
Abstract
Oncolytic virotherapy is a kind of antitumor therapy using viruses with natural or engineered tumor-selective replication to intentionally infect and kill tumor cells. An early clinical trial has been performed in the 1950s using wild-type and non-engineered in vitro-passaged virus strains and vaccine strains (first generation oncolytic viruses). Because of the advances in biotechnology and virology, the field of virotherapy has rapidly evolved over the past two decades and innovative recombinant selectivity-enhanced viruses (second generation oncolytic viruses). Nowadays, therapeutic transgene-delivering “armed” oncolytic viruses (third generation oncolytic viruses) have been engineered using many kinds of viruses. In this chapter, the history, mechanisms, rationality, and advantages of oncolytic virotherapy by herpes simplex virus (HSV) are mentioned. Past and ongoing clinical trials by oncolytic HSVs (G207, HSV1716, NV1020, HF10, Talimogene laherparepvec (T-VEC, OncoVEXGM-CSF)) are also summarized. Finally, the way of enhancement of oncolytic virotherapy by gene modification or combination therapy with radiation, chemotherapy, or immune checkpoint inhibitors are discussed.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Herpes simplex virus
- HSV
- Oncolytic virotherapy
- G207
- HSV1716
- NV1020
- HF10
- Talimogene laherparepvec
- T-VEC
- OncoVEXGM-CSF
1 Introduction
Cancer is the second worldwide cause of death, exceeded only by cardiovascular diseases (Pérez-Herrero and Fernández-Medarde 2015). For local and nonmetastatic cancers, surgery is the most effective and valuable treatment but is inefficient when the cancer has spread throughout the body. For advanced and metastatic cancers, systemic cytotoxic chemotherapy and/or radiation therapy has been used, but in some cancers such as malignant melanoma, these therapies are ineffective. Recent advances in understanding of cancer biology and immunology have spurred the development of numerous targeted therapies including molecular-targeted therapies or immunotherapies. In particular, a class of immune modulatory drugs targeting the immune checkpoint pathways like anti-PD-1 antibody (nivolumab) has demonstrated remarkable durable remissions in a part of advanced malignant melanoma patients (Tang et al. 2016). However, these agents cause many systemic adverse reactions, such as pneumonitis, colitis, and autoimmune diseases (Dossett et al. 2015; Spain et al. 2016). The high prices of these drugs are also problematic.
Besides these agents, another immunotherapy with tumor destruction using oncolytic virus (oncolytic virotherapy) has been studied for several decades and showed significant progress in recent years. This review focuses on the progress of oncolytic virotherapy, especially by using herpes simplex virus (HSV) for malignant internal tumors and brain tumors based on the mechanisms and clinical development. We also discuss the attempts for enhancing the effectiveness of oncolytic virotherapy by gene modification or combination therapies.
2 What Is Oncolytic Virotherapy?
Oncolytic virotherapy is a kind of antitumor therapy using viruses with natural or engineered tumor-selective replication to intentionally infect and kill tumor cells. The phenomenon that tumor regression following naturally acquired virus infections has been known over 100 years before. In 1904, a patient with chronic myelogenous leukemia had a dramatic decrease in white blood cells during a “flu-like” illness (Dock 1904). In 1912, a woman with cervical carcinoma responded to repeated rabies vaccinations (DePace 1912). An early clinical trial has been performed in the 1950s using wild-type and non-engineered in vitro-passaged virus strains and vaccine strains (first generation oncolytic viruses). For example, a clinical trial was performed using 30 patients with cervical cancer treated with different adenovirus serotypes (Huebner et al. 1956). More than 50% of patients showed a marked to moderate local tumor response; no systemic responses were reported, but the prolongation of survival was not significantly significant.
Because of the advances in biotechnology and virology, the field of virotherapy has rapidly evolved over the past two decades and innovative recombinant selectivity-enhanced viruses (second generation oncolytic viruses). In 1991, Martuza et al. first reported oncolytic virotherapy against mice glioma (malignant brain tumor) model using genetically engineered HSV. In that report, intraneoplastic inoculation of a thymidine kinase-negative mutant of herpes simplex virus-1 (dlsptk) prolonged survival of nude mice with intracranial U87 gliomas (Martuza et al. 1991). Nowadays, therapeutic transgene-delivering “armed” oncolytic viruses (third generation oncolytic viruses) have been engineered using many kinds of viruses. Table 4.1 shows the viruses studying as oncolytic virus today.
3 Mechanisms of Oncolytic Virotherapy
Mainly, there are two kinds of mechanisms for oncolytic viruses to kill cancer cells (Sze et al. 2013). The first and direct oncolytic effects are caused by viral infection itself and tumor cell lysis. By infection into tumor cells, viruses can produce viral proteins that are antigenic. After the lysis of infected cell, new virions are released and will infect neighboring cancer cells.
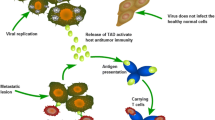
By releasing tumor antigens and triggering an immune response by infection, viruses can act as immunomodulators or tumor vaccines. By viral infection, inflammation occurs, and innate and acquired immune cells including cytotoxic T lymphocytes, natural killer cells, dendritic cells, and phagocytic cells will eliminate cancer cells. In addition, development of memory against tumor antigens will begin by these immune responses and will act on distant metastases as well (Fig. 4.1).

