
Abstract
The propanediol utilization bacterial microcompartments are specialized protein-based organelles in Salmonella that facilitate the catabolism of 1,2-propanediol when available as the sole carbon source. This smart prokaryotic cell organelle compartmentalizes essential enzymes and substrates in a volume of a few attoliters compared to the femtoliter volume of a bacterial cell thereby enhancing the enzyme kinetics and properly orchestrating the downstream pathways. A shell or coat, which is composed of a few thousand protein subunits, wraps a chain of consecutively acting enzymes and serves as ducts for the diffusion of substrates, cofactors, and products into and out of the core of the microcompartment. In this article we bring together the properties of the wrappers of the propanediol utilization bacterial microcompartments to update our understanding on the mechanism of the formation of these unique wraps, their assembly, and interaction with the encapsulated enzymes.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Bacterial microcompartments (MCPs) are interesting genre of prokaryotic inclusions which were discovered 75 years ago by electron microscopy, and initially these structures were thought to be viruses inside bacteria. Till date, however, the MCPs have not shown any resemblance to any known viral capsid protein. Several genetic and molecular biology studies have confirmed that these structures are specialized chambers inside bacteria that aid metabolism by concentrating substrates and enzymes in a confined space (Cheng et al. 2011; Kerfeld et al. 2005; Shively et al. 1973a; Yeates et al. 2011, 2013). MCPs were first discovered in cyanobacteria and were named as carboxysomes because of their role in CO2 fixation by the Calvin cycle (G Drews 1956; Shively et al. 1973b). Later on, homologues of these carboxysomes were discovered in several bacteria, and the literature witnessed the reports on the characterization of at least seven different genres of MCPs in 16% of bacteria across 23 different phyla in 30 loci, and more are being discovered (Axen et al. 2014; Beeby et al. 2009; Farah Abdul-Rahman and Jeffrey 2013; Jorda et al. 2013; Sutter et al. 2017). All these different MCPs have similar structure which comprises of a protein shell that encapsulates a cluster of related enzymes for a particular function. Genomic and structural data indicate that the shell proteins are present in a wide range of bacteria, and there exists a high level of structural sophistication leading to the development of these wrappers of a definite size and shape in vivo. Genetic, biochemical, and microbiological studies on the MCPs indicate that the general function of these prokaryotic organelles is to optimize metabolic pathways having intermediates that are toxic to the cell or are volatile which can diffuse out freely through the cell membrane (Jorda et al. 2013, 2015; Kerfeld et al. 2010; Yeates et al. 2011, 2013). Recent biophysical studies have shed light on the topological and mechanical features of these organelles. Using the β-carboxysome as a paradigm, it has been shown that these organelles have a flexible organization and soft mechanical properties as compared to rigid viruses (Faulkner et al. 2017).
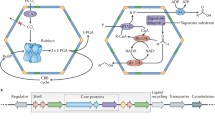
The general scheme for a MCP action involves movement of a metabolic substrate from the cytoplasm to the MCP core where it is acted upon by a series of enzymes along with the formation of a toxic intermediate (Havemann et al. 2002; Penrod and Roth 2006; Rondon et al. 1995). The outer wrapper proteins of the MCPs play a crucial role in trapping this intermediate inside and channelizing it to downstream enzymes for further processing (Dou et al. 2008; Havemann et al. 2002; Price and Badger 1989; Rondon et al. 1995). As and when required, the product(s) move out of the MCP, where they enter the central metabolism of the cell thereby providing energy for cell growth. The basic function of the MCPs thus is to segregate molecular processes leading to proper channelization of the nutrients in an energy-efficient approach. Among the common MCPs reported till date, the carboxysomes play a major role in carbon fixation, while the Eut (ethanolamine utilization) and the Pdu (1,2-propanediol utilization) MCPs act as metabolosomes for the utilization of specific substrates and are crucial for development and dissemination of enteric pathogens. The carboxysomes account for close to 25% of the carbon fixation of the nature and are present in almost all bacteria using the Calvin cycle for carbon utilization (Kerfeld et al. 2010; Rae et al. 2013). Carboxysomes encapsulate two enzymes: carbonic anhydrase and RuBisCO. The carbonic anhydrase transforms bicarbonate to produce CO2 which is then fixed to ribulose-1,5-bisphosphate by the enzyme RuBisCO (Chen et al. 2013). Sequestration by the carboxysomes allows the concentration of CO2 to be elevated in the immediate vicinity of RuBisCO, improving its catalytic efficiency (Dou et al. 2008; Price et al. 2008; Rae et al. 2013). The other main MCPs, Pdu-MCP and Eut-MCP found in the enteric pathogen Salmonella, help the organism to grow in the infected intestine leading to their propagation in new hosts (Thiennimitr et al. 2011; Winter and Baumler 2011; Winter et al. 2010). Both 1,2-propanediol and ethanolamine are the by-products of the incomplete utilization of fucose or rhamnose in the intestine (Badía et al. 1985; Baldoma and Aguilar 1988). The Pdu- and Eut-MCPs operate by sequestering toxic/volatile catabolic intermediate like propionaldehyde during 1,2-propanediol utilization (in Pdu-MCPs) or formaldehyde during ethanolamine utilization (in Eut-MCPs). A schematic representation of the action of the Pdu-MCPs is shown in Fig. 23.1. Apart from enhancing the enzyme activity by compartmentalization, the MCPs also help to reduce cytotoxicity, DNA damage, and the loss of valuable nutrient source.

