
Abstract
Solution-processed organic semiconductor devices are expected to provide flexible, large-area, light-weight, low-cost, and environment-friendly electronic products for next-generation devices. As small molecular crystals have intrinsically high electrical properties, this chapter focuses on the fabrication technique of single crystalline and polycrystalline organic electronic devices using solution processes. It starts with a description of the relationship between the crystal structure and electrical properties of organic semiconductors. Then, several techniques for preparing single-crystalline organic field effect transistors are discussed. Next, a precursor approach to preparing polycrystalline films is addressed. With this approach, well-soluble and stable precursor compounds are deposited on substrates and then quantitatively converted into target materials by applying external stimuli, such as heat or light. The recent progress of organic field effect transistors and organic photovoltaics with this approach is summarized.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Organic semiconductor
- Solution process
- Crystalline film
- Organic field effect transistor
- Organic photovoltaics
9.1 Introduction
Development of high-performance printed organic semiconductor devices, such as organic field-effect transistors (OFETs), organic photovoltaics (OPVs), and organic light emitting diodes (OLEDs), is highly desired for the next-generation technologies of ‘printable electronics’. Organic semiconductors are expected to provide flexible, large-area, light-weight, low-cost, and environment-friendly electronic products, including wearable and/or transparent sheet-type solar cells, flexible displays, and so on. The performances of organic electronic devices largely depend on the charge transport efficiency in the organic active layers and carrier injection at the interface of organic/metal electrodes.
The organic semiconducting materials can be classified as crystalline small molecules; soluble small molecules, including oligomers and liquid crystalline materials; and π-conjugated conductive polymers. The key structure of these materials is the π-electron conjugation over the molecule frameworks. The π-conjugated polymers have π-electron conjugation through the polymer backbones. Thus, the charge carriers can travel along the main polymer chains. Small molecular semiconducting materials have aromatic frameworks over which the π-electrons spread; however, the electrons are localized in each molecule. For efficient charge carrier transport, charge carriers must efficiently move between the molecules. An immense number of compounds have been developed since the 1990s for small-molecule organic semiconductor materials [1,2,3].
For efficient intermolecular charge transport between molecules, a π-π interaction with neighbouring molecules is necessary, and a crystalline film with a proper packing structure is desirable. Single-crystal devices are optimal devices with high intrinsic electrical properties; however, the preparation of numerous large single crystals was not practical. Recently several noteworthy solution-processing techniques for single-crystal devices have been reported. It will be surveyed in Sect. 9.4.
Polycrystalline film is the second choice, while vacuum deposition is a well-known technique for the preparation of polycrystalline films. It enables the very pure polycrystalline film to have good electrical properties. Nevertheless, the preparation of large devices on plastic film is difficult with this method, and molecules with a larger molecular weight cannot be deposited on account of the decomposition before sublimation. To apply the polycrystalline organic electronic devices, a preparation technique of polycrystalline films by a solution process is required. Recently, high-performance devices with solution-processed polycrystalline films have been reported. In Sects. 9.5 and 9.6, the preparation and morphology control of crystalline organic semiconductor layers by a solution process are addressed.
9.2 Organic Devices for Evaluating Charge Transport Efficiency
To evaluate the intrinsic charge transport abilities of organic semiconducting materials, the charge carrier mobilities are estimated by various techniques. These techniques include the time-of-flight (TOF), space-charge-limited current (SCLC), time-resolved microwave conductivity (TRMC), OFETs, etc. [4]. In this section, the morphology and charge carrier mobilities of the prepared crystalline films are discussed in terms of OFET and OPV performances. The basic structures and mechanisms of the OFET and OPV devices are first described.
9.2.1 Organic Field-Effect Transistors
OFETs are crucial components for organic electronic devices using organic semiconductors in the channel. An OFET is a three-electrode system with gate, source, and drain electrodes. There are four OFET device structure types: top-contact bottom-gate (TCBG), bottom-contact bottom-gate (BCBG), top-contact top-gate (TCTG), and bottom-contact top-gate (BCTG) structures, depending on the relative structure of electrodes and organic semiconductors (Fig. 9.1).

Four OFET architecture types: TCBG, top-contact bottom-gate (a); BCBG, bottom-contact bottom-gate (b); TCTG, top-contact top-gate (c); and BCTG, bottom-contact top-gate (d) structures
The amplitude of the drain–source current (I DS) is controlled by the applied gate voltage (V G) and drain–source voltage (V DS). The field applied to the gate electrode induces a charge (channel) in the semiconducting layer. The polarity of the gate bias determines the channel polarity. When the hole (electron) is a main charge carrier, it is called a p-type (n-type) OFET. The polarity depends on the electronic properties of the semiconductor materials. The carrier travels horizontally between source and drain electrodes in the channel, and a large π–π interaction between neighbouring semiconductor molecules in a horizontal direction is desirable.
The carrier mobility (μ) indicates the rate of the carrier moving through the channel in the semiconductor layer under an electrical field. The μ value is calculated using Eq. (9.1):
where I SD is the drain current, W and L are the channel width and length, respectively, C i is the capacitance per unit area of the gate dielectric, and V G and V th are the gate voltage and threshold voltage, respectively. In Fig. 9.2, an example of output curves of I SD at several V G’s (a) and a transfer curve at a certain V SD (b) are shown. The OFET properties are discussed mainly in terms of μ, V th, and the on/off current ratio (I ON/I OFF). V th is the voltage required for driving charges that are mobile; a value close to zero is desirable. I ON/I OFF is the ratio of the drain current at the on and off states. The larger the I ON/I OFF is, the better it is, and at least I ON/I OFF > 103 is required.

Example of FET data: (a) Output curves of I SD at each V G and (b) transfer curve at a certain V SD. The μ value is calculated from the inclination of the tangential line (red dotted line) and V th from the intercept of the tangential line and x-axis
9.2.2 Organic Photovoltaics
A two-layer OPV fabricated by vacuum deposition using copper phthalocyanine for the p-material and a perylene tetracarboxylic derivative (PV) for the n-material (Fig. 9.3a) with a photocurrent conversion efficiency (PCE) of 0.95% was reported by Tang in 1986 [5]. In 1991, Hiramoto et al. reported a three-layered OPV with an interlayer of co-deposited pigments of p-type metal-free phthalocyanine (H2Pc) and n-type perylene tetracarboxylic derivative (Me-PTC) between respective p- and n-pigment layers (Fig. 9.3b) [6]. The 0.7% PCE performance was two times higher than that of the two-layered device on account of the efficient carrier photo-generation in the interlayer. The single p- and n-type pigment layers, which sandwich the interlayer, effectively provide the built-in potential to the photo-active interlayer. The single p- and n-layers also play an important role as carrier transport layers.

Another key advancement was reported by Heeger et al. in 1995 as a monolayer bulk-heterojunction (BHJ) OPV using a mixture of semiconducting polymer (MEH-PPV) for p-type material and PCBM for n-type material with PCE = 2.9% (Fig. 9.3c) [7]. Efficient charge separation resulted from a bicontinuous network of an internal donor-acceptor heterojunction, and the PCE performance surpassed 11%.
Typical current–voltage (J-V) curves with and without photo-irradiation are shown in Fig. 9.4. The OPV performance is evaluated by power conversion efficiency (PCE), as shown in Eq. 9.2.
where P out is the output power, P in is the input power, J SC is the short-circuit current density, V OC is the open-circuit voltage, FF is the fill factor, and P light is the power of the incident light. In addition, J SC is the maximum current that can be obtained under conditions of a zero-resistance load. The larger absorption coverage of organic molecules and the larger charge transport efficiency of the organic layer increase J SC. Moreover, V OC is the maximum voltage with no load applied. V OC largely depends on the difference of energy level of the highest occupied molecular orbital (HOMO) of the donor material and the lowest unoccupied molecular orbital (LUMO) of the acceptor material. The crystal packing structure of the materials in the active layer also influences V OC by changing the ionization energy of the materials. FF is the ratio between the maximum power and the full square spanned by the J SC and V OC values, as described in Eq. 9.3. FF is influenced by the electrical properties of the active layers and depends on the competition between charge carrier recombination and transport processes.

Typical current–voltage curves. Solid line, photocurrent; dashed line, dark current
The fundamental mechanism to generate photocurrent in OPV is summarized in Fig. 9.5. Conversion of light energy to electric energy can be classified into five steps:
-
Step 1: Absorption of light by donor and/or acceptor materials generates an exciton (a coulombically bound electron-hole pair).
-
Step 2: The exciton diffuses into the interface of the donor and acceptor materials.
-
Step 3: The charge separates from the exciton geminate hole-electron pair.
-
Step 4: The free charge carrier is transported toward the electrodes.
-
Step 5: The charge carrier is injected into the electrodes.