An oncolytic virus, represented by black hexagons, attacks a cancerous cell (a). As a result of a natural tropism for the cell type or a specificity for a tumor-related cell surface antigen or receptor, the virus enters the malignant cell more readily than it would enter into a normal cell (e), which may not exhibit the same receptivity to infection. Upon infecting the malignant cell, the permissive nature of the cell that allows malignant genetic material to propagate also allows unchecked replication of the virus (b). An infection of a normal cell (f) is abortive as a result of the cell’s ability to recognize and to destroy abnormal genetic material. The infected cell (b) will produce viral proteins that are antigenic and may alert the immune system, and, if the virus is genetically armed, the cell may produce cytokines and other signaling chemicals to activate the immune system. The infected cancer cell is eventually overwhelmed by the viral infection and lyses, releasing new viral particles locally to infect neighboring malignant cells (c). Lysis releases new viral and tumor- related antigens, which may be recognized and attacked by the immune system, represented here by a lymphocyte (L). Lysis-related viremia may result in infection of distant metastases (d), transforming a locoregional effect into a systemic effect. Activation of the immune system with elevation of systemic cytokine levels and activated leukocytes further enhances the systemic effect. Parts of the immune system may also develop memory and learn to recognize tumor antigens, potentially providing a more durable defense against residual and recurrent disease. (Adapted from Sze et al. 2013)
So far, many types of viruses, including HSV, adenoviruses, and adeno-associated viruses, have been engineered and evaluated for their potential as therapeutic agents in the treatment of malignant neoplasm
4 HSV as Oncolytic Virus
Herpes simplex virus type 1 (HSV-1) and type 2 (HSV-2) are important human pathogens that cause a variety of skin diseases from recurrent herpes labialis, herpes genitalis, and Kaposi’s varicelliform eruption (eczema herpeticum) to life-threatening diseases such as herpes encephalitis and neonatal herpes (Nishiyama 2004). Especially among immunocompromised patients, the virus can be systemically disseminated and cause fatal infection (Witt et al. 2009). HSV was the first of the human herpes viruses to be discovered and has been the most intensively studied. Since the publication of the complete genomic DNA sequence of HSV-1 in 1988 (McGeoch et al. 1988), a number of studies have focused on elucidating the roles of individual HSV genes in viral replication and pathogenicity. These studies also identified the HSV gene products involved in the regulation of gene expression, interaction with the host cell, and evasion from the host immune system. The resulting depth of knowledge of HSV has allowed the development of potential therapeutic agents and vectors for several applications in human diseases.
4.1 Structure, Natural History, and Gene Function of HSV
HSV is an enveloped, double-stranded linear DNA virus about 100 nm in diameter. The viral particle is an icosahedral capsid containing the viral DNA with a genome of 152 kb encoding over 74 distinct genes. The capsid is surrounded by an amorphous layer known as the tegument, which contains viral structural and regulatory proteins, and external envelope containing numerous glycoproteins (Fig. 4.2a). Following primary infection from skin or mucosa, HSV enters into nerve endings and is then transported to the dorsal root and trigeminal ganglia where the virus establishes latent infection. The virus is reactivated by stimuli such as UV irradiation, mental or physical stress, or menstruation and causes symptomatic or asymptomatic recurrent infection (Mori and Nishiyama 2005).

(a) Structure of HSV. HSV is an enveloped, double-stranded linear DNA virus which diameter is about 100 nm. The virus particle comprehends an icosahedral capsid, which contains the viral DNA with a genome of 152 kb encoding over 74 distinct genes. Around the capsid, there is an amorphous layer known as the tegument, containing viral structural and regulatory proteins, surrounded by external envelope glycoproteins (b) HSV genome. HSV has genome of 152 kb encoding over 74 distinct genes. The HSV genome consists of two long structures of unique sequences (designated long (UL) and short (Us)), both of which are flanked by a pair of inverted repeat regions (TRL-IRL and IRs-TRs). There is a single copy of the “a” sequence at each terminus and one at the junction between IRL and IRs
The HSV genome consists of two long structures of unique sequences (designated long (UL) and short (US)), both of which are flanked by a pair of inverted repeat regions (TRL–IRL and IRS–TRS). There is a single copy of the “a” sequence, which contains the specific signals for packaging of viral DNA into capsids (Taylor et al. 2002), at each terminus and one at the junction between IRL and IRS (Fig. 4.2b).
HSV genes are classified into three groups by the regulation of their expression: immediate early (IE), early (E), and late (L). The IE gene products regulate gene transcription and include the US12 gene product, ICP47 which is responsible for decreasing MHC class I expression in infected cells via inhibition of the transporter associated with antigen presentation (TAP). The E gene products promote viral DNA synthesis in either viral DNA replication or in nucleic acid metabolism. Most of the L gene products are virion components such as capsid proteins, tegument proteins, and envelope glycoproteins (Nishiyama 1996).
HSV genes can also be divided into two groups according to whether or not they are essential for viral replication in cultured cells. Approximately half of the genes are essential genes that are necessary for viral replication and which encode capsid proteins, viral DNA replication proteins, viral DNA cleavage/packaging proteins, and some envelope glycoproteins. HSV is unable to replicate with even a single dysfunction in an essential gene. The remaining genes have been shown to be dispensable for replication. These accessory genes encode enzymes involved in nucleic acid metabolism, regulatory proteins required for efficient viral replication, proteins for protecting the virus and infected cells from the host immunity, and genes with other undetermined functions. Although the accessory genes are not necessary for viral replication in cell culture, the expression of these genes enables the virus to replicate effectively in a variety of cell types under different conditions, resulting in the replication and survival of HSV in humans (Mori and Nishiyama 2006). The functions of HSV gene products, their expression phase, and whether they are essential or accessory genes are summarized in Table 4.2.
4.2 Rationality of Using HSV for Oncolytic Virotherapy
HSV offers a number of advantages as an oncolytic agent.
-
1.
Unlike many other viruses that only bind to a single receptor, HSV has four cellar receptors and has broad host range that allows the virus to infect and replicate almost all cell lines. As a result, oncolytic viruses derived from HSV can be applied therapeutically to many different types of tumors. In addition, in contrast to other oncolytic viruses, the property might protect against the rapid development of resistance to virotherapy using HSV.
-
2.
HSV can infect both in replicating and non-replicating cells such as neuronal cells. This property enables oncolytic HSV to applying brain tumors such as glioblastoma.
-
3.
HSV has the potential for incorporating a large size of foreign DNA. It is useful when making therapeutic transgene-delivering “armed” oncolytic viruses.
-
4.
Undesired infection or toxicity from the virus replication can be controlled by effective anti-herpetic agents such as acyclovir and famciclovir .
-
5.
Compared to adenoviruses, lytic infection by HSV usually kills target cells much more rapidly and effectively. For example, HSV can form visible plaques in cultured cells in 2 days, in contrast to 7 to 9 days for an adenovirus. In in vitro studies, it has also shown that HSV can kill almost 100% of cultured cancer cells at a multiplicity of infection (MOI) of 0.01 (Fu and Zhang 2002).
-
6.
HSV can infect many kinds of animals. Due to the similarity in the viral pathogenicity in mice, guinea pigs, and monkeys, to that in humans, preclinical studies of oncolytic HSV can be performed relatively easily by using these animal models.
-
7.
The determination of complete open reading frames and identification of disease-related viral genes (Marconi et al. 2008; Todo 2008).