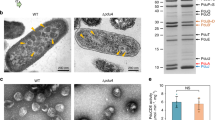
Pdu-MCP. (a) Schematic representation of the pdu operon in Salmonella enterica Serovar Typhimurium LT2. (b) Schematic representation of the Pdu-MCP structure and functional pathway indicating the role of the enzymes. (c) Electron micrographs and (d) SDS-PAGE of purified Pdu-MCP from LT2 strain [Sinha Lab]
All the MCPs reported till date have shown an icosahedral structure, though deviations from a perfect icosahedra have been observed in some cases (Bobik 2006; Cannon et al. 2001; Cheng et al. 2008; Shively et al. 1973a). The core elements in assembly of a MCP include the formation of the external shell or wrap with the concurrent encapsulation of the enzyme cluster inside. The wrap or the envelope is made up of two different kinds of proteins: hexagonal proteins that make up the faces of the polyhedral structure and pentagonal proteins that make up the vertices of the polyhedra. The hexagonal shell proteins belong to the family of bacterial microcompartment domain (BMC) which is typically about 90 amino acids long and have an α/β fold, while the vertex proteins belong to the BMV (bacterial microcompartment vertex) family of proteins (Kerfeld et al. 2010; Yeates et al. 2011). The BMC domain hexagonal proteins self-assemble into a flat disc-shaped hexagonal structure that forms the basic building block of the external wrap. In most of the shell proteins, the two sides of the disc have different properties with respect to their hydrophobicity and curvature. These discs typically have a concave hydrophobic surface which homes the C-termini and the N-termini of the proteins and has been suggested to be facing the MCP lumen (Tanaka et al. 2009). These C-termini and N-termini are involved in the interactions with the luminal enzymes leading to the proper orchestration of the MCP functions (Fan et al. 2010; Lehman et al. 2017). Although there is no direct evidence on the arrangement of the shell proteins of the outer wrap of the MCPs, several studies hypothesize that the wrap is made up of mixed sheets composed of the multiple BMC proteins. A single shell thus is composed of few thousand different proteins of 4–8 types (depending on the type of MCP), and each type bears a special functional role. The pentagonal vertex proteins belong to the family of BMV (bacterial microcompartment vertex) proteins and are crucial for the closure of the polyhedra. This unique architecture of the MCPs helps in efficient encapsulation of enzymes, substrates, and cofactors in a volume as low as an attoliter compared to the femtoliter volume of the cell and offers an energy-efficient paradigm for bacteria to survive under special conditions. Lessons from the MCP structure-function relationship have of late become very intriguing in developing novel synthetic bioreactors.
Followed by carboxysomes, the most studied MCPs are the Pdu-MCPs in Salmonella which aid the vitamin B12-dependent metabolism of 1,2-propanediol (1,2-PD). The Pdu-MCPs grow only in the presence of 1,2-PD as the carbon source. The Pdu operon in Salmonella encodes all the shell proteins and the associated 1,2-PD-degrading enzymes (Chen et al. 1994) (Fig. 23.1a). In this chapter we will focus on the Pdu-MCPs with special emphasis on the properties of the protein wraps and their mechanism of interaction with the internal enzyme cluster.
2 Pdu-MCP Envelope
The present understanding of the Pdu-MCP is that the outer wrap is composed of eight structural proteins, PduA, PduB, PduB’, PduJ, PduK, PduT, PduU, and PduN. PduM has also been suggested to be a part of the shell, although its role is unclear (Sinha et al. 2012). The shell proteins PduA, PduB, PduB’, PduJ, PduK, PduT, and PduU belong to the family of bacterial microcompartment (BMC) domain and are hexagonal in shape. PduN belongs to the class of pentagonal BMV proteins and forms the vertices of MCPs. While PduA, PduJ, and PduU are hexamers of six monomeric BMC domains, PduB/B’ and PduT are trimers of two tandem BMC domains. The BMC domain hexagonal proteins have pores at the center which are thought to play crucial roles in the selective transport of enzyme substrates, products, and cofactors (Kerfeld et al. 2005). 2D-IEF-SDS-PAGE have identified PduA, PduB, PduB’, and PduJ as major components of the Pdu-MCP shell while PduK, PduN, PduT, and PduU as minor components (Havemann and Bobik 2003). Genetic studies suggest that among the shell proteins, PduB/B’, PduJ, and PduN are crucial for the assembly of the Pdu-MCP, and deletion of any of these genes leads to the formation of nonfunctional Pdu-MCPs (Cheng et al. 2011). In the following few paragraphs, we will discuss each of the shell proteins and its role in wrapping the internal enzyme cluster.