Five-step mechanism to generate photocurrent in a typical planar p-n-type OPV device
To improve the PCE performance, all five steps should efficiently progress. The film morphology plays important roles in each step.
In Step 1, the excited state (exciton) of the donor and/or acceptor molecules is due to incident light absorption. The exciton generation efficiency strongly relates to the molar extinction coefficient of the donor and acceptor materials. By the π–π interaction between molecules in the film, the absorption is broadened and the absorption peaks shift from the peak of the unimolecular spectrum in solution. With H-aggregation (parallel orientation) of the molecules, the absorption shows a hypsochromic (blue) shift; the J-aggregation (head-to-tail orientation) shows a bathochromic (red) shift.
The exciton is deemed an electron and hole pair. The charges are located in the same molecule and strongly bound by a Coulombic interaction. An energetic barrier to transferring the charge from the exciton to the neighbouring molecules exists. In Step 2, the exciton must diffuse to the donor-acceptor interface to encounter the partner for transferring an electron from the donor to acceptor molecule. The exciton diffusion length of organic semiconductors is approximately 10 nm in crystalline films, depending on the packing structure. The packing structure influences the diffusion direction. In p-n-type OPV systems, excitons less than 10 nm from the interface of the two layers can contribute to carrier generation, while excitons far from the interface cannot reach it. The BHJ structure is superior to the p-n structure owing to the increased interface area. The morphology control of the mixed layer to improve the charge separation probability is important.
For Step 3 charge separation, the ionization potential (IP) and electron affinity (EA) of donor and acceptor materials directly influence the OPV performance. V OC is closely related to the difference between the donor-material IP and acceptor material EA. Donor IP and acceptor EA in crystalline film are different from those for the gas-phase-isolated molecule (the difference is called polarization energy; see Sect. 9.3.3). The difference is greatly influenced by the molecular packing structure.
After charge separation, positive and negative charges travel to anode and cathode electrodes, respectively (Step 4). Charge carrier mobilities in crystalline films are influenced by many factors, including the molecular packing structure, grain size, disorder, presence of impurities, temperature, electric field, charge carrier density, molecule size/molecular weight, and pressure [4]. Molecular packing in single crystals influences the charge transport efficiency and anisotropy, especially by electron coupling (or transfer integral), reorganization energy, and polarization energies [8]. These three key factors are overviewed in Sect. 9.3. Even for the bulk hetero layer from combined polymers and fullerene derivatives, the partial crystallinity of the materials was reported as being crucial for good carrier mobilities and stability [9].
For a good electrical property and effective charge injection at the interface of the active layer and electrodes, the crystalline structure of the p- and n-layers is favourable (Step 5). The crystalline p- and n-layers in the p-i-n structure prevent a current leak and support charge injection to electrodes [6].
9.3 Charge Transport Mechanism of Organic Semiconductors
9.3.1 Organic Versus Inorganic Semiconductors
Compared to inorganic semiconducting materials, the charge transport properties of organic semiconductors are intrinsically low. The exciton nature of organic materials (Frenkel exciton) is very different from those of inorganic semiconductors (Wannier exciton) [10]. In inorganic semiconductors (such as doped silicon, GaAs, ZnS, etc.), atoms are covalently bound, and the exciton electron is only weakly bound to the hole with minimal binding energy. Thus, the average exciton orbital radius (∼16 nm) is larger than the lattice unit. The carrier diffusion length is ∼100 μm; the charge carrier mobilities are over the order of 100 cm2 V−1 s−1.
For small molecular crystalline organic semiconductors (Fig. 9.6), the lattice unit is based on molecules. The average exciton radius is approximately 1 nm, the carrier diffusion length is approximately several tens of nanometres, and the charge carrier mobilities are less than the order of 100 cm2 V−1 s−1. To increase the charge carriermobility of organic semiconductors, the morphologies of the organic molecules are very important. The crystalline molecules are weakly bound by CH–π, π–π, and van der Waals interactions.

Structures of acene-type semiconductors discussed in this chapter
Figure 9.7 shows typical packing structures observed for organic semiconducting materials: the herringbone and 1D π-stacking structures of pentacene [11]. In the herringbone arrangement, an aromatic edge-to-face (CH-π) interaction dominates, yielding 2D electronic interactions. In the 1D π-stacking arrangement, a face-to-face (π–π) interaction of electron-rich faces dominates, yielding a coplanar-stacked structure with some degree of displacement along molecule long and short axes. In the 1D π-stacking arrangement, the electron coupling in the stacking direction tends to be large. Substituents on π-frames efficiently influence the packing structure by van der Waals interactions between substituents, steric hindrance, and the π-frame electronic structure.

Pentacene herringbone (top) and 1D π-stacking (bottom) structures. (Reproduced with permission from [11]. Copyright 2006 American Chemical Society)
9.3.2 Transfer Integral and Reorganization Energy
Charge transport efficiency depends on the transfer integral among neighbouring molecules. The transfer integral is a theoretically estimated electronic coupling using the single-crystal structure molecular configuration. It strongly depends on the molecule electronic structure, neighbouring-molecule mutual distances and orientations, and displacement degree along long and short molecule axes.
In 2012, Watanabe et al. were successful in isolating hexacene for the first time [12] (Sect. 9.5.1 for synthesis). They compared its single-crystal structures, transfer integrals, reorganization energies, and hole mobilities with those of naphthalene, anthracene, tetracene, and pentacene. All acenes showed a herringbone motif single-crystal structure [13,14,15], and Fig. 9.8 shows the hexacene packing structure. A higher electronic coupling (t +) along both T1 and T2 directions is observed by increasing the number of benzene rings (Table 9.1). A significantly higher hole mobility (μ +) of hexacene than the other acenes was theoretically estimated: hexacene, 1.461 cm2 V−1 s−1; pentacene, 0.832 cm2 V−1 s−1; tetracene, 0.470 cm2 V−1 s−1; anthracene, 0.158 cm2 V−1 s−1; and naphthalene, 0.0511 cm2 V−1 s−1. The hole mobilities are well fitted by an equation relating to the number (x) of aromatic rings, axb (x = 2–6), where a = 0.00584 and b = 3.09. The calculation suggests that hexacene μ + is most efficient along the a–b plane. The best single-crystal (SC)-FET performance of hexacene was 4.28 cm2 V−1 s−1 with an on/off ratio of 1 × 105 and threshold of 37 V, while pentacene SC-FET prepared in the same manner showed a hole mobility of 1.2 cm2 V−1 s−1 with an on/off ratio of 3 × 106 and a threshold at −7 V.

X-ray hexacene crystallographic analysis: (a) two adjacent hexacene molecules (the Oak Ridge Thermal Ellipsoid Plot (ORTEP) drawing), (b) hexacene molecule layer arrangement on a–b plane, and (c) hexacene arrays along a-axis. Transfer-integral computation distances: T 1 and T 2 transverse, P parallel, L longitudinal. (Reproduced with permission from [12]. Copyright 2012 Nature Publishing Group)
Reorganization energy (λ) is an important quantity associated with local electron–phonon coupling [16]. It is described in Marcus theory as energy consumption during molecular-level carrier transfer. High mobility can be attained by small reorganization energy of the hopping model [2]. Reorganized energy is divided into intramolecular (inner) and intermolecular (outer) types. In a solution, the outer contribution from surroundings is greater than the inner one. In molecular crystals, intramolecular reorganization energy ranges from 100 to 200 meV, while the intermolecular type is <10 meV. With π-expanded and delocalized molecules, reorganization energy tends to be small because the generated charge is delocalized in the molecule. Hexacene reorganization energy (λ +) is much smaller than smaller acenes (Table 9.1). Fullerene has a 3D spherical or rugby ball-type shape. The reorganization energy is small owing to the spherical-surface delocalized electronic structure. This is an important reason why fullerene derivatives are commonly used for OPV n-materials.
9.3.3 Polarization Energy
Polarization energy is charge energetic stabilization through interaction with the electrostatic environment surrounding the charged molecule in the crystal. Lichtenberger et al. measured high-resolution gas-phase and solid-phase ultraviolet photoelectron spectroscopy (UPS) of pentacene and TIPS-PEN to obtain intra- and intermolecular electronic effects (Fig. 9.9) [17]. By gas-phase UPS measurement, ionization energy at the pentacene single-molecule level and TIPS-PEN can be obtained. In the solid phase, the cation is stabilized by polarizing the surrounding medium, and ionization energies are lowered from the gas phase to solid phase. Solid-phase pentacene ionization energy is lowered by 1.7 eV from the gas phase; that of TIPS-PEN is lowered by 0.4 eV from the gas phase to the solid phase. The pentacene ionization order and TIPS-PEN are reversed in these phases.

He I UPS: first ionization energy bands of pentacene (red) and TIPS-PEN (black) (solid lines, gas phase; dotted lines, solid phase). (Adapted with permission from [17]. Copyright 2010 American Chemical Society)
A larger polarization of the surrounding medium leads to a larger intermolecular reorganization energy. Ryno et al. described the relationship of polarization energy to crystal structure [18]. Figure 9.10 shows the magnitudes of the dipole moments induced by a positive charge on the central molecule of five-molecule clusters for pentacene (herringbone packing) and TIPS-PEN (brickwork packing). A slight asymmetry in induced dipole moments (0.063 D vs. 0.059 D) is evident in the pentacene herringbone packing configuration, while the TIPS-PEN brickwork configuration shows a larger induced dipole (0.21 D) moment with no asymmetry. These differences indicate that induced dipole moments are very important to charge carrier stabilization in the brickwork-packed TIPS-pentacene. These results elucidate those of Fig. 9.9, where the TIPS-PEN brickwork configuration shows a fundamental change in the quadrupole and induced dipole interactions, resulting in smaller bulk polarization energy compared to pentacene.

Induced dipoles on the nearest neighbours of a positively charged pentacene (a) and TIPS-PEN (b), respectively, determined by the parameterised atomic multipole optimized energetics for biological applications (AMOEBA) force field. Dark red molecules in (a) have induced dipoles of 0.059 D; those in light red have induced dipoles of 0.063 D. All nearest neighbours in (b) have induced dipoles of 0.214 D. (Adapted with permission from [18]. Copyright 2014 American Chemical Society)
9.4 Deposition of Crystalline Structure by a Solution Process
Charge carrier mobility of organic semiconductors is influenced by the film crystal structure, as described in Sect. 9.3. Fundamental research on the large single-crystal (SC) device is important; development of a preparation method of a flat, regular crystalline film by a solution process is critical. In this section, approaches to preparing SC-FET devices by a solution process are outlined.
9.4.1 Single-Crystal FET by a Solution Process
The FET device size has decreased owing to technological advancements. A smaller FET enables more sophisticated devices. To address this size demand, SC-FET has been studied because the single-crystalline device has good charge transport performance (20–40 cm2 V−1 s−1) with few grain boundaries compared to polycrystalline film [19]. To realize solution-processed SC-FETs, Briseno et al. patterned organic SC-FET arrays [20]. Figure 9.11 shows the preparation of patterned SC-FET devices, as well as scanning electron microscope (SEM) images of patterned pentacene, rubrene, and C60 arrays. Rubrene- and pentacene-based transistor arrays yielded μ = ∼0.6 ± 0.5 cm2 V−1 s−1 with I ON/I OFF > 107 and μ = 0.3 cm2 V−1 s−1 with I ON/I OFF > 105, respectively. Devices from patterning n-channel materials, C60 and tetracyanoquinodimethane (TCNQ), exhibited mobilities of 0.03 and 10−4 cm2 V−1 s−1, respectively.