-
8.
The risk of introducing an insertional mutation during HSV oncolytic therapy appears minimal because HSVs rarely integrate into cellular DNA. While strong immunogenicity and cell toxicity induced by HSV infection are major disadvantages for developing gene delivery vectors using HSV, they are beneficial when developing vaccine vectors or anticancer agent by HSV recombination.
4.3 Important Genes for Making Effective Oncolytic HSVs
4.3.1 Immediate Early Genes
ICP0 (infected cell polypeptide 0 ) is the RL2 gene product. It belongs to the immediate early proteins (IE) that it required for effective initiation of viral lytic infection and reactivation from latent infection of HSV (Bringhurst and Schaffer 2006). ICP0 is a 775-amino acid really interesting new gene (RING)-finger-containing protein that possesses E3 ubiquitin ligase activity, which is required for ICP0 to activate HSV-1 gene expression; disrupt nuclear domain (ND) 10 structures; mediate the degradation of cellular proteins including cdc34, Sp100, and PML; and evade the host cell’s intrinsic and innate antiviral defenses. This protein degradation may create a favorable microenvironment for viral replication (Boehmer and Nimonkar 2003; Lilley et al. 2005). ICP0 also prevents cellular rRNA degradation (Sobol and Mossman 2006). It has been reported that ICP0 mutation impairs viral replication in normal cells. On the other hand, the ICP0 mutant KM100 virus exhibits an oncolytic effect on tumor cells, causing tumor regression and increased survival in experimental breast cancer models in mice (Hummel et al. 2005).
ICP4 is also a regulator of viral transcription that is required for productive infection. Since viral genes are transcribed by cellular RNA polymerase II (RNA pol II), ICP4 must interact with components of the pol II machinery to regulate viral gene expression. It has been shown previously that ICP4 interacts with TATA box-binding protein (TBP), TFIIB, and the TBP-associated factor 1 (TAF1) in vitro (Zabierowski and Deluca 2008).
NV1066, which has only one of two originally present copies of both ICP0 and ICP4, has an antitumor effect against breast cancer, pleural cancer, bladder cancer, and esophageal cancer (Mullerad et al. 2005; Stiles et al. 2006a; b). Interestingly, this virus not only destroys tumor cells but also induces apoptosis in uninfected cells via the cellular bystander pathway. This apoptosis hinders viral spread from cell to cell. Previous studies showed that pharmaceutically inhibiting apoptosis can improve the oncolytic viral proliferation and the antitumor effect (Stanziale et al. 2004).
ICP47 also belongs to IE proteins. It inhibits the transporter associated with antigen presentation (TAP), decreasing MHC class I expression and preventing infected cells from presenting viral to CD8+ cells (Hill et al. 1995). The lack of ICP47 increases MHC class I expression, which might induce an enhanced antitumor immune response. The bovine herpesvirus 1 (BHV-1) TAP-inhibitor (UL49.5)-expressing oncolytic virus showed superior efficacy treating bladder and breast cancer in murine preclinical models that was dependent upon a CD8+ T-cell response. In addition to treating directly injected, subcutaneous tumors, UL49.5-oncolytic virotherapy reduced untreated, contralateral subcutaneous tumor size and naturally occurring metastasis (Pourchet et al. 2016).
4.3.2 Early and Late Genes
Ribonucleotide reductase (RR) catalyzes the reduction of ribonucleotides to deoxyribonucleotides. As a result, it provides sufficient precursors for the de novo synthesis of DNA. Because HSV has its own RR, replication of the virus is independent of the host cell cycle. By inactivating the viral RR gene, viral replication is completely under the control of host cell dividing conditions. RR-deficient HSV-1 such as hrR3 was expected to exhibit selective oncolytic effects and increase their potency when combined with radiation; however, complementary toxicity was seen between radiation and hrR3, without evidence of viral replication (Spear et al. 2000).
The γ34.5 gene product (ICP34.5 ) enables the virus to replicate in neurons and spread within the brain. When this gene is deleted, HSV-1 cannot complete a lytic infection in neurons and thus cannot cause encephalitis (Kanai et al. 2012). Attenuated viruses that are mutated in the γ34.5 may be useful for malignant tumors in the central nervous system.
5 How to Make Oncolytic HSVs?
There are several established techniques for generating recombinant HSV. Traditionally, recombinant HSV mutants have been generated by homologous recombination between purified HSV DNA and a recombination plasmid in co-transfected cells (Bataille and Epstein 1995). An alternative procedure is the transfection of cells with overlapping cosmids containing appropriate insertions or deletions. Expression of genes contained in cosmids leads, through recombination, to the construction of full-length viral genome (Kong et al. 1999). In these methods, there are several problems such as the inefficiency of recombination and the need to screen or select plaques for the correct recombinant. This has hampered the development of new recombinant HSV vectors.
Recently, novel recombinant technique using bacterial artificial chromosome (BAC) has enabled the cloning of the whole HSV genome as a BAC plasmid and its subsequent manipulation in E. coli (Stavropoulos and Strathdee 1998). BAC cloning requires the insertion of mini F plasmid sequences and antibiotic resistance genes into the viral genome. The total length of these BAC backbone sequences is usually greater than 6 kb. Insertion of BAC sequences into the wild-type HSV genome (152 kb) increases the genome length to approximately 158 kb, leaving insufficient space for the insertion of additional sequences. To avoid deleterious effects of the BAC sequences, including growth defects and potential transmission between bacteria and man, some herpes virus BAC clones have been constructed with loxP site-flanked BAC sequences that can be removed by Cre recombinase (Tanaka et al. 2003). One potential disadvantage of the BAC system is the potential for higher rates of error in DNA replication in bacteria than eukaryotic cells, whether or not this will prove to be a problem is not yet known.
6 Oncolytic Viruses Derived from HSV-1 that Have Reached Clinical Testing
There have been several clinical trials with HSV-1 mutants as oncolytic agents using gene deletion described before. These mutants have been applied for the treatment of malignant brain tumors or malignant melanoma and other solid tumors (Table 4.3).
