Appl. Phys. 43, 445001 (2010)
Ma, Z.Y., Guan, Y.P., Liu, H.Z.: Synthesis and characterization of micron-sized monodisperse superparamagnetic polymer particles with amino groups. J. Polym. Sci. Part A Polym. Chem. 43(15), 3433–3439 (2005)
Lu, C.H., Qi, L.M., Yang, J.H., et al.: Simple template-free solution route for the controlled synthesis of Cu(OH)2 and CuO nanostructures. J. Phys. Chem. B 108(46), 17825–17831 (2004)
Hua, R., Li, Z.K.: Sulfhydryl functionalized hydrogel with magnetism: synthesis, characterization, and adsorption behavior study for heavy metal removal. Chem. Eng. J. 249, 189–200 (2014)
Martinson, C.A., Reddy, K.J.: Adsorption of arsenic(III) and arsenic(V) by cupric oxide nanoparticles. J. Colloid Interface Sci. 336(2), 406–411 (2009)
Wang, X.L., Liu, Y.K., Zheng, J.T.: Removal of As(III) and As(V) from water by chitosan and chitosan derivative: a review. Environ. Sci. Pollut. Res. 23(14), 13789–13801 (2016)
Zhang, G.S., Ren, Z.M., Zhang, X.W., et al.: Nanostructured iron(III)-copper(II) binary oxide: a novel adsorbent for enhanced arsenic removal from aqueous solutions. Water Res. 47(12), 4022–4031 (2013)
Maeda, K., Domen, K.: New non-oxide photocatalysts designed for overall water splitting under visible light. J. Phys. Chem. C 111(22), 7851–7861 (2007)
Huang, M.R., Ding, Y.B., Li, X.G.: Lead-ion potentiometric sensor based on electrically conducting microparticles of sulfonic phenylenediamine copolymer. Analyst 138(13), 3820–3829 (2013)
Huang, M.R., Rao, X.W., Li, X.G., et al.: Lead ion-selective electrodes based on polyphenylenediamine as unique solid ionophores. Talanta 85(3), 1575–1584 (2011)
Huang, J.Y., Li, S.H., Ge, M.Z., et al.: Robust superhydrophobic TiO2@fabrics for UV shielding, self-cleaning and oil–water separation. J. Mater. Chem. A 3(6), 2825–2832 (2015)
Li, X.G., Ma, X.L., Sun, J., et al.: Powerful reactive sorption of silver(I) and mercury(II) onto poly(o-phenylenediamine) microparticles. Langmuir 25(3), 1675–1684 (2009)
Wang, J.J., Jiang, J., Hu, B., et al.: Uniformly shaped poly(p-phenylenediamine) microparticles: shape-controlled synthesis and their potential application for the removal of lead ions from water. Adv. Func. Mater. 18(7), 1105–1111 (2008)
Huang, M.R., Lu, H.J., Song, W.D., et al.: Dynamic reversible adsorption and desorption of lead ions through a packed column of poly(m-phenylenediamine) spheroids. Soft Mater. 8(2), 149–163 (2010)
Zhang, L.Y., Wang, T., Wang, H.Y., et al.: Graphene@poly(m-phenylenediamine) hydrogel fabricated by a facile post-synthesis assembly strategy. Chem. Commun. 49(85), 9974–9976 (2013)
Yu, W.T., Zhang, L.Y., Meng, Y., et al.: High conversion synthesis of functional poly(m-phenylenediamine) nanoparticles by Cu-OH-assisted method and its superior ability toward Ag+ adsorption. Synth. Met. 176, 78–85 (2013)
Wang, H.Y., Chai, L.Y., Hu, A.J., et al.: Self-assembly microstructures of amphiphilic polyborate in aqueous solutions. Polymer 50(13), 2976–2980 (2009)
Harris, J.K., Rose, G.D., Bruening, M.L.: Spontaneous generation of multilamellar vesicles from ethylene oxide/butylene oxide diblock copolymers. Langmuir 18(14), 5337–5342 (2002)
Li, H.Q., Lai, Y.K., Huang, J.Y., et al.: Multifunctional wettability patterns prepared by laser processing on superhydrophobic TiO2 nanostructured surfaces. J. Mater. Chem. B 3(3), 342–347 (2015)
Dey, J., Kumar, S., Nath, S., et al.: Additive induced core and corona specific dehydration and ensuing growth and interaction of Pluronic F127 micelles. J. Colloid Interface Sci. 415, 95–102 (2014)