(a) Procedure for growing organic single crystals on substrates patterned by micro-contact printing, which uses polydimethylsiloxane (PDMS) stamps with relief features inked with thick octadecyltriethoxysilane (OTS) film and then pressed onto the substrates. To grow the patterned single crystals, the patterned substrate is placed in a glass tube with the organic source material, vacuum-sealed (0.38 mm Hg), and placed in a temperature gradient furnace tube. (b–d) Patterned single-crystal arrays of different organic semiconductor materials. Dotted squares denote the size and location of the OTS-stamped domain. The organic material molecular structure is shown beside the image of its single-crystal array. (b) Pentacene (SEM, 4 × 4-μm stamped domain size), (c) rubrene (SEM, 25 × 25 μm), and (d) C60 (optical micrograph, 8 × 8 μm). (Reproduced with permission from [20]. Copyright 2006 Nature Publishing Group)
9.4.2 TIPS-PEN
Another strategy for solution-processed SC-FET is introducing substituents to improve compound solubility in the organic solvents. TIPS-PEN was reported as a soluble pentacene derivative by Anthony in 2001 [21]. The performance of field-effect mobility of TIPS-PEN fabricated by thermal deposition was 0.4 cm2 V−1 s−1, [22]. The BGBC FET prepared by blading TIPS-PEN toluene solution and evaporating solvent produced a 0.17 cm2 V−1 s−1 hole mobility with I ON/I OFF of 105 [23].
Kim et al. produced 1D single-crystalline micro-ribbons through TIPS-PEN self-assembly by injecting a minimum volume of TIPS-PEN concentrated toluene solution in acetonitrile [24]. The typical micro-ribbon dimensions are as follows: 100–600 nm (height), 4–13 μm (width), and 40–800 μm (length). The average field-effect mobility (0.75 cm2 V−1 s−1) and I ON/I OFF (ca. 105) were obtained for 18 devices from 4 different batches. The highest TIPS-PEN FET mobility was 1.42 cm2 V−1 s−1.
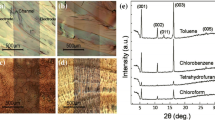
In 2007, Park et al. compared carrier mobilities of simple bottom-contact TIPS-PEN FETs by drop casting, dip coating, and spin depositing [25]. X-ray diffraction analysis showed similar peak patterns of a well-organized molecular structure with vertical intermolecular spacing of 16.8 Å. However, the peak intensity suggested the drop-cast films had the best ordering, while the spin-cast film had the worst. The improved molecular ordering and device performance related to the film formation speed. The device with the drop-cast film prepared from 1 wt% toluene solution (b.p. 110.6 °C) had FET mobilities of 0.2–1.8 cm2 V−1 s−1 (73 devices, average 0.65 cm2 V−1 s−1) with V th of 0–10 V (3.4 V average). The dip-coated film from 1 wt% chlorobenzene solution was 0.1–0.6 cm2 V−1 s−1, and the spin-cast film from 0.2 wt% film was 0.05–0.2 cm2 V−1 s−1.
Giri et al. reported a solvent shearing method to control the TIPS-PEN π–π stacking distance from 3.33 to 3.08 Å by changing the film-preparation shearing speed [26]. The film lattice strain correlated with the nearest-neighbour charge transfer integral change from 11.7 meV to −36.9 meV. The hole carrier mobility was 0.8 cm2 V−1 s−1 for unstrained film and 4.6 cm2 V−1 s−1 for strained film.
For solution-process enlargement of TIPS-PEN single crystal, Diao et al. used a fluid-enhanced crystal engineering method (Fig. 9.12) [27]. By controlling film thickness and solvent, nucleation and crystal growth was controlled, resulting in preparation of millimetre-wide, centimetre-long enlargement of TIPS-PEN single-crystalline domains. By excellent domain alignment and omitting grain boundaries, a 11 cm2 V−1 s−1 maximum mobility was attained.

(a) Solution shearing using a micropillar-patterned blade (micropillars are not drawn to scale; arrow, shearing direction). (b) Micropillar-patterned blade (SEM): top view of micropillars (35 μm wide; 42 μm high) under optical microscope (inset). (c) Simulated fluid flow streamlines around micropillars (arrow, flow direction); colour coding indicates velocity scale (mm s−1) from 0 (deep blue) to 1.3 mm s−1 (dark red). (d–f) Cross-polarized optical micrograph of TIPS-PEN film coated from mesitylene solution with (d, right; f) and without micropillars (d, left; e) at a 0.6 mm s−1 shearing speed. (Reproduced with permission from [27]. Copyright 2013 Nature Publishing Group)
9.4.3 Thioacene Derivatives
Takimiya et al. controlled the crystalline structure by adding alkyl groups on the thienoacene derivatives. Alkyl chains influence the crystalline-film packing motif via the van der Waals interaction between alkyl chains. They are soluble in organic solvents; hence, various solution processes were developed. In 2007, Eibata et al. prepared 2,7-dialkyl-substituted [1]benzothieno[3,2-b][1]-benzothiophene (Cn-BTBT: n = 5∼14) and compared the solubility, interlayer spacing (d-spacing), and FET performance (Fig. 9.13) [28, 29]. The spin-coated film showed (00 l) reflections in the in-plane XRD pattern, indicating the spun-cast film of Cn-BTBT yielded an end-on orientation on the substrates. The best performance of the spun-cast film was obtained for C13-BTBT film as μ = 1.20–2.75 cm2 V−1 s−1. This C8-BTBT crystalline film performance was improved by preparing a terrace and step structure in micrometre scale using molecule self-assembly with edge–casting method. The FET mobility improved as high as 5 cm2 V−1 s−1. OFET mass production with an average mobility of 3.5 cm2 V−1 s−1 at specified positions on substrates were possible with this method [30, 31].

Minemawari et al. achieved inkjet printing of SC films of C8-BTBT. The anti-solvent ink (pure anhydrous dimethylformamide (DMF)) was first printed and then overprinted with the solution ink (C8-BTBT solution in dichlorobenzene (DCB)), yielding TCTG thin-film transistors with average carrier mobility as high as 16.4 cm2 V−1 s−1 (31.3 cm2 V−1 s−1 as maximum) (Fig. 9.14) [32]. Phase separation between organic semiconducting molecules and insulating polymers caused by the difference in the surface energies have also been utilized for preparing highly ordered single crystals for SC-FETs. C8-BTBT single crystals have been prepared from polymer (poly(methyl methacrylate) (PMMA) or polystyrene (PS)) blends by solvent annealing, affording SC-FETs showing high mobility (9.1 cm2 V−1 s−1) and band-like transport behaviours [33].

(a) Inkjet printing process of SC films of C8-BTBT. Anti-solvent ink (A) is the first inkjet-printed (Step 1). Solution ink (B) is sequentially overprinted to form intermixed droplets confined to a predefined area (Step 2). Semiconducting thin films grown at droplet liquid–air interfaces (Step 3). Solvent fully evaporates (Step 4). (b) Micrographs of a 20 × 7 array of inkjet-printed C8-BTBT single-crystal thin films. (c) Film crossed-Nicol polarized micrographs. (d) Expanded micrograph of the film with stripes caused by molecular-layer steps. (e) Atomic-force microscopy image and height profile (bottom) showing the step-and-terrace structure on the film surfaces. (Reproduced with permission from [32]. Copyright 2011 Nature Publishing Group)
Dinaphtho[2,3-b:2′,3′-f]thieno[3,2-b]thiophene (DNTT) showed herringbone packing in a single crystal (Fig. 9.13). Non-substituted DNTT top-contact FET was prepared by vacuum deposition on octyltrichlorosilane (OTS)-treated substrate at T sub = 60 °C to give 2.0 cm2 V−1 s−1 and I ON/I OFF of 107 [34]. Alkyl-substituted Cn-DNTTs were not adequately soluble for the solution process, in contrast to Cn-BTBT. However, vapour-deposited C10-DNTT yielded a high crystalline film with FET mobility close to 8.0 cm2 V−1 s−1 [35]. C10-DNTT was used for patterning solution-crystallized BGTC-OFET. C10-DNTT in hot o-dichlorobenzene (100 °C) was set and gradually dried with crystal growth, and 7 cm2 V−1 s−1 average mobility was obtained.
In 2013, V-shaped DNT-V and its alkylated compounds were reported by Okamoto and Takeya et al. (Fig. 9.13) [36]. Depending on the alkylated position and the alkyl chain length, transfer integral changed, and the mobility in vapour-deposited polycrystals and solution-processed single crystals changed. The best performance of C6-DNT-VW single crystal was 9.5 cm2 V−1 s−1 [36].
DNDBT was reported in 2014 with a high carrier mobility of 16 cm2 V−1 s−1 by edge–casting method (Fig. 9.13) [37]. All solution-processed DNDBT SC-FET with a mobility of 6.6 cm2 V−1 s−1 was reported using POMIRAN™ substrate and EPRIMA™AL insulator [38].
Dithieno[3,2-b:2′,3′-d]thiophene derivatives (DTT-8, DTT-12, DTT-NP8, and DTT-BT8) as high-performance p-type organic semiconductors were reported in 2013 by Yasuda and Adachi et al. [39, 40]. The drop-casting deposition of DTT-8 in 1,2-dichlorobenzene solution followed by drying for 12 h gave micro-ribbons, extending uniformly in one dimension up to several hundreds of micrometres or even longer, with widths ranging from 4 to 50 mm and thicknesses of 100–300 nm. The DTT-8 molecules form a nanosegregated lamellar structure consisting of alternately stacked π-conjugated cores and aliphatic chains along the a-axis. In each layer, the molecules are densely packed in a herringbone arrangement, which is cooperatively reinforced by intermolecular S•••S and S•••C(π) interactions. The hole mobility (μ) of the DTT-8 micro-ribbon was found to reach 10.2 cm2 V−1 s−1 with a high on/off current ratio on the order of 107.
9.5 Precursor Approach for Field-Effect Transistors
Aromatic semiconducting materials without substituents are often low soluble in common organic solvents. Therefore, it is difficult to deposit them on the substrate by a solution process, except for some examples described in Sect. 9.4. In the so-called precursor approach, well-soluble, stable precursor compounds are used as starting materials, and the precursors are quantitatively converted into target materials with released leaving groups in gas phase, solution, film, powders, and crystals by applying external stimuli, such as heat or light. The polyacetylene precursor was first reported in 1980, then tert-butoxycarbonyl (Boc)-DPP, a latent pigment, was reported in 1997 (Fig. 9.15) [41, 42].

