
Abstract
Protein-protein interactions (PPIs) provide a rich source of drug targets for the development of new generation of clinical therapeutics. However, targeting PPIs by small-molecule inhibitors remains a significant challenge due to large, flat, and hydrophobic features of the PPI interfaces. Recently, important advances have been made in the discovery and development of small-molecule PPI inhibitors. This chapter aims to give an overview of the structural features of PPIs as well as the design strategies of small-molecule PPI inhibitors. Moreover, PPI inhibitors under clinical development will be briefly introduced and two successful examples in PPI-based drug development (venetoclax and lifitegrast) will be highlighted.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Protein-protein interactions
- Small-molecule inhibitors
- Structural epitopes
- Hot spots
- Fragment-based drug design
- Bcl-2 inhibitor venetoclax
- LFA-1/ICAM-1 inhibitor lifitegrast
1 Introduction
Protein-protein interactions (PPIs) regulate a number of biological processes both in normal and disease states [1]. It is estimated that human interactome, the complex network of PPIs, contains about 130,000–650,000 types of PPI [2, 3]. The pivotal importance of PPIs makes them a rich source of targets for the development of novel therapeutic drugs. There are several ways to modulate PPI complexes including inhibition, stabilization, direct binding, and allosteric binding. A direct PPI modulator binds to the interaction surface of one protein, thereby sterically preventing or stabilizing the binding to its protein partner. In contrast, an allosteric modulator binds at a distant region outside of the protein interaction interface and remotely acts on the protein binding by triggering conformational change. For the type of binding effect, PPI inhibitors compete with one of the protein partners and prevent its binding, which represent a major approach in current PPI-based drug discovery. Another way to interfere with the PPI-associated biological functions is the stabilization of PPI complexes. PPI stabilizers bind to the regions at or near the PPI interface and promote the binding without competing with any of the protein partners [4]. Also, the therapeutic effects of PPI stabilizers are attracting increasing research interests [4,5,6]. Currently, most PPI modulators in clinical development are small-molecule inhibitors [7] and this chapter will mainly focus on design strategies and case studies of small-molecule PPI inhibitors.
2 Challenges in the Discovery of Small-Molecule PPI Inhibitors
Although PPIs represent promising targets for the development of new generation of clinical therapeutics, the design of selective and potent small-molecule inhibitors is rather changeling as compared to that for traditional targets (e.g., proteinase, kinases, G protein-coupled receptors) [8,9,10]. The nature of the PPI interfaces is significantly different from that of traditional drug targets, which have well-defined pockets for binding small molecules [11]. The PPI interface is generally large (about 1,500–3,000 Å2), flat (often exposed to solvent) [12], and dominated with hydrophobic and charged characteristics [13, 14]. Such features bring difficulties to discover small molecules that can effectively interrupt PPIs. First, a potent PPI inhibitor is required to have large molecular weight (MW) and high hydrophobicity so that it can cover a large and hydrophobic surface area. However, such a binder may have poor solubility and face pharmacokinetic problems. Second, the natural binder of a specific PPI interface is the protein counterpart itself, whose amino acids involved in PPIs are not contiguous. Thus, the protein or peptide involved in PPI cannot be used as a good starting point for the design and identification of small-molecule inhibitors. Third, existing compound libraries are mainly collected or constructed for traditional drug targets, which cannot effectively cover the chemical space of PPI inhibitors. Therefore, it is highly challenging to find a high-quality hit or lead through high-throughput screening (HTS) of PPI inhibitors. Moreover, validation of a PPI inhibitor from artifactual binding requires more biological assays than that for traditional targets.
3 Structures and Classifications of PPIs
Despite these challenges, remarkable progress has been achieved in the discovery and development of small-molecule PPI inhibitors [15,16,17]. The knowledge of the topological features of PPI interfaces is critically important for the identification of small-molecule inhibitors. Generally, PPI interface consists of a core region and a rim region [18]. According to the PPI buried surface area and binding affinity, PPI interfaces can be generally classified into four categories: “tight and wide,” “tight and narrow,” “loose and narrow,” and “loose and wide” [19]. The properties and examples of the four classes of PPIs are depicted in Table 1.1 and Fig. 1.1. Among them, the “narrow and tight” PPIs are more druggable to design small-molecule inhibitors, whereas the “loose and wide” is the most difficult to be targeted by small molecules.

Topological features of four classes of PPIs according to the PPI contact area and binding affinity
In Arkin’s review, PPIs can be classified into primary peptide epitopes, secondary structure epitopes, and tertiary structural epitopes according to the complexity of epitopes (Table 1.2) [20]. The difficulties in identifying small-molecule inhibitors increase as the interface becomes more complex (from primary to tertiary epitopes). The primary peptide epitope consists of a primary linear protein as one side of the interface sequence. This type of PPI interface is particularly amenable to be targeted by drug-like small molecules. The secondary structure epitopes mainly include α-helix, β-sheet, and extended peptides. Key residues on the peptide are not continuous in the primary sequence, which are centered on two to three subpockets. The secondary structural epitopes have also been proven tractable to small-molecule inhibition. Tertiary structural epitopes require multiple sequences with discontinuous binding sites, which are the most challenging targets with limited successful examples.
More recently, Skidmore et al. divided PPIs into a series of structural classes including globular protein–globular protein interactions, globular protein–peptide interactions, and peptide–peptide interactions [21]. These structural classes can be further differentiated depending on whether the peptides have continuous epitope or undergo substantial conformational changes upon binding (Fig. 1.2). PPIs between globular proteins are highly challenging for small-molecule drug discovery [22]. In contrast, globular protein–peptide interactions have been proven to be more druggable. The difficulty in targeting peptide–peptide interactions depends on whether there is a defined binding site.

