
Abstract
Prokaryotes have various defense systems, such as clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR associated (Cas) adaptive immune systems, to protect themselves from invading foreign DNA, in particular mobile genetic elements (MGEs). In prokaryotic genomes, various classes of the genes encoding prokaryotic defense systems often cluster in specific genomic regions, referred to as defense islands, which are involved in the evolution and diversification of prokaryotic defense systems. In this chapter, we review the functions of prokaryotic defense systems, their evolutionary dynamics, and their co-evolutionary arms race with invading foreign DNA. We also introduce our previous works related to the comparative genomic analyses of Streptococcus species and oral bacterial species, in particular focusing on restriction-modification (R-M) systems and CRISPR-Cas adaptive immune systems.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Prokaryotic defense systems
- Clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR associated (Cas) adaptive immune systems
- Invading foreign DNA
- Mobile genetic elements (MGEs)
- Defense islands
- Prokaryotic genome evolution
- Co-evolutionary arms race
3.1 The Roles of Mobile Genetic Elements in Prokaryotic Genome Evolution
Prokaryotes (bacteria and archaea) are ubiquitous in the natural environment, and encounter invading foreign DNA, in particular mobile genetic elements (MGEs), such as plasmids, DNA transposons, and bacteriophages (or simply phages) (Figs. 3.1a–c) [1, 2]. Plasmids are self-replicating extrachromosomal DNA molecules, which can be transmitted horizontally (referred to as horizontal gene transfer) between prokaryotic cells by direct cell-to-cell contact, referred to as conjugative transfer [3, 4]. The conjugative transfer of transmissible plasmids, such as F plasmids (Fig. 3.1a; the F plasmids enable prokaryotes to transfer DNA from the donor cells harboring the F plasmids (referred to as F+ strains) to the recipient cells (referred to as F− strains)) [5, 6], is thought to be one of the most important pathways for the transmission of virulence genes and antibiotic resistance genes in prokaryotes [7, 8]. Several studies have reported that virulence genes and antibiotic resistance genes of Enterobacteriaceae, including Escherichia coli, Enterococcus faecalis, and Enterococcus faecium, could be transferred through plasmids [8,9,10].


Schematic description of the horizontal gene transfer by mobile gene elements (MGEs); (a) the F plasmid-mediated conjugation between F+ and F− prokaryotic cells; (b) typical Class II DNA transposons by a “cut and paste” mechanism (site-specific transposition is shown); (c) lytic and lysogenic phage life cycles (solid bold arrows indicate general lytic and lysogenic phage life cycles and dotted arrows indicate prophage induction under specific conditions, such as environmental stimulation or stress)
DNA transposons, which are self-transmissible DNA elements that excise themselves and transfer into another genomic location, also mediate the horizontal gene transfer of antibiotic resistance genes in prokaryotes through the mating of prokaryotic cells [11, 12] (see also Chap. 8). The mobilization of DNA transposons is typically facilitated by transposable elements composed of a transposase-encoding gene and two terminal inverted repeats (TIRs) flanked by target site duplications (TSDs) (Fig. 3.1b) [13]. A transposase-encoding gene in transposable elements is first transcribed and translated to a transposase, which recognizes and binds to TIRs of the transposable elements and then catalyzes the excision of the transposable elements from the donor site flanked by TSDs [13]. The transposable elements bound to the transposase integrate themselves into another genomic location, and finally are flanked by other TSDs [13]. Transposons can be divided into two major classes (Class I and Class II) [14] and other distinct classes, including self-synthesizing DNA transposons [15], based on the nature of the transposition intermediate. Class I transposons, known as retrotransposons, move through an RNA intermediate and are reverse transcribed by a transposable element-encoded reverse transcriptase (RT) before their integration at another genomic location by a “copy and paste” mechanism [14]. Class II DNA transposons encode a transposase as described above, and simply move as DNA segments into another genomic location by a “cut and paste” mechanism [14]. Self-synthesizing DNA transposons (alternatively, called Polintons [15, 16] or Mavericks [17]), first discovered in eukaryotic genome sequences [15], encode a protein-primed family B DNA polymerase and a retroviral-like integrase, and are more likely involved in transposon replication [18, 19]. DNA transposons in bacteria, particularly in Gram-negative bacteria, often contain integrons, which were first characterized in 1989 [20] and are currently classified into five classes, class 1, class 2, class 3, class 4, and class 5, according to their integrase (intI) gene sequences [21, 22]. Among the five classes of integrons, the class 1 integrons are common in Gram-negative bacteria, and are captured by a Tn402 transposon to generate a transposon/integron hybrid carrying an antibiotic and antiseptic resistance-encoding cassette [22]; hence, the integrons are thought to be one of the key players in the dissemination of antibiotic resistance genes within a bacterial community [23]. Domingues et al. [24] have experimentally demonstrated that exposure of Acinetobacter baylyi to integron-containing Salmonella DNA led to the horizontal gene transfer of the integron by natural genetic transformation facilitated by the transposition of a Tn21-like transposon that contains the integron [24]. In Domingues et al. [24], exposure of A. baylyi to integron-containing Acinetobacter baumannii DNA also led to the horizontal gene transfer of the integron by natural genetic transformation, indicating that the interspecies transfer of transposons and integrons is not limited by the genetic relatedness of donor and recipient cells [24]. These findings strongly support that natural genetic transformation provides the widespread of MGEs, including transposons, in divergent species of bacteria [24].
Phages, which are obligate intracellular parasites of prokaryotes, present a serious threat to the life cycles of prokaryotes, and invade their host cells and propagate themselves in several ways, through lytic, lysogenic, pseudolysogenic, and chronic infection (Fig. 3.1c; only lytic and lysogenic life cycles are shown) [25, 26]. Lytic phages (alternatively, called virulent phages) use their host cell’s translational machinery to synthesize their own proteins, and can kill the host cells to release their progenies by cell lysis [27]. Lysogenic phages (alternatively, called temperate phages) integrate their DNA into their host chromosomes and replicate passively along with the hosts [28]. In general, the infection of lysogenic phages does not initially cause cell lysis and remains in a dormant state in their host genomes (referred to as prophages) [26, 28]. However, in response to specific environmental stimulation or stress, the prophages are excised from the host genomes and can start reproducing progeny phages (referred to as prophage induction) [28, 29]. Very recently, Erez et al. [30] have experimentally demonstrated that Bacillus subtilis phage phi3T encodes various communication peptides, denoted as the arbitrium system, for a lysis-lysogeny switch [30]. In the arbitrium system, the probability that B. subtilis phage phi3T enters into a lysogenic cycle is proportional to the increase in the concentration of arbitrium peptides produced during its lytic cycle as host cell disruption, and is then inversely proportional to the number of living host cells [30]. The findings indicate that the arbitrium system enables the progenies of B. subtilis phage phi3T to prevent the overkill of their host cells. Pseudolysogeny (alternatively, called a phage carrier state) may be another survival strategy of phages in starved prokaryotic cells, in which, unlike the lysogenic infection, lytic phages postpone their lytic life cycles, until the infected host cells are exposed to nutrient-rich environment [26, 31]. Most of the pseudolysogeny remains unclear, yet may be important to better understand the complexity of phage life cycles and their roles in the natural environment. In chronic infection, rod-shaped single-stranded DNA phages, such as filamentous bacteriophage M13, can replicate and release their progenies for a long period without killing their host cells [25, 32]. Their ability to chronically replicate the progenies without host cell disruption has been used for laboratory experiments, such as M13 cloning [33] and phage display technology [34].
Among the phage life cycles described above, the horizontal gene transfer between prokaryotic cells typically occurs via their lysogenic cycles [27, 35] (see also Chap. 5). During the lysogenic cycles, the prophages occasionally package various pieces of the donor genomes, and allow the recipients to acquire new functions in a process, referred to as lysogenic conversion [28, 35, 36]. In the early 1950s, Freeman [37] has experimentally demonstrated that non-virulent strains of Corynebacterium diphtheriae infected with specific phages could be converted to virulent strains [37]. Other researchers discovered phage-inducible chromosomal islands (PICIs) in Gram-positive bacteria, in particular Staphylococcus aureus [35, 38,39,40]. S. aureus pathogenicity islands (SaPIs), which are known as the prototypical members of PICI family, are widely spread among staphylococcal species [41] and their mobility has been experimentally demonstrated [38, 42]. Recently, the PICIs are defined as a family of MGEs that might be responsible for the horizontal gene transfer of antibiotic resistance, and phage resistance, and in particular virulence genes [35, 40, 41]. Hence, phage-mediated transduction is considered as one of the important contributors to the widespread of virulence genes, antibiotic resistance genes, and phage resistance genes in the environment [43, 44].
Since the integration of foreign DNA into prokaryotic genomes makes an impact on prokaryotic life cycles, prokaryotes have several defense systems to protect themselves from invasion by foreign DNA. For instance, restriction-modification (R-M) systems, DNA phosphorothioate (PT) modification, toxin-antitoxin (TA) systems, phage abortive infection (Abi) systems, Argonaute proteins, bacteriophage exclusion (BREX) systems, and clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR associated (Cas) adaptive immune systems (henceforth called CRISPR-Cas systems) have been identified in the genomes of many prokaryotes (Figs. 3.2a–f and 3.3) [46]. These defense systems are roughly divided into relatively non-specific innate immune systems (R-M systems, DNA PT modification, TA systems, Abi systems, pAgo proteins, and BREX systems) and highly specific adaptive immune systems (CRISPR-Cas systems) [46]. This chapter introduces a brief overview of the principle of the innate immune systems in prokaryotes, followed by CRISPR-Cas systems, their evolutionary dynamics, and finally our investigation of CRISPR-Cas systems in Streptococcus species and oral bacterial species.