In synthetic terms, leaving groups improve precursor solubility; thus, purification and derivatisation of insoluble and/or unstable compounds becomes possible. After precursor purification, the final conversion step yields pure target compounds without further purification. The precursor approach can be applied to insoluble-crystalline-film deposition and morphology control in a solution process. FET and OPV performances via the precursor approach are now addressed [43,44,45].
A typical fabrication of the BGTC-OFET device with a precursor approach is shown in Fig. 9.16. After precursor-solution spin-coating, the precursor film is annealed or photo-irradiated to convert the precursor into the target semiconductor materials in situ. The film morphology can be controlled by film-preparation conditions: a selection of solvent to dissolve the precursor, precursor-solution concentration, spin-coating rotation rate and time, substrate temperature during spin-coating and post-annealing, solvent-annealing condition, etc. The polycrystalline film enables preparation of stable devices compared to amorphous film.

Device fabrication of BGTC-FET by the precursor approach
9.5.1 Pentacene Precursors
Several thermally convertible pentacene precursors were prepared by adding leaving groups (dienophile) to pentacene (diene) by the Diels–Alder reaction. This reaction is reversible. The precursor can be quantitatively reconverted to pentacene by heating in film. FET performance of pentacene film prepared by the precursor approach is shown in Fig. 9.17a [46,47,48,49,50,51]. Yamada et al. reported in 2005 6,13–dihydro-6,13-ethanopentacene-15,16-dione (α-diketone precursor: PDK) as a photo-convertible precursor of pentacene (Fig. 9.17b) [52, 53]. PDK is thermally stable over 300 °C but is converted to pentacene by 460 nm irradiation or UV light. This reaction was used for synthesizing substituted pentacene and higher acenes (hexacene, heptacene, octacene, nonacene) by Yamada et al. [54, 55], Neckers et al. [56,57,58,59], and Bettinger et al. [60,61,62].

Acene FET performance using a photo-precursor approach was studied [63,64,65,66]. Pentacene using PDK achieved a hole mobility of 0.86 cm2 V−1 s−1, V th = −1.4 V, and I ON/I OFF = 4.3 × 106 [67]. Photo-converted-film high FET mobility was attributed to continuously connected grain boundaries from partial crystallinity obtained by deposition condition optimisation (1% addition of high boiling point solvent to spin-coating solvent, irradiation intensity and duration, and substrate temperature during irradiation).
Watanabe et al. reported a pentacene monoketone precursor (CO-PEN) that extrudes a CO unit upon heating at 150 °C or UV irradiation (Fig. 9.17c) [68]. CO-PEN is unsuitable for FET device fabrication by the precursor approach because it reacts from heating and UV irradiation. Nevertheless, Watanabe et al. prepared pure hexacene single crystal by direct crystal growth on OcTS/SiO2/silicon substrate by heating the precursor in a nitrogen atmosphere using the physical vapour-transport (PVT) method (Sect. 3.2, Fig. 9.17d) [69]. Table 9.1 outlines the FET performance.
9.5.2 DNTT Precursors
DNTT is a promising material for use in organic semiconductors because DNTT exhibits high carrier mobility and heat resistance as a result of its molecule packing structure and high ionization potential. Since DNTT has a very low solubility in organic solvents, a soluble DNTT precursor was developed [70]. Soeda et al. reported a thermal reaction in ionic liquid to produce DNTT single crystals as large as 200 μm square with a thickness of 50–200 nm hole mobility as high as 2.4 cm2 V−1 s−1 in the saturation region, which is comparable to recently reported results for DNTT single-crystal transistors based on vapour-grown crystals (Fig. 9.18) [71]. It was a comparable value with the pentacene OFET prepared with this method: a hole mobility as high as 2.1 cm2 V−1 s−1 in the saturation region, and 0.9 cm2 V−1 s−1 in the linear region. This method yields a ‘single crystal’ phase, the most thermodynamically stable structure below 190 °C for pentacene.

Thermal reaction of DNTT precursor
High-performance short channel (20 μm) OTFTs with a maximum mobility of 4.7 cm2 V−1 s−1 were fabricated by solution process using the DNTT precursor/PS blend, followed by annealing at 200 °C for 10 min [72]. Changing the stereoisomer ratio of the DNTT precursors in the blend, in combination with the surface treatment of substrates, can control the position of the DNTT layer and the crystal size after the thermal conversion of the precursor to DNTT [73]. By using solution phosphorous-doped Si nanoparticle floating gate in combination with DNTT precursor/PS blend, the solution-processed transistor memory was attained [74].
9.5.3 Tetrabenzoporphyrin
Tetrabenzoporphyrin (BP) is a p-type organic semiconductor. Because of BP insolubility in common organic solvents, soluble BP precursors were developed for BP synthesis [43, 75,76,77]. Bicyclo[2.2.2]octadiene-fused porphyrin (CP) can be quantitatively converted to BP by releasing four ethylene equivalents as a solid and in film by heating over 150 °C (Fig. 9.19). With the quantitative conversion from CP to BP, pure BP can be obtained from purified CP without further purification. The amorphous spun-cast film of CP is changed to polycrystalline BP film during thermal conversion. BP film morphology and grain size can be controlled by the annealing temperature and time [78]. Okujima et al. reported more soluble precursors (MeCP) that release four isoprene equivalents by heating [77].

The BP packing structure of single crystals is the herringbone motif (Fig. 9.20) [80, 81]. Aramaki et al. reported in 2004 a BP FET by the precursor approach with 0.017 cm2 V−1 s−1 mobility [78] and Noguchi et al. in 2010 with 0.070 cm2 V−1 s−1 [81]. The mobility was better than that prepared by BP direct vacuum deposition (10−4 to 10−8 cm2 V−1 s−1 order). The annealing temperature and presence of an underlying BP layer affected the phase-transition behaviour. Grain size was determined by the balance between nucleation and growth rate. A higher annealing temperature tended to increase them. High growth rates caused rough surfaces. At 150 °C, although the nucleation rate was low, the grains had adequate time to grow up to 5–8 μm with smooth surfaces. At 210 °C, the grains were small and the surface was rough because many nucleates arose [81].

BP herringbone packing structure with (a) π–π stacking distance and (b) transfer integrals. Rewritten using the crystallographic information file data of [80]
Metalation has a primary influence on the BP electronic structure and film morphologies [82], although the framework of the porphyrin is the same. Kanicki et al. and Aramaki et al. reported FET performances of polycrystalline films of BP and its metal complexes (Ni, Cu, Zn) prepared by the thermal precursor approach [43, 78, 83,84,85,86]. The limiting factors in electronic devices fabricated from polycrystalline organic semiconductors are the crystalline grain size and grain boundary density. However, the polycrystalline CuBP thin film has crystalline domains of approximately 20 μm wide (Fig. 9.21) [86]. This was confirmed by the CuBP parallel-aligned nanorods of 55 nm wide, 300 nm long, and 100 nm tall. This parallel nanorod orientation was evident with a higher coverage density. CuBP OFETs demonstrated hole mobilities typically on the order of 0.1 cm2 V−1 s−1, V th around 5 V, I ON/I OFF near 104. NiBP showed a similar rod with an average rod height of 110 nm, width of 200 nm, and lengths up to 2 μm [84]. Close to the gate insulator surface, or when there was a low density of molecules, NiBP rods aligned parallel to the interface. These rod piles with random orientations in thicker film produced rough surfaces. The OFETs demonstrated hole mobilities typically on the order of 0.22 cm2 V−1 s−1, V th around −13 V, I ON/I OFF near 103.

Polarized optical micrograph of a continuous, spun-cast CuBP thin film on thermally oxidized c-Si. The electrodes shown in the figure are 20 μm wide. Colour variations in the film indicate individual nanorod domains. (Reproduced with permission from [86]. Copyright 2007 Elsevier)
BP can be deposited on pre-patterned trenches for precise control and placement of long- and short-range ordering of the organic semiconductor [85]. Shea et al. prepared BP film from CP on the substrates pre-patterned by photoresisting. The best dimensions observed for the preferred molecular alignment were obtained for substrates with widths of 100 nm, depths of 40 nm, and a nanogroove periodicity of 460 nm. Grooves with smaller periodicity appeared to prevent nucleation within the trenches; crystals instead randomly formed on the top surfaces of the grooves. The height should be adequately large to induce a molecular orientation yet sufficiently small to prevent film roughness. A charge carrier field-effect mobility at V GS = −40 V of 2.0 × 10−3 cm2 V−1 s−1 was extracted for trenches parallel to L; for trenches aligned perpendicular to L, μ = 2.5 × 10−4 cm2 V−1 s−1.
9.5.4 TMS-BP and TIPS-BP
As mentioned above, TIPS-PEN is a good semiconducting material with 1D or 2D π-stacking, while pentacene shows a herringbone packing structure. Introducing triisopropylsilylethyny groups to pentacene affects the packing structure in a single-crystal and solution-processed nanocrystalline film. Yamada et al. thus prepared 5,15-bistrimethylsilylethynybenzoporphyrin (TMS-BP) and 5,15-bistriisopropylsilylethynybenzoporphyrin (TIPS-BP) [87,88,89,90]. TMS-BP and TIPS-BP X-ray single-crystal structures are shown in Fig. 9.22. TMS-BP shows a herringbone motif. The π–π stacking between BP frameworks is 3.21 Å.

TMS-BP and TIPS-BP molecular and packing structures with transfer integrals of TMS-BP herringbone (a), 1D-stacked (b), and TISP-BP 2D-stacked (brickwork) structures (c). (Adapted with permission from [90] Copyright 2015 World Scientific Publishing Co. and [91] Copyright 2017 American Chemical Society)
Takahashi et al. found that TIPS-H2BP has two different packing structures in the single-crystalline state [91]. The first polymorph consists of a 1D extended columnar π-stacking motif (Fig. 9.22b). The second polymorph is arranged in a brickwork motif; π-stacking extends in 2D (Fig. 9.22c). The charge transfer integral based on the TMS-BP and TIPS-BP single-crystal structure is shown in Fig. 9.22. The calculated transfer integral (t) in the TMS-BP crystal is higher than those of TIPS-BP.
TMS-BP and TIPS-BP BGTC-FET was fabricated by the precursor approach. The best hole mobility, μ = 0.11 cm2 V−1 s−1, was obtained for TMS-H2BP, as shown in Table 9.2 [90]. As predicted by the transfer integrals, TMS-BP showed better μ than TIPS-BP.
TIPS-BP is soluble in organic solvents; thus, an intrinsic TIPS-BP solution process is possible. Takahashi et al. compared FET performances of TIPS-BP films prepared by drop casting and dip coating. The performance and packing structure were strongly affected by the use of a suitable deposition method (dip coating), proper solvent (dichloromethane/chloroform, 9:1), and an appropriate self-assembled monolayer (SAM) material as the substrate surfactant [92, 93]. Using CH2Cl2 or a mixture of CH2Cl2 and CHCl3 as solvents for dip coating, the TIPS-BP films with a 2D π-stack-rich motif were obtained. The maximum field-effect hole mobility of 1.1 cm2 V−1 s−1 was achieved with 12-cyclohexyldodecylphosphonic acid (CDPA) as SAM (Table 9.3). This value is higher by orders of magnitude than the record mobility for pristine freebase BP (0.070 cm2 V−1 s−1) [81].
9.5.5 Other Precursors for OFET
Murphy et al. reported in 2004 a sexithiophene (T6) solution-processed OFET using thermolysis of diester-functionalized oligothiophene derivative (Fig. 9.23) [94, 95]. Near-edge X-ray absorption fine structure spectroscopy tracked the thermal processing effects on the oligothiophene precursor film chemistry and structure [96]. Thermolysis began at 150 °C for T6. The mobility increased from 150 to 200 °C. During conversion, vertical molecule orientation on substrate occurred.

