
Abstract
Although de-novo neuroendocrine prostate cancer (NEPC) is rare, with increasing use of potent androgen receptor (AR) pathway inhibitors, the incidence of treatment-related NEPC (t-NEPC) is rapidly rising. Since NEPC is an aggressive disease with poor prognosis, novel therapeutic strategies are urgently needed. Recent genomic and molecular analysis have identified key oncogenes (MYCN, AURKA) and tumor suppressor genes (TP53, RB1) to play key roles in driving NEPC. Novel in vivo and in vitro research models of NEPC were developed to serve as valuable resource to study functional relevance of the key genes in NEPC development. Upon AR pathway inhibition, these genomic alterations seem to facilitate epithelial plasticity by upregulating the genes implicated in maintaining pluripotency (SOX2, EZH2), resulting in development of divergent tumor including NEPC from castration resistant prostate cancer. Further understanding of the molecular biology is required to identify novel molecular targets and biomarkers that would help rescue patients from this lethal variant.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others
Keywords
- Neuroendocrine prostate cancer
- Anaplastic prostate cancer
- Aggressive variant prostate cancer
- Epithelial plasticity
39.1 Introduction
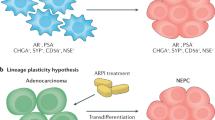
Neuroendocrine prostate cancer (NEPC) is a rare form of aggressive prostate cancer which grows independently of androgen receptor (AR) signaling pathway. Typically, the tumor shows small cell carcinoma morphology, expresses neuroendocrine(NE) markers such as Chromogranin A, Synaptophysin, NCAM1, and NSE, and does not express AR or PSA. Clinically, it is characterized by exclusive visceral or predominantly lytic bone metastases, bulky tumor masses, sensitivity to platinum containing chemotherapy regimen, and poor survival [1]. The incidence of de-novo NEPC was reported to be 0.5–2% [2]. However, NEPC is also known to arise in patients who have been heavily treated with AR pathway targeting therapy [3]. This form of NEPC is known as treatment-related neuroendocrine prostate cancer (t-NEPC) [4]. The incidence of t-NEPC has been rising rapidly due to increasing use of potent AR pathway inhibitors such as Abiraterone and Enzalutamide. A recent autopsy series showed that up to 25% of the patients dying from CRPC demonstrated some signs of t-NEPC [5]. With its increasing incidence and lack of appropriate treatment, NEPC is emerging as an imminent threat to treatment of prostate cancer patients, and it is imperative to understand its disease biology and develop novel treatment strategy for this lethal disease [6].
39.2 Classification of NEPC
In 2013, a working group assembled by Prostate Cancer Foundation proposed a new pathologic classification of NEPC [7]. The new classification consisted of (1) usual prostate adenocarcinoma with NE differentiation, (2) adenocarcinoma with Paneth cell NE differentiation, (3) carcinoid tumor, (4) small cell carcinoma (SCC), (5) large cell neuroendocrine carcinoma (LCNEC), and (6) mixed NE carcinoma-acinar adenocarcinoma. In addition, CRPC with small cell carcinoma-like clinical presentation was defined as an independent entity. The word “treatment-related neuroendocrine prostate cancer (t-NEPC)” has been used interchangeably with “anaplastic prostate carcinoma” [1] and “aggressive variant prostate carcinoma (AVPC)” [6]; however, the latter two are defined entirely based on clinical factors and may encompass a broader range of AR independent CRPC. Prior to the proposal of the new pathologic classification, the clinical impact of NEPC was confounded by contradictory results [8, 9]. Since the clinical implication of NEPC other than SCC and t-NEPC is unclear, the working group recommended against routine IHC examination of prostate cancer specimen for NE markers. Currently, it is recommended that PCa mixed with NE marker positive cells be treated with androgen deprivation therapy unless the tumor shows morphologically distinct SCC [6].