Classification of PPIs and examples. a Bcl-XL–BAD (PDB ID: 2XA0); b XIAP–SMAC (PDB ID: 1G73); c KEAP1 (Kelch-like ECH-associated protein 1)-NRF2 (nuclear factor erythroid 2-related factor 2) (PDB ID: 2DYH); d bromodomains (PDB ID: 3UVW); e IL-2–IL-2R (PDB ID: 1Z92); f MYC–MAX (PDB ID: 1NKP)
4 “Hot Spots” as Structural Basis for the Design of Small-Molecule PPI Inhibitors
The concept of “hot spots” in PPI interfaces was introduced by Clackson and Wells in 1995 [23], which means a few key residues are responsible for the majority of the binding free energy in PPI [18, 23]. Notably, the surface area of “hot spots” is significantly smaller than the entire PPI interface [24]. Recent studies indicated that the existence of “hot spots” was prevalent in PPI interfaces [18]. They account for an average of 9.5% of the interfacial residues and are generally located in the core regions [25]. Another important feature of “hot spots” is the conformational change upon binding small molecules [26, 27]. When a small molecule binds to the PPI interface, the opening of so-called transient pockets that facilitate the ligand binding can be observed [28, 29]. “Hot spot” residues are highly adaptive with low energy barriers for conformational changes. The flexibility of “hot spots” for small-molecules binding has been observed in a number of PPI targets such as IL-2 receptor α (IL-2Rα) [28], Bcl-XL [30], HDM2 [31], and HPV-18-E2 [32]. “Hot spot” residues are often enriched in tryptophan (21%), arginine (13.3%), and tyrosine (12.3%), which allow adaptive conformational change and form various interactions to accommodate small molecules [25]. The existence and dynamic features of “hot spots” offer an opportunity to underscore the challenge to identify small-molecule PPI inhibitors.
The “hot spots” of PPI interfaces can be determined by alanine scanning mutagenesis [23, 25, 33] in combination with structural biology studies. The former can measure the contribution of each residue to the binding affinity with the partner protein by serially mutating each residue to alanine. Moreover, X-ray crystallography enables to provide key structural information about the distribution and orientation of these hot spot residues in PPI interfaces. Also, solving crystal structures of the target protein in free state and in complex with different ligands is helpful to understand the dynamic properties of the “hot spots,” which is highly valuable for inhibitor design. Computational methods, such as molecular dynamics (MD) simulations, are complementary tools to investigate the dynamic features of the “hot spots” [34,35,36,37,38,39].
5 Overview of Strategies for the Design of Small-Molecule PPI Inhibitors
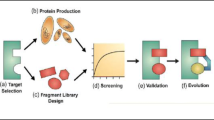
Recently, important progress has been made in the discovery and development of small-molecule PPI inhibitors [20, 39,40,41,42,43,44,45,46,47,48,49,50,51]. Herein, state-of-the-art strategies of PPI-based drug discovery will be briefly introduced. More details about the advantages and limitations of the methodologies and successful examples can refer to our recent review [52]. The first step for the discovery of small-molecule PPI inhibitors is to determine the structure of the PPI interface (Fig. 1.3) [53]. Due to the flexibility of PPI interface, the availability of structures from different statuses (unbound protein, protein-protein complex, protein–inhibitors complex) can significantly improve the efficiency of inhibitor design. Then, druggability assessment is necessary to evaluate whether the protein has well-defined binding sites or pockets to accommodate small molecules because the success in designing PPI inhibitors largely depends on the target type [54]. The next important step is the identification of the “hot spots” on the PPI interface [55, 56]. After establishing assays for biological evaluation, the strategy to discover initial hits depends on the properties of PPI hot spots. Currently, screening strategies, designing strategies, and synthetic strategies represent three major methods for small-molecule PPI inhibitor discovery. Screening strategies mainly include HTS, fragment screening, and virtual screening [39], which aim to discover PPI inhibitors from known compound libraries. Among them, fragment screening in combination with fragment-based drug design (FBDD) has the advantages of higher hit rate and better ligand efficiency (LE). Designing strategies focus on building novel small molecules to mimic the key interactions of the hot spot residues, which are used as the starting points for substructure search, bioisostere design, and de novo design. Besides hot spot residues, key secondary structure motif (i.e., α-helix, β-turn, and β-strand) involved in PPI interface can also be for inhibitor design. A new scaffold decorated with the side chains of hot spot residues is designed to mimic spatial orientation and interactions of the original secondary structure. Synthetic strategies aim to explore new chemical space for PPI inhibitor screening by developing efficient synthetic methods to construct new libraries with chemical diversity and complexity. When initial hits are available, validation studies are necessary to exclude false positives. Secondary assays to determine the kinetic and thermodynamic parameters (e.g., association and dissociation rates) as well as solving the structures of protein–hit complexes are important for selecting suitable hits for further optimization. Structural optimization of the hits and leads aims to improve the binding affinity, therapeutic effects, and drug-likeness, and the strategies are similar to those for traditional targets. Finally, drug candidates can be obtained for preclinical and clinical trials until they are marked for therapeutic application.

Current strategies for the design and development of small-molecule PPI inhibitors
6 Small-Molecule PPI Inhibitors Under Clinical Development
The discovery and development of PPIs inhibitors have been greatly accelerated with better understanding of the structure and functions of PPIs and numerous medicinal chemistry efforts in this field [57]. Up to now, a great number of highly potent small-molecule PPI inhibitors have been identified and several of them are marketed or under different stages of clinical evaluations. According to a recent review by Abell and Skidmore [21], small-molecule PPI inhibitors in clinical development are summarized in Table 1.3. On April 11, 2016, venetoclax (ABT-199) was approved by FDA for the treatment of chronic lymphocytic leukemia (CLL) with 17p deletion, which represents the first marketed small-molecule PPI inhibitor [58]. Subsequently, lifitegrast (SAR 1118) [59], a small-molecule inhibitor of LFA-1/ICAM-1, was approved for the treatment of dry eye syndrome on July 11, 2016. Here, the drug discovery and medicinal chemistry optimization process of venetoclax and lifitegrast were briefly introduced.