Oligothiophene T4–T7 thermal reaction and FET performance by the precursor approach [96]
Hirao et al. prepared thermal precursors (Pc-pre and Nc-pre) of phthalocyanine and naphthalocyanine and deposited BGTC-FET (Fig. 9.24) [97, 98], marking the first examples of solution-processed FETs of phthalocyanines.

9.6 Organic Photovoltaics
The precursor approach can be used for OPV devices. In Fig. 9.25, the preparation process of p-n-, BHJ-, and p-i-n-type OPVs by the thermal precursor approach is described. Precursors of p-materials have been mainly developed to date, while few precursors of n-materials have been reported. BHJ layer can be prepared by spin coating a mixed solution of a p-material precursor and a soluble n-material, such as PCBM. It was converted to a p- and n-material mixed film by heating or irradiating. The layered structure was fabricated by repeating spin coating and converting. The film morphology and surface roughness can be controlled by the condition of the conversion from the precursors to the target materials, thermal or solvent annealing, etc.

Fabrication of p-n, BHJ, and p-i-n OPV devices by the thermal precursor approach using BP for the p-material and PCBM for the n-material. For the photo-precursors, photo-irradiation instead of heating was applied for the conversion
9.6.1 Solution-Processed p-n Structure Using BP Precursors
BP has been studied as a p-material for solution-processed OPV. As a counterpart of BP, PCBM, a common acceptor, was the first choice. However, the OPV performances of p-n junction solar cells were not satisfactory, as shown in Table 9.4 [99]. With the combination of BP and PCBM (run 1 in Table 9.4), V OC value is small owing to the small energy gap between the highest occupied molecular orbital (HOMO) energy level of BP and the lowest unoccupied molecular orbital (LUMO) energy level of PCBM. J SC is not adequately large because BP cannot absorb sufficient light at 500–600 nm and over 700 nm. Owing to the high crystallinity of BP, the precursor approach yields polycrystalline BP film with grain sizes of 100 nm, which is larger than the average exciton diffusion length in organic materials (Fig. 9.30a in Sect. 9.6.3) [100,101,102]. In spite of the drawbacks, various attempts have been tried to use BP derivatives for p-material in OPVs.
Guide et al. reported the effect of copper metalation of BP (CuBP) donor material in a p-n-type OPV combined with PCBM for the acceptor material (run 2 in Table 9.4) [99]. Compared with the BP/PCBM system, the CuBP/PCBM system has a lower J SC, and, in turn, a lower PCE. The authors investigated the cause of the difference between the two materials. The overall average current under the same applied bias of +0.01 V (relative to the substrate) was an order of magnitude higher for CuBP films (260 ± 100 pA) than for BP films (14 ± 7 pA) of very similar thicknesses. The charge carrier mobility of CuBP was sufficiently high both in and out of the plane. These data suggest that the charge transport is not a limiting factor in CuBP devices. Despite similar light absorption characteristics, the external quantum efficiency (EQE) showed that the conversion of incident photons to collected electrons was extremely inefficient precisely in the spectral region where CuBP absorption dominated (650–690 nm). Differences in the dependence of EQE and J SC on the donor film thickness for bilayer OPVs with the two different donors suggested a short effective exciton diffusion length (L D) in CuBP as the factor limiting solar cell performance. That is, an effective L D of 14.6 ± 2.2 nm for BP and 5.5 ± 1.1 nm for PCBM were calculated for BP/PCBM system, while an effective L D of 2.0 ± 0.7 nm for BP and 4.3 ± 1.5 nm for PCBM were calculated for CuBP/PCBM system.
9.6.2 Solution-Processed p-i-n Structure
The p-i-n device of BP and PCBM showed a comparable performance with the p-n structure as PCE = 2.0% owing to V OC = 0.55 V, J SC = 7.0 mA/cm, and FF = 0.51 (run 3 in Table 9.4) [100]. With bis(dimethylphenylsilylmethyl)[60]fullerenes (SIMEF) as an acceptor (Fig. 9.26), OPV performance was improved to PCE = 5.2% owing to V OC = 0.75 V; J SC = 10.5 mA/cm2; FF = 0.6 (run 4 in Table 9.4) [101]. The important difference between PCBM and SIMEF is the crystallization temperature: SIMEF crystallizes from an amorphous state at 149 °C; for PCBM, it occurs at 195 °C [100]. To deposit the i-layer, a homogeneous mixture (a typical weight ratio of 3:7) of CP and an acceptor SIMEF in chloroform/chlorobenzene is spin coated. CP is converted in situ to BP at 180 °C to form an interdigitated i-layer. The choice of 180 °C is crucial for conversion from CP to BP because SIMEF is crystalline at this temperature. In the i-layer, the desired interdigitated BHJ is achieved with the average size of the columns at 65 nm in height and 26 nm in diameter (Fig. 9.27a, b). This is comparable to the exciton diffusion length of common organic semiconducting separation and carrier transport. When PCBM is used in place of SIMEF, the BP crystal morphology in the i-layer is far less uniform in size than that in the SIMEF device. In addition, the BP crystals are rectangular (a usual bulk morphology of BP crystals) rather than round columnar (Fig. 9.27c). This difference is due to the difference of crystallization temperatures of SIMEF (149 °C) and PCBM (195 °C). BP crystal formation at 180 °C in the SIMEF device occurs in the matrix of SIMEF crystals, whereas it occurs in amorphous PCBM in the PCBM device.


SEM images of the (a) side view of p-i-n structure made from BP and SIMEF (glass/ITO/PEDOT-PSS/BP/BP-SIMEF/SIMEF), (b) column/canyon region of the p-i structure after toluene washing, and (c) side view of the toluene-washed BP/PCBM device, where rectangular BP crystals irregularly grow (Adapted with permission from [101]. Copyright 2009 American Chemical Society)
Tamura and Matsuo theoretically analysed the exciton diffusion length and charge mobility in BP and SIMEF crystals using density functional theory (DFT) and Fermi’s golden rule [103]. Because of the herringbone packing structure of the BP crystal, the value of the intermolecular electronic coupling of the hole transfer, V hole, and the exciton coupling, V XT, of the parallel H-aggregate were relatively large compared with other directions. Because the charge transfer coupling of the parallel H-aggregate was large, the electron exchange—i.e. Dexter transfer—significantly contributed to the exciton coupling. For other directions, the electron exchange was small. Thus, the Coulomb coupling, i.e. the Förster transfer, was dominant. Intermolecular electronic coupling for the electron transfers, V el, of SIMEF was also anisotropic because V el was affected by the steric hindrance of the functional groups in SIMEF. As a result, the electron transport was efficient only along a direction that did not cross the functional groups.
Nguyen et al. reported the nanoscale morphology and conductivity/photoconductivity of a recently developed OPV system based on BP and either PCBM or [6,6]-phenyl-C61-butyric acid n-butyl ester (PCBNB) using conducting atomic force microscopy (C-AFM) and photoconductive atomic force microscopy (PC-AFM) (runs 5 and 6 in Table 9.4 and Fig. 9.26) [104]. Although the published values for the (LUMO energies of both PCBM and PCBNB are nearly identical, the p-i-n device fabricated using PCBNB as an acceptor consistently exhibits higher PCEs than the device with PCBM (2.8% versus 1.5%) owing to an increase in V OC from 0.44 to 0.60 V; in J SC from 5.66 to 7.22 mA cm−2; and in FF from 0.61 to 0.64. The BP in the i-layer forms columnar structures with a diameter of approximately 70 nm; however, the slight difference in the alkyl chains of PCBM and PCBNB has a large impact on the film morphology, phase separation, and device performance. For devices with PCBM, unfavourable vertical morphology, large-phase separation, and thus lower PCE are observed. When PCBNB is used, a more homogeneous morphology, finer-phase separation, and higher PCE are observed.
Zhen et al. found a solid solution of 75:25 mixture of BP and CABP produced a higher PCE value than the devices using the single donor BP and CABP, respectively (Fig. 9.26) [105]. CABP has three important features for improving the PCE performance: structural similarity with BP to be intercalated in BP crystals, an acenaphthylene fragment that decreases the band gap, and two chlorine atoms that lower the HOMO energy level, which may otherwise not match well with that of BP. Different ratios of two soluble donor precursors (CP and CACP) were tested in combination with SIMEF2 for the i-layer of p-i structure OPV. Then a very smooth surface reminiscent of a molten solid was achieved by the combination of 75:25 mixture of BP and CABP in a SIMEF2 matrix (porphyrin: SIMEF2 = 3:7), shown by a low-landing-voltage high-resolution SEM (Fig. 9.28). These characteristic nanoscale morphologies occurred only when the CP:CACP mixture was thermolysed in the presence of SIMEF2 and the BP undercoating layer. The 100:0 or 0:100 BP:CABP showed a nano-columnar morphology of considerable regularity. The 75:25 mixture showed the highest PCE with the increase of J SC (runs 7–9 in Table 9.4). The most noticeable feature in EQE spectrum was the remarkable enhancement at 350–700 nm, where both BP and CABP were strongly absorbed (Soret bands at 440 and 420 nm, respectively). This enhanced EQE was attributed to the improvement of the internal quantum efficiency (IQE) rather than the absorption efficiency. Even though the HOMO (donor)-LUMO (acceptor) energy level differences were the smallest for the solid solution cell among the three cells (BP, BP/CABP, and CABP), the highest V OC was obtained. This is explained by the suppressed recombination current, J SO. CABP molecules intercalated in the BP crystal lattice induced partial disordering of the structure, which decreased J SO and hence increased V OC [106].