3 Shell Proteins of Pdu-MCP
3.1 PduA
PduA protein comprises 16% of the entire shell of the Pdu-MCP and has been the most studied shell protein in the Pdu-MCP system (Havemann and Bobik 2003; Havemann et al. 2002). This protein is 94 amino acid long and is encoded by the pduA gene of the pdu operon. It is a hexamer with the monomers containing only one BMC domain. The crystal structure of PduA shows adjacent hexamers interacting together to form an extended two-dimensional molecular sheet. These sheets are stabilized by specific interactions of a number of conserved edge amino acids (K26, N29, and R79) and are crucial for the architecture of the whole MCP envelope (Pang et al. 2014; Sinha et al. 2014). The mutation K26A in PduA leads to the complete abolition of the Pdu-MCP (Fig. 23.2), while the mutations N29A and R79A lead to the formation of enlarged and porous Pdu-MCPs. The crystal structure of the PduA-K26A mutant protein hexamer assemblies displays two crystal forms 1 and 2. These crystal forms when superimposed on the wild-type PduA hexamer structure showed RMSDs of 1.0 Å and 0.9–1.2 Å, respectively, suggesting that PduA-K26A protein had no major conformational differences compared to wild-type PduA. However, the PduA-K26A mutant did not form extended protein sheets in crystals in contrast to the wild-type PduA (Fig. 23.2). One crystal form of PduA-K26A forms one-dimensional sheets leading to the formation of a three-dimensional shell protein assembly that cannot form the MCP facet. Hence, the mutant proteins cannot be incorporated in the MCPs leading to the non-formation of the polyhedral structures. It is also envisaged that PduA-K26A protein may interact with the internal enzyme cluster similar to the wild-type protein leading to non-closure of the shell and eventually a damaged architecture of the MCPs. Stability studies done at the individual protein level also demonstrate lower transition temperature for the K26A mutant compared to the wild type (Sinha Lab, unpublished results). It has also been shown that these K26 and R79 residues stabilize the PduA assembly in bacterial cytoplasm as well (Pang et al. 2014). PduA protein presents a central pore of 0.6 nm diameter which is lined by the backbone amides of the residues G39 and S40 and side chain of S40 from each of the six monomers. This pore has been shown to be conduit for transport of 1,2-PD across the MCP envelope (Chowdhury et al. 2015). Molecular dynamics simulations study has shown that the pore of PduA hexamer has a lower-energy barrier for the movement of 1,2-PD, compared to the movement of toxic metabolite propionaldehyde (Park et al. 2017). The anchoring of 1,2-PD in the pore occurs by the formation of two hydrogen bonds between the 1,2-PD and the pore-lining atoms, which is less probable for propionaldehyde. A ΔpduA developed in Salmonella shows enlarged Pdu-MCPs compared to the wild type along with growth arrest due to propionaldehyde toxicity and an increased growth rate at limiting vitamin B12 concentrations. These studies suggest the importance of PduA protein in the integrity and function of the Pdu-MCPs (Cheng et al. 2011; Havemann et al. 2002; Sinha et al. 2014).

Crystal structure of PduA (PDB ID-3NGK) showing interactions of adjacent hexamers to form an extended two-dimensional molecular sheet. The mutation K26A (PDB ID-4PPD) disrupts these edge-to-edge interactions, preventing the formation of an extended sheet
3.2 PduB/PduB'
Close to one-half of the Pdu shell is made up of PduB (28%) and PduB’ (25%). These two proteins are formed by a pair of overlapping genes pduB/B’ in the pdu operon with their start sites 111 base pairs apart, resulting in extra 37 amino acids at the N-terminal of PduB’. The role of this extra 37 amino acid is still unclear. It has been suggested by Lehman et al. that these 37 amino acids play a role in the association of the shell with the internal enzyme cluster (Lehman et al. 2017). The PduB/B' proteins have tandem BMC domains, and the biological assembly is a trimer containing six BMC domains. These oligomeric assemblies in the shell proteins are known as pseudohexamers. Our understanding of the PduB is based on the crystal structure of PduB from Lactobacillus reuteri, which is the only solved crystal structure (PDB ID-4FAY) of the protein so far (Pang et al. 2012). PduB protein is related to those BMC domain proteins that have a gated pore like the EutL protein or the CsoS1D in Eut-MCP and α-carboxysome, respectively (Klein et al. 2009; Sagermann et al. 2009; Takenoya et al. 2010). Both the EutL and CsoS1D proteins display two conformations in the crystal structures: open central pore and closed central pore which implicates crucial roles of these tandem BMC repeats in gated transport of materials in and out of the MCPs. The domain duplication allows greater flexibility involved in the dynamics of pore activity. The crystal structure of PduB from L. reuteri, however, shows only a closed pore, and each unit of the trimer binds to one molecule of glycerol suggesting an involvement in substrate transport. PduB from the Lactobacillus reuteri also forms extended two-dimensional sheets in crystals, though the intersheet contact is reported to be out of plane, indicating a lower stability. Modelling studies have confirmed that PduB and PduA form mixed sheets where the edges completely align suggesting better packing of the proteins as expected in the physiological shell. Salmonella strains with deleted pduB genes from the pdu operon are unable to form the MCPs. Interestingly, the electron micrographs of pduB-deleted Salmonella cells show protein deposits without the presence of polyhedral MCPs. This suggests that in the absence of the PduB/B' proteins, the shell cannot be formed, and enzyme clusters with fragments of the shell remain as protein inclusions inside the cell. These deletion mutants also show the phenotype of porous shell with aldehyde toxicity and retarded growth, indicating a nonfunctional shell. A recent study shows that N-terminal 37 amino acids of the PduB shell protein are crucial in binding the central enzyme core to the shell and provide insights into the sidedness (Lehman et al. 2017).