6.1 Fragment-Based Discovery of Bcl-2 Inhibitor Venetoclax
The discovery and development of Bcl-2 inhibitor venetoclax represent one of the most successful examples of PPI-based drug discovery [91]. Bcl (B-cell lymphoma) family of proteins (e.g., Bcl-XL, Bcl-2, Bcl-w, and Mcl-1) is anti-apoptotic proteins, whose interactions with pro-apoptotic proteins such as Bak, Bax, and Bad play key roles in both normal and abnormal apoptotic processes [92]. Initial drug discovery efforts were focused on the non-selective Bcl-2/Bcl-XL/Bcl-w inhibitor navitoclax (ABT-263) [93]. NMR structural studies revealed that the Bcl-XL/BAK interface was a long and hydrophobic groove [94]. The “hot spots” include several hydrophobic and charged residues (e.g., Ile85, Leu78, and Asp83). ABT-263 was discovered by a combination of NMR-based fragment screening, parallel synthesis and structure-based design and ADME optimization. Initially, the research group from Abbot screened a library containing 10,000 fragments using 15N HSQC NMR spectroscopy [95]. Weak fragment hits 31 (Ki = 300 μM) and 32 (Ki = 4,300 μM) were found to occupy two different subsites of Bcl-XL (Fig. 1.4) [96]. Guided by the structural information of Bcl-XL/fragment complexes, the two fragment hits were linked by the acylsulfonamide group to maintain the key interactions and followed by optimization of the substitution of acylsulfonamide to yield inhibitor 33 (Ki = 36 μM) with improved affinity with Bcl-XL. Further structure-based optimization of lead compound 33 led to the discovery of ABT-737 (34) as a highly potent Bcl-XL inhibitor (Ki ≤ 1 nM), which successfully mimicked the α-helical BH3 domain of BAK [30, 97, 98]. However, ABT-737 is not orally bioavailable and subsequent medicinal chemistry optimizations generated clinical candidate ABT-263 (35) [99]. ABT-263 showed subnanomolar affinities for Bcl-2, Bcl-XL, and Bcl-W with improved bioavailability [100]. Surprisingly, ABT-263 performed poorly in clinical trials probably due to its non-selective profile. Thus, further clinical evaluations were performed on the selective Bcl-2 inhibitor venetoclax [101, 102]. Venetoclax selectively blocks Bcl-2 protein, leading to programmed cell death of CLL cells.

Fragment-based design of Bcl-2 inhibitor venetoclax (a). The binding modes of the Bcl-XL inhibitors (b–d) were generated from the crystal structures in PDB database (PDB codes: 1YSG, 1YSI, and 2YXJ)
6.2 Discovery of LFA-1/ICAM-1 Inhibitor Lifitegrast for the Treatment of Dry Eye Syndrome
The PPI between LFA-1 and ICAM-1 is essential in lymphocyte and immune system function. Small-molecule LFA-1/ICAM-1 inhibitors can be used to develop novel drugs for the treatment of dry eye. The ICAM-1 epitope containing discontinuous residues Glu34, Lys39, Met64, Tyr66, Asn68, and Gln73 was identified as “hot spots.” Gadek et al. designed compounds 36 (IC50 = 47 nM) and 37 (IC50 = 1.4 nM) as potent LFA-1 inhibitors. Their structures were embedded with the carboxylic acid, sulfide, phenol, and carboxamide groups to mimic the ICAM-1 hot spots (Fig. 1.5) [103]. Structure–activity relationship (SAR) analysis revealed that the inhibitors shared a “left-wing”–“central scaffold”–“right-wing” structural mode. Based on this assumption, Zhong et al. designed bicyclic tetrahydroisoquinoline (THIQ) as the central scaffold and discovered a highly active LFA-1 inhibitor 38 (IC50 = 9 nM) with good in vivo efficacy [104]. Further optimization studies were focused on improving pharmacokinetic profiles and successfully discovered lifitegrast (9) as a new drug for the treatment of dry eye [59, 105].

Drug design process of LFA-1/ICAM-1 inhibitor lifitegrast
7 Conclusions
With the progress of structural biology studies of PPIs, the identification of “hot spots” for inhibitor design as well as numerous medicinal chemistry efforts, the development of small-molecule PPI inhibitors has come into reality with two marketed drugs and a number of clinical candidates. The encouraging success has attracted increasing interests and activities from both pharmaceutical industry and academia. Deeper understanding of the structures, functions, and dynamics of PPIs is highly desirable to improve the efficiency of PPI-based drug discovery. Also, the drug design principles and drug-like criteria for PPI inhibitors need to be further investigated. Taken together, with increasing knowledge and experience gained for small-molecule PPI inhibitors, more challenging PPI targets will become accessible to drug discovery. It is expected that more PPI inhibitors will come into clinical application in the near future.