Schematic description of prokaryotic innate immune systems; (a) restriction-modification (R-M) systems (typical Type II R-M systems are shown); (b) DNA phosphorothioate (PT) modification; (c) toxin-antitoxin (TA) systems (a hok/sok system is shown as Type I TA system); (d) phage abortive infection (Abi) systems (Rex system, which is carried out in ascending order of numbers); (e) prokaryotic Argonaute (pAgo) proteins (typical long pAgo proteins are shown); (f) bacteriophage exclusion (BREX) systems

Schematic description of clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR associated (Cas) adaptive immune systems (referring to Barrangou and Horvath [45] with slight modification)
3.2 Prokaryotic Innate Immune Systems Against Foreign DNA Invasion
R-M systems (Fig. 3.2a), first discovered in the early 1950s [47, 48], are one of the best-characterized prokaryotic defense systems. The defense mechanism of R-M systems allows prokaryotes to distinguish between their own DNA (methylated DNA) and invading foreign DNA (non-methylated DNA) by a pair of enzymes, a restriction endonuclease (REase) and a DNA methyltransferase (MTase) [49, 50]. An REase recognizes methylated bases (5-methylcytosine (m5C), N4-methylcytosine (m4C), and N6-methyladenine (m6A)) in host DNA, and cleaves non-methylated DNA as non-self DNA at specific sites [51]. Meanwhile, a DNA MTase acts as a host DNA methylation enzyme, which transfers a methyl group (CH3) from methyl donating compounds, such as S-adenosylmethionine (SAM), into the potential REase target sites in the host DNA for the prevention of self DNA cleavage [52,53,54]. R-M systems are classified into four types, Type I, Type II, Type III, and Type IV, based on their enzyme composition, sequence recognition, cleavage position, co-factor requirements, and substrate specificity [53]. Among the four types of R-M systems, Type II R-M systems are the most typical and most studied R-M systems [46]. The Type II R-M enzymes, such as EcoRI isolated from E. coli species [55], have been commonly used for laboratory experiments, such as restriction fragment length polymorphisms (RFLPs) and DNA cloning [56], because of its highly specific DNA cleavage ability. Meanwhile, Type IV R-M systems are composed of only an REase (referred to as a Type IV restriction enzyme), which can recognize and cleave only methylated DNA, unlike other Type R-M systems [57, 58].
DNA PT modification (Fig. 3.2b) is a sulfur modification of self DNA, in which a non-bridging oxygen atom in self DNA is substituted with a sulfur atom, and unmodified non-self DNA is cleaved [59]. The defense mechanism of DNA PT modification is based on self/non-self DNA discrimination, like R-M systems. The DNA degradation (Dnd) phenomenon of DNA PT modification was initially observed during electrophoresis of DNA from the Gram-positive bacterium Streptomyces lividans [60], and is typically associated with a family of proteins (DndA-E) encoded by a five-gene dnd cluster (dndA-E) [59]. DndA (cysteine desulfurase), DndC (phosphoadenosine phosphosulfate reductase), DndD (ATPase), and DndE (DNA-binding protein with a distinct fold) proteins form a complex to replace an oxygen atom with sulfur, whereas a DndB protein acts as a negative transcriptional regulator for PT-modifying genes, and is not essential for DNA PT modification [46, 59, 61, 62]. A DndD protein acts as an ATPase possibly associated with DNA structure alteration or DNA nicking during sulfur incorporation [63,64,65]. Xu et al. [66] have reported that Salmonella enterica serovar Cerro 87 possesses a four-gene cluster homologous to dndB-E genes, termed dptB-E, and its cognate three-gene dnd cluster (dndF-H) (alternatively, called dptF-H genes) [59, 66]. The DndF-H proteins encoded by dndF-H genes make double-stranded DNA damage as a trigger for SOS response, cell filamentation, and prophage induction [59]. The combination of the DNA PT modification-related genes found in the S. enterica serovar Cerro 87 has been proposed as DNA PT modification-dependent R-M systems [67], which are composed of modification-related genes (dndB-E) and restriction-related genes (dndF-H) [67], and have been experimentally demonstrated as temperature-dependent defense systems [59].
TA systems (Fig. 3.2c) are also one of the most studied prokaryotic defense systems, as well as R-M systems, and are closely linked to dormancy induction or programmed cell death (PCD) [68]. The genetic modules of TA systems are composed of stable toxin genes and unstable antitoxin genes in multiple copies, and activated when prokaryotic cells possessing the genetic modules are exposed to stress, such as antibiotic treatment and phage infection [68, 69]. TA system-related genes, known as post-segregational killing (PSK) systems, were first discovered in the 1980s [70,71,72], such as a hok/sok system composed of hok (host killing), sok (suppression of killing), and mok (modulation of killing) in plasmid R1 of E. coli [70]. In the hok/sok system (Fig. 3.2c), the activity of hok as stable toxin-encoding mRNA is suppressed by being bound to sok as non-coding unstable neutralize antisense RNA that is complementary to hok, resulting in the formation of an RNA duplex that is degraded by RNase III [73, 74]. However, when the plasmids encoding TA systems are not transmitted to daughter cells, the sok is rapidly degraded and highly toxic Hok proteins are synthesized from the hok, ultimately leading to cell death by depolarization of the cell membrane [73, 74]. TA systems are classified into six types, Type I, Type II, Type III, Type IV, Type V, and Type VI, based on the nature and the mechanism of antitoxins [75]. Among the six types, Type II TA systems are well-characterized TA systems, in which protein antitoxins neutralize the toxin activity of TA systems by directly binding to their cognate toxins [76]. In most Type II TA systems, the Type II toxins are a potent endoribonuclease and cleave cellular mRNA at specific sequences to inhibit translation in response to stress [75]. For instance, drug-tolerant Mycobacterium tuberculosis, which possesses a large number of TA systems [77], shows antibiotic tolerance by a remarkable downregulation of genes associated with growth, metabolism, and lipid synthesis, and an upregulation of stress-associated sigma factors, transcription factors, drug efflux pumps, and toxin-antitoxin genes during a prolonged antibiotic exposure [78]. Another example of toxin-antitoxin genes in Type II TA systems is higher eukaryotes and prokaryotes nucleotide-binding (HEPN)-minimal nucleotidyltransferase (MNT) genetic modules, which are the most abundant genes in hyperthermophilic archaea [79,80,81]. The HEPN-MNT genetic modules are composed of HEPN as RNA-cleaving toxins and MNT as predicted antitoxins, and are predicted as antibiotic resistance systems [80]. These TA systems are widely present in chromosomes or plasmids of prokaryotes as TA loci, and are stably maintained in the populations of viable cells [68, 74, 76].
Abi systems (Fig. 3.2d) lead to the death of infected cells (referred to as altruistic suicide), to terminate the production of progeny phages, and then protect clonal cells in the prokaryotic populations [82, 83]. Many of Abi phenotypes, denoted by “Abi” and capital alphabet letters (e.g., AbiA), have been found in the plasmids of Lactococcus lactis [84], which is one of the model microorganisms for Abi system studies. Although Abi systems are thought to be distinct from TA systems, many of Abi systems share the genetic modules with TA systems, such as HEPN domains described above [46, 85]. A recent study has shown that Type III TA systems, ToxINpa composed of ToxN as endoribonuclease toxins and ToxIpa as RNA antitoxins, are employed by the phytopathogenic bacterium Pectobacterium atrosepticum for anti-phage activity [82, 86]. On the other hand, the Rex system found in λ-lysogenic E. coli strains [83, 87] is a well-studied Abi system that has the different mechanism from ToxINpa system. The Rex system is composed of two proteins, RexA protein as an intracellular sensor and RexB protein as an ion channel [83, 88]. When phage infection occurs, RexA protein is first activated by recognizing a phage protein-DNA complex, followed by RexB protein activation [88, 89]. The activated RexB protein induces a drop in the cellular ATP level, thereby stopping cell multiplication and aborting the lytic growth of the infecting phages [88, 89].
Prokaryotic Argonaute (pAgo) proteins (Fig. 3.2e) are nucleic acid-guided proteins, which use RNA or DNA guides and provide specific cleavage of complementary nucleic acid targets as a host defense mechanism [90]. Argonaute proteins, first discovered in eukaryotes [91], are essential components of an RNA-induced silencing complex (RISC) responsible for RNA interference (RNAi) (alternatively, called RNA silencing) [92,93,94]. Small non-coding RNA, such as microRNA (miRNA), small interfering RNA (siRNA), and P-element induced wimpy testis (PIWI)-interacting RNA (piRNA), guides Argonaute proteins to complementary RNA targets in RNAi [92, 95, 96], leading to translational inhibition, mRNA destabilization, or RNA target cleavage [97]. In contrast to eukaryotic Argonaute (eAgo) proteins, some pAgo proteins are key players in specific DNA target cleavage, referred to as DNA interference (DNAi) [98, 99]. Based on the domain structure, pAgo proteins are roughly divided into two groups, long and short pAgo proteins [100]. Among the two groups of pAgo proteins, the domain structure of long pAgo proteins is structurally similar to that of eAgo proteins [101]. Both eAgo proteins and long pAgo proteins are composed of a PIWI endonuclease domain, a PIWI-Argonaute-Zwille (PAZ) domain, a middle (MID) domain, and an amino-terminal (N-terminal) domain, along with two domain linkers (L1 and L2) [46, 100]. Meanwhile, short pAgo proteins are composed of only a PIWI domain and a MID domain [100]. Instead of a PAZ domain and a N-terminal domain, short pAgo proteins are associated with the genes encoding an analogue of PAZ (APAZ) domain, which is fused to a putative nuclease domain, such as Sir2, Mrr, or TIR proteins [90, 101]. It is assumed that short pAgo proteins are responsible for nucleic acid-guided target recognition, and their associated nuclease-APAZ domain is responsible for guide generation and/or target degradation, respectively [101]. Recent bioinformatic analysis has revealed that pAgo proteins are encoded in ~32% and ~9% of the sequenced archaeal and bacterial genomes, respectively [100], and the phylogenetic tree of pAgo-encoding genes does not follow the prokaryotic phylogeny based on ribosomal RNA and other universal genes [100]. These indicated that horizontal gene transfer results in the widespread of the pAgo protein-encoding genes in prokaryotes. Other recent studies have demonstrated that most of the characterized long pAgo proteins, including Thermus thermophilus (TtAgo) [99] and Pyrococcus furiosus (PfAgo) [102], can target DNA using either DNA or RNA guides [101], and can act as prokaryotic defense systems against invading foreign DNA as well as RNAi in eukaryotes. Moreover, it has been proposed that the features of long pAgo proteins described above can be used as genome-editing tools, because they can selectively cleave double-stranded DNA targets [101].