6.1 G207
G207 has been credited as the first oncolytic virus generated by genetic engineering technology. The virus was constructed from HSV-1 by deleting both copies of the γ34.5 gene and an insertional mutation in the ICP6 gene (Mineta et al. 1995). First, the safety and efficacy of G207 were demonstrated in preclinical animal models (Mineta et al. 1995, Sundaresan et al. 2000. Todo et al. 2000). Then G207 was tested in the treatment of malignant glioma in a phase I clinical trial. Up to 3 × 109 plaque-forming units (pfu) of virus was injected into tumors of 21 patients and revealed that the virus was well tolerated (Markert et al. 2009). Because of a lack of convincing evidence of clinical efficacy, G207 clinical development has not yet reached the phase II stage of testing.
6.2 HSV1716
HSV1716 is a spontaneous mutant of a replication selective HSV-1 that bears a deletion of 759 bp in each copy of the γ34.5 gene. Two separate phase I clinical trials have evaluated the safety of HSV1716 with high-grade glioma (HGG) and with stage IV melanoma (Harrow et al. 2004; MacKie et al. 2001). In the HGG study, HSV1716 DNA was detected by PCR at the sites of inoculation. In several patients, an immune response to the virus was detected. Although it remains unclear whether the immune response to the virus contributes to the eradication of cancer cells infected by the virus, a significant increase in long-term survival following surgery was also observed. In the melanoma trial, immunohistochemical staining of injected nodules revealed that virus replication was confined to tumor cells and had no toxicity in patients.
6.3 NV1020
NV1020 has deletions of both UL56 genes and one copy of the γ34.5 gene. In preclinical studies, NV1020 was evaluated as oncolytic agents in several solid tumors outside the brain (Advani et al. 1999; Cozzi et al. 2001; Ebright et al. 2002). In these studies, the virus showed effectiveness in treating several tumors in both mouse and rat models. Then NV1020 was evaluated against liver metastases from colorectal cancer. Followed by chemotherapy, NV1020 was tested in 12 patients with colorectal cancer hepatic metastases. It was well tolerated when NV1020 was given through hepatic arterial infusion. Reported side reactions were mainly transient febrile reactions and transient lymphopenia. Over half of the treated patients showed partial responses or stable disease, indicating therapeutic efficacy (Geevarghese et al. 2010; Kemeny et al. 2006; Sze et al. 2012).
6.4 Talimogene Laherparepvec (T-VEC)
T-VEC, formerly known as OncoVEX GM-CSF , has deletion of the genesγ34.5 and US12 which encodes ICP47 and contains the gene encoding human granulocyte macrophage colony-stimulating factor (GM-CSF) (Liu et al. 2003). Gene modification of this virus was intended to increase the lytic activity of the virus (over deletion of the ICP47 gene) and to potentiate the ability of virotherapy to induce antitumor immunity (deletion of ICP47 combined with insertion of GM-CSF). Preclinical studies of OncoVEXGM-CSF showed that this virus can effectively reduced injected tumors and also induced antitumor immunity that could protect animals against tumor rechallenge (Liu et al. 2003). The safety of OncoVEXGM-CSF was evaluated in a phase I study in patients with metastatic breast, head/neck and gastrointestinal cancers, and malignant melanoma. Overall, intralesional administration of the virus was well tolerated by patients (Hu et al. 2006). In phase II study, OncoVEXGM-CSF was injected in patients with metastatic melanoma (Senzer et al. 2009). The overall response rate was 26%. Surprisingly, all responding patients showed regressions of both injected and noninjected lesions (Fig. 4.3). An increase in CD8+ T-cells and a reduction in CD4+FoxP3+ regulatory T-cells were detected in biopsy samples of regressing lesions (Kaufman et al. 2010).

Breast cancer patient treated with T-VEC. The injection was made into tumor 1 (arrow). Tumor regressions were observed both in injected and in noninjected lesions, consistent with both direct oncolytic and immune-mediated antitumor effects. (Adapted from Hu et al. 2006)
A randomized phase III trial was performed in 291 patients with unresected stage IIIB to IV melanoma, with 127 patients receiving subcutaneous GM-CSF as the control arm (OPTiM; NCT00769704) (Andtbacka et al. 2015). T-VEC was administered at a concentration of 108 plaque-forming units (pfu)/mL injected into 1 or more skin or subcutaneous tumors on days 1 and 15 of each 28-day cycle for up to 12 months, while GM-CSF was administered at a dose of 125 μg/m2/day subcutaneously for 14 consecutive days followed by 14 days of rest, in 28-day treatment cycles for up to 12 months. At the primary analysis, 290 deaths had occurred (T-VEC, n = 189; GM-CSF, n = 101). T-VEC treatment produced a significant improvement in (1) durable response rate (TVEC 16% vs. GM-CSF control arm 2%), (2) objective response rate (26% vs. 6%), and (3) complete response rate (11% vs. 1%). The difference of the median overall survival rate, a secondary end point of this trial, between T-VEC and GM-CSF treatment groups was 4.4 months. The most common adverse events with T-VEC were fatigue, chills, and pyrexia, but the only grade 3 or 4 treatment-related adverse event, occurring in over 2% of patients, was cellulitis (T-VEC, n = 6; GM-CSF, n = 1). There were no fatal treatment-related adverse events. Median overall survival (OS) was 23.3 months for the T-VEC arm versus 18.9 months for the GM-CSF arm (hazard ratio, 0.79; P = 0.051), but the difference in OS became significant (P = 0.049) by the time of drug application (Andtbacka et al. 2015). This phase III trial was the first to prove that local intralesional injections with an oncolytic virus can not only suppress the growth of injected tumors, and in 2015, the US Food and Drug Administration (FDA) approved T-VEC as a first oncolytic HSV for the treatment of advanced inoperable malignant melanoma.
6.5 HF10
HSV-1 mutant strain HF10, derived from an in vitro-passaged laboratory strain of HSV-1, is an alternative candidate for an oncolytic HSV. Previous studies have shown that HF10 does not cause any neurological symptoms in mice when inoculated into the peripheral tissues and organs due to its inability to invade the central nervous system (Nishiyama et al. 1991). The HF10 genome has a deletion of 3832 bp to the right of the UL and UL/IRL junction. Sequences from 6025 to 8319 bp have also been deleted from the TRL, and 6027 bp of DNA has been inserted in an inverted orientation. Sequence analysis revealed that HF10 lacks the expression of functional UL43, UL49.5, UL55, UL56, and LAT (Ushijima et al. 2007). Although the detailed mechanisms of the HF10 phenotype are not clear, the lack of the UL56 gene and LAT may play an important role. UL56 associates with the kinesin motor protein KIF1A, and the absence of UL56 reduces the neuroinvasiveness of HSV without affecting viral replication in vitro (Koshizuka et al. 2005). The LAT promoter region is also known to be associated with neurovirulence (Jones et al. 2005). The mechanisms of HF10 in tumor selectivity are also unknown, but the differences in the IFN pathway between normal cells and cancer cells may be involved (Nawa et al. 2008).