References
Arkin MR, Wells JA (2004) Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat Rev Drug Discov 3(4):301–317
Stumpf MP, Thorne T, de Silva E, Stewart R, An HJ, Lappe M, Wiuf C (2008) Estimating the size of the human interactome. Proc Natl Acad Sci USA 105(19):6959–6964
Venkatesan K, Rual JF, Vazquez A, Stelzl U, Lemmens I, Hirozane-Kishikawa T, Hao T, Zenkner M, Xin X, Goh KI, Yildirim MA, Simonis N, Heinzmann K, Gebreab F, Sahalie JM, Cevik S, Simon C, de Smet AS, Dann E, Smolyar A, Vinayagam A, Yu H, Szeto D, Borick H, Dricot A, Klitgord N, Murray RR, Lin C, Lalowski M, Timm J, Rau K, Boone C, Braun P, Cusick ME, Roth FP, Hill DE, Tavernier J, Wanker EE, Barabasi AL, Vidal M (2009) An empirical framework for binary interactome mapping. Nat Methods 6(1):83–90
Thiel P, Kaiser M, Ottmann C (2012) Small-molecule stabilization of protein-protein interactions: an underestimated concept in drug discovery? Angew Chem Int Ed Engl 51(9):2012–2018
Milroy LG, Bartel M, Henen MA, Leysen S, Adriaans JM, Brunsveld L, Landrieu I, Ottmann C (2015) Stabilizer-Guided Inhibition of Protein-Protein Interactions. Angew Chem Int Ed Engl 54(52):15720–15724
Zarzycka B, Kuenemann MA, Miteva MA, Nicolaes GA, Vriend G, Sperandio O (2016) Stabilization of protein-protein interaction complexes through small molecules. Drug Discov Today 21(1):48–57
Mullard A (2012) Protein-protein interaction inhibitors get into the groove. Nat Rev Drug Discov 11(3):173–175
Whitty A, Kumaravel G (2006) Between a rock and a hard place? Nat Chem Biol 2(3):112–118
Chene P (2006) Drugs targeting protein-protein interactions. ChemMedChem 1(4):400–411
Fry DC (2008) Drug-like inhibitors of protein-protein interactions: a structural examination of effective protein mimicry. Curr Protein Pept Sci 9(3):240–247
Cheng AC, Coleman RG, Smyth KT, Cao Q, Soulard P, Caffrey DR, Salzberg AC, Huang ES (2007) Structure-based maximal affinity model predicts small-molecule druggability. Nat Biotechnol 25(1):71–75
Blundell TL, Burke DF, Chirgadze D, Dhanaraj V, Hyvonen M, Innis CA, Parisini E, Pellegrini L, Sayed M, Sibanda BL (2000) Protein-protein interactions in receptor activation and intracellular signalling. Biol Chem 381(9–10):955–959
Jones S, Thornton JM (1996) Principles of protein-protein interactions. Proc Natl Acad Sci USA 93(1):13–20
Lo Conte L, Chothia C, Janin J (1999) The atomic structure of protein-protein recognition sites. J Mol Biol 285(5):2177–2198
Wells JA, McClendon CL (2007) Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature 450(7172):1001–1009
Buchwald P (2010) Small-molecule protein-protein interaction inhibitors: therapeutic potential in light of molecular size, chemical space, and ligand binding efficiency considerations. IUBMB Life 62(10):724–731
Jubb H, Higueruelo AP, Winter A, Blundell TL (2012) Structural biology and drug discovery for protein-protein interactions. Trends Pharmacol Sci 33(5):241–248
Moreira IS, Fernandes PA, Ramos MJ (2007) Hot spots–a review of the protein-protein interface determinant amino-acid residues. Proteins 68(4):803–812
Smith MC, Gestwicki JE (2012) Features of protein-protein interactions that translate into potent inhibitors: topology, surface area and affinity. Expert Rev Mol Med 14:e16
Arkin MR, Tang Y, Wells JA (2014) Small-molecule inhibitors of protein-protein interactions: progressing toward the reality. Chem Biol 21(9):1102–1114
Scott DE, Bayly AR, Abell C, Skidmore J (2016) Small molecules, big targets: drug discovery faces the protein-protein interaction challenge. Nat Rev Drug Discov 15(8):533–550
Jubb H, Blundell TL, Ascher DB (2015) Flexibility and small pockets at protein-protein interfaces: new insights into druggability. Prog Biophys Mol Biol 119(1):2–9
Clackson T, Wells JA (1995) A hot spot of binding energy in a hormone-receptor interface. Science 267(5196):383–386
Keskin O, Gursoy A, Ma B, Nussinov R (2008) Principles of protein-protein interactions: what are the preferred ways for proteins to interact? Chem Rev 108(4):1225–1244
Bogan AA, Thorn KS (1998) Anatomy of hot spots in protein interfaces. J Mol Biol 280(1):1–9
DeLano WL, Ultsch MH, de Vos AM, Wells JA (2000) Convergent solutions to binding at a protein-protein interface. Science 287(5456):1279–1283
Atwell S, Ultsch M, De Vos AM, Wells JA (1997) Structural plasticity in a remodeled protein-protein interface. Science 278(5340):1125–1128
Arkin MR, Randal M, DeLano WL, Hyde J, Luong TN, Oslob JD, Raphael DR, Taylor L, Wang J, McDowell RS, Wells JA, Braisted AC (2003) Binding of small molecules to an adaptive protein-protein interface. Proc Natl Acad Sci USA 100(4):1603–1608
Eyrisch S, Helms V (2009) What induces pocket openings on protein surface patches involved in protein-protein interactions? J Comput Aided Mol Des 23(2):73–86
Bruncko M, Oost TK, Belli BA, Ding H, Joseph MK, Kunzer A, Martineau D, McClellan WJ, Mitten M, Ng SC, Nimmer PM, Oltersdorf T, Park CM, Petros AM, Shoemaker AR, Song X, Wang X, Wendt MD, Zhang H, Fesik SW, Rosenberg SH, Elmore SW (2007) Studies leading to potent, dual inhibitors of Bcl-2 and Bcl-xL. J Med Chem 50(4):641–662