BREX systems (Fig. 3.2f) are a relatively novel defense mechanism against phage infection, and are composed of a combination of six genes among 15 BREX-related genes [103]. The BREX-related genes include five pgl genes (pglW, pglX, pglXI, pglY, and pglZ) and ten brx genes (brxA, brxB, brxC, brxD, brxE, brxF, brxHI, brxHII, brxL, and brxP) [103]. Each of these genes encodes a serine/threonine kinase (pglW), an adenine-specific DNA methyltransferase (pglX, pglXI), an ATP-binding P-loop protein posessing ATPase activity (pglY), an alkaline phosphatase (pglZ), an NusB-like RNA-binding anti-termination protein (brxA), a protein of unknown function (brxB, brxE, brxF), an ATP-binding protein (brxC, brxD), an Lhr-like helicase (brxHI), a DNA/RNA helicase (brxHII), a Lon-like protease (brxL), and a phosphoadenosine phosphosulfate (PAPS) reductase (brxP), respectively [103,104,105]. In the early 1980s, BREX-like systems have been already discovered in Streptomyces coelicolor A3(2), denoted as “phage growth limitation (Pgl) systems” [106], which are composed of pglWXYZ genes [107]. Moreover, two of the five pgl genes in BREX systems, pglXZ genes, show sequence homology to genes in Pgl systems [103]. The phage resistance mechanism of BREX systems is based on self/non-self DNA discrimination, like R-M systems [103]. Prokaryotes possessing BREX systems methylate their own genomes, typically at the fifth position of a non-palindromic 5′-TAGGAG-3′ hexameric sequence, and can prevent phage DNA replication, but they do not cleave phage DNA, unlike R-M systems [103]. BREX systems are currently classified into six types, Type 1, Type 2, Type 3, Type 4, Type 5, and Type 6, based on the six-gene combination and the order of the six genes, all of which contain a pglZ gene [103]. Goldfarb et al. [103] analyzed the genomes of approximately 1500 bacteria and archaea and revealed that pglZ genes as a putative member of an alkaline phosphatase superfamily are present in approximately 10% of these microbial genomes [103], suggesting that BREX systems are widely distributed as phage resistance systems.
3.3 CRISPR-Cas Adaptive Immune Systems
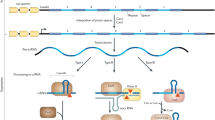
CRISPR-Cas is protein-encoding genes located in hypervariable genetic loci (referred to as CRISPR-cas loci), which are composed of multiple repeat-spacer arrays and the CRISPR-associated (cas) genes encoding CRISPR-associated (Cas) proteins located in close-proximity to the repeat-spacer arrays [45]. CRISPR-cas loci in prokaryotes memorize past encounters with invading foreign DNA as CRISPR spacers and act as prokaryotic adaptive immune systems [45]. The repeat-spacer arrays, later referred to as CRISPR arrays [108, 109], were first discovered in the 3′-end flanking region of alkaline phosphatase isozyme (iap) genes in E. coli [110]. Soon after, similar repeats were discovered in the genome sequences of many prokaryotes [111]. In 2000, the prokaryotic repeat-spacer arrays were named as short regularly spaced repeats (SRSR) [112]; in 2002, the arrays were renamed as spacers interspersed direct repeats (SPIDR) [108, 109], and finally as clustered regularly interspaced short palindromic repeats (CRISPR) on the basis of the structural feature [108, 109]. At the same time, four cas genes (cas1, cas2, cas3, and cas4 genes encoding Cas1, Cas2, Cas3, and Cas4 proteins, respectively) were also identified nearby CRISPR arrays, and the structural coherence of the CRISPR arrays and the cas genes was characterized as CRISPR-cas loci [108, 109]. In 2005, the involvement of CRISPR-cas loci in adaptive immune systems against invading foreign DNA began to be speculated on the basis of some scientific evidence; (1) CRISPR spacers derive from the genomic fragments of invading foreign DNA including MGEs (later referred to as protospacers) [113,114,115,116], (2) phages and plasmids harboring the protospacers fail to infect their hosts yielding antisense RNA (later referred to as CRISPR RNA (crRNA)) [114, 115, 117]. The experimental demonstration of CRISPR-Cas systems against phage infection was carried out by Barrangou et al. [118], in which phage-sensitive Streptococcus thermophilus strains became resistant to Streptococcal phages after acquiring new CRISPR spacers identical to the protospacer sequences in the phage DNA [118]. Soon after, the adaptive immune activity of CRISPR-Cas systems against plasmid transfer was experimentally demonstrated by Marraffini and Sontheimer [119], in which the CRISPR-cas loci in Staphylococcus epidermidis strains acted as DNAi machinery, and prevented the transfer of staphylococcal conjugative plasmids, as well as RNAi in eukaryotes [119].
CRISPR-Cas systems act as DNA-encoded, RNA-guided, and nucleic acid-targeting interference (CRISPR interference) in prokaryotes (Fig. 3.3) [45]. The first step of CRISPR-Cas systems is to detect the motifs associated with protospacers of invading foreign DNA, referred to as protospacer adjacent motifs (PAMs) [120], and cut the protospacers at the site a few base pairs upstream or downstream from the PAMs by nuclease and integrase activity of Cas proteins, such as a Cas1–Cas2 protein complex or Cas9 proteins, respectively [121,122,123]. The shortly cut protospacers as new CRISPR spacers are integrated between multiple repeats of CRISPR arrays adjacent to a leader sequence in CRISPR-cas loci, and the CRISPR arrays are transcribed into a long precursor, referred to as precursor CRISPR RNA (pre-crRNA) [121]. The pre-crRNA is further trimmed into multiple short segments of crRNA by the endonuclease activity of Cas proteins, such as Cas6 proteins [122, 124]. The crRNA binds to the complementary nucleic acid sequences, and the crRNA-guided CRISPR interference is then carried out by the crRNA-guided nuclease activity of Cas proteins (referred to as an effector), such as Cas9 proteins [125], which cleave the crRNA-guided nucleic acid targets.
To date, CRISPR-cas loci are present in approximately 50% of bacterial genomes and approximately 90% of archaeal genomes [122]. CRISPR-Cas systems are currently classified into two classes (class 1 and class 2), including six major types (Type I, Type II, Type III, Type IV, Type V, and Type VI) and 28 subtypes (e.g., Type I-A), based on the CRISPR-Cas machinery, in particular the combination of Cas proteins, and its mode of action (Table 3.1) [126]. A recent report has described that approximately 90% of all identified CRISPR-cas loci belong to class 1 CRISPR-Cas systems, and the remaining 10% belong to class 2 CRISPR-Cas systems [128]. Class 1 CRISPR-Cas systems with multi-protein effector complexes include Type I, Type III, and Type IV CRISPR-Cas systems [126]. Among the three types in class 1 CRISPR-Cas systems, Type I and Type III CRISPR-Cas systems are the most common CRISPR-Cas systems and are distributed in many archaea and a few bacteria, whereas Type IV CRISPR-Cas systems are a relatively rare [127, 129]. The multi-protein effector complexes of class 1 CRISPR-Cas systems are composed of multiple Cas proteins, such as CRISPR-associated complex for antiviral defense (Cascade) in Type I CRISPR-Cas systems [117, 130] and a CRISPR-Cas subtype Mtube (Csm)-CRISPR RAMP module (Cmr) complex in Type III CRISPR-Cas systems [131, 132]. Both of the multiple Cas proteins, such as Cas5, Cas6, Cas7, and Cmr proteins, include paralogous repeat-associated mysterious proteins (RAMPs) responsible for RNA binding and/or ribonuclease activity [133]. Several structural analyses have revealed that RAMPs contain single- or multi-domains of RNA recognition motif (RRM), known as a ferredoxin-like fold, which is a common protein fold often found in the structure of nucleic acid-binding proteins [133,134,135]. Recently, RT-associated CRISPR-cas loci encoding an RT-Cas1 fusion protein have been found by several bioinformatic analyses [136,137,138], and were classified as Type III CRISPR-Cas systems [139]. Soon after, it was experimentally demonstrated that RT-associated Type III CRISPR-Cas systems can acquire CRISPR spacers directly from RNA [140], implying that the Type III CRISPR-Cas multi-protein effector modules with an RT-Cas1 fusion protein may have the ability to adapt to invaders with both DNA and RNA [141].
Class 2 CRISPR-Cas systems with single and long protein effector modules include Type II, Type V, and Type VI CRISPR-Cas systems [126, 128]. Among the three types in class 2 CRISPR-Cas systems, Type II CRISPR-Cas systems with Cas9 proteins as a multi-domain effector are one of the best-characterized CRISPR-Cas systems [142], and are widely used for genome editing [143,144,145,146]. In Type II CRISPR-Cas systems, pre-crRNA is bound to trans-acting CRISPR RNA (tracrRNA), which is complementary to the repeat sequence in the pre-crRNA, and the pre-crRNA-tracrRNA duplex is processed into a crRNA-tracrRNA duplex by a double-stranded RNA-specific ribonuclease RNase III [147,148,149]. The crRNA-tracrRNA duplex forms a complex with Cas9 proteins, and the Cas9-crRNA-tracrRNA complex finally introduces site-specific double-stranded DNA cleavage at the specific target site [148, 150]. Cas9 proteins contain two nuclease domains, RuvC and HNH, to generate paired double nicks [151], which can be re-programmed by single-guide RNA (sgRNA) to cleave its specific DNA target [145, 146, 148]. In Type V and Type VI CRISPR-Cas systems, Cas12 and Cas13 proteins are predicted as multi-domain protein effector, respectively [126, 128]. All Cas12 proteins contain RuvC-like endonuclease domains, and in some cases a putative novel nuclease (Nuc) domain [152]. Very recently, new CRISPR-Cas systems, referred to as CRISPR-CasX and CRISPR-CasY, were discovered from uncultivated bacteria [153], and were putatively classified as new subtypes of Type V CRISPR-Cas systems, because both CasX and CasY proteins contain RuvC-like domains (renamed as Cas12e (CasX) and Cas12d (CasY) proteins, respectively) [127]. All Cas13 proteins contain two HEPN domains, which may possess RNase activity, as well as HEPN toxins in Type II TA systems (see Sect. 3.2) [80, 81], suggesting that Cas13 proteins would have evolved from HEPN toxins [127].