39.3 Cell of Origin of NEPC
Normal prostate gland contains foci of NE cells scattered within the prostatic epithelium [7]. These cells are known to release various peptide hormones including chromogranin A, calcitonin, and NSE and affect the surrounding cells. However, in normal prostate gland, these cells are quiescent. Whether NEPC arises from these NE cells or from epithelial cells has been under long debate. Although the origin of de-novo NEPC is still not clear, recent genomic and molecular studies have shown that t-NEPC arises from adenocarcinoma by transdifferentiation [6, 10,11,12,13]. TMPRSS-ERG gene fusion is the most frequent structural variation seen in prostate cancer, and is reported to be observed in nearly half of PCa cases [14]. TMPRSS-ERG translocation is known to be an early event in PCa carcinogenesis [15]. Intriguingly, the reported frequency of the translocation is similar in t-NEPC compared to that in adenocarcinoma [16], and a recent study reported that there was a large overlap in the overall somatic copy-number landscape between CRPC and t-NEPC [13]. Epithelial plasticity is a phenomenon in which cells treated with specific molecular targeting therapy acquire phenotypic characteristics of a cell lineage whose survival no longer depends on the targeted pathway [17]. A recent molecular study has shown that MYCN and AKT1 could transform human epithelial cells to both PCa and NEPC [18], and another study demonstrated that TP53 and RB1 silenced prostate cancer cells could give rise to both CRPC and NEPC [19, 20]. Supported by these robust genetic and molecular biology data, it is now considered that upon potent AR pathway inhibition, NEPC develops from adenocarcinoma as a result of epithelial plasticity [13].
39.4 Research Models of NEPC
Until recently, LNCaP cell line has been studied extensively as a model of NEPC transdifferentiation, since the cells start to take “neuronal” cell morphology and express NE markers under various stress including androgen depletion [21, 22] and treatments with cAMP [23,24,25], cytokines [26, 27], and growth factors [28]. However, the morphology of the cells is completely distinct from those of SCC, and the cells are generally slower growing than the untreated cells [29]. Even though some researchers have claimed that these cells promote growth of the surrounding cells in a paracrine manner [30], it is more likely that these cells represent quiescent NE cells seen in some CRPC specimen and not the clinically aggressive NEPC. It has recently been reported that dual knockdown of TP53 and RB1 in LNCaP cells facilitates lineage plasticity and some of the cells transdifferentiate into NEPC. Considering the critical role of these major tumor suppressors discussed later in this chapter, this may be a more appropriate model to study NEPC development in vitro.
To date, only one cell line has been established from clinical NEPC. NCI-H660 was initially described as a small cell lung carcinoma [31], however, later corrected to be derived from the prostate, and the cell line harbors TMPRSS-ERG translocation [32]. The cell line also harbors TP53 mutation and RB1 deletion. Interestingly, the cell line grows as floating cells similar to most other cell lines derived from SCC of the lung, and is easier to grow in vivo than in vitro. Considering the origin of the cell line, NCI-H660 represents the best model to study t-NEPC; however, its slow growth in vitro and difficulty of transfection raises the bar in terms of its use in many molecular biology experiments.
Several patient-derived xenograft (PDX) models of NEPC have been reported [33,34,35]. Of those, LTL-331/LTL-331R is a unique model of transdifferentiation [11]. LTL-331, which was established by grafting a Gleason score 9 adenocarcinoma from a patient into mouse sub-renal capsule, regresses upon castration, however, later regrows as a PSA negative NEPC (LTL-331R). Even though LTL-331 shows normal adenocarcinoma morphology and expresses AR and PSA, LTL-331R is consistent with SCC, does not express AR or PSA, and expresses NE markers including Chromogranin A and Synaptophysin. The rapid growth of LTL-331R is consistent with aggressive behavior of clinical NEPC, and at the transcriptome level, LTL-331R is highly similar to clinical NEPC. At the DNA level, LTL-331 and LTL-331R show very similar copy-number profile and fusion gene profile, suggesting transdifferentiation from adenocarcinoma to NEPC rather than clonal selection of preexisting minor NEPC cells. Even though the transdifferentiation from LTL-331 to LTL-331R is highly reproducible, the exact mechanism or genetic signature that predispose to NEPC transdifferentiation is unclear. Intriguingly, the LTL-331 harbors a single-copy loss of TP53 and functional C277G mutation in the remaining allele [10]. In addition, there is a single-copy loss of RB1. Since dual alteration of TP53 and RB1 is known to facilitate lineage plasticity, and there is significant alteration in the Rb pathway genetic signature upon transdifferentiation from LTL-331 to LTL-331R, these baseline alterations of TP53 and RB1 may be one of the factors which predispose to transdifferentiation. To date, LTL-331 model serves as the only model of transdifferentiation.