Grasberger BL, Lu T, Schubert C, Parks DJ, Carver TE, Koblish HK, Cummings MD, LaFrance LV, Milkiewicz KL, Calvo RR, Maguire D, Lattanze J, Franks CF, Zhao S, Ramachandren K, Bylebyl GR, Zhang M, Manthey CL, Petrella EC, Pantoliano MW, Deckman IC, Spurlino JC, Maroney AC, Tomczuk BE, Molloy CJ, Bone RF (2005) Discovery and cocrystal structure of benzodiazepinedione HDM2 antagonists that activate p53 in cells. J Med Chem 48(4):909–912
Wang Y, Coulombe R, Cameron DR, Thauvette L, Massariol MJ, Amon LM, Fink D, Titolo S, Welchner E, Yoakim C, Archambault J, White PW (2004) Crystal structure of the E2 transactivation domain of human papillomavirus type 11 bound to a protein interaction inhibitor. J Biol Chem 279(8):6976–6985
Cunningham BC, Wells JA (1989) High-resolution epitope mapping of hGH-receptor interactions by alanine-scanning mutagenesis. Science 244(4908):1081–1085
Kozakov D, Hall DR, Chuang GY, Cencic R, Brenke R, Grove LE, Beglov D, Pelletier J, Whitty A, Vajda S (2011) Structural conservation of druggable hot spots in protein-protein interfaces. Proc Natl Acad Sci USA 108(33):13528–13533
Beglov D, Hall DR, Brenke R, Shapovalov MV, Dunbrack RL Jr, Kozakov D, Vajda S (2012) Minimal ensembles of side chain conformers for modeling protein-protein interactions. Proteins 80(2):591–601
Yang CY, Wang S (2011) Hydrophobic binding hot spots of Bcl-xL protein-protein interfaces by cosolvent molecular dynamics simulation. ACS Med Chem Lett 2(4):280–284
Klepeis JL, Lindorff-Larsen K, Dror RO, Shaw DE (2009) Long-timescale molecular dynamics simulations of protein structure and function. Curr Opin Struct Biol 19(2):120–127
Fuller JC, Burgoyne NJ, Jackson RM (2009) Predicting druggable binding sites at the protein-protein interface. Drug Discov Today 14(3–4):155–161
Villoutreix BO, Bastard K, Sperandio O, Fahraeus R, Poyet JL, Calvo F, Deprez B, Miteva MA (2008) In silico-in vitro screening of protein-protein interactions: towards the next generation of therapeutics. Curr Pharm Biotechnol 9(2):103–122
Zinzalla G, Thurston DE (2009) Targeting protein-protein interactions for therapeutic intervention: a challenge for the future. Future Med Chem 1(1):65–93
Meireles LM, Mustata G (2011) Discovery of modulators of protein-protein interactions: current approaches and limitations. Curr Top Med Chem 11(3):248–257
Jin L, Wang W, Fang G (2014) Targeting protein-protein interaction by small molecules. Annu Rev Pharmacol Toxicol 54:435–456
Aeluri M, Chamakuri S, Dasari B, Guduru SK, Jimmidi R, Jogula S, Arya P (2014) Small molecule modulators of protein-protein interactions: selected case studies. Chem Rev 114(9):4640–4694
Milroy LG, Grossmann TN, Hennig S, Brunsveld L, Ottmann C (2014) Modulators of protein-protein interactions. Chem Rev 114(9):4695–4748
Higueruelo AP, Jubb H, Blundell TL (2013) Protein-protein interactions as druggable targets: recent technological advances. Curr Opin Pharmacol 13(5):791–796
Villoutreix BO, Labbe CM, Lagorce D, Laconde G, Sperandio O (2012) A leap into the chemical space of protein-protein interaction inhibitors. Curr Pharm Des 18(30):4648–4667
Valkov E, Sharpe T, Marsh M, Greive S, Hyvonen M (2012) Targeting protein-protein interactions and fragment-based drug discovery. Top Curr Chem 317:145–179
Azzarito V, Long K, Murphy NS, Wilson AJ (2013) Inhibition of alpha-helix-mediated protein-protein interactions using designed molecules. Nat Chem 5(3):161–173
London N, Raveh B, Schueler-Furman O (2013) Druggable protein-protein interactions–from hot spots to hot segments. Curr Opin Chem Biol 17(6):952–959
Ivanov AA, Khuri FR, Fu H (2013) Targeting protein-protein interactions as an anticancer strategy. Trends Pharmacol Sci 34(7):393–400
Nero TL, Morton CJ, Holien JK, Wielens J, Parker MW (2014) Oncogenic protein interfaces: small molecules, big challenges. Nat Rev Cancer 14(4):248–262
Sheng C, Dong G, Miao Z, Zhang W, Wang W (2015) State-of-the-art strategies for targeting protein-protein interactions by small-molecule inhibitors. Chem Soc Rev 44(22):8238–8259
Guo W, Wisniewski JA, Ji H (2014) Hot spot-based design of small-molecule inhibitors for protein-protein interactions. Bioorg Med Chem Lett 24(11):2546–2554
Wanner J, Fry DC, Peng Z, Roberts J (2011) Druggability assessment of protein-protein interfaces. Future Med Chem 3(16):2021–2038
Eyrisch S, Helms V (2007) Transient pockets on protein surfaces involved in protein-protein interaction. J Med Chem 50(15):3457–3464
Villoutreix BO, Kuenemann MA, Poyet JL, Bruzzoni-Giovanelli H, Labbe C, Lagorce D, Sperandio O, Miteva MA (2014) Drug-Like protein-protein interaction modulators: challenges and opportunities for drug discovery and chemical biology. Mol Inform 33(6–7):414–437
Berg T (2008) Small-molecule inhibitors of protein-protein interactions. Curr Opin Drug Discov Devel 11(5):666–674
Ashkenazi A, Fairbrother WJ, Leverson JD, Souers AJ (2017) From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. Nat Rev Drug Discov 16(4):273–284