The CRISPR-Cas systems described above show a remarkable diversity in terms of the combination of Cas proteins and its mode of action [126]. Meanwhile, recent comparative genomic and structural analyses have revealed that Cas1 and Cas2 proteins, in particular Cas1 proteins, are universally conserved, and are thought to play key roles in almost all CRISPR-Cas systems [19, 127]. Krupovic et al. [19] discovered “genomic islands” in the genome sequences of many archaea and some bacteria, in which some cas1 genes encoding homologous Cas1 proteins are not associated with CRISPR-cas loci, referred to as cas1-solo genes [19]. The cas1-solo genes are classified into two groups, group 1 and group 2, based on cas1 gene phylogeny [19]. Among the two groups, group 2 cas1-solo genes are co-localized with protein-primed family B DNA polymerase-encoding genes (henceforth called polB genes) in genomic islands [19], as well as self-synthesizing DNA transposons in eukaryotes [15]. The MGEs composed of cas1-solo genes, polB genes, TIRs, and TSDs (see Sect. 3.1) were denoted as “casposons,” [19] which employ Cas1-solo proteins as an integrase (alternatively, called a casposase) [154]. Casposons are currently classified into four families, family 1, family 2, family 3, and family 4, based on the gene composition and phylogeny of Cas1 proteins [155]. The experimental demonstration of casposon mobilization has not been performed yet, but comparative genomic analysis of various strains of Methanosarcina mazei has shown the recent mobility of casposons as potentially active MGEs [155]. Béguin et al. [156] have revealed the close similarities between the insertion mechanisms of casposons and CRISPR spacers facilitated by a casposase and a Cas1–Cas2 protein complex, respectively [156]. These findings strongly support the evolutionary relationship between the adaptive modules of CRISPR-Cas systems and casposons [156].
The evolution and diversification of CRISPR-Cas systems appear to be driven by the co-evolutionary arms race between prokaryotes and phages. Phages possess the genes encoding anti-CRISPR proteins, referred to as Acr genes, which encode anti-CRISPR proteins that show the ability to inhibit the functions of diverse effector complexes in CRISPR-Cas systems [157]. As described above, lysogenic phages integrate their own genomes into their host chromosomes, and then form prophage regions [27], and might express the anti-CRISPR genes to maintain the prophage regions during lysogeny [158]. To date, more than 20 families of Acr genes have been identified in previously published reports, and can inhibit the anti-phage activity in some subtypes of Type I and Type II CRISPR-Cas systems [157]. Bondy-Denomy et al. [159] have experimentally demonstrated the two mechanisms of anti-CRISPR activity by AcrF1, AcrF2, and AcrF3 proteins, which specifically inhibit the activity of Type I-F CRISPR-Cas system [159]. AcrF1 and AcrF2 proteins could inhibit the DNA-binding activity of the multi-protein effector complex in Type I-F CRISPR-Cas system, whereas AcrF3 proteins directly bind to Cas3 proteins and prevent their recruitment to the DNA-bound multi-protein effector complex [159]. It has also been known that phages adapt and evolve against some prokaryotic innate immune systems, in particular R-M systems [160]. Several phages possess MTase-encoding genes, and modify their own genomes by the MTase activity to protect themselves from the REase activity of R-M systems [161]. Another mechanism is that the phages possessing overcome classical restriction (ocr) genes produce Ocr proteins, which directly bind to both MTase and REase of Type I R-M systems, and then inhibit the endonuclease activity of the R-M systems [160, 161]. The co-evolution by the prokaryotic host-phage arms race remains unclear, but it could be the result of a diversification of prokaryotic defense systems [162].
3.4 Effect of the Evolutionary Dynamics of Defense Systems on Prokaryotic Genome Evolution
Makarova et al. [163] have performed the comparative genomic analysis of 1055 completely sequenced prokaryotic genomes focused on defense systems, and have indicated the involvement of transposable elements in the gain, loss, and exchange of the defense system-encoding genes (henceforth called defense genes) [163]. The results suggested that many species have intraspecific variation in defense systems through the horizontal gene transfer of defense genes. The authors also discovered specific genomic locations in prokaryotic genomes, referred to as defense islands [163], which are enriched in not only the genes encoding deferent classes of defense systems, in particular R-M systems, T-A systems, and BREX systems, but also uncharacterized genes as candidates for new types of defense systems [103, 163]. After that, the comparative analysis of 35 groups of closely related bacterial genomes and one group of archaeal genomes has demonstrated that the defense genes in more than half of the analyzed genomes tend to be co-localized in defense islands [164]. In some species, MGEs in defense islands are frequently involved in the evolution and diversification of defense systems, suggesting that defense islands are responsible for the enhanced dynamics of the evolution in prokaryotic defense systems [164].
Very recently, Ofir et al. [165] have reported new defense systems, denoted as defense island system associated with restriction modification (DISARM), as multi-gene R-M systems [165]. DISARM systems are composed of five genes: four genes encode a DNA MTase, a helicase domain, a phospholipase D (PLD) domain, a domain of unknown function (DUF) 1998 (a helicase-associated domain), and one remaining gene has an unknown function [165]. Among the five genes, at least the four genes encoding a DNA MTase, a helicase domain, a DUF1998, and a gene of unknown function are essential for the anti-phage activity of DISARM systems. Meanwhile, the genes encoding a PLD domain are not always essential, but are required for the anti-phage activity against myophage SPO1 and podophages Nf. Among the five genes, a DUF1998 is enriched in defense islands, and is then thought to be a part of anti-phage defense systems [163], also indicating that DISARM systems are widely spread prokaryotic defense systems in defense islands.
Alternative DNA methylation levels have been shown to cause changes in gene expression in bacteria [166], because bacteria use a DNA MTase as a switch to systematically change their transcriptome [167, 168]. It is conceivable that the variation in R-M systems may affect the DNA methylation patterns and also may ultimately split clonal populations into epigenetic lineages [169]. In fact, clustering based on the presence/absence of R-M systems in genomes accurately reproduced the core genome phylogenetic structure in Neisseria meningitidis, and each phylogenetic clade harbored a unique repertoire of R-M systems [170]. A genomic clade-distinctive combination of the R-M system enzymes was also observed in Burkholderia pseudomallei, which is the causative agent for melioidosis [171]. Furthermore, clade-specific DNA methylation patterns in B. pseudomallei were observed, suggesting that R-M systems contribute to the limiting intraspecific exchange of genetic material, and the genomic clades may represent functional units of genetic isolation in this species [171]. In Streptococcus pneumoniae, a genomic region, where different lineages code for variable R-M systems, was found [172]. This locus has been proposed to play a role in the fine-tuning of the extent of genomic plasticity [172]. Therefore, these findings suggested that the intraspecific variation in R-M systems plays a key role in the genome diversification. Prokaryotic defense systems other than R-M systems also appear to have additional functions besides the limitation/prevention against invasion by foreign DNA. A role in stabilizing genomic islands was proposed for TA systems [76, 166]. CRISPR-Cas systems are also able to control transcription endogenously, and regulate important lifestyle-based bacterial phenotypes, such as pathogenicity [173].
To analyze the intraspecific diversification of bacterial populations associated with prokaryotic defense systems, we have investigated 47 genome sequences of Streptococcus suis, an important swine pathogen and an emerging zoonotic agent, with a focus on the defense genes [174]. Our comparative genome analysis of S. suis indicated similar or identical profiles of the defense genes related to R-M systems, TA systems, Abi systems, and CRISPR-Cas systems in the same genomic clusters and several cassette-like defense system loci [174]. Among the defense system loci, one locus found in S. suis was a variable region. In the region, not only genetic elements in R-M systems but also those in Abi systems and CRISPR-Cas systems, prophages, and/or other genes were replaced with each other [174]. In addition, the shift of prokaryotic defense systems at the locus was coincident with the branching of the genomic clusters in many cases [174]. Taken together with our investigation and previously published researches, it is possible to hypothesize that some prokaryotic defense systems, in particular R-M systems, affect the extent of genomic plasticity and the intraspecific diversification of bacterial populations in certain species, and in some cases deferent classes of defense genes are located on the same locus. However, it remains unclear to what extent such prokaryotic defense systems are involved in the intraspecific speciation. Future investigation is needed to evaluate the authenticity of this hypothesis.
3.5 Functions of CRISPR-Cas Adaptive Immune Systems in Streptococcus Species and Oral Bacterial Species
We have investigated how CRISPR-Cas systems act in prokaryotic cells by the use of Streptococcus species and oral bacterial species, and are suspecting that CRISPR-Cas systems play key roles in the genomic evolution of the bacterial species investigated. Streptococcus pyogenes, also known as group A beta-hemolytic Streptococci (GAS), is one of the most virulent pathogens causing a broad spectrum of infectious diseases, such as pharyngitis as the most common bacterial disease in children and streptococcal toxic shock syndrome (STSS) as life-threatening illness [175]. Our investigation found numerous prophage regions in the genome sequences of S. pyogenes, although their genome size is a relatively small (approximately 1.9 mega base pairs) [176]. In addition, the streptococcal virulence genes encoding superantigens, a hyaluronidase, and a streptodornase were also found in the prophage regions of the S. pyogenes genome sequences investigated [176]. These results suggested that S. pyogenes takes advantage of prophages within its genome for survival. Subsequently, we have investigated the association between the number of prophages and CRISPR-cas loci in the genomic sequences of 13 S. pyogenes strains available in a public nucleotide database [177]. As a result, CRISPR-cas loci were found in 10 of the 13 strains investigated, and interestingly the number of CRISPR spacers was inversely proportional to the number of prophages in the ten strains [177], suggesting that S. pyogenes is able to control the acquisition of prophages based on the contents of CRISPR-cas loci, and also contribute to strain-specific pathogenesis of streptococcal phages (Fig. 3.4a).