3.3 PduJ
PduJ protein covers close to 22% of the Pdu shell and shares greater than 80% sequence identity with PduA protein. The crystal structures of PduA and PduJ overlap with same pore-lining and edge contact residues. Interestingly, however, it has been demonstrated that PduJ has no role in the transport of 1,2-PD across the shell. By elegant gene replacement studies in the chromosome locus, Choudhury et al. have shown that when pduA is replaced by pduJ, the latter acquires the function of the former (Chowdhury et al. 2016). These studies suggest that the locus of the gene on the pdu operon decides the function of the gene under question. This study is one of the first studies in the field that relates the function of a gene with its location. The pduJ deletion in the MCPs leads to the formation of elongated MCPs in contrast to the porous MCPs in case of pduA deletion. The elongated MCP advocates have faulty closure of faces of the MCP polyhedra, and hence it is hypothesized that PduJ forms the edges that join the facets of the Pdu-MCPs. However, studies on the effect of the replacement of the pduJ gene by pduA gene on the joining of facets of Pdu-MCPs have not yet been reported.
3.4 PduK
PduK is an interesting shell protein with an N-terminal (~90 amino acids) consisting of the MCP domain fold and a C-terminal (~70 amino acids) of unknown function. This protein is known to bind iron (Cheng et al. 2011). The C-terminal has a Fe–S-binding motif (residues 130–152) which suggests that this protein might be involved in some redox or electron transfer reactions at the Pdu-MCP surface (Crowley et al. 2010). However, there are no experimental evidences to support this hypothesis. Unlike the pduB/B’ deletion which leads to a nonfunctional strain, the pduK deletion leads to a functionally active MCP with an aggregated morphology (Cheng et al. 2011). Based on these observations, it has been inferred that PduK is involved in the segregation of MCPs in vivo especially during cell division. However, more experimental evidences are required to strengthen this hypothesis.
3.5 PduT
The PduT has two interesting characteristics: it is a pseudohexameric protein consisting of 184-residue-long tandem domains of the canonical (i.e., non-permuted) variety of the α/β BMC fold connected by a short linker sequence. The first domain is made up of residues 1–88, while the second domain comprises of residues 94–184 (Pang et al. 2011). The two tandem domains of PduT although conform closely to the canonical BMC fold and align with each other with RMSD = 1.14, their sequences are only 24% identical to each other. PduT forms only 3% of the entire shell, and unlike the other shell proteins, it does not form an extended sheet as observed in the crystal structures. Another interesting feature of the PduT protein is the presence of 4[Fe–S] cluster at the central pore (Crowley et al. 2010). The other proteins in the Pdu-MCP having the [Fe–S] centers are PduS and PduK (Chowdhury et al. 2014). EPR studies on a PduT homologue from C. freundii have confirmed the presence of a [4Fe–4S] cluster. The pore region of PduT has three cysteine residues C38, one from each subunit. The [4Fe–4S] cluster has an essentially cubic structure, and the modeled placement of the cluster orients it with its cubic body diagonal along the threefold axis of symmetry down the center of the pore in the PduT trimer. The metal cluster and its three cysteine ligands obey this symmetry. In addition, the [4Fe–4S] cluster calls for a fourth S-ligand to an iron atom lying on the threefold symmetry. This fourth S-atom in the 4[Fe–S] center has not yet been identified. The[4Fe–4S] cluster in PduT is reported to be accessible from both the luminal and the cytoplasmic side of the MCP and suggests a possible role in assisting redox chemistry. In the crystal structure of PduT, C-108 and C-136 are located close enough to form a disulfide bond. However the side chains are positioned in such a way that the disulfide bond is not formed. It is suggested that the formation of this disulfide bond due to rotamer conformational changes might be resulting in a redox-sensitive change in the pore properties. These cysteine positions are conserved in a sequence alignment of PduT homologues, while they are absent in other Pdu shell proteins. As of now the role of redox coupling in the Pdu-MCP function is not obvious; however, meticulous studies on the structure-activity relationship of PduT might suggest crucial phenomenon. Mutations in PduT have been reported to marginally weaken the growth of Salmonella on 1,2-PD and do not significantly affect MCP structure or function. All these taken together suggest that PduT might be important for Salmonella under different physiochemical conditions. Like PduB, PduT is also a protein with tandem BMC domains; however, unlike PduB, PduT does not form extended sheets in crystal structures (Crowley et al. 2010).
3.6 PduU
PduU is a minor component of the Pdu shell. This hexameric protein displays a circularly permuted BMC domain fold and fails to form hexagonal layers in crystal structures. PduU forms the blocked pore, bendable type of the shell protein (Crowley et al. 2008) and bears structural similarity with the EutS protein of the Eut-MCP (Tanaka et al. 2010). On the convex side of the central pore, PduU is capped by a parallel six-stranded β-barrel structure which is composed of one β-sheet from each of the monomers. Due to the presence of the β-barrel, the central pore of PduU is predicted to be not involved in any transport mechanism. However, Jorda et al. (2015) have shown that PduV, a Pdu enzyme with unknown function, binds to this β-barrel structure of PduU through its N-terminal region. This binding interaction might play a role in targeting PduV to Pdu-MCPs (Jorda et al. 2015). The concave face of PduU has a deep cavity lined by several hydrophobic residues. These residues are suggested to interact with luminal proteins, though it is not certain if this side of the PduU protein faces the lumen of the Pdu-MCP.