The loss of HF10 neuroinvasiveness and its high potency of replication in tumor cells contribute to the usefulness of HF10 as an oncolytic virotherapy for non-brain malignancy. HF10 therapy exhibited striking antitumor efficacy of peritoneally disseminated internal malignancies of immunocompetent mice models (Kimata et al. 2003; Kohno et al. 2005; Teshigahara et al. 2004; Watanabe et al. 2008). In a BALB/c mouse model of disseminated peritoneal colon carcinoma, 100% of intraperitoneally HF10-treated mice survived without remarkable side effects (Takakuwa et al. 2003). HF10 virotherapy using a mouse melanoma model was also studied. In the intraperitoneal melanoma model, all mice survived when given intraperitoneal injections of HF10 compared to none of the control mice (Fig. 4.4a, b). In the subcutaneous melanoma model, intratumoral inoculation of HF10 showed not only tumor growth inhibition at the injected site but also the induction of systemic antitumor immune responses in mice (Watanabe et al. 2008).

Tumor growth reduction by HF10 in a murine intraperitoneal melanoma model. DBA/2 mice were injected intraperitoneally with 1 × 105 clone M3 cells and then were injected with PBS (control) or 1 × 107 pfu of HF10 at days 6,7, and 8. Representative clinical pictures of control (a) and HF10-treated (b) mice at day 14
Several clinical trials have been done with HF10 thus (Nakao et al. 2004; Kimata et al. 2006; Fujimoto et al. 2006). Six patients with recurrent breast cancer who were treated with HF10 showed no serious adverse effects, and distinct tumor regression was observed in all patients (Kimata et al. 2006). Another clinical trial was carried out in three patients with advanced head and neck squamous cell carcinoma (Fujimoto et al. 2006). Although no significant tumor regression was found after injection with HF10 at a low dose, pathological examination revealed extensive tumor cell death and fibrosis, with marked infiltration of CD4+ or CD8+ T-cells. Moreover, a number of HSV antigen-positive cells were detected within the tumor even at 2 weeks postinjection. These studies suggest that HF10 is safe and effective for oncolytic virotherapy. Currently, phase I/II trial of HF10 in patients with solid cutaneous tumors, including melanomas, has been completed (NCT01017185) in the USA.
7 Enhancement of Oncolytic Virotherapy by Gene Modification
7.1 Receptor Retargeted Mutants
HSV has several receptors to enter into host cells. For instance, glycoproteins gC and gB binds to heparan sulfate and glycoprotein gD binds to herpesvirus entry mediator (HVEM) (Salameh et al. 2012). There have been many attempts to make effective infection of oncolytic HSVs into cancer cells with limiting cell specificity by altering the receptors of the viruses.
IL-13 is the ligand of the IL-13 receptor 2α, expressed in glioblastoma and high-grade astrocytoma (Sengupta et al. 2014). IL-13 insertion mutants into gC or gD has been constructed as oncolytic HSVs against these tumors (Zhou et al. 2002; Zhou and Roizman 2006).
Another example is HER-2 . HER-2 is a member of the EGFR (epidermal growth factor receptor) family. This protein is overexpressed in breast and ovarian cancers, gastric carcinomas, glioblastomas, and so on (Jackson et al. 2013). R-LM249 was created by replacing gD dispensable region with the sequence for the single-chain antibody trastuzumab, which targets human epidermal HER-2 (Menotti et al. 2008). In preclinical study, a therapeutic effect of R-LM249 against a murine model of HER2 glioblastoma has been reproted (Gambini et al. 2012). R-LM249 also showed a therapeutic effect against peritoneal and brain metastases of ovarian and breast cancers by intraperitoneal injections (Nanni et al. 2013).
7.2 Modified (Armed and Targeted) Oncolytic HSV
To enhance antitumor responses of oncolytic HSVs, many studies have been conducted. One strategy of this is to generate insert immunostimulatory genes into oncolytic HSVs. Numerous immune-stimulating genes have been inserted into various oncolytic HSVs including IL-2, IL-12, IL-15, IL-18, tumor necrosis factor alpha, CD80 (B7.1), and GM-CSF like T-VEC (Nakashima and Chiocca 2014). These genes have many functions to activate, proliferate, differentiate, and maturate innate and acquired immune cells important for antitumor responses such as macrophage, dendritic cells, natural killer cells, cytotoxic T-cells, helper T-cells, and B cells.
Another way is to generate HSVs expressing therapeutic genes, including those that can activate prodrugs. There are many reports of oncolytic HSVs that have been modified to code for enzymes that catalyze prodrugs into active substrates, for example, HSV1yCD codes for the yeast cytosine deaminase (CD) enzyme that converts the nontoxic 5- uorocytosine into 5-FU (Nakamura et al. 2001).
7.3 Oncolytic HSVs as Amplicon Vector
Amplicon vectors are HSV-1 particles that carry a concatemeric form of a DNA plasmid, named the amplicon plasmid, instead of the viral genome. An amplicon plasmid has one origin of replication (generally ori-S) and one packaging signal (pac or a) from HSV-1, in addition to the transgenic sequences of interest. The vector has identical structure to wild-type HSV-1, so it has same immunological and host-range as wild-type virus.
HF10 has been investigated as a helper virus. An HF10-packaged mouse GM-CSF-expressing amplicon (mGM-CSF amplicon) was used to infect subcutaneously inoculated murine colorectal tumor cells (CT26 cells), and the antitumor effects were compared to tumors treated only with HF10. When mice subcutaneously inoculated with CT26 cells were intratumorally injected with HF10 or mGM-CSF amplicon, greater tumor regression and prolonged survival was seen in mGM-CSF amplicon-treated animals (Kohno et al. 2007). This amplicon system might be one of the good tools to used for tailor-made therapy.