Functions of CRISPR-Cas systems in Streptococcus species and oral bacterial species; (a) Streptococcus pyogenes; (b) Streptococcus mutans; (c) three red complex species (Porphyromonas gingivalis, Tannerella forsythia, and Treponema denticola), and their competitive and cooperative interaction
As for other Streptococcus species, we have analyzed the complete genome sequences of several Streptococcus mutans serotype c strains, which are known as a major cause of dental caries (tooth decay) [178, 179]. As a result, CRISPR-cas loci were found in almost all of the analyzed S. mutans serotype c strains, where no prophage regions were found in all the S. mutans serotype c strains [178, 179]. In addition, the CRISPR spacer sequences in the analyzed S. mutans serotype c strains exhibited high similarity to the genome sequences from several streptococcal phages, including S. mutans phage M102 [179]. Our investigation suggested that Streptococcus species possess species-specific survival strategy; S. mutans may be attacked by streptococcal phages and then protected by CRISPR-Cas systems (Fig. 3.4b), whereas S. pyogenes acquires virulence-encoding genes by phage-mediated transduction.
We have also investigated the functions of CRISPR-Cas systems in three oral bacterial species referred to as “red complex,” Porphyromonas gingivalis, Tannerella forsythia, and Treponema denticola [180,181,182,183]; they have been frequently detected in human periodontal pockets [184]. We have first sequenced the complete genome of P. gingivalis strain TDC60 isolated from a severe periodontal lesion in a Japanese patient for subsequent comparative genome analysis [181]. In the comparative genome analysis of the 3 P. gingivalis complete genome sequences including the complete genome of P. gingivalis strain TDC60, multiple CRISPR-Cas subtypes were identified in each of their genome sequences, although the potential targets were not determined for the CRISPR-Cas subtypes identified [181]. To identify the targets, our further investigation of CRISPR-Cas systems in P. gingivalis was carried out [182, 183]. In Watanabe et al. [182], 60 P. gingivalis isolates were used for genetic typing and intraspecific diversity analysis [182]. As a result, a total of 2150 CRISPR spacers were identified in the 60 P. gingivalis isolates, and only 29 of the 2150 CRISPR spacers exhibited high sequence similarity to the genome sequences available in public nucleotide databases [182]. Of the 29 CRISPR spacers analyzed, 19 CRISPR spacers exhibited high sequence similarity to the three P. gingivalis genome sequences available in the databases; hence, we hypothesized that genetic recombination and rearrangement within P. gingivalis strains might be regulated by CRISPR-Cas systems [182]. The comparative genome analysis was then performed in Watanabe et al. [183] using the draft genome sequences of 51 P. gingivalis isolates, and the publicly available genome sequences of 13 P. gingivalis and 46 other Porphyromonas species [183]. As a result, the CRISPR spacers (identified from the 41 P. gingivalis isolates) with potential targets in the genus Porphyromonas were approximately 23 times more abundant than those with potential targets in other genus taxa (1720/6896 CRISPR spacers vs. 74/6896 CRISPR spacers) [183]. These results strongly suggested that CRISPR-Cas systems in P. gingivalis are able to limit genetic recombination and rearrangement by acquiring the genome fragments of other P. gingivalis strains as self-targeting CRISPR spacers.
As for other red complex species, T. forsythia and T. denticola, we have analyzed the draft genome sequences of 19 T. forsythia strains and 14 T. denticola strains for comparison [180]. In the comparative genome analysis of the 19 T. forsythia strains and the 14 T. denticola strains, 106/1631 and 7/78 CRISPR spacers exhibited high sequence similarity to the genome sequences available in public nucleotide databases, respectively [180]. In the 19 T. forsythia strains, 16/106 and 3/106 CRISPR spacers exhibited significant sequence similarity to the genome sequences of P. gingivalis and T. denticola, respectively [180]. In addition, the CRISPR spacers found in four of the 19 T. forsythia strains exhibited high sequence similarity to the MTase-encoding genes in P. gingivalis [180]. These results suggested that CRISPR-Cas systems in T. forsythia may attack MGEs including the genes encoding defense systems in other red complex species [180]. Meanwhile, 6/7 CRISPR spacers found in the 14 T. denticola strains exhibited significant sequence similarity to hypothetical genes in the genome sequences of T. denticola, suggesting that T. denticola limits genetic recombination and rearrangement within T. denticola strains [180].
Further comparative genome analysis was performed to reveal the association between the three red complex species [180]. As a result, gene deficiencies were mutually compensated in metabolic pathways when the genes of all the three red complex species were taken into account, suggesting that there is cooperative relationship among the three red complex species (Fig. 3.4c) [180]. Meanwhile, the three red complex species may have competitive interaction via CRISPR-Cas systems as described above (Fig. 3.4c) [180]. The association between the three red complex species may allow them to have different genomic evolutionary strategy to survive in the dental environment.
3.6 Future Perspectives
In this chapter, the functions of prokaryotic defense systems and their co-evolutionary arms race with invading foreign DNA were discussed. The prokaryote-phage co-evolution is thought to be one of the drivers of phenotypic and genotypic diversification [162]. A recent study has described that, on average, the origins of approximately 7% of the CRISPR spacers found in prokaryotic genome sequences were identifiable, whereas the remaining 93% have not yet been unidentifiable, termed “CRISPR dark matter” [185]. It is noteworthy that identifying the CRISPR dark matter would provide blueprints for better understanding of microbial complexity and robustness in the biosphere. Moreover, it has also been discussed that several phages may be able to control the community structure in the prokaryotic populations. For instance, CRISPR-cas loci have been found in two prophage regions in Clostridium difficile genome sequences [186], suggesting that the phages possessing CRISPR-cas loci enable their hosts to prevent the infection of other phage types for their interspecific competition.
In our previous investigation, the time-course metagenomic analysis of microbes in artificially polluted soils with four harmful aromatic compounds, 3-chlorobenzoate (3CB), phenanthrene, biphenyl, and carbazole, showed another potential capability of CRISPR-Cas systems (unpublished data). The time-course metagenomic analysis showed that the number of identified CRISPR repeats rapidly and proportionally changed according to those of bacteria in the polluted soil bacterial community. Meanwhile, the number of identified CRISPR spacers in the polluted soil bacterial community was stable throughout the analysis period. The time-course metagenomic analysis also showed that the species diversity and functions of the bacterial community were resilient against the chemical disturbance, whereas the contents of CRISPR arrays were altered and did not return to their original states. These results strongly suggested that the bacteria exposed to the chemicals newly inserted unidentifiable CRISPR spacers between the identifiable repeats for recording the environmental fluctuation; hence, we may call the phenomenon associated with the CRISPR arrays in the bacterial community as “memory of bacterial communities.” From all the information presented in this chapter, we conclude that studying the prokaryotic defense systems allows us not only to understand the co-evolutionary arms race between prokaryotes and invading foreign DNA but also to predict microbial complexity and robustness.
References
Fineran PC, Charpentier E. Memory of viral infections by CRISPR-Cas adaptive immune systems: acquisition of new information. Virology. 2012;434:202–9.
García-Aljaro C, Ballesté E, Muniesa M. Beyond the canonical strategies of horizontal gene transfer in prokaryotes. Curr Opin Microbio. 2017;38:95–105.
Actis LA, Tolmasky ME, Crosa JH. Bacterial plasmids: replication of extrachromosomal genetic elements encoding resistance to antimicrobial compounds. Front Biosci. 1999;4:D43–62.
Burrus V. Mechanisms of stabilization of integrative and conjugative elements. Curr Opin Microbiol. 2017;38:44–50.
Gubbins MJ, Will WR, Frost LS. The F-plasmid, a paradigm for bacterial conjugation. In: Mullany P, editor. The dynamic bacterial genome. Cambridge: Cambridge University Press; 2005. p. 151–206.
Lederberg J, Cavalli LL, Lederberg EM. Sex compatibility in Escherichia coli. Genetics. 1952;37:720–30.
Bedhomme S, Perez Pantoja D, Bravo IG. Plasmid and clonal interference during post-horizontal gene transfer evolution. Mol Ecol. 2017;26:1832–47.
Schubert S, Darlu P, Clermont O, et al. Role of intraspecies recombination in the spread of pathogenicity islands within the Escherichia coli species. PLoS Pathog. 2009;5:e1000257.
García-Solache M, Lebreton F, McLaughlin RE, et al. Homologous recombination within large chromosomal regions facilitates acquisition of β-lactam and vancomycin resistance in Enterococcus faecium. Antimicrob Agents Chemother. 2016;60:5777–86.
Manson JM, Hancock LE, Gilmore MS. Mechanism of chromosomal transfer of Enterococcus faecalis pathogenicity island, capsule, antimicrobial resistance, and other traits. Proc Natl Acad Sci. 2010;107:12269–74.
Marosevic D, Kaevska M, Jaglic Z. Resistance to the tetracyclines and macrolide-lincosamide-streptogramin group of antibiotics and its genetic linkage—a review. Ann Agric Environ Med. 2017;24:338–44.
Martínez JL, Coque TM, Lanza VF, et al. Genomic and metagenomic technologies to explore the antibiotic resistance mobilome. Ann N Y Acad Sci. 2017;1388:26–41.