SEM images (a–f) for blended BP and CABP on the BP layer after removing SIMEF2 by washing with toluene. Top view images at the BP:CABP blend ratios of 100:0 (a), 75:25 (b), 50:50 (c), 25:75 (d), and 0:100 (e), and a cross-sectional view of the 75:25 blend (f). Scale bar, 200 nm (Reproduced with permission from [105]. Copyright 2015 American Chemical Society)
9.6.3 BP-C60 for BHJ Layer
Donor–acceptor (D–A) dyads have been widely studied to understand charge separation and recombination processes between donor and acceptor units. Various dyad compounds have also been examined for use as active materials in BHJ OPVs. This is because the direct connection of donor and acceptor components can be beneficial for forming a maximal D–A interface within a film, thereby providing high charge separation efficiency [107,108,109]. Tamura et al. reported a p-i-n-type OPV with a covalently linked BP-fullerene (D–A) compound for the i-layer, while BP was used for the p-layer, and PC61BM was used for the n-layer. It was compared with the p-n-type and BHJ-type solar cell (Table 9.5) [102, 110]. The comparison of flexible versus rigid D–A compounds (f-BP–C60 and r-BP–C60, respectively, in Fig. 9.29) as i-layer materials suggest that flexible f-BP–C60 is suitable for the i-layer on the highly crystalline BP layer because the BP film roughness is levelled by the f-BP–C60 and the device electrical properties are improved by the smooth interface (Fig. 9.30).


9.6.4 BP-DPP for BHJ
BP has a high photo-absorption coefficient of more than 105 M−1 cm−1 around 450–500 nm, where the solar light intensity is strongest. However, it has rather narrow absorption bands at Soret and Q bands (600–700 nm). It is transparent at 500–600 nm and over 700 nm. To overcome this drawback, DPP-linked BP with different alkyl chains on the DPP unit was synthesized to control the film morphology [111]. They prepared BHJ OPV of Cn-DPP-BP (n = 4, 6, 8, or 10, depending on the alkyl chain length) in combination with PCBM by the precursor approach because of the insolubility of Cn-DPP-BP in common organic solvents (Fig. 9.31). The PCE performance drastically changed in accordance with the alkyl chain length because of the drastic change of J SC (Table 9.6). The grazing-incidence wide-angle X-Ray diffraction (GIWAXD) measurements suggested that C4-DPP–BP in blended films tends to have larger amounts of crystallites derived from π–π stacking in the out-of-plane direction. This is more suitable for charge carrier transport of OPV than the C10-DPP–BP oriented to the edge against the substrate. In addition, PCBM tended to produce a more crystalline structure in combination with C4-DPP–BP than C10-DPP–BP.

Thermal conversion from Cn-DPP–CP to Cn-DPP–BP [111]
9.6.5 Photo-Precursor Approach
The photo-precursor approach was applied to OPV fabrication [112,113,114,115]. In this approach, in situ photoconversion reaction of precursors to the target materials is performed instead of thermal conversion in the previous sections (Fig. 9.32). When 2,6-dithienylanthracene (DTA) and a fullerene derivative PC71BM were used as the donor and acceptor, respectively, the best p-i-n OPV produced a higher photovoltaic efficiency than the corresponding p-n device by 24% and bulk-heterojunction device by 67% (run 1–3 in Table 9.7). A systematic comparison of different device types indicated that the neat p- and n-layers enhanced charge extraction and carrier injection and the blended i-layer improved charge photo-generation [114].

Furthermore, the prototype hetero p-i-n system was examined with 2,6-bis(5′-(2-ethylhexyl)-(2,2′-bithiophen)-5-yl)anthracene (EH-DBTA) for the i-layer and DTA for the p-layer (run 4). The resulting devices showed significantly enhanced photovoltaic performances compared to the homo p-i-n devices. The PCE enhancement was attributed to the better miscibility with PC71BM and higher photo-absorption capability inherent in EH-DBTA compared to DTA. This three-component system, employing the proper materials for each layer, was enabled by the precursor approach for the solution process.
Suzuki et al. designed the improved materials for the photo-precursor approach [115]. PhBADT is suitable for p-layer because of the transparency in a wide range of the visible region and forming three-dimensionally extended π–π contacts in the solid state, which is favourable in achieving effective hole-transport paths. AtD is suitable for i-layer because of the efficient absorption of the sunlight and good miscibility with PC71BM. Compared to BHJ structure (run 5), the p-in structure after solvent annealing afforded the 5.9% PCE (run 7), which was the highest PCE value reported for p-i-n-type OPVs prepared by solution process.
9.6.6 Other p-type Materials for OPV
Benzoporphycene (BPc) is a structural isomer of benzoporphyrin (BP) (Fig. 9.33) [116]. Owing to its lower symmetry compared to BP, the absorption of the Q-band region is stronger than in BP. Saeki et al. investigated BPc as a p-material for BHJ OPV combined with PCBM. The precursor CPc molecules were converted to BPc by annealing above 125 °C in the film. When the CPc/PCBM mixed film was annealed at 200 °C, a phase-separated structure composed of BPc and PCBM crystals was formed. In the presence of CPc/PCBM, PCBM played the role of both a matrix and an aggregation inhibitor of BPc under the thermal conversion process [117]. The process of spin coating of the CPc/PCBM solution and heating at 200 °C for 10 min was repeated five times to produce the multi-stacked film. The BPc homo-epitaxial crystallization was observed with a height of 100 nm, which was approximately five times the height of a single layer of BPc grains [118]. Thus, the precursor approach enabled the preparation of thick film by repeating the deposition process of spin coating and converting.

(a) Synthetic scheme of BPc from precursor CPc. (b) UV-vis absorption spectra of multi-stacked BPc/PCBM films. Inset: dependence of absorbance at 660 nm on the number of alternating spin-coating/annealing cycles. (c) Topological SEM image of a BPc/PCBM film containing five layers prepared at 200 °C after rinsing of PCBM. The long axis of the crystal in the upper layer is aligned to be parallel to that of the lower layer, as indicated by arrows. (d) Schematic representation of the homo-epitaxial growth of BPc. (e) Cross-sectional SEM image of a BPc/PCBM film with five layers after rinsing (Adapted with permission from [118]. Copyright 2013, Japan Society of Applied Physics)
A dialkoky-substituted copper Pc precursor, CuPc(OMe)2 was reported in 2011 (Fig. 9.34) [119]. The solution-processed OPV with thermal conversion of the precursors showed a solvent-dependent performance, which can be explained by the film morphology. The flat surface morphology obtained using a solvent with a low b.p., such as toluene or chloroform, resulted in a device with better photovoltaic performance because of a lower charge leakage (i.e. a higher FF) [120]. The best PCE performance of the p-n structure with mix-PCBM (a mixture of PC61BM and PC71BM) was 1.35%.

Chen et al. reported a solution-processed p-i-n OPV device with quinacridone (QA) and PC61BM using Boc–QA as a soluble precursor of QA [121]. The p-i-n trilayer structure PCE performance was 0.83%; it was 0.61 and 0.57% for the p-n bilayer and BHJ structures, respectively (Table 9.8). V OC of the p-in system was better than that in the BHJ system. It was achieved by reducing the electron leakage current through introduction of carrier blocking layers (QA and PC61BM) between the organic layers and electrodes.
9.6.7 Precursors for n-type Materials
A few precursors of n-materials have been reported to date. Lee et al. reported a thermal precursor C60-CpCO2R (R = hexyl, octyl and ethylhexyl groups), which can be converted to C60 by annealing at 120 °C (Fig. 9.35). BHJ OPV performance of P3HT/C60 using C60-CpCO2R as a precursor was improved more than that of direct spin coating of P3HT/C60. The best performance was obtained from P3HT/C60-CpCO2Hex at 120 °C annealing (PCE = 2%), which was twice that of the intrinsic P3HT/C60 device. An AFM image showed that as-prepared P3HT/C60 had micrometre-order grains, whereas the film from P3HT/C60-CpCO2Hex was smooth [122].