8 Combination Therapy
8.1 Combination with Radiation
There is a report about intratumoral HSV G207 injection to glioma patients prior to a single palliative fraction of radiotherapy (Markert et al. 2014). The combination therapy showed some synergistic activity. Combination with chemoradiotherapy was also considered. Combined chemoradiotherapy with cisplatin and intratumoral injection of T-VEC for stage III/IV head and neck cancer patients showed 93% of complete response (CR) (Harrington et al. 2010).
8.2 Combination with Chemotherapy
Combination therapy with oncolytic HSV and chemotherapy was first evaluated with HSV1716 and four standard chemotherapeutic drugs: methotrexate, cisplatin, mitomycin C, and doxorubicin (Toyoizumi et al. 1999). Since then, there have been many studies reporting the increased efficacy of oncolytic HSV in combination with a many kinds of existing and potentially new anticancer drugs including cyclophosphamide, docetaxol, etoposide, 5- uorouracil (5-FU), and so on. In our laboratory, combination therapy with HF10 and chemotherapy has been studied (Braidwood et al. 2013).
For instance, we have shown enhanced antitumoral activity was shown in murine colorectal cancer model by HF10 inoculation following GEM treatment even in the distal tumor (Esaki et al. 2013). In murine subcutaneous melanoma model, intratumoral HF10 inoculation significantly inhibited tumor growth. When mice were treated with HF10 and dacarbazine (DTIC), the combination therapy induced a robust systemic antitumor immune response and prolonged survival. IFN-γ secretion from splenocytes of the HF10-DTIC combination therapy group showed more IFN-γ secretion than did the other groups (Tanaka et al. unpublished data).
8.3 Combination with Immune Checkpoint Inhibitors
Because oncolytic virotherapy has an aspect of cancer immunotherapy, combination therapy with oncolytic virotherapy and with immune checkpoint inhibitors is promising.
A phase Ib study of T-VEC and the anti-CTLA-4 antibody ipilimumab were administrated to patients with untreated, advanced cutaneous melanoma. The overall response rate by immune-related response criteria was 50% (Puzanov et al. 2016). The result was higher than would be expected from ipilimumab alone (10%). High regression rates were observed. The phase I study of combination therapy with pembrolizumab (NCT02263508) is ongoing. With regard to HF10, a phase II study of combination treatment with ipilimumab in patients with unresectable or metastatic melanoma is ongoing both in USA (NCT02272855) and in Japan.
9 Conclusion
In summary, HSV has many advantages for cancer therapy, and significant progress has been made in generating more effective oncolytic HSVs. Although much more work is required to better understand the efficacy and safety issues of these oncolytic HSVs before clinical use, the results from extensive preclinical and clinical trials have clearly demonstrated the potential of HSV recombinants for oncolytic viruses. Moreover, combination therapy with oncolytic HSVs and conventional chemotherapy radiotherapy and immune checkpoint inhibitors will expand the potential of oncolytic virotherapy.
References
Advani SJ, Chung SM, Yan SY et al (1999) Replication-competent, nonneuroinvasive genetically engineered herpes virus is highly effective in the treatment of therapy-resistant experimental human tumors. Cancer Res 59:2055–2058
Andtbacka RH, Kaufman HL, Collichio F et al (2015) Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J Clin Oncol 33:2780–2788
Bataille D, Epstein AL (1995) Herpes simplex virus type 1 replication and recombination. Biochimie 77:787–795
Boehmer PE, Nimonkar AV (2003) Herpes virus replication. IUBMB Life 55:3–22
Braidwood L, Graham SV, Graham A et al (2013) Oncolytic herpes viruses, chemotherapeutics, and other cancer drugs. Oncolytic Virother 2:57–74
Bringhurst RM, Schaffer PA (2006) Cellular stress rather than stage of the cell cycle enhances the replication and plating efficiencies of herpes simplex virus type 1 ICP0- viruses. J Virol 80:4528–4537
Cozzi PJ, Malhotra S, Mcauliffe P et al (2001) Intravesical oncolytic viral therapy using attenuated, replication-competent herpes simplex viruses G207 and Nv1020 is effective in the treatment of bladder cancer in an orthotopic syngeneic model. FASEB J 15:1306–1308
DePace NG (1912) Sulla Scomparsa di un enorme cancro begetante del callo dell’utero senza cura chirurgica [Italian]. Ginecol 9:82
Dock G (1904) Influence of complicating diseases upon leukemia. Am J Med Sci 127:563–592
Dossett LA, Kudchadkar RR, Zager JS (2015) BRAF and MEK inhibition in melanoma. Expert Opin Drug Saf 14:559–570
Ebright MI, Zager JS, Malhotra S et al (2002) Replication-competent herpes virus NV1020 as direct treatment of pleural cancer in a rat model. J Thorac Cardiovasc Surg 124:123–129
Esaki S, Goshima F, Kimura H et al (2013) Enhanced antitumoral activity of oncolytic herpes simplex virus with gemcitabine using colorectal tumor models. Int J Cancer 132:1592–1601
Fu X, Zhang X (2002) Potent systemic antitumor activity from an oncolytic herpes simplex virus of syncytial phenotype. Cancer Res 62:2306–2312
Fujimoto Y, Mizuno T, al SS (2006) Intratumoral injection of herpes simplex virus HF10 in recurrent head and neck squamous cell carcinoma. Acta Otolaryngol 126:1115–1117
Gambini E, Reisoli E, Appolloni I et al (2012) Replication-competent herpes simplex virus retargeted to HER2 as therapy for high-grade glioma. Mol Ther 20:994–1001
Geevarghese SK, Geller DA, De Haan HA et al (2010) Phase I/II study of oncolytic herpes simplex virus NV1020 in patients with extensively pretreated refractory colorectal cancer metastatic to the liver. Hum Gene Ther 21:1119–1128
Harrington KJ, Hingorani M, Tanay MA et al (2010) Phase I/II study of oncolytic HSV GM-CSF in combination with radiotherapy and cisplatin in untreated stage III/IV squamous cell cancer of the head and neck. Clin Cancer Res 16:4005–4015