Jangam D, Feschotte C, Betrán E. Transposable element domestication as an adaptation to evolutionary conflicts. Trends Genet. 2017;33:817–31.
Wicker T, Sabo F, Hua-Van A, et al. Reply: a unified classification system for eukaryotic transposable elements should reflect their phylogeny. Nat Rev Genet. 2009;10:276.
Kapitonov VV, Jurka J. Self-synthesizing DNA transposons in eukaryotes. Proc Natl Acad Sci. 2006;103:4540–5.
Krupovic M, Koonin EV. Polintons: a hotbed of eukaryotic virus, transposon and plasmid evolution. Nat Rev Microbiol. 2015;13:105–15.
Pritham EJ, Putliwala T, Feschotte C. Mavericks, a novel class of giant transposable elements widespread in eukaryotes and related to DNA viruses. Gene. 2007;390:3–17.
Krupovic M, Koonin EV. Self-synthesizing transposons: unexpected key players in the evolution of viruses and defense systems. Curr Opin Microbiol. 2016;31:25–33.
Krupovic M, Makarova KS, Forterre P, et al. Casposons: a new superfamily of self-synthesizing DNA transposons at the origin of prokaryotic CRISPR-Cas immunity. BMC Biol. 2014;12:36.
Stokes HT, Hall RM. A novel family of potentially mobile DNA elements encoding site-specific gene-integration functions: integrons. Mol Microbiol. 1989;3:1669–83.
AlAmri AM. Integron in Gram negative bacteria: classes and fitness cost. Asian J Med Sci. 2017;8:1–4.
Gillings MR. Integrons: past, present, and future. Microbiol Mol Biol Rev. 2014;78:257–77.
Yu G, Li Y, Liu X, et al. Role of integrons in antimicrobial resistance: a review. Afr J Microbiol Res. 2013;7:1301–10.
Domingues S, Harms K, Fricke WF, et al. Natural transformation facilitates transfer of transposons, integrons and gene cassettes between bacterial species. PLoS Pathog. 2012;8:e1002837.
Clokie MRJ, Millard AD, Letarov A, et al. Phages in nature. Bacteriophage. 2011;1:31–45.
Weinbauer MG. Ecology of prokaryotic viruses. FEMS Microbiol Rev. 2004;28:127–81.
Salmond GPC, Fineran PC. A century of the phage: past, present and future. Nat Rev Microbiol. 2015;13:777–86.
Howard-Varona C, Hargreaves KR, Abedon ST, et al. Lysogeny in nature: mechanisms, impact and ecology of temperate phages. ISME J. 2017;11:1511–20.
Lemire S, Figueroa-Bossi N, Bossi L. Bacteriophage crosstalk: coordination of prophage induction by trans-acting antirepressors. PLoS Genet. 2011;7:e1002149.
Erez Z, Steinberger-Levy I, Shamir M, et al. Communication between viruses guides lysis–lysogeny decisions. Nature. 2017;541:488–93.
Ripp S, Miller RV. The role of pseudolysogeny in bacteriophage-host interactions in a natural freshwater environment. Microbiology. 1997;143:2065–70.
Rakonjac J, Bennett NJ, Spagnuolo J, et al. Filamentous bacteriophage: biology, phage display and nanotechnology applications. Curr Issues Mol Biol. 2011;13:51–76.
Messing J, Gronenborn B, Müller-Hill B, et al. Filamentous coliphage M13 as a cloning vehicle: insertion of a HindII fragment of the lac regulatory region in M13 replicative form in vitro. Proc Natl Acad Sci. 1977;74:3642–6.
Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228:1315–7.
Penadés JR, Chen J, Quiles-Puchalt N, et al. Bacteriophage-mediated spread of bacterial virulence genes. Curr Opin Microbiol. 2015;23:171–8.
Bondy-Denomy J, Davidson AR. When a virus is not a parasite: the beneficial effects of prophages on bacterial fitness. J Microbiol. 2014;52:235–42.
Freeman VJ. Studies on the virulence of bacteriophage-infected strains of Corynebacterium diphtheriae. J Bacteriol. 1951;61:675.
Lindsay JA, Ruzin A, Ross HF, et al. The gene for toxic shock toxin is carried by a family of mobile pathogenicity islands in Staphylococcus aureus. Mol Microbiol. 1998;29:527–43.
Martínez-Rubio R, Quiles-Puchalt N, Martí M, et al. Phage-inducible islands in the Gram-positive cocci. ISME J. 2017;11:1029–42.
Novick RP, Christie GE, Penadés JR. The phage-related chromosomal islands of Gram-positive bacteria. Nat Rev Microbiol. 2010;8:541–51.
Penadés JR, Christie GE. The phage-inducible chromosomal islands: a family of highly evolved molecular parasites. Annu Rev Virol. 2015;2:181–201.
Úbeda C, Maiques E, Knecht E, et al. A suggested nomenclature for bacterial host modification and restriction systems and their enzymes. Mol Microbiol. 2005;56:836–44.
Colavecchio A, Cadieux B, Lo A, et al. Bacteriophages contribute to the spread of antibiotic resistance genes among foodborne pathogens of the Enterobacteriaceae family—a review. Front Microbiol. 2017;8:1108.
Touchon M, de Sousa JAM, Rocha EPC. Embracing the enemy: the diversification of microbial gene repertoires by phage-mediated horizontal gene transfer. Curr Opin Microbiol. 2017;38:66–73.
Barrangou R, Horvath P. A decade of discovery: CRISPR functions and applications. Nat Microbiol. 2017;2:17092.
Koonin EV, Makarova KS, Wolf YI. Evolutionary genomics of defense systems in archaea and bacteria. Annu Rev Microbiol. 2017a;71:233–61.
Luria SE. Host-induced modifications of viruses. Cold Spring Harb Symp Quant Biol. 1953;18:237–44.
Luria SE, Human ML. A nonhereditary, host-induced variation of bacterial viruses. J Bacteriol. 1952;64:557.
Blow MJ, Clark TA, Daum CG, et al. The epigenomic landscape of prokaryotes. PLoS Genet. 2016;12:e1005854.
Smith HO, Nathans D. A suggested nomenclature for bacterial host modification and restriction systems and their enzymes. J Mol Biol. 1973;81:419–23.
Pingoud A, Jeltsch A. Recognition and cleavage of DNA by type-II restriction endonucleases. FEBS J. 1997;246:1–22.
Loenen WAM, Dryden DTF, Raleigh EA, et al. Type I restriction enzymes and their relatives. Nucleic Acids Res. 2014;42:20–44.
Roberts RJ, Belfort M, Bestor T, et al. A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res. 2003;31:1805–12.
Smith RM, Jacklin AJ, Marshall JJT, et al. Organization of the BcgI restriction–modification protein for the transfer of one methyl group to DNA. Nucleic Acids Res. 2013;41:405–17.
Hedgpeth J, Goodman HM, Boyer HW. DNA nucleotide sequence restricted by the RI endonuclease. Proc Natl Acad Sci. 1972;69:3448–52.
Mesapogu S, Jillepalli CM, Arora DK. Restriction enzymes and their role in microbiology. In: Dilip KA, Surajit D, Mesapogu S, editors. Analyzing microbes manual of molecular biology techniques. Berlin: Springer; 2013. p. 31–6.
Ishikawa K, Fukuda E, Kobayashi I. Conflicts targeting epigenetic systems and their resolution by cell death: novel concepts for methyl-specific and other restriction systems. DNA Res. 2010;17:325–42.
Loenen WA, Raleigh EA. The other face of restriction: modification-dependent enzymes. Nucleic Acids Res. 2013;42:56–69.
Chen C, Wang L, Chen S, et al. Convergence of DNA methylation and phosphorothioation epigenetics in bacterial genomes. Proc Natl Acad Sci. 2017b;114:4501–6.
Zhou X, Deng Z, Firmin JL, et al. Site-specific degradation of Streptomyces lividans DNA during electrophoresis in buffers contaminated with ferrous iron. Nucleic Acids Res. 1988;16:4341–52.
Lan W, Hu Z, Shen J, et al. Structural investigation into physiological DNA phosphorothioate modification. Sci Rep. 2016;6:25737.
Zheng T, Jiang P, Cao B, et al. DndEi exhibits helicase activity essential for DNA phosphorothioate modification and ATPase activity strongly stimulated by DNA substrate with a GAAC/GTTC motif. J Biol Chem. 2016;291:1492–500.
Hu W, Wang C, Liang J, et al. Structural insights into DndE from Escherichia coli B7A involved in DNA phosphorothioation modification. Cell Res. 2012;22:1203–6.
Wang L, Chen S, Vergin KL. DNA phosphorothioation is widespread and quantized in bacterial genomes. Proc Natl Acad Sci. 2011;108:2963–8.
Yao F, Xu T, Zhou X, et al. Functional analysis of spfD gene involved in DNA phosphorothioation in Pseudomonas fluorescens Pf0-1. FEBS Lett. 2009;583:729–33.
Xu T, Yao F, Zhou X, et al. A novel host-specific restriction system associated with DNA backbone S-modification in Salmonella. Nucleic Acids Res. 2010;38:7133–41.
Cao B, Cheng Q, Gu C, et al. Pathological phenotypes and in vivo DNA cleavage by unrestrained activity of a phosphorothioate-based restriction system in Salmonella. Mol Microbiol. 2014;93:776–85.
Yamaguchi Y, Park JH, Inouye M. Toxin-antitoxin systems in bacteria and archaea. Annu Rev Genet. 2011;45:61–79.
Pandey DP, Gerdes K. Toxin–antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res. 2005;33:966–76.
Gerdes K, Larsen JE, Molint S. Stable inheritance of plasmid R1 requires two different loci. J Bacteriol. 1985;161:292–8.
Gerdes K, Rasmussen PB, Molin S. Unique type of plasmid maintenance function: postsegregational killing of plasmid-free cells. Proc Natl Acad Sci. 1986;83:3116–20.
Ogura T, Hiraga S. Mini-F plasmid genes that couple host cell division to plasmid proliferation. Proc Natl Acad Sci. 1983;80:4784–8.