3.7 PduN
PduN is a bacterial microcompartment vertex protein which is a pentamer, and the closest analogues are the CcmL protein in carboxysomes, the EutN protein in the Eut-MCPs, or the GrpN protein in Grp-MCP (Wheatley et al. 2013). These vertex proteins form the vertices of the MCPs and help to form a closed polyhedral structure. PduN is the least abundant protein in the Pdu-MCP and is detected only by Western blotting or in vivo fluorescent tagging. This less abundance of PduN is expected, as a perfect icosahedra will have only 12 vertices. It has been reported that the copy numbers of the vertex proteins in a MCP is ~60 which is very less as compared to a few thousand copies of flat facet proteins. Although less abundant, PduN plays a crucial role in delivering the closed structure of the MCPs, and the deletion of pduN gene results in the formation of totally abnormal Pdu-MCPs with completely altered functions during growth on 1,2-PD (Cheng et al. 2011). This observation holds true for related vertex proteins in the β-carboxysomes as well (Cai et al. 2009).
3.8 PduM
PduM is the only shell protein that is devoid of a BMC or a BMV domain and yet is crucial for the assembly of the Pdu-MCPs. This protein does not have any analogue in the carboxysome of the Eut-MCP system. It is a 163-amino acid-long protein of unknown function, the deletion of which leads to the formation of abnormal nonfunctional Pdu-MCPs. Electron micrographs demonstrate abnormal protein aggregates in a pduM-deleted Salmonella strain, while biochemical assays suggest a nonfunctional MCP with phenotype of propionaldehyde toxicity (Sinha et al. 2012).
4 Interactions Between Pdu Shell Proteins and Core Enzymes
The Pdu-MCPs encapsulate at least five different enzymes inside the lumen (PduCDE, PduP, PduO, PduGH, and PduS) (Crowley et al. 2008; Sinha et al. 2012). The last decade has shed a lot of light on the underlying principles for the self-assembly of MCPs especially on how the enzymes are encapsulated inside the protein shell. The role of N-terminal extensions or the encapsulation peptides of the core enzymes has been shown to be very significant in this regard (Aussignargues et al. 2015). For the Pdu-MCP, the first report demonstrated that the N-terminal of PduP (propionaldehyde dehydrogenase) protein is crucial for its encapsulation within the MCP. The N-terminal extension of PduP when tagged to heterologous proteins such as green fluorescent protein (GFP) resulted in their successful entrapment within Pdu microcompartment (Fan et al. 2010). Scanning mutagenesis studies have revealed that the residues E7, I10, and L14 in the N-terminal of PduP are essential for the encapsulation of PduP. Further, in silico modelling has shown that the N-terminal of PduP interacts with the C-terminal of PduA, a shell protein (Fan et al. 2012). In another study, it has been shown that the first 18 amino acids in the N-terminal of the medium subunit (PduD) are necessary and sufficient for the encapsulation of PduCDE (diol dehydratase) complex (Chowdhury et al. 2014). Impaired encapsulation of this enzyme has been observed upon deletion of N-terminal extension of PduD subunit (Fan and Bobik 2011). However, specific binding interaction between PduCDE and any of the shell proteins needs more experimental evidences.
Beside molecular biology approach, computational and bioinformatics approaches have enabled us to look into the depth of the protein assembly in microcompartments. Computational modelling has predicted various interactions between shell proteins and core enzymes of Pdu microcompartment. The Pdu interactome model prepared using coevolution-based methods suggests that the shell protein PduA acts as a “universal hub” for directing the core enzymes PduC, PduD, PduE, PduL, and PduP into the microcompartment. These enzymes have N-terminal extensions which bind to a cleft on the concave face of PduA hexamer (Jorda et al. 2015). Three important predictions have been made in this model that needs to be analyzed carefully. First, the enzyme PduP binds to only two of the shell proteins, namely, PduA and PduJ. This has been confirmed earlier by the co-elution experiments conducted by Fan and co-workers, where only PduA and PduJ co-eluted with His-tagged PduP (Fan et al. 2012). Second, all the three subunits of PduCDE diol dehydratase binds to PduA hexamer through their N-terminal extensions. This explains the impaired encapsulation of PduCDE within the microcompartment upon deletion of N-terminal of PduD subunit (Fan and Bobik 2011). However, it has also been observed that the chromosomal deletion of PduA or PduJ has no effect on the diol dehydratase activity of the purified microcompartments (Sinha et al. 2014), which suggests an association of PduCDE with the Pdu microcompartment even in the absence of PduA or PduJ. This can be explained by the fact that PduA is 80% identical to PduJ in terms of amino acid sequence (Crowley et al. 2010). It is plausible that the void created by the deletion of PduA is taken up by architecturally similar PduJ; but there is a lack of evidence in this regard. Also, deletion of PduJ results in the formation of an impaired and elongated microcompartment (Cheng et al. 2011), and therefore, increased diol dehydratase activity of purified microcompartments in case of chromosomal deletion of PduJ must be studied in this context. Third, the shell protein PduB doesn’t bind to any of the core enzymes. This is indeed thought-provoking, as a recent study has shown the importance of N-terminal of PduB' in binding shell protein to the core enzymes. Substituting isoleucine with a hydrophilic amino acid, threonine (IΔT), at the tenth position in the N-terminal of PduB' has a deleterious effect on the encapsulation of core enzymes, including PduCDE and PduP (Lehman et al. 2017). This severe depletion in the concentration of core enzyme within microcompartments in IΔT mutants indicates the importance of hydrophobic residue isoleucine in core–shell interaction. It can be hypothesized that the shell protein PduB' has no specific interaction with any of the core enzymes but has an overall global impact in retaining the core enzymes within Pdu microcompartment through hydrophobic interactions. This hypothesis is based on the fact that PduB and PduB' constitute 50% of the Pdu shell proteins (Havemann and Bobik 2003). Therefore, deletion of isoleucine at the tenth residue of PduB' may critically reduce the hydrophobic environment within the microcompartment as PduB accounts for almost 50% of the shell. This may result in a diminished retention of core enzymes within the microcompartment. However, experimental evidences are needed to support this hypothesis. Although it appears that the encapsulation strategies for the enzymes inside the Pdu-MCPs follow a common mechanism, apart from PduCDE and PduP, there are no reports on the encapsulation of other enzymes like PduG, PduH, PduO, PduS, and PduQ. An interesting area of exploration would be identification of the interacting shell protein-enzyme pair. This would lead to a better understanding of the packaging principles of the megadalton-sized MCPs.