Zhong M, Hanan EJ, Shen W, Bui M, Arkin MR, Barr KJ, Evanchik MJ, Hoch U, Hyde J, Martell JR, Oslob JD, Paulvannan K, Prabhu S, Silverman JA, Wright J, Yu CH, Zhu J, Flanagan WM (2011) Structure-activity relationship (SAR) of the alpha-amino acid residue of potent tetrahydroisoquinoline (THIQ)-derived LFA-1/ICAM-1 antagonists. Bioorg Med Chem Lett 21(1):307–310
Cervantes-Gomez F, Lamothe B, Woyach JA, Wierda WG, Keating MJ, Balakrishnan K, Gandhi V (2015) Pharmacological and protein profiling suggests venetoclax (ABT-199) as optimal partner with ibrutinib in chronic lymphocytic leukemia. Clin Cancer Res 21(16):3705–3715
Konopleva M, Watt J, Contractor R, Tsao T, Harris D, Estrov Z, Bornmann W, Kantarjian H, Viallet J, Samudio I, Andreeff M (2008) Mechanisms of antileukemic activity of the novel Bcl-2 homology domain-3 mimetic GX15-070 (obatoclax). Cancer Res 68(9):3413–3420
Ding Q, Zhang Z, Liu JJ, Jiang N, Zhang J, Ross TM, Chu XJ, Bartkovitz D, Podlaski F, Janson C, Tovar C, Filipovic ZM, Higgins B, Glenn K, Packman K, Vassilev LT, Graves B (2013) Discovery of RG7388, a potent and selective p53-MDM2 inhibitor in clinical development. J Med Chem 56(14):5979–5983
Sun D, Li Z, Rew Y, Gribble M, Bartberger MD, Beck HP, Canon J, Chen A, Chen X, Chow D, Deignan J, Duquette J, Eksterowicz J, Fisher B, Fox BM, Fu J, Gonzalez AZ, Gonzalez-Lopez De Turiso F, Houze JB, Huang X, Jiang M, Jin L, Kayser F, Liu JJ, Lo MC, Long AM, Lucas B, McGee LR, McIntosh J, Mihalic J, Oliner JD, Osgood T, Peterson ML, Roveto P, Saiki AY, Shaffer P, Toteva M, Wang Y, Wang YC, Wortman S, Yakowec P, Yan X, Ye Q, Yu D, Yu M, Zhao X, Zhou J, Zhu J, Olson SH, Medina JC (2014) Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2-p53 inhibitor in clinical development. J Med Chem 57(4):1454–1472
Jeay S, Gaulis S, Ferretti S, Bitter H, Ito M, Valat T, Murakami M, Ruetz S, Guthy DA, Rynn C, Jensen MR, Wiesmann M, Kallen J, Furet P, Gessier F, Holzer P, Masuya K, Wurthner J, Halilovic E, Hofmann F, Sellers WR, Graus Porta D (2015) A distinct p 53 target gene set predicts for response to the selective p53-HDM2 inhibitor NVP-CGM097. eLife e06498
Perez VL, Pflugfelder SC, Zhang S, Shojaei A, Haque R (2016) Lifitegrast, a novel integrin antagonist for treatment of dry eye disease. Ocul Surf 14(2):207–215
Tabernero J, Dirix L, Schoffski P, Cervantes A, Lopez-Martin JA, Capdevila J, van Beijsterveldt L, Platero S, Hall B, Yuan Z, Knoblauch R, Zhuang SH (2011) A phase I first-in-human pharmacokinetic and pharmacodynamic study of serdemetan in patients with advanced solid tumors. Clin Cancer Res 17(19):6313–6321
Vu B, Wovkulich P, Pizzolato G, Lovey A, Ding Q, Jiang N, Liu JJ, Zhao C, Glenn K, Wen Y, Tovar C, Packman K, Vassilev L, Graves B (2013) Discovery of RG7112: a small-molecule MDM2 inhibitor in clinical development. ACS Med Chem Lett 4(5):466–469
Tovar C, Graves B, Packman K, Filipovic Z, Higgins B, Xia M, Tardell C, Garrido R, Lee E, Kolinsky K, To KH, Linn M, Podlaski F, Wovkulich P, Vu B, Vassilev LT (2013) MDM2 small-molecule antagonist RG7112 activates p53 signaling and regresses human tumors in preclinical cancer models. Cancer Res 73(8):2587–2597
Wang S, Sun W, Zhao Y, McEachern D, Meaux I, Barriere C, Stuckey JA, Meagher JL, Bai L, Liu L, Hoffman-Luca CG, Lu J, Shangary S, Yu S, Bernard D, Aguilar A, Dos-Santos O, Besret L, Guerif S, Pannier P, Gorge-Bernat D, Debussche L (2014) SAR405838: an optimized inhibitor of MDM2-p53 interaction that induces complete and durable tumor regression. Cancer Res 74(20):5855–5865
Potin D, Launay M, Monatlik F, Malabre P, Fabreguettes M, Fouquet A, Maillet M, Nicolai E, Dorgeret L, Chevallier F, Besse D, Dufort M, Caussade F, Ahmad SZ, Stetsko DK, Skala S, Davis PM, Balimane P, Patel K, Yang Z, Marathe P, Postelneck J, Townsend RM, Goldfarb V, Sheriff S, Einspahr H, Kish K, Malley MF, DiMarco JD, Gougoutas JZ, Kadiyala P, Cheney DL, Tejwani RW, Murphy DK, McIntyre KW, Yang X, Chao S, Leith L, Xiao Z, Mathur A, Chen BC, Wu DR, Traeger SC, McKinnon M, Barrish JC, Robl JA, Iwanowicz EJ, Suchard SJ, Dhar TG (2006) Discovery and development of 5-[(5S,9R)-9-(4-cyanophenyl)-3-(3,5-dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4.4]non-7-yl-methyl]-3-thiophenecarboxylic acid (BMS-587101)—a small molecule antagonist of leukocyte function associated antigen-1. J Med Chem 49(24):6946–6949
Watterson SH, Xiao Z, Dodd DS, Tortolani DR, Vaccaro W, Potin D, Launay M, Stetsko DK, Skala S, Davis PM, Lee D, Yang X, McIntyre KW, Balimane P, Patel K, Yang Z, Marathe P, Kadiyala P, Tebben AJ, Sheriff S, Chang CY, Ziemba T, Zhang H, Chen BC, DelMonte AJ, Aranibar N, McKinnon M, Barrish JC, Suchard SJ, Murali Dhar TG (2010) Small molecule antagonist of leukocyte function associated antigen-1 (LFA-1): structure-activity relationships leading to the identification of 6-((5S,9R)-9-(4-cyanophenyl)-3-(3,5-dichlorophenyl)-1-methyl-2,4-dioxo-1,3,7-tria zaspiro[4.4]nonan-7-yl)nicotinic acid (BMS-688521). J Med Chem 53(9):3814–3830