Gerdes K, Helin K, Christensen OW, et al. Translational control and differential RNA decay are key elements regulating postsegregational expression of the killer protein encoded by the parB locus of plasmid R1. J Mol Biol. 1988;203:119–29.
Harms A, Brodersen DE, Mitarai N, et al. Toxins, targets, and triggers: an overview of toxin-antitoxin biology. Mol Cell. 2018;70:768.
Page R, Peti W. Toxin-antitoxin systems in bacterial growth arrest and persistence. Nat Chem Biol. 2016;12:208–14.
Mruk I, Kobayashi I. To be or not to be: regulation of restriction–modification systems and other toxin–antitoxin systems. Nucleic Acids Res. 2014;42:70–86.
Ramage HR, Connolly LE, Cox JS. Comprehensive functional analysis of Mycobacterium tuberculosis toxin-antitoxin systems: implications for pathogenesis, stress responses, and evolution. PLoS Genet. 2009;5:e1000767.
Walter ND, Dolganov GM, Garcia BJ, et al. Transcriptional adaptation of drug-tolerant Mycobacterium tuberculosis during treatment of human tuberculosis. J Infect Dis. 2015;212:990–8.
Grynberg M, Erlandsen H, Godzik A. HEPN: a common domain in bacterial drug resistance and human neurodegenerative proteins. Trends Biochem Sci. 2003;28:224–6.
Makarova KS, Wolf YI, Koonin EV. Comprehensive comparative-genomic analysis of type 2 toxin-antitoxin systems and related mobile stress response systems in prokaryotes. Biol Direct. 2009a;4:19.
Yao J, Guo Y, Zeng Z, et al. Identification and characterization of a HEPN-MNT family type II toxin–antitoxin in Shewanella oneidensis. Microb Biotechnol. 2015;8:961–73.
Chen B, Akusobi C, Fang X, et al. Environmental T4-family bacteriophages evolve to escape abortive infection via multiple routes in a bacterial host employing “altruistic suicide” through type III toxin-antitoxin systems. Front Microbiol. 2017a;8:1006.
Labrie SJ, Samson JE, Moineau S. Bacteriophage resistance mechanisms. Nat Rev Microbiol. 2010;8:317–27.
Chopin MC, Chopin A, Bidnenko E. Phage abortive infection in lactococci: variations on a theme. Curr Opin Microbiol. 2005;8:473–9.
Anantharaman V, Makarova KS, Burroughs AM, et al. Comprehensive analysis of the HEPN superfamily: identification of novel roles in intra-genomic conflicts, defense, pathogenesis and RNA processing. Biol Direct. 2013;8:15.
Blower TR, Pei XY, Short FL, et al. A processed noncoding RNA regulates an altruistic bacterial antiviral system. Nat Struct Mol Bio. 2011;18:185–90.
Benzer S. Fine structure of a genetic region in bacteriophage. Proc Natl Acad Sci. 1955;41:344–54.
Parma DH, Snyder M, Sobolevski S, et al. The Rex system of bacteriophage lambda: tolerance and altruistic cell death. Genes Dev. 1992;6:497–510.
Snyder L. Phage-exclusion enzymes: a bonanza of biochemical and cell biology reagents? Mol Microbiol. 1995;15:415–20.
Makarova KS, Wolf YI, van der Oost J, et al. Prokaryotic homologs of Argonaute proteins are predicted to function as key components of a novel system of defense against mobile genetic elements. Biol Direct. 2009b;4:29.
Bohmert K, Camus I, Bellini C, et al. AGO1 defines a novel locus of Arabidopsis controlling leaf development. EMBO J. 1998;17:170–80.
Hutvagner G, Simard MJ. Argonaute proteins: key players in RNA silencing. Nat Rev Mol Cell Biol. 2008;9:22–32.
Wang Y, Juranek S, Li H, et al. Structure of an argonaute silencing complex with a seed-containing guide DNA and target RNA duplex. Nature. 2008a;456:921–6.
Wang Y, Sheng G, Juranek S, et al. Structure of the guide-strand-containing argonaute silencing complex. Nature. 2008b;456:209–13.
Peters L, Meister G. Argonaute proteins: mediators of RNA silencing. Mol Cell. 2007;26:611–23.
Shabalina SA, Koonin EV. Origins and evolution of eukaryotic RNA interference. Trends Ecol Evol. 2008;23:578–87.
Ketting RF. The many faces of RNAi. Dev Cell. 2011;20:148–61.
Kaya E, Doxzen KW, Knoll KR, et al. A bacterial Argonaute with noncanonical guide RNA specificity. Proc Natl Acad Sci. 2016;113:4057–62.
Swarts DC, Jore MM, Westra ER, et al. DNA-guided DNA interference by a prokaryotic Argonaute. Nature. 2014a;507:258–61.
Swarts DC, Makarova K, Wang Y, et al. The evolutionary journey of Argonaute proteins. Nat Struct Mol Biol. 2014b;21:743–53.
Hegge JW, Swarts DC, van der Oost J. Prokaryotic Argonaute proteins: novel genome-editing tools? Nat Rev Microbiol. 2018;16:5–11.
Swarts DC, Hegge JW, Hinojo I, et al. Argonaute of the archaeon Pyrococcus furiosus is a DNA-guided nuclease that targets cognate DNA. Nucleic Acids Res. 2015;43:5120–9.
Goldfarb T, Sberro H, Weinstock E, et al. BREX is a novel phage resistance system widespread in microbial genomes. EMBO J. 2015;34:169–83.
Barrangou R, van der Oost J. Bacteriophage exclusion, a new defense system. EMBO J. 2014;34:134–5.
Hoskisson PA, Sumby P, Smith MC. The phage growth limitation system in Streptomyces coelicolor A(3)2 is a toxin/antitoxin system, comprising enzymes with DNA methyltransferase, protein kinase and ATPase activity. Virology. 2015;477:100–9.
Chinenova TA, Mkrtumian NM, Lomovskaia ND. Genetic characteristics of a new phage resistance trait in Streptomyces coelicolor A3 (2). Genetika. 1982;18:1945–52.
Sumby P, Smith MCM. Genetics of the phage growth limitation (Pgl) system of Streptomyces coelicolor A3(2). Mol Microbiol. 2002;44:489–500.
Jansen R, van Embden JDA, Gaastra W, et al. Identification of a novel family of sequence repeats among prokaryotes. OMICS. 2002a;6:23–33.
Jansen R, Van Embden JDA, Gaastra W, et al. Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol. 2002b;43:1565–75.
Ishino Y, Shinagawa H, Makino K, et al. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol. 1987;169:5429–33.
Mojica FJM, Rodriguez-Valera F. The discovery of CRISPR in archaea and bacteria. FEBS J. 2016;283:3162–9.
Mojica FJ, Díez-Villaseñor C, Soria E, et al. Biological significance of a family of regularly spaced repeats in the genomes of Archaea, Bacteria and mitochondria. Mol Microbiol. 2000;36:244–6.
Deveau H, Barrangou R, Garneau JE, et al. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J Bacteriol. 2008;190:1390–400.
Bolotin A, Quinquis B, Sorokin A, et al. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiology. 2005;151:2551–61.
Mojica FJ, Díez-Villaseñor C, García-Martínez J, et al. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J Mol Evol. 2005;60:174–82.
Pourcel C, Salvignol G, Vergnaud G. CRISPR elements in Yersinia pestis acquire new repeats by preferential uptake of bacteriophage DNA, and provide additional tools for evolutionary studies. Microbiology. 2005;151:653–63.
Brouns SJJ, Jore MM, Lundgren M, et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321:960–4.
Barrangou R, Fremaux C, Deveau H, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–12.
Marraffini LA, Sontheimer EJ. CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science. 2008;322:1843–5.
Mojica FJM, Díez-Villaseñor C, García-Martínez J, et al. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology. 2009;155:733–40.
Amitai G, Sorek R. CRISPR-Cas adaptation: insights into the mechanism of action. Nat Rev Microbiol. 2016;14:67–76.
Marraffini LA. CRISPR-Cas immunity in prokaryotes. Nature. 2015;526:55–61.
Nuñez JK, Lee AS, Engelman A, et al. Integrase-mediated spacer acquisition during CRISPR-Cas adaptive immunity. Nature. 2015;519:193–8.
Carte J, Wang R, Li H, et al. Cas6 is an endoribonuclease that generates guide RNAs for invader defense in prokaryotes. Genes Dev. 2008;22:3489–96.
Heler R, Samai P, Model JW, et al. Cas9 specifies functional viral targets during CRISPR-Cas adaptation. Nature. 2015;519:199–202.
Koonin EV, Makarova KS. Mobile genetic elements and evolution of CRISPR-Cas systems: all the way there and back. Genome Biol Evol. 2017;9:2812–25.
Koonin EV, Makarova KS, Zhang F. Diversity, classification and evolution of CRISPR-Cas systems. Curr Opin Microbiol. 2017b;37:67–78.
Shmakov S, Smargon A, Scott D, et al. Diversity and evolution of class 2 CRISPR-Cas systems. Nat Rev Microbiol. 2017b;15:169–82.
Zhang Q, Ye Y. Not all predicted CRISPR-Cas systems are equal: isolated cas genes and classes of CRISPR like elements. BMC Bioinf. 2017;18:92.
Sinkunas T, Gasiunas G, Waghmare SP, et al. In vitro reconstitution of Cascade-mediated CRISPR immunity in Streptococcus thermophilus. EMBO J. 2013;32:385–94.
Haft DH, Selengut J, Mongodin EF, et al. A guild of 45 CRISPR-associated (Cas) protein families and multiple CRISPR/Cas subtypes exist in prokaryotic genomes. PLoS Comput Biol. 2005;1:e60.
Tamulaitis G, Venclovas Č, Siksnys V. Type III CRISPR-Cas immunity: major differences brushed aside. Trends Microbiol. 2017;25:49–61.