5 Conclusions and Perspectives
The literature till date has a virtuous amount of information regarding the overall shape, size, and structure of the bacterial microcompartments. Genetic and biochemical studies on the Pdu-MCPs have enhanced our understanding regarding the interaction of the shell proteins and internal enzyme clusters. However, these understandings can be reinforced using co-crystallization experiments with the shell proteins and the enzymes. Further, in vitro binding studies between the shell proteins and the enzymes will shed light on the intensity and dynamics of these interactions. Strategies to mimic the in vivo shell protein assemblies in vitro also require some attention. At present only one substrate-shell protein pair (1,2-propanediol-PduA) has been identified for the Pdu-MCPs. The entry routes for vitamin B12 and other chemicals like NADH and ATP still remain unexplored. Computational studies with respect to modelling and docking the shell protein and cofactors may provide useful insights into the structure-function activity of the individual shell proteins. With respect to translational approaches, these genre of organelles have a vast prospective to be applied as nanoreactors for biofuel or pharmaceuticals production or as containers for storage of useful biomaterials once the evolution, transport, and dynamics are comprehensively demonstrated.
References
Aussignargues C, Paasch BC, Gonzalez-Esquer R, Erbilgin O, Kerfeld CA (2015) Bacterial microcompartment assembly: the key role of encapsulation peptides. Commun Integr Biol 8:e1039755
Axen SD, Erbilgin O, Kerfeld CA (2014) A taxonomy of bacterial microcompartment loci constructed by a novel scoring method. PLoS Comput Biol 10:e1003898
Badía J, Ros J, Aguilar J (1985) Fermentation mechanism of fucose and rhamnose in Salmonella typhimurium and Klebsiella pneumoniae. J Bacteriol 161:435–437
Baldoma L, Aguilar J (1988) Metabolism of L-fucose and L-rhamnose in Escherichia coli: aerobic-anaerobic regulation of L-lactaldehyde dissimilation. J Bacteriol 170:416–421
Beeby M, Bobik TA, Yeates TO (2009) Exploiting genomic patterns to discover new supramolecular protein assemblies. Protein Sci 18:69–79. https://doi.org/10.1002/pro.1
Bobik TA (2006) Polyhedral organelles compartmenting bacterial metabolic processes. Appl Microbiol Biotechnol 70:517–525. https://doi.org/10.1007/s00253-005-0295-0
Cai F, Menon BB, Cannon GC, Curry KJ, Shively JM, Heinhorst S (2009) The pentameric vertex proteins are necessary for the icosahedral carboxysome shell to function as a CO2 leakage barrier. PLoS One 4:e7521
Cannon GC, Bradburne CE, Aldrich HC, Baker SH, Heinhorst S, Shively JM (2001) Microcompartments in prokaryotes: carboxysomes and related polyhedra. Appl Environ Microbiol 67:5351–5361
Chen P, Andersson DI, Roth JR (1994) The control region of the pdu/cob regulon in Salmonella typhimurium. J Bacteriol 176:5474–5482
Chen AH, Robinson-Mosher A, Savage DF, Silver PA, Polka JK (2013) The bacterial carbon-fixing organelle is formed by shell envelopment of preassembled cargo. PLoS One 8:e76127
Cheng S, Liu Y, Crowley CS, Yeates TO, Bobik TA (2008) Bacterial microcompartments: their properties and paradoxes. BioEssays 30:1084–1095
Cheng S, Sinha S, Fan C, Liu Y, Bobik TA (2011) Genetic analysis of the protein shell of the microcompartments involved in coenzyme B12-dependent 1,2-propanediol degradation by Salmonella. J Bacteriol 193:1385–1392
Chowdhury C, Sinha S, Chun S, Yeates TO, Bobik TA (2014) Diverse bacterial microcompartment organelles. Microbiol Mol Biol Rev 78:438–468
Chowdhury C, Chun S, Pang A, Sawaya MR, Sinha S, Yeates TO, Bobik TA (2015) Selective molecular transport through the protein shell of a bacterial microcompartment organelle. Proc Natl Acad Sci U S A 112:2990–2995. https://doi.org/10.1111/mmi.13423
Chowdhury C, Chun S, Sawaya MR, Yeates TO, Bobik TA (2016) The function of the PduJ microcompartment shell protein is determined by the genomic position of its encoding gene. Mol Microbiol 101:770–783. https://doi.org/10.1128/jb.00785-16
Crowley CS, Sawaya MR, Bobik TA, Yeates TO (2008) Structure of the PduU shell protein from the Pdu microcompartment of Salmonella. Structure 16:1324–1332