Kelly TA, Kim JML, R.M. (2004) Preparation of [6,7-dihydro-5H-imidazo[1,2-a]imidazole-3-sulfonylamino]propionamide derivatives for treatment of inflammatory disease. WO 2004041827
Polishchuk PG, Samoylenko GV, Khristova TM, Krysko OL, Kabanova TA, Kabanov VM, Kornylov AY, Klimchuk O, Langer T, Andronati SA, Kuz’min VE, Krysko AA, Varnek A (2015) Design, virtual screening, and synthesis of antagonists of alphaIIbbeta3 as antiplatelet agents. J Med Chem 58(19):7681–7694
Gowda RM, Khan IA, Vasavada BC, Sacchi TJ (2004) Therapeutics of platelet glycoprotein IIb/IIIa receptor antagonism. Am J Ther 11(4):302–307
Zablocki JA, Rico JG, Garland RB, Rogers TE, Williams K, Schretzman LA, Rao SA, Bovy PR, Tjoeng FS, Lindmark RJ et al (1995) Potent in vitro and in vivo inhibitors of platelet aggregation based upon the Arg-Gly-Asp sequence of fibrinogen. (Aminobenzamidino)succinyl (ABAS) series of orally active fibrinogen receptor antagonists. J Med Chem 38(13):2378–2394
Feldstein CA (1999) Sibrafiban (Genentech). IDrugs 2(5):460–465
Sugiura T, Kageyama S, Andou A, Miyazawa T, Ejima C, Nakayama A, Dohi T, Eda H (2013) Oral treatment with a novel small molecule alpha 4 integrin antagonist, AJM300, prevents the development of experimental colitis in mice. J Crohns Colitis 7(11):e533–e542
Halland N, Blum H, Buning C, Kohlmann M, Lindenschmidt A (2014) Small macrocycles as highly active integrin alpha2beta1 antagonists. ACS Med Chem Lett 5(2):193–198
Davenport RJ, Munday JR (2007) Alpha4-integrin antagonism—an effective approach for the treatment of inflammatory diseases? Drug Discov Today 12(13–14):569–576
Astles PC, Harris NV, Morley AD (1999) Preparation of substituted β-alanines as integrin-mediated cell adhesion inhibitors. WO 9933789
Sircar I, Gudmundsson KS, Martin R, Liang J, Nomura S, Jayakumar H, Teegarden BR, Nowlin DM, Cardarelli PM, Mah JR, Connell S, Griffith RC, Lazarides E (2002) Synthesis and SAR of N-benzoyl-L-biphenylalanine derivatives: discovery of TR-14035, a dual alpha(4)beta(7)/alpha(4)beta(1) integrin antagonist. Bioorg Med Chem 10(6):2051–2066
Cai J, Hu Y, Li W, Li L, Li S, Zhang M, Li Q (2011) The neuroprotective effect of propofol against brain ischemia mediated by the glutamatergic signaling pathway in rats. Neurochem Res 36(10):1724–1731
Derakhshan A, Chen Z, Van Waes C (2017) Therapeutic small molecules target inhibitor of apoptosis proteins in cancers with deregulation of extrinsic and intrinsic cell death pathways. Clin Cancer Res 23(6):1379–1387
Benetatos CA, Mitsuuchi Y, Burns JM, Neiman EM, Condon SM, Yu G, Seipel ME, Kapoor GS, Laporte MG, Rippin SR, Deng Y, Hendi MS, Tirunahari PK, Lee YH, Haimowitz T, Alexander MD, Graham MA, Weng D, Shi Y, McKinlay MA, Chunduru SK (2014) Birinapant (TL32711), a bivalent SMAC mimetic, targets TRAF2-associated cIAPs, abrogates TNF-induced NF-kappaB activation, and is active in patient-derived xenograft models. Mol Cancer Ther 13(4):867–879
Wong H, Gould SE, Budha N, Darbonne WC, Kadel EE 3rd, La H, Alicke B, Halladay JS, Erickson R, Portera C, Tolcher AW, Infante JR, Mamounas M, Flygare JA, Hop CE, Fairbrother WJ (2013) Learning and confirming with preclinical studies: modeling and simulation in the discovery of GDC-0917, an inhibitor of apoptosis proteins antagonist. Drug Metab Dispos 41(12):2104–2113
Flygare JA, Beresini M, Budha N, Chan H, Chan IT, Cheeti S, Cohen F, Deshayes K, Doerner K, Eckhardt SG, Elliott LO, Feng B, Franklin MC, Reisner SF, Gazzard L, Halladay J, Hymowitz SG, La H, LoRusso P, Maurer B, Murray L, Plise E, Quan C, Stephan JP, Young SG, Tom J, Tsui V, Um J, Varfolomeev E, Vucic D, Wagner AJ, Wallweber HJ, Wang L, Ware J, Wen Z, Wong H, Wong JM, Wong M, Wong S, Yu R, Zobel K, Fairbrother WJ (2012) Discovery of a potent small-molecule antagonist of inhibitor of apoptosis (IAP) proteins and clinical candidate for the treatment of cancer (GDC-0152). J Med Chem 55(9):4101–4113
Bailey D, Jahagirdar R, Gordon A, Hafiane A, Campbell S, Chatur S, Wagner GS, Hansen HC, Chiacchia FS, Johansson J, Krimbou L, Wong NC, Genest J (2010) RVX-208: a small molecule that increases apolipoprotein A-I and high-density lipoprotein cholesterol in vitro and in vivo. J Am Coll Cardiol 55(23):2580–2589
Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, Chandwani R, Marazzi I, Wilson P, Coste H, White J, Kirilovsky J, Rice CM, Lora JM, Prinjha RK, Lee K, Tarakhovsky A (2010) Suppression of inflammation by a synthetic histone mimic. Nature 468(7327):1119–1123
Albrecht BK, Gehling VS, Hewitt MC, Vaswani RG, Cote A, Leblanc Y, Nasveschuk CG, Bellon S, Bergeron L, Campbell R, Cantone N, Cooper MR, Cummings RT, Jayaram H, Joshi S, Mertz JA, Neiss A, Normant E, O’Meara M, Pardo E, Poy F, Sandy P, Supko J, Sims RJ 3rd, Harmange JC, Taylor AM, Audia JE (2016) Identification of a Benzoisoxazoloazepine Inhibitor (CPI-0610) of the Bromodomain and Extra-Terminal (BET) family as a candidate for human clinical trials. J Med Chem 59(4):1330–1339