A thermal precursor of C60, C60(9MA)2, was reported in 2016 by Umeyama et al. (Fig. 9.35) [123]. The PCE value of the OPV devices based on PCDTBT:C60–9MA adducts improved from 0.69% to 3.51% by a factor of approximately five, which was associated with a significant enhancement of J SC and FF when the device was annealed at 140 °C. A factor that hampered the achievement of excellent PCE was the recovery of the p-conjugation and the consequently lowered LUMO levels of fullerenes by the thermal annealing.
Fullerene derivatives, N601 and N602, were quantitatively converted from diketone precursors, N601DK and N602 DK, by irradiation of 460 nm of light [124]. N601 film prepared by the photo-precursor approach showed comparable electron mobility with PCBM film. The PCE performance of BHJ OPV of P3HT/N601 was 2.43%, which of P3HT/PCBM was 2.16%.
9.7 Summary
In this chapter, solution-process procedures for single crystalline and polycrystalline films for organic semiconductor devices were summarized. The organic crystalline films have intrinsic potential for high-performance devices; however, the solution process was not easy on account of their high crystallinity, namely, the low solubility in common organic solvents. One breakthrough in addressing this problem was the development of soluble materials with high crystallinity. The soluble compounds with alkyl substituents were believed to have low electrical properties on account of the insulating substituents. Nevertheless, some materials were found to have good electrical properties with assistance from the substituents for a good packing motif from the van der Waals interaction. The soluble materials encouraged the progress of the deposition technique to attain a 102 cm2 V−1 s−1 order of mobilities. The precursor approach showed potential for improving device performance by controlling the deposition condition, such as the annealing temperature, photo-irradiation condition, solvents, post-annealing, solvent annealing, etc. Further development for new materials can advance organic electronic devices.
References
J.E. Anthony, Angew. Chem. Int. Ed. 47, 452 (2008)
K. Takimiya, S. Shinamura, I. Osaka, E. Miyazaki, Adv. Mater. 23, 4347 (2011)
J. Mei, Y. Diao, A.L. Appleton, L. Fang, Z. Bao, J. Am. Chem. Soc. 135, 6724 (2013)
V. Coropceanu, J. Cornil, D.A. da Silva Filho, Y. Olivier, R. Silbey, J.-L. Brédas, Chem. Rev. 107, 926 (2007)
C.W. Tang, Appl. Phys. Lett. 48, 183 (1986)
M. Hiramoto, H. Fujiwara, M. Yokoyama, Appl. Phys. Lett. 58, 1062 (1991)
G. Yu, J. Gao, J.C. Hummelen, F. Wudl, A.J. Heeger, Science 270, 1789 (1995)
J.-L. Brédas, D. Beljonne, V. Coropceanu, J. Cornil, Chem. Rev. 104, 4971 (2004)
V. Vohra, K. Kawashima, T. Kakara, T. Koganezawa, I. Osaka, K. Takimiya, H. Murata, Nat. Photonics 9, 403 (2015)
S. Blumstengel, S. Sadofev, C. Xu, J. Puls, F. Henneberger, Phys. Rev. Lett. 97, 237401 (2006)
J.E. Anthony, Chem. Rev. 106, 5028 (2006)
M. Watanabe, Y.J. Chang, S.-W. Liu, T.-H. Chao, K. Goto, M.M. Islam, C.-H. Yuan, Y.-T. Tao, T. Shinmyozu, T.J. Chow, Nat. Chem. 4, 574 (2012)
S.C. Capelli, A. Albinati, S.A. Mason, B.T.M. Willis, J. Phys. Chem. A 110, 11695 (2006)
C.P. Brock, J.D. Dunitz, Acta Cryst B46, 795 (1990)
D. Holmes, S. Kumaraswamy, A.J. Matzger, K.P.C. Vollhardt, Chem. Eur. J. 5, 3999 (1999)
V. Coropceanu, O. Kwon, B. Wex, B.R. Kaafarani, N.E. Gruhn, J.C. Durivage, D.C. Neckers, J.-L. Brédas, Chem. Eur. J. 12, 2073 (2006)
O.L. Griffith, J.E. Anthony, A.G. Jones, D.L. Lichtenberger, J. Am. Chem. Soc. 132, 580 (2010)
S.M. Ryno, C. Risko, J.-L. Brédas, J. Am. Chem. Soc. 136, 6421 (2014)
T. Hasegawa, J. Takeya, Sci. Technol. Adv. Mater. 10, 024314 (2009)
A.L. Briseno, S.C.B. Mannsfeld, M.M. Ling, S. Liu, R.J. Tseng, C. Reese, M.E. Roberts, Y. Yang, F. Wudl, Z. Bao, Nature 444, 913 (2006)
J.E. Anthony, J.S. Brooks, D.L. Eaton, S.R. Parkin, J. Am. Chem. Soc. 123, 9482 (2001)
C.D. Sheraw, T.N. Jackson, D.L. Eaton, J.E. Anthony, Adv. Mater. 15, 2009 (2003)
M.M. Payne, S.R. Parkin, J.E. Anthony, C.-C. Kuo, T.N. Jackson, J. Am. Chem. Soc. 127, 4986 (2005)
D.H. Kim, D.Y. Lee, H.S. Lee, W.H. Lee, Y.H. Kim, J.I. Han, K. Cho, Adv. Mater. 19, 678 (2007)
S.K. Park, T.N. Jackson, J.E. Anthony, D.A. Mourey, Appl. Phys. Lett. 91, 063514 (2007)
G. Giri, E. Verploegen, S.C.B. Mannsfeld, S. Atahan-Evrenk, D.H. Kim, S.Y. Lee, H.A. Becerril, A. Aspuru-Guzik, M.F. Toney, Z. Bao, Nature 480, 504 (2011)
Y. Diao, B.C.-K. Tee, G. Giri, J. Xu, D.H. Kim, H.A. Becerril, R.M. Stoltenberg, T.H. Lee, G. Xue, S.C.B. Mannsfeld, Z. Bao, Nat. Mater. 12, 665 (2013)
H. Ebata, T. Izawa, E. Miyazaki, K. Takimiya, M. Ikeda, H. Kuwabara, T. Yui, J. Am. Chem. Soc. 129, 15732 (2007)
T. Izawa, E. Miyazaki, K. Takimiya, Adv. Mater. 20, 3388 (2008)
T. Uemura, Y. Hirose, M. Uno, K. Takimiya, J. Takeya, Appl. Phys. Express 2, 111501 (2009)
J. Soeda, Y. Hirose, M. Yamagishi, A. Nakao, T. Uemura, K. Nakayama, M. Uno, Y. Nakazawa, K. Takimiya, J. Takeya, Adv. Mater. 23, 3309 (2011)
H. Minemawari, T. Yamada, H. Matsui, J. Tsutsumi, S. Haas, R. Chiba, R. Kumai, T. Hasegawa, Nature 475, 364 (2011)
C. Liu, T. Minari, X. Lu, A. Kumatani, K. Takimiya, K. Tsukagoshi, Adv. Mater. 23, 523 (2010)
T. Yamamoto, K. Takimiya, J. Am. Chem. Soc. 129, 2224 (2007)
M.J. Kang, I. Doi, H. Mori, E. Miyazaki, K. Takimiya, M. Ikeda, H. Kuwabara, Adv. Mater. 23, 1222 (2011)
T. Okamoto, C. Mitsui, M. Yamagishi, K. Nakahara, J. Soeda, Y. Hirose, K. Miwa, H. Sato, A. Yamano, T. Matsushita, T. Uemura, J. Takeya, Adv. Mater. 25, 6392 (2013)
C. Mitsui, T. Okamoto, M. Yamagishi, J. Tsurumi, K. Yoshimoto, K. Nakahara, J. Soeda, Y. Hirose, H. Sato, A. Yamano, T. Uemura, J. Takeya, Adv. Mater. 26, 4546 (2014)
S. Sakai, J. Soeda, R. Häusermann, H. Matsui, C. Mitsui, T. Okamoto, M. Ito, K. Hirose, T. Sekiguchi, T. Abe, M. Uno, J. Takeya, Org. Electron. 22, 1 (2015)
Y.S. Yang, T. Yasuda, H. Kakizoe, H. Mieno, H. Kino, Y. Tateyama, C. Adachi, Chem. Commun. 49, 6483 (2013)
H. Mieno, T. Yasuda, Y. Seok, C. Adachi, Chem. Lett. 43, 294 (2014)
J.H. Edwards, W.J. Feast, Polymer 21, 595 (1980)
J.S. Zambounis, Z. Hao, A. Iqbal, Nature 388, 131 (1997)
H. Yamada, T. Okujima, N. Ono, Chem. Commun. 2957 (2008)
M. Watanabe, K.-Y. Chen, Y.J. Chang, T.J. Chow, Acc. Chem. Res. 46, 1606 (2013)
M. Suzuki, T. Aotake, Y. Yamaguchi, N. Noguchi, H. Nakano, K.-I. Nakayama, H. Yamada, J. Photochem. Photobiol. C Photochem. Rev. 18, 50 (2014)
A.R. Brown, A. Pomp, D.M. De Leeuw, D.B.M. Klaassen, E.E. Havinga, P. Herwig, K. Müllen, J. Appl. Phys. 79, 2136 (1996)
P.T. Herwig, K. Müllen, Adv. Mater. 11, 480 (1999)
A. Afzali, C.D. Dimitrakopoulos, T.L. Breen, J. Am. Chem. Soc. 124, 8812 (2002)
A. Afzali, C.D. Dimitrakopoulos, T.O. Graham, Adv. Mater. 15, 2066 (2003)
A. Afzali, C.R. Kagan, G.P. Traub, Synth. Met. 155, 490 (2005)
G.S. Tulevski, Q. Miao, A. Afzali, T.O. Graham, C.R. Kagan, C. Nuckolls, J. Am. Chem. Soc. 128, 1788 (2006)
H. Uno, Y. Yamashita, M. Kikuchi, H. Watanabe, H. Yamada, T. Okujima, T. Ogawa, N. Ono, Tetrahedron Lett. 46, 1981 (2005)
H. Yamada, Y. Yamashita, M. Kikuchi, H. Watanabe, T. Okujima, H. Uno, T. Ogawa, K. Ohara, N. Ono, Chem. Eur. J. 11, 6212 (2005)
S. Katsuta, H. Yamada, T. Okujima, H. Uno, Tetrahedron Lett. 51, 1397 (2010)
S. Katsuta, H. Saeki, K. Tanaka, Y. Murai, D. Kuzuhara, M. Misaki, N. Aratani, S. Masuo, Y. Ueda, H. Yamada, J. Mater. Chem. C 2, 986 (2014)
R. Mondal, B.K. Shah, D.C. Neckers, J. Photochem. Photobiol. A Chem. 192, 36 (2007)
R. Mondal, R.M. Adhikari, B.K. Shah, D.C. Neckers, Org. Lett. 9, 2505 (2007)
R. Mondal, A.N. Okhrimenko, B.K. Shah, D.C. Neckers, J. Phys. Chem. B 112, 11 (2008)