Sang, P.L., Wang, Y.Y., Zhang, L.Y., et al.: Effective adsorption of sulfate ions with poly(m-phenylenediamine) in aqueous solution and its adsorption mechanism. Trans. Nonferrous Met. Soc. China 23(1), 243–252 (2013)
Li, X.G., Wang, L.X., Jin, Y., et al.: Preparation and identification of a soluble copolymer from pyrrole and o-toluidine. J. Appl. Polym. Sci. 82(2), 510–518 (2001)
Chai, L.Y., Zhang, L.Y., Wang, H.Y., et al.: An effective and scale-up self-assembly route to prepare the rigid and smooth oligo(o-phenylenediamine) microfibers in acidic solution by NaClO2. Mater. Lett. 64(21), 2302–2305 (2010)
Li, X.G., Huang, M.R., Duan, W.: Novel multifunctional polymers from aromatic diamines by oxidative polymerizations. Chem. Rev. 102(9), 2925–3030 (2002)
Liu, M.L., Ye, M., Yang, Q., et al.: A new method for characterizing the growth and properties of polyaniline and poly(aniline-co-o-aminophenol) films with the combination of EQCM and in situ FTIR spectroelectrochemisty. Electrochim. Acta 52(1), 342–352 (2006)
Huang, M.R., Li, X.G., Yang, Y.L.: Oxidative polymerization of o-phenylenediamine and pyrimidylamine. Polym. Degrad. Stab. 71(1), 31–38 (2000)
Yu, W.T., Zhang, L.Y., Wang, H.Y., et al.: Adsorption of Cr(VI) using synthetic poly(m-phenylenediamine). J. Hazard. Mater. 260, 789–795 (2013)
Zhang, L.Y., Chai, L.Y., Liu, J., et al.: pH manipulation: a facile method for lowering oxidation state and keeping good yield of poly(m-phenylenediamine) and its powerful Ag+ adsorption ability. Langmuir 27(22), 13729–13738 (2011)
Losito, I., Malitesta, C., De Bari, I., et al.: X-ray photoelectron spectroscopy characterization of poly(2,3-diaminophenazine) films electrosynthesised on platinum. Thin Solid Films 473(1), 104–113 (2005)
Frost, R.L., Xi, Y.F., Wood, B.J.: Thermogravimetric analysis, PXRD, EDX and XPS study of chrysocolla (Cu, Al)2H2Si2O5(OH)4·nH2O-structural implications. Thermochim. Acta 545, 157–162 (2012)
Pelissier, B., Beaurain, A., Fontaine, H., et al.: Investigations on HCl contaminated Cu 200 mm wafers using parallel angle resolved XPS. Micro Microelectron. Eng. 86(4–6), 1013–1016 (2009)
Ayad, M.M., Amer, W.A., Stejskal, J.: Effect of iodine solutions on polyaniline films. Thin Solid Films 517(21), 5969–5973 (2009)
Han, J., Dai, J., Guo, R.: Highly efficient adsorbents of poly(o-phenylenediamine) solid and hollow sub-microspheres towards lead ions: a comparative study. J. Colloid Interface Sci. 356(2), 749–756 (2011)
Stejskal, J., Trchová, M., Brožová, L., et al.: Reduction of silver nitrate by polyaniline nanotubes to produce silver-polyaniline composites. Chem. Pap. 63(1), 77–83 (2009)
Izumi, C.M.S., Brito, H.F., Ferreira, A.M.D.C., et al.: Spectroscopic investigation of the interactions between emeraldine base polyaniline and Eu(III) ions. Synth. Met. 159(5–6), 377–384 (2009)
Huang, M.R., Huang, S.J., Li, X.G.: Facile synthesis of polysulfoaminoanthraquinone nanosorbents for rapid removal and ultrasensitive fluorescent detection of heavy metal ions phys. J. Phys. Chem. C 115(13), 5301–5315 (2011)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Chai, LY., Li, QZ., Wang, QW., Wang, YY., Yang, WC., Wang, HY. (2019). Arsenic Behaviors and Pollution Control Technologies in Aqueous Solution. In: Chai, LY. (eds) Arsenic Pollution Control in Nonferrous Metallurgy. Springer, Singapore. https://doi.org/10.1007/978-981-13-6721-2_3
Download citation
DOI: https://doi.org/10.1007/978-981-13-6721-2_3
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-6720-5
Online ISBN: 978-981-13-6721-2
eBook Packages: Earth and Environmental ScienceEarth and Environmental Science (R0)