Crowley CS, Cascio D, Sawaya MR, Kopstein JS, Bobik TA, Yeates TO (2010) Structural insight into the mechanisms of transport across the Salmonella enterica Pdu microcompartment shell. J Biol Chem 285:37838–37846
Dou Z, Heinhorst S, Williams EB, Murin CD, Shively JM, Cannon GC (2008) CO2 fixation kinetics of Halothiobacillus neapolitanus mutant carboxysomes lacking carbonic anhydrase suggest the shell acts as a diffusional barrier for CO2. J Biol Chem 283:10377–10384. https://doi.org/10.1074/jbc.M709285200
Fan C, Bobik TA (2011) The N-terminal region of the medium subunit (PduD) packages adenosylcobalamin-dependent diol dehydratase (PduCDE) into the Pdu microcompartment. J Bacteriol 193:5623–5628
Fan C et al (2010) Short N-terminal sequences package proteins into bacterial microcompartments. Proc Natl Acad Sci 107:7509–7514
Fan C, Cheng S, Sinha S, Bobik TA (2012) Interactions between the termini of lumen enzymes and shell proteins mediate enzyme encapsulation into bacterial microcompartments. Proc Natl Acad Sci 109:14995–15000
Farah Abdul-Rahman EP, Jeffrey LB (2013) The distribution of polyhedral bacterial microcompartments suggests frequent horizontal transfer and operon reassembly. J Phylogenet Evol Biol 1:1–7. https://doi.org/10.4172/2329-9002.1000118
Faulkner M et al (2017) Direct characterization of the native structure and mechanics of cyanobacterial carboxysomes. Nanoscale 9:10662–10673. https://doi.org/10.1039/c7nr02524f
Drews G, Niklowitz W (1956) Beiträge zur Cytologie der Blaualgen. II. Zentroplasma und granulare Einschlüsse von Phormidium uncinatum. Arch Mikrobiol 24(2):147–162
Havemann GD, Bobik TA (2003) Protein content of polyhedral organelles involved in coenzyme B12-dependent degradation of 1,2-propanediol in Salmonella enterica serovar typhimurium LT2. J Bacteriol 185:5086–5095
Havemann GD, Sampson EM, Bobik TA (2002) PduA is a shell protein of polyhedral organelles involved in coenzyme B(12)-dependent degradation of 1,2-propanediol in Salmonella enterica serovar typhimurium LT2. J Bacteriol 184:1253–1261
Jorda J, Lopez D, Wheatley NM, Yeates TO (2013) Using comparative genomics to uncover new kinds of protein-based metabolic organelles in bacteria. Protein Sci 22:179–195. https://doi.org/10.1002/pro.2196
Jorda J, Liu Y, Bobik TA, Yeates TO (2015) Exploring bacterial organelle interactomes: a model of the protein-protein interaction network in the Pdu microcompartment. PLoS Comput Biol 11:e1004067
Kerfeld CA, Sawaya MR, Tanaka S, Nguyen CV, Phillips M, Beeby M, Yeates TO (2005) Protein structures forming the shell of primitive bacterial organelles. Science 309:936–938. https://doi.org/10.1126/science.1113397
Kerfeld CA, Heinhorst S, Cannon GC (2010) Annu Rev Microbiol 64:391–408. https://doi.org/10.1146/annurev.micro.112408.134211
Klein MG et al (2009) Identification and structural analysis of a novel carboxysome shell protein with implications for metabolite transport. J Mol Biol 392:319–333. https://doi.org/10.1016/j.jmb.2009.03.056
Lehman BP, Chowdhury C, Bobik TA (2017) The N terminus of the PduB protein binds the protein shell of the Pdu microcompartment to its enzymatic core. J Bacteriol 199:e00785–e00716
Pang A, Warren MJ, Pickersgill RW (2011) Structure of PduT, a trimeric bacterial microcompartment protein with a 4Fe–4S cluster-binding site. Acta Crystallogr D Biol Crystallogr 67:91–96
Pang A, Liang M, Prentice MB, Pickersgill RW (2012) Substrate channels revealed in the trimeric lactobacillus reuteri bacterial microcompartment shell protein PduB. Acta Crystallogr D Biol Crystallogr 68:1642–1652. https://doi.org/10.1107/s0907444912039315
Pang A, Frank S, Brown I, Warren MJ, Pickersgill RW (2014) Structural insights into higher order assembly and function of the bacterial microcompartment protein PduA. J Biol Chem 289:22377–22384. https://doi.org/10.1074/jbc.M114.569285
Park J, Chun S, Bobik TA, Houk KN, Yeates TO (2017) Molecular dynamics simulations of selective metabolite transport across the propanediol bacterial microcompartment shell. J Phys Chem B 121:8149–8154. https://doi.org/10.1021/acs.jpcb.7b07232
Penrod JT, Roth JR (2006) Conserving a volatile metabolite: a role for carboxysome-like organelles in Salmonella enterica. J Bacteriol 188:2865–2874. https://doi.org/10.1128/JB.188.8.2865-2874.2006