R. Mondal, C. Tönshoff, D. Khon, D.C. Neckers, H.F. Bettinger, J. Am. Chem. Soc. 131, 14281 (2009)
C. Tönshoff, H.F. Bettinger, Angew. Chem. Int. Ed. 49, 4125 (2010)
C. Tönshoff, H.F. Bettinger, Chem. Eur. J. 18, 1789 (2012)
H.F. Bettinger, R. Mondal, M. Krasowska, D.C. Neckers, J. Org. Chem. 78, 1851 (2013)
H. Yamada, C. Ohashi, T. Aotake, S. Katsuta, Y. Honsho, H. Kawano, T. Okujima, H. Uno, N. Ono, S. Seki, K.-I. Nakayama, Chem. Commun. 48, 11136 (2012)
C. Ohashi, H. Yamada, K. Nakayama, Mol. Cryst. Liq. Cryst. 580, 103 (2013)
S. Masuo, K. Tanaka, M. Oe, H. Yamada, Phys. Chem. Chem. Phys. 16, 13483 (2014)
C. Quinton, M. Suzuki, Y. Kaneshige, Y. Tatenaka, C. Katagiri, Y. Yamaguchi, D. Kuzuhara, N. Aratani, K.-I. Nakayama, H. Yamada, J. Mater. Chem. C 3, 5995 (2015)
K.-I. Nakayama, C. Ohashi, Y. Oikawa, T. Motoyama, H. Yamada, J. Mater. Chem. C 1, 6244 (2013)
K.-Y. Chen, H.-H. Hsieh, C.-C. Wu, J.-J. Hwang, T.J. Chow, Chem. Commun., 1065 (2007)
R.A. Laudise, C. Kloc, P.G. Si, pkins, and T. Siegrist. J. Crysst. Growth 187, 449 (1998)
J. Soeda, T. Okamoto, A. Hamaguchi, Y. Ikeda, H. Sato, A. Yamano, J. Takeya, Org. Electron. 14, 1211 (2013)
M. Uno, Y. Tominari, M. Yamagishi, I. Doi, E. Miyazaki, K. Takimiya, J. Takeya, Appl. Phys. Lett. 94, 223308 (2009)
Y. Kimura, T. Nagase, T. Kobayashi, A. Hamaguchi, Y. Ikeda, T. Shiro, K. Takimiya, H. Naito, Adv. Mater. 27, 727 (2014)
A. Hamaguchi, T. Negishi, Y. Kimura, Y. Ikeda, K. Takimiya, S.Z. Bisri, Y. Iwasa, T. Shiro, Adv. Mater. 27, 6606 (2015)
W. Li, M. Yi, H. Ling, F. Guo, T. Wang, T. Yang, L. Xie, W. Huang, J. Phys. D: Appl. Phys. 49, 125104 (2016)
C.M.B. Carvalho, T.J. Brocksom, K.T. de Oliveira, Chem. Soc. Rev. 42, 3302 (2013)
S.-D. Jeong, B. Min, S.Y. Cho, C. Lee, B.K. Park, K.-S. An, J. Lim, J. Org. Chem. 77, 8329 (2012)
T. Okujima, Y. Hashimoto, G. Jin, H. Yamada, H. Uno, N. Ono, Tetrahedron 64, 2405 (2008)
S. Aramaki, Y. Sakai, N. Ono, Appl. Phys. Lett. 84, 2085 (2004)
S. Ito, T. Murashima, N. Ono, H. Uno, Chem. Commun., 1661 (1998)
S. Aramaki, J. Mizuguchi, Acta Cryst B59, o1556 (2003)
N. Noguchi, S. Junwei, H. Asatani, M. Matsuoka, Cryst. Growth Des. 10, 1848 (2010)
P.B. Shea, J. Kanicki, J. Appl. Phys. 111, 073709 (2012)
P.B. Shea, J. Kanicki, N. Ono, J. Appl. Phys. 98, 014503 (2005)
P.B. Shea, J. Kanicki, L.R. Pattison, P. Petroff, M. Kawano, H. Yamada, N. Ono, J. Appl. Phys. 100, 034502 (2006)
P.B. Shea, C. Chen, J. Kanicki, L.R. Pattison, P. Petroff, H. Yamada, N. Ono, Appl. Phys. Lett. 90, 233107 (2007)
P.B. Shea, L.R. Pattison, M. Kawano, C. Chen, J. Chen, P. Petroff, D.C. Martin, H. Yamada, N. Ono, J. Kanicki, Synth. Met. 157, 190 (2007)
H. Yamada, K. Kushibe, T. Okujima, H. Uno, N. Ono, Chem. Commun., 383 (2006)
H. Yamada, K. Kushibe, S. Mitsuogi, T. Okujima, H. Uno, N. Ono, Tetrahedron Lett. 49, 4731 (2008)
K. Takahashi, D. Kuzuhara, N. Aratani, H. Yamada, J. Photopolym. Sci. Tech. 26, 213 (2013)
K. Takahashi, N. Yamada, D. Kumagai, D. Kuzuhara, M. Suzuki, Y. Yamaguchi, N. Aratani, K. Nakayama, H. Yamada, J. Porphyrins Phthalocyanines 19, 465 (2015)
K. Takahashi, B. Shan, X. Xu, S. Yang, T. Koganezawa, D. Kuzuhara, N. Aratani, M. Suzuki, Q. Miao, H. Yamada, ACS Appl. Mater. Interfaces 9, 8211 (2017)
D. Liu, X. Xu, Y. Su, Z. He, J. Xu, Q. Miao, Angew. Chem. Int. Ed. 52, 6222 (2013)
D. Liu, Z. He, Y. Su, Y. Diao, S.C.B. Mannsfeld, Z. Bao, J. Xu, Q. Miao, Adv. Mater. 26, 7190 (2014)
A.R. Murphy, J.M.J. Fréchet, P. Chang, J. Lee, V. Subramanian, J. Am. Chem. Soc. 126, 1596 (2004)
A.R. Murphy, P.C. Chang, P. VanDyke, J. Liu, J.M.J. Fréchet, V. Subramanian, D.M. DeLongchamp, S. Sambasivan, D.A. Fischer, E.K. Lin, Chem. Mater. 17, 6033 (2005)
D.M. DeLongchamp, S. Sambasivan, D.A. Fischer, E.K. Lin, P. Chang, A.R. Murphy, J.M.J. Fréchet, V. Subramanian, Adv. Mater. 17, 2340 (2005)
T. Akiyama, A. Hirao, T. Okujima, H. Yamada, H. Uno, N. Ono, Heterocycles 74, 835 (2007)
A. Hirao, T. Akiyama, T. Okujima, H. Yamada, H. Uno, Y. Sakai, S. Aramaki, N. Ono, Chem. Commun., 4714 (2008)
M. Guide, J.D.A. Lin, C.M. Proctor, J. Chen, C. García-Cervera, T.-Q. Nguyen, J. Mater. Chem. A 2, 7890 (2014)
Y. Matsuo, A. Iwashita, Y. Abe, C.-Z. Li, K. Matsuo, M. Hashiguchi, E. Nakamura, J. Am. Chem. Soc. 130, 15429 (2008)
Y. Matsuo, Y. Sato, T. Niinomi, I. Soga, H. Tanaka, E. Nakamura, J. Am. Chem. Soc. 131, 16048 (2009)
Y. Tamura, H. Saeki, J. Hashizume, Y. Okazaki, D. Kuzuhara, M. Suzuki, N. Aratani, H. Yamada, Chem. Commun. 50, 10379 (2014)
H. Tamura, Y. Matsuo, Chem. Phys. Lett. 598, 81 (2014)
M. Guide, X.-D. Dang, T.-Q. Nguyen, Adv. Mater. 23, 2313 (2011)
Y. Zhen, H. Tanaka, K. Harano, S. Okada, Y. Matsuo, E. Nakamura, J. Am. Chem. Soc. 137, 2247 (2015)
M.D. Perez, C. Borek, S.R. Forrest, M.E. Thompson, J. Am. Chem. Soc. 131, 9281 (2009)
J.L. Segura, N. Martín, D.M. Guldi, Chem. Soc. Rev. 34, 31 (2005)
M. Wang, F. Wudl, J. Mater. Chem. 22, 24297 (2012)
H. Imahori, T. Umeyama, K. Kurotobi, Y. Takano, Chem. Commun. 48, 4032 (2012)
Y. Tamura, D. Kuzuhara, M. Suzuki, H. Hayashi, N. Aratani, H. Yamada, J. Mater. Chem. A 4, 15333 (2016)
K. Takahashi, D. Kumagai, N. Yamada, Y. Yamaguchi, T. Koganezawa, D. Kuzuhara, M. Suzuki, N. Aratani, S. Masuo, K.-I. Nakayama, and H. Yamada, Proceeding of International Conference on Hybrid and Organic Photovoltaics (HOPV2016) (2016)
T. Motoyama, T. Kiyota, H. Yamada, K.-I. Nakayama, Solar Energy Mater. Sol. Cells 114, 156 (2013)
H. Yamada, Y. Yamaguchi, R. Katoh, T. Motoyama, T. Aotake, D. Kuzuhara, M. Suzuki, T. Okujima, H. Uno, N. Aratani, K. Nakayama, Chem. Commun. 49, 11638 (2013)
Y. Yamaguchi, M. Suzuki, T. Motoyama, S. Sugii, C. Katagiri, K. Takahira, S. Ikeda, H. Yamada, K.-I. Nakayama, Sci. Rep. 4, 7151 (2014)
M. Suzuki, Y. Yamaguchi, K. Uchinaga, K. Takahira, C. Quinton, S. Yamamoto, N. Nagami, M. Furukawa, K. Nakayama, H. Yamada, Chem. Sci. 9, 6614 (2018). https://doi.org/10.1039/C8SC01799A
D. Kuzuhara, J. Mack, H. Yamada, T. Okujima, N. Ono, N. Kobayashi, Chem. Eur. J. 15, 10060 (2009)
H. Saeki, O. Kurimoto, M. Misaki, D. Kuzuhara, H. Yamada, Y. Ueda, Appl. Phys. Express 6, 035601 (2013)
H. Saeki, M. Misaki, D. Kuzuhara, H. Yamada, Y. Ueda, Jpn. J. Appl. Phys. 52, 111601 (2013)
Y. Kikukawa, T. Fukuda, A. Fuyuhiro, N. Ishikawa, N. Kobayashi, Chem. Commun. 47, 8518 (2011)
H. Wang, N. Su, L.-C. Wang, X. Wu, S. Bui, A. Nielsen, H.-T. Vo, Y. Luo, X.-J. Ma, Methods Mol. Biol. 1211, 139 (2014)
T.L. Chen, J.J.-A. Chen, L. Catane, B. Ma, Org. Electron. 12, 1126 (2011)
J.K. Lee, Y.-M. Wang, S. Cho, F. Wudl, A.J. Heeger, Org. Electron. 10, 1223 (2009)
T. Umeyama, S. Shibata, H. Imahori, RSC Adv. 6, 83758 (2016)
K. Kawajiri, T. Kawanoue, M. Yamato, K. Terai, M. Yamashita, M. Furukawa, N. Aratani, M. Suzuki, K. Nakayama, H. Yamada, ECS J. Solid State Sci. Technol. 6, M3068 (2017)
Acknowledgment
This work was supported by CREST, Japan Science and Technology Agency (JST), Grants-in-Aid for Scientific Research (Nos. 16H02286, 26105004, 26620167), and the Program for Promoting the Enhancement of Research Universities in NAIST sponsored by the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Yamada, H. (2019). Nanostructure Control of Crystalline Organic Thin Films by Solution Processes. In: Kubozono, Y. (eds) Physics and Chemistry of Carbon-Based Materials. Springer, Singapore. https://doi.org/10.1007/978-981-13-3417-7_9
Download citation
DOI: https://doi.org/10.1007/978-981-13-3417-7_9
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-3416-0
Online ISBN: 978-981-13-3417-7
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)