Harrow S, Papanastassiou V, Harland J et al (2004) HSV1716 injection into the brain adjacent to tumour following surgical resection of high-grade glioma: safety data and long-term survival. Gene Ther 11:1648–1658
Hill A, Jugovic P, York I et al (1995) Herpes simplex virus turns off the TAP to evade host immunity. Nature 375:411–415
Hu JCC, Coffin RS, Davis CJ (2006) A phase I study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin Cancer Res 12:6737–6747
Huebner RJ, Rowe WP, Schatten WE et al (1956) Studies on the use of viruses in the treatment of carcinoma of the cervix. Cancer 9:1211–1218
Hummel JL, Safroneeva E, Mossman KL (2005) The role of ICP0-Null HSV-1 and interferon signaling defects in the effective treatment of breast adenocarcinoma. Mol Ther 12:1101–1110
Jackson C, Browell D, Gautrey H et al (2013) Clinical significance of HER-2 splice variants in breast cancer progression and drug resistance. Int J Cell Biol 2013:973584
Jones C, Inman M, Peng W et al (2005) The herpes simplex virus type 1 locus that encodes the latency-associated transcript enhances the frequency of encephalitis in male BALB/c mice. J Virol 79:14465–14469
Kanai R, Zaupa C, Sgubin D et al (2012) Effect of γ34.5 deletions on oncolytic herpes simplex virus activity in brain tumors. J Virol 86:4420–4431
Kaufman HL, Kim DW, DeRaffele G et al (2010) Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann Surg Oncol 17:718–730
Kemeny N, Brown K, Covey A et al (2006) Phase I, open-label, dose-escalating study of a genetically engineered herpes simplex virus, NV1020, in subjects with metastatic colorectal carcinoma to the liver. Hum Gene Ther 17:1214–1224
Kimata H, Takakuwa H, Goshima F et al (2003) Effective treatment of disseminated peritoneal colon cancer with new replication-competent herpes simplex viruses. Hepato-Gastroenterology 50:961–966
Kimata H, Imai T, Kikumori T et al (2006) Pilot study of oncolytic viral therapy using mutant herpes simplex virus (HF10) against recurrent metastatic breast cancer. Ann Surg Oncol 13:1078–1084
Kohno S, Luo C, Goshima F et al (2005) Herpes simplex virus type 1 mutant HF10 oncolytic viral therapy for bladder cancer. Urology 66:1116–1121
Kohno SI, Luo C, Nawa A et al (2007) Oncolytic virotherapy with an HSV amplicon vector expressing granulocyte-macrophage colony-stimulating factor using the replication-competent HSV type 1 mutant HF10 as a helper virus. Cancer Gene Ther 14:918–926
Kong Y, Yang T, Geller AI (1999) An efficient in vivo recombination cloning procedure for modifying and combining HSV-1 cosmids. J Virol Methods 80:129–136
Koshizuka T, Kawaguchi Y, Nishiyama Y (2005) Herpes simplex virus type 2 mem- brane protein UL56 associates with the kinesin motor protein KIF1A. J Gen Virol 86:527–533
Lilley CE, Carson CT, Muotri AR et al (2005) DNA repair proteins affect the lifecycle of herpes simplex virus 1. Proc Natl Acad Sci U S A 102:5844–5849
Liu BL, Robinson M, Han ZQ et al (2003) ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther 10:292–303
MacKie RM, Stewart B, Brown SM (2001) Intralesional injection of herpes simplex virus 1716 in metastatic melanoma. Lancet 357:525–526
Markert JM, Liechty PG, Wang W et al (2009) Phase Ib trial of mutant herpes simplex virus G207 inoculated pre-and post-tumor resection for recurrent GBM. Mol Ther 17:199–207
Markert JM, Razdan SN, Kuo HC et al (2014) A phase 1 trial of oncolytic HSV-1, G207, given in combination with radiation for recurrent GBM demonstrates safety and radiographic responses. Mol Ther 22:1048–1055
Martuza RL, Malick A, Markert JM et al (1991) Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science 252:854–885
Marconi P, Argnani R, Berto E et al (2008) HSV as a vector in vaccine development and gene therapy. Hum Vaccin 4:91–105
McGeoch DJ, Dalrymple MA, Davison AJ et al (1988) The complete DNA sequence of the long unique region in the genome of herpes simplex virus type 1. J Gen Virol 69:1531–1574
Menotti L, Cerretani A, Hengel H et al (2008) Construction of a fully retargeted herpes simplex virus 1 recombinant capable of entering cells solely via human epidermal growth factor receptor 2. J Virol 82(20):10153–10161
Mineta T, Rabkin SD, Yazaki T et al (1995) Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat Med 1:938–943
Mori I, Nishiyama Y (2005) Herpes simplex virus and varicella-zoster virus: why do these human alphaherpesviruses behave so differently from one another? Rev Med Virol 15:393–406
Mori I, Nishiyama Y (2006) Accessory genes define the relationship between the herpes simplex virus and its host. Microbes Infect 8:2556–2562
Mullerad M, Bochner BH, Adusumilli PS et al (2005) Herpes simplex virus based gene therapy enhances the efficacy of mitomycin C for the treatment of human bladder transitional cell carcinoma. J Urol 174:741–746
Nakamura H, Mullen JT, Chandrasekhar S et al (2001) Multimodality therapy with a replication-conditional herpes simplex virus 1 mutant that expresses yeast cytosine deaminase for intratumoral conversion of 5-fluorocytosine to 5-fluorouracil. Cancer Res 61:5447–5452
Nakao A, Kimata H, al IT (2004) Intratumoral injection of herpes simplex virus HF10 in recurrent breast cancer. Ann Oncol 15:988–989
Nakashima H, Chiocca EA (2014) Modification of HSV-1 to an oncolytic virus. Methods Mol Biol 1144:117–127
Nanni P, Gatta V, Menotti L et al (2013) Preclinical therapy of disseminated HER-2+ ovarian and breast carcinomas with a HER-2-retargeted oncolytic herpesvirus. PLoS Pathog 9:e1003155
Nawa A, Luo C, Zhang L et al (2008) Non- engineered, naturally oncolytic herpes simplex virus HSV1 HF-10: applications for cancer gene therapy. Curr Gene Ther 8:208–221
Nishiyama Y (1996) Herpesvirus genes: molecular basis of viral replication and pathogenicity. Nagoya J Med Sci 59:107–119