Boi M, Gaudio E, Bonetti P, Kwee I, Bernasconi E, Tarantelli C, Rinaldi A, Testoni M, Cascione L, Ponzoni M, Mensah AA, Stathis A, Stussi G, Riveiro ME, Herait P, Inghirami G, Cvitkovic E, Zucca E, Bertoni F (2015) The BET Bromodomain Inhibitor OTX015 affects pathogenetic pathways in preclinical B-cell tumor models and synergizes with targeted drugs. Clin Cancer Res 21(7):1628–1638
Schenk RL, Strasser A, Dewson G (2017) BCL-2: long and winding path from discovery to therapeutic target. Biochem Biophys Res Commun 482(3):459–469
Adams JM, Cory S (1998) The Bcl-2 protein family: arbiters of cell survival. Science 281(5381):1322–1326
Rudin CM, Hann CL, Garon EB, Ribeiro de Oliveira M, Bonomi PD, Camidge DR, Chu Q, Giaccone G, Khaira D, Ramalingam SS, Ranson MR, Dive C, McKeegan EM, Chyla BJ, Dowell BL, Chakravartty A, Nolan CE, Rudersdorf N, Busman TA, Mabry MH, Krivoshik AP, Humerickhouse RA, Shapiro GI, Gandhi L (2012) Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin Cancer Res 18(11):3163–3169
Sattler M, Liang H, Nettesheim D, Meadows RP, Harlan JE, Eberstadt M, Yoon HS, Shuker SB, Chang BS, Minn AJ, Thompson CB, Fesik SW (1997) Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science 275(5302):983–986
Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O’Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH (2005) An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435(7042):677–681
Petros AM, Dinges J, Augeri DJ, Baumeister SA, Betebenner DA, Bures MG, Elmore SW, Hajduk PJ, Joseph MK, Landis SK, Nettesheim DG, Rosenberg SH, Shen W, Thomas S, Wang X, Zanze I, Zhang H, Fesik SW (2006) Discovery of a potent inhibitor of the antiapoptotic protein Bcl-xL from NMR and parallel synthesis. J Med Chem 49(2):656–663
Wendt MD, Shen W, Kunzer A, McClellan WJ, Bruncko M, Oost TK, Ding H, Joseph MK, Zhang H, Nimmer PM, Ng SC, Shoemaker AR, Petros AM, Oleksijew A, Marsh K, Bauch J, Oltersdorf T, Belli BA, Martineau D, Fesik SW, Rosenberg SH, Elmore SW (2006) Discovery and structure-activity relationship of antagonists of B-cell lymphoma 2 family proteins with chemopotentiation activity in vitro and in vivo. J Med Chem 49(3):1165–1181
Park CM, Oie T, Petros AM, Zhang H, Nimmer PM, Henry RF, Elmore SW (2006) Design, synthesis, and computational studies of inhibitors of Bcl-XL. J Am Chem Soc 128(50):16206–16212
Park CM, Bruncko M, Adickes J, Bauch J, Ding H, Kunzer A, Marsh KC, Nimmer P, Shoemaker AR, Song X, Tahir SK, Tse C, Wang X, Wendt MD, Yang X, Zhang H, Fesik SW, Rosenberg SH, Elmore SW (2008) Discovery of an orally bioavailable small molecule inhibitor of prosurvival B-cell lymphoma 2 proteins. J Med Chem 51(21):6902–6915
Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P, Roberts L, Tahir SK, Xiao Y, Yang X, Zhang H, Fesik S, Rosenberg SH, Elmore SW (2008) ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res 68(9):3421–3428
Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, Huang DC, Hymowitz SG, Jin S, Khaw SL, Kovar PJ, Lam LT, Lee J, Maecker HL, Marsh KC, Mason KD, Mitten MJ, Nimmer PM, Oleksijew A, Park CH, Park CM, Phillips DC, Roberts AW, Sampath D, Seymour JF, Smith ML, Sullivan GM, Tahir SK, Tse C, Wendt MD, Xiao Y, Xue JC, Zhang H, Humerickhouse RA, Rosenberg SH, Elmore SW (2013) ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med 19(2):202–208
Anderson MA, Deng J, Seymour JF, Tam C, Kim SY, Fein J, Yu L, Brown JR, Westerman D, Si EG, Majewski IJ, Segal D, Heitner Enschede SL, Huang DC, Davids MS, Letai A, Roberts AW (2016) The BCL2 selective inhibitor venetoclax induces rapid onset apoptosis of CLL cells in patients via a TP53-independent mechanism. Blood 127(25):3215–3224
Gadek TR, Burdick DJ, McDowell RS, Stanley MS, Marsters JC Jr, Paris KJ, Oare DA, Reynolds ME, Ladner C, Zioncheck KA, Lee WP, Gribling P, Dennis MS, Skelton NJ, Tumas DB, Clark KR, Keating SM, Beresini MH, Tilley JW, Presta LG, Bodary SC (2002) Generation of an LFA-1 antagonist by the transfer of the ICAM-1 immunoregulatory epitope to a small molecule. Science 295(5557):1086–1089
Zhong M, Shen W, Barr KJ, Arbitrario JP, Arkin MR, Bui M, Chen T, Cunningham BC, Evanchik MJ, Hanan EJ, Hoch U, Huen K, Hyde J, Kumer JL, Lac T, Lawrence CE, Martell JR, Oslob JD, Paulvannan K, Prabhu S, Silverman JA, Wright J, Yu CH, Zhu J, Flanagan WM (2010) Discovery of tetrahydroisoquinoline (THIQ) derivatives as potent and orally bioavailable LFA-1/ICAM-1 antagonists. Bioorg Med Chem Lett 20(17):5269–5273
Zhong CX, Wu JX, Liang JX, Wu QH (2012) Laparoscopic and gasless laparoscopic sigmoid colon vaginoplasty in women with vaginal agenesis. Chin Med J (Engl) 125(2):203–208
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Dong, G., Sheng, C. (2018). Overview of Protein-Protein Interactions and Small-Molecule Inhibitors Under Clinical Development. In: Sheng, C., Georg, G. (eds) Targeting Protein-Protein Interactions by Small Molecules. Springer, Singapore. https://doi.org/10.1007/978-981-13-0773-7_1
Download citation
DOI: https://doi.org/10.1007/978-981-13-0773-7_1
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-0772-0
Online ISBN: 978-981-13-0773-7
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)