Price GD, Badger MR (1989) Expression of human carbonic anhydrase in the cyanobacterium Synechococcus PCC7942 creates a high CO(2)-requiring phenotype : evidence for a central role for Carboxysomes in the CO(2) concentrating mechanism. Plant Physiol 91:505–513
Price GD, Badger MR, Woodger FJ, Long BM (2008) Advances in understanding the cyanobacterial CO2-concentrating-mechanism (CCM): functional components, ci transporters, diversity, genetic regulation and prospects for engineering into plants. J Exp Bot 59:1441–1461. https://doi.org/10.1093/jxb/erm112
Rae BD, Long BM, Badger MR, Price GD (2013) Functions, compositions, and evolution of the two types of carboxysomes: polyhedral microcompartments that facilitate CO2 fixation in cyanobacteria and some proteobacteria. Microbiol Mol Biol Rev 77:357–379
Rondon MR, Horswill AR, Escalante-Semerena JC (1995) DNA polymerase I function is required for the utilization of ethanolamine, 1,2-propanediol, and propionate by Salmonella typhimurium LT2. J Bacteriol 177:7119–7124
Sagermann M, Ohtaki A, Nikolakakis K (2009) Crystal structure of the EutL shell protein of the ethanolamine ammonia lyase microcompartment. Proc Natl Acad Sci U S A 106:8883–8887
Shively JM, Ball F, Brown DH, Saunders RE (1973a) Functional organelles in prokaryotes: polyhedral inclusions (carboxysomes) of Thiobacillus neapolitanus. Science 182:584–586
Shively JM, Ball FL, Kline BW (1973b) Electron microscopy of the carboxysomes (polyhedral bodies) of Thiobacillus neapolitanus. J Bacteriol 116:1405–1411
Sinha S, Cheng S, Fan C, Bobik TA (2012) The PduM protein is a structural component of the microcompartments involved in coenzyme B(12)-dependent 1,2-propanediol degradation by Salmonella enterica. J Bacteriol 194:1912–1918
Sinha S, Cheng S, Sung YW, McNamara DE, Sawaya MR, Yeates TO, Bobik TA (2014) Alanine scanning mutagenesis identifies an asparagine–arginine–lysine triad essential to assembly of the shell of the Pdu microcompartment. J Mol Biol 426:2328–2345
Sutter M, Greber B, Aussignargues C, Kerfeld CA (2017) Assembly principles and structure of a 6.5-MDa bacterial microcompartment shell. Science 356:1293–1297. https://doi.org/10.1126/science.aan3289
Takenoya M, Nikolakakis K, Sagermann M (2010) Crystallographic insights into the pore structures and mechanisms of the EutL and EutM shell proteins of the ethanolamine-utilizing microcompartment of Escherichia coli. J Bacteriol 192:6056–6063
Tanaka S, Sawaya MR, Phillips M, Yeates TO (2009) Insights from multiple structures of the shell proteins from the beta-carboxysome. Protein Sci 18:108–120
Tanaka S, Sawaya MR, Yeates TO (2010) Structure and mechanisms of a protein-based organelle in. E coli Sci 327:81–84. https://doi.org/10.1126/science.1179513
Thiennimitr P et al (2011) Intestinal inflammation allows Salmonella to use ethanolamine to compete with the microbiota. Proc Natl Acad Sci U S A 108:17480–17485
Wheatley NM, Gidaniyan SD, Liu Y, Cascio D, Yeates TO (2013) Bacterial microcompartment shells of diverse functional types possess pentameric vertex proteins. Protein Sci 22:660–665
Winter SE, Baumler AJ (2011) A breathtaking feat: to compete with the gut microbiota, Salmonella drives its host to provide a respiratory electron acceptor. Gut Microbes 2:58–60
Winter SE et al (2010) Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature 467:426–429
Yeates TO, Thompson MC, Bobik TA (2011) The protein shells of bacterial microcompartment organelles. Curr Opin Struct Biol 21:223–231. https://doi.org/10.1016/j.sbi.2011.01.006
Yeates TO, Jorda J, Bobik TA (2013) The shells of BMC-type microcompartment organelles in bacteria. J Mol Microbiol Biotechnol 23:290–299. https://doi.org/10.1159/000351347
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Bari, N.K., Kumar, G., Sinha, S. (2018). The Wrappers of the 1,2-Propanediol Utilization Bacterial Microcompartments. In: Chattopadhyay, K., Basu, S. (eds) Biochemical and Biophysical Roles of Cell Surface Molecules. Advances in Experimental Medicine and Biology, vol 1112. Springer, Singapore. https://doi.org/10.1007/978-981-13-3065-0_23
Download citation
DOI: https://doi.org/10.1007/978-981-13-3065-0_23
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-3064-3
Online ISBN: 978-981-13-3065-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)