Nishiyama Y (2004) Herpes simplex virus gene products: the accessories reflect her lifestyle well. Rev Med Virol 14:33–46
Nishiyama Y, Kimura H, Daikoku T (1991) Complementary lethal invasion of the central nervous system by nonneuroinvasive herpes simplex virus types 1 and 2. J Virol 65:4520–4524
Pérez-Herrero E, Fernández-Medarde A (2015) Advanced targeted therapies in cancer: drug nanocarriers, the future of chemotherapy. Eur J Pharm Biopharm 93:52–79
Pourchet A, Fuhrmann SR, Pilones KA et al (2016) CD8(+) T-cell immune evasion enables oncolytic virus immunotherapy. EBioMedicine 5:59–67
Puzanov I, Milhem MM, Minor D et al (2016) Talimogene laherparepvec in combination with ipilimumab in previously untreated, unresectable stage IIIB-IV melanoma. J Clin Oncol 34:2619–2626
Salameh S, Sheth U, Shukla D (2012) Early events in herpes simplex virus lifecycle with implications for an infection of lifetime. Open Virol J 6:1–6
Sengupta S, Thaci B, Crawford AC et al (2014) Interleukin-13 receptor alpha 2-targeted glioblastoma immunotherapy. Biomed Res Int 2014:952128
Senzer NN, Kaufman HL, Amatruda T et al (2009) Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J Clin Oncol 27:5763–5771
Sobol PT, Mossman KL (2006) ICP0 prevents RNase L-independent rRNA cleavage in herpes simplex virus type 1-infected cells. J Virol 80:218–225
Spain L, Diem S, Larkin J (2016) Management of toxicities of immune checkpoint inhibitors. Cancer Treat Rev 44:51–60
Spear MA, Sun F, Eling DJ et al (2000) Cytotoxicity, apoptosis, and viral replication in tumor cells treated with oncolytic ribonucleotide reductase-defective herpes simplex type 1 virus (hrR3) combined with ionizing radiation. Cancer Gene Ther 7:1051–1059
Stanziale SF, Petrowsky H, Adusumilli PS et al (2004) Infection with oncolytic herpes simplex virus-1 induces apoptosis in neighboring human cancer cells: a potential target to increase anticancer activity. Clin Cancer Res 10:3225–3232
Stavropoulos TA, Strathdee CA (1998) An enhanced packaging system for helper- dependent herpes simplex virus vectors. J Virol 72:7137–7143
Stiles BM, Adusumilli PS, Bhargava A et al (2006a) Minimally invasive localization of oncolytic herpes simplex viral therapy of metastatic pleural cancer. Cancer Gene Ther 13:53–64
Stiles BM, Adusumilli PS, Stanziale SF et al (2006b) Estrogen enhances the efficacy of an oncolytic HSV-1 mutant in the treatment of estrogen receptor-positive breast cancer. Int J Oncol 28:1429–1439
Sundaresan P, Hunter WD, Martuza RL et al (2000) Attenuated, replication-competent herpes simplex virus type 1 mutant G207: safety evaluation in mice. J Virol 74:3832–3841
Sze DY, Iagaru AH, Gambhir SS et al (2012) Response to intra-arterial oncolytic virotherapy with the herpes virus NV1020 evaluated by [18F]fluorodeoxyglucose positron emission tomography and computed tomography. Hum Gene Ther 23:91–97
Sze DY, Reid TR, Rose SC (2013) Oncolytic virotherapy. J Vasc Interv Radiol 24:1115–1122
Takakuwa H, Goshima F, Nozawa N et al (2003) Oncolytic viral therapy using a spontaneously generated herpes simplex virus type 1 variant for disseminated peritoneal tumor in immunocompetent mice. Arch Virol 148:813–825
Tanaka M, Kagawa H, Yamanashi Y et al (2003) Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J Virol 77:1382–1391
Tang T, Eldabaje R, Yang L (2016) Current status of biological therapies for the treatment of metastatic melanoma. Anticancer Res 36:3229–3241
Taylor TJ, Brockman MA, McNamee EE et al (2002) Herpes simplex virus. Front Biosci 7:d752–d764
Teshigahara O, Goshima F, Takao K et al (2004) Oncolytic viral therapy for breast cancer with herpes simplex virus type 1 mutant HF 10. J Surg Oncol 85:42–47
Todo T (2008) Oncolytic virus therapy using genetically engineered herpes simplex viruses. Front Biosci 200813:2060–2064
Todo T, Feigenbaum F, Rabkin SD et al (2000) Viral shedding and biodistribution of G207, a multimutated, conditionally replicating herpes simplex virus type 1, after intracerebral inoculation in aotus. Mol Ther 2:588–595
Toyoizumi T, Mick R, Abbas AE et al (1999) Combined therapy with chemotherapeutic agents and herpes simplex virus type 1 ICP34.5 mutant (HSV-1716) in human non- small cell lung cancer. Hum Gene Ther 10:3013–3029
Ushijima Y, Luo C, Goshima F et al (2007) Determination and analysis of the DNA sequence of highly attenuated herpes simplex virus type 1 mutant HF10, a potential oncolytic virus. Microbes Infect 9:142–149
Watanabe D, Goshima F, Mori I et al (2008) Oncolytic virotherapy for malignant melanoma with herpes simplex virus type 1 mutant HF10. J Dermatol Sci 50:185–196
Witt MN, Braun GS, Ihrler S et al (2009) Occurrence of HSV-1-induced pneumonitis in patients under standard immunosuppressive therapy for rheumatic, vasculitic, and connective tissue disease. BMC Pulm Med 18:22
Zabierowski SE, Deluca NA (2008) Stabilized binding of TBP to the TATA box of herpes simplex virus type 1 early (tk) and late (gC) promoters by TFIIA and ICP4. J Virol 82:3546–3554
Zhou G, Roizman B (2006) Construction and properties of a herpes simplex virus 1 designed to enter cells solely via the IL-13alpha2 receptor. Proc Natl Acad Sci U S A 103:5508–5513
Zhou G, Ye GJ, al DW (2002) Engineered herpes simplex virus 1 is dependent on IL13Ralpha 2 receptor for cell entry and independent of glycoprotein D receptor interaction. Proc Natl Acad Sci U S A 99:15124–15129
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Watanabe, D., Goshima, F. (2018). Oncolytic Virotherapy by HSV. In: Kawaguchi, Y., Mori, Y., Kimura, H. (eds) Human Herpesviruses. Advances in Experimental Medicine and Biology, vol 1045. Springer, Singapore. https://doi.org/10.1007/978-981-10-7230-7_4
Download citation
DOI: https://doi.org/10.1007/978-981-10-7230-7_4
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-7229-1
Online ISBN: 978-981-10-7230-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)