Reeks J, Naismith JH, White MF. CRISPR interference: a structural perspective. Biochem J. 2013;453:155–66.
Maris C, Dominguez C, Allain FHT. The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS J. 2005;272:2118–31.
Wang R, Li H. The mysterious RAMP proteins and their roles in small RNA-based immunity. Protein Sci. 2012;21:463–70.
Kojima KK, Kanehisa M. Systematic survey for novel types of prokaryotic retroelements based on gene neighborhood and protein architecture. Mol Biol Evol. 2008;25:1395–404.
Makarova KS, Grishin NV, Shabalina SA, et al. A putative RNA-interference-based immune system in prokaryotes: computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol Direct. 2006;1:7.
Simon DM, Zimmerly S. A diversity of uncharacterized reverse transcriptases in bacteria. Nucleic Acids Res. 2008;36:7219–29.
Toro N, Nisa-Martínez R. Comprehensive phylogenetic analysis of bacterial reverse transcriptases. PLoS One. 2014;9:e114083.
Silas S, Mohr G, Sidote DJ, et al. Direct CRISPR spacer acquisition from RNA by a natural reverse transcriptase-Cas1 fusion protein. Science. 2016;351:aad4234.
Silas S, Makarova KS, Shmakov S, et al. On the origin of reverse transcriptase-using CRISPR-Cas systems and their hyperdiverse, enigmatic spacer repertoires. MBio. 2017;8:e00897–17.
Sontheimer EJ, Barrangou R. The bacterial origins of the CRISPR genome-editing revolution. Hum Gene Ther. 2015;26:413–24.
Doudna JA, Charpentier E. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096.
Mali P, Yang L, Esvelt KM, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–6.
Ran FA, Hsu PD, Lin C-Y, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013a;154:1380–9.
Ran FA, Hsu PD, Wright J, et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013b;8:2281–308.
Deltcheva E, Chylinski K, Sharma CM, et al. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471:602–7.
Jinek M, Chylinski K, Fonfara I, et al. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–21.
Karvelis T, Gasiunas G, Miksys A, et al. crRNA and tracrRNA guide Cas9-mediated DNA interference in Streptococcus thermophilus. RNA Biol. 2013;10:841–51.
Chylinski K, Le Rhun A, Charpentier E. The tracrRNA and Cas9 families of type II CRISPR-Cas immunity systems. RNA Biol. 2013;10:726–37.
Gasiunas G, Barrangou R, Horvath P, et al. Cas9–crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci. 2012;109:E2579–86.
Yamano T, Nishimasu H, Zetsche B, et al. Crystal structure of Cpf1 in complex with guide RNA and target DNA. Cell. 2016;165:949–62.
Burstein D, Harrington LB, Strutt SC, et al. New CRISPR-Cas systems from uncultivated microbes. Nature. 2017;542:237–41.
Hickman AB, Dyda F. The casposon-encoded Cas1 protein from Aciduliprofundum boonei is a DNA integrase that generates target site duplications. Nucleic Acids Res. 2015;43:10576–87.
Krupovic M, Shmakov S, Makarova KS, et al. Recent mobility of casposons, self-synthesizing transposons at the origin of the CRISPR-Cas immunity. Genome Biol Evol. 2016;8:375–86.
Béguin P, Charpin N, Koonin EV, et al. Casposon integration shows strong target site preference and recapitulates protospacer integration by CRISPR-Cas systems. Nucleic Acids Res. 2016;44:10367–76.
Sontheimer EJ, Davidson AR. Inhibition of CRISPR-Cas systems by mobile genetic elements. Curr Opin Microbiol. 2017;37:120–7.
Chaudhary K, Chattopadhyay A, Pratap D. Anti-CRISPR proteins: counterattack of phages on bacterial defense (CRISPR/Cas) system. J Cell Physiol. 2017;233:57–9.
Bondy-Denomy J, Garcia B, Strum S, et al. Multiple mechanisms for CRISPR-Cas inhibition by anti-CRISPR proteins. Nature. 2015;526:136–9.
Samson JE, Magadán AH, Sabri M, et al. Revenge of the phages: defeating bacterial defences. Nat Rev Microbiol. 2013;11:675–87.
Krüger DH, Bickle TA. Bacteriophage survival: multiple mechanisms for avoiding the deoxyribonucleic acid restriction systems of their hosts. Microbiol Rev. 1983;47:345–60.
Koskella B, Brockhurst MA. Bacteria–phage coevolution as a driver of ecological and evolutionary processes in microbial communities. FEMS Microbiol Rev. 2014;38:916–31.
Makarova KS, Wolf YI, Snir S, et al. Defense islands in bacterial and archaeal genomes and prediction of novel defense systems. J Bacteriol. 2011;193:6039–56.
Puigbò P, Makarova KS, Kristensen DM, et al. Reconstruction of the evolution of microbial defense systems. BMC Evol Biol. 2017;17:94.
Ofir G, Melamed S, Sberro H, et al. DISARM is a widespread bacterial defence system with broad anti-phage activities. Nat Microbiol. 2018;3:90–8.
Vasu K, Nagaraja V. Diverse functions of restriction-modification systems in addition to cellular defense. Microbiol Mol Biol Rev. 2013;77:53–72.
Sánchez-Romero MA, Cota I, Casadesús J. DNA methylation in bacteria: from the methyl group to the methylome. Curr Opin Microbiol. 2015;25:9–16.
Srikhanta YN, Maguire TL, Stacey KJ, et al. The phasevarion: a genetic system controlling coordinated, random switching of expression of multiple genes. Proc Natl Acad Sci. 2005;102:5547–51.
Casadesus J, Low D. Epigenetic gene regulation in the bacterial world. Microbiol Mol Biol Rev. 2006;70:830–56.
Budroni S, Siena E, Hotopp JCD, et al. Neisseria meningitidis is structured in clades associated with restriction modification systems that modulate homologous recombination. Proc Natl Acad Sci. 2011;108:4494–9.
Nandi T, Holden MTG, Didelot X, et al. Burkholderia pseudomallei sequencing identifies genomic clades with distinct recombination, accessory, and epigenetic profiles. Genome Res. 2015;25:129–41.
Eutsey RA, Powell E, Dordel J, et al. Genetic stabilization of the drug-resistant PMEN1 Pneumococcus lineage by its distinctive DpnIII restriction-modification system. MBio. 2015;6:e00173–15.
Barrangou R. The roles of CRISPR-Cas systems in adaptive immunity and beyond. Curr Opin Immunol. 2015;32:36–41.
Okura M, Nozawa T, Watanabe T, et al. A locus encoding variable defense systems against invading DNA identified in Streptococcus suis. Genome Biol Evol. 2017;9:1000–12.
Ralph AP, Carapetis JR. Group a streptococcal diseases and their global burden. In: Chhatwal GS, editor. Host-pathogen interactions in streptococcal diseases. Berlin: Springer; 2012. p. 1–27.
Nakagawa I, Kurokawa K, Yamashita A, et al. Genome sequence of an M3 strain of Streptococcus pyogenes reveals a large-scale genomic rearrangement in invasive strains and new insights into phage evolution. Genome Res. 2003;13:1042–55.
Nozawa T, Furukawa N, Aikawa C, et al. CRISPR inhibition of prophage acquisition in Streptococcus pyogenes. PLoS One. 2011;6:e19543.
Aikawa C, Furukawa N, Watanabe T, et al. Complete genome sequence of the serotype k Streptococcus mutans strain LJ23. J Bacteriol. 2012;194:2754–5.
Maruyama F, Kobata M, Kurokawa K, et al. Comparative genomic analyses of Streptococcus mutans provide insights into chromosomal shuffling and species-specific content. BMC Genomics. 2009;10:358.
Endo A, Watanabe T, Ogata N, et al. Comparative genome analysis and identification of competitive and cooperative interactions in a polymicrobial disease. ISME J. 2015;9:629–42.
Watanabe T, Maruyama F, Nozawa T, et al. Complete genome sequence of the bacterium Porphyromonas gingivalis TDC60, which causes periodontal disease. J Bacteriol. 2011;193:4259–60.
Watanabe T, Nozawa T, Aikawa C, et al. CRISPR regulation of intraspecies diversification by limiting IS transposition and intercellular recombination. Genome Biol Evol. 2013;5:1099–114.
Watanabe T, Shibasaki M, Maruyama F, et al. Investigation of potential targets of Porphyromonas CRISPRs among the genomes of Porphyromonas species. PLoS One. 2017;12:e0183752.
Paster BJ, Olsen I, Aas JA, et al. The breadth of bacterial diversity in the human periodontal pocket and other oral sites. Periodontol 2000. 2006;42:80–7.
Shmakov SA, Sitnik V, Makarova KS, et al. The CRISPR spacer space is dominated by sequences from species-specific mobilomes. MBio. 2017a;8:e01397–17.
Sebaihia M, Wren BW, Mullany P, et al. The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat Genet. 2006;38:779–86.
Acknowledgements
This work was supported by the Ministry of Education, Culture, Sports, Science and Technology of Japan and the Japan Society for the Promotion of Science under Grants-in-Aid for Scientific Research (KAKENHI) (grant numbers 16H05830/16H05501/16H01782/16H02767), a Kurita Water and Environment Foundation Grant, an Ichiro Kanehara Foundation Scholarship Grant for Research in Basic Medical Sciences and Medical Care, a Senri Life Science Foundation Kishimoto Grant, a grant from the Japan Agency for Medical Research and Development (project number 965144), and a grant by JST/JICA, SATREPS.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Ito, T., Okura, M., Maruyama, F. (2019). Acquired and Innate Immunity in Prokaryotes Define Their Evolutionary Story. In: Nishida, H., Oshima, T. (eds) DNA Traffic in the Environment. Springer, Singapore. https://doi.org/10.1007/978-981-13-3411-5_3
Download citation
DOI: https://doi.org/10.1007/978-981-13-3411-5_3
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-3410-8
Online ISBN: 978-981-13-3411-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)