With recent identification of key oncogenes and tumor suppressor genes in NEPC development, several genetically engineered mouse models of NEPC have been developed. Next generation sequencing studies have identified amplification of MYCN in NEPC [2], and a murine model expressing N-myc specifically in the prostate was generated [36]. In the model, N-myc overexpression, in cooperation with Pten knockout, resulted in large invasive prostate tumors with a variety of morphologies including foci of AR positive adenocarcinoma and SCC. This likely represents the NEPC formation as a result of lineage plasticity. Another genomic hallmark of NEPC is aberration of p53 and Rb pathway, which is also common in SCC of the lung. Conditional double knockout of Pten and Rb1 in the murine prostate resulted in development of heterogenous tumor, and additional p53 knockout conferred de-novo resistance to hormone therapy [19]. The double and triple knockout models showed gene signature similar to clinical NEPC. Overall, these genetically engineered mouse models could serve as ideal models to study development of NEPC which occurs as a result of epithelial plasticity.
A classic murine model of spontaneous prostate carcinogenesis is transgenic adenocarcinoma mouse prostate (TRAMP) [37]. TRAMP model is a genetically engineered murine model driven by conditional expression of SV40 large T antigen in the prostate, and p53 and Rb pathways are inactivated. TRAMP male mouse develops PCa with distant metastasis by 24–30 weeks of age, and subsequently some tumors progresses to NEPC, in line with lineage plasticity [38]. Cell lines have also been established from TRAMP tumors for in vitro use [39].
The research models discussed in this section are mainly for studies of t-NEPC. Currently, there is no specific model for de-novo NEPC, and whether the research models for t-NEPC could also be used to study de-novo NEPC is not clear.
39.5 Molecular Basis of NEPC
Next generation sequencing of clinical NEPC samples have opened the door to understanding the genomic and molecular features of NEPC. Here we specifically focus on the major pathways and genes involved in t-NEPC development, and how these findings contributed to the current concept that t-NEPC develops from adenocarcinoma as a result of epithelial plasticity.
39.5.1 MYCN, AURKA, and NEPC
MYCN and AURKA amplifications were among the first genomic aberrations identified using next generation sequencing of NEPC [2]. These alterations were discovered by RNA-sequencing and oligonucleotide array of a cohort of NEPC and PCa clinical samples followed by validation using a large patient cohort. The study showed MYCN and AURKA overexpression/ gene amplification in 40% of NEPC and 5% of PCa. MYCN and AURKA are oncogenes that are known to interact with each other. Interestingly, in nearly all AURKA amplification positive case of NEPC, there was concurrent amplification of MYCN. Aurora kinase A and N-myc protein interacted in vitro and enhanced Aurora kinase A stability. N-myc overexpressed LNCaP cells were sensitive to Aurora kinase A inhibitor in vitro. In vivo, NCI-H660 xenograft model was sensitive to Auroka kinase A inhibitor in contrast to LNCaP xenograft which showed no response. These findings have led to an ongoing multicenter phase II clinical trial using Auroka kinase A inhibitor MLN8237 in NEPC patients (ClinicalTrials.gov Identifier: NCT01799278). Early results showed modest response; however, two patients achieved exceptional response with complete resolution of liver metastasis. Additional biomarker to predict responders is likely to be required.
The critical role of MYCN in NEPC development has prompted generation of the murine model discussed above. Gene set enrichment analysis of the tumor that developed in the model showed enrichment of PRC2/EZH2 targets and suppression of AR signaling [36]. EZH2 is a component of PRC2 complex that primarily methylates H3K27 to suppress transcription and is implicated in maintaining pluripotency. EZH2 cooperatively suppress expression of N-Myc targets including AR and drives NEPC. EZH2 silencing as well as EZH2 inhibition using GSK503 restored Enzalutamide sensitivity of PTEN and RB1 double knockout mouse in vivo. Another EZH2 inhibitor (GSK343) preferentially decreased the viability of NCI-H660 cells, as compared to that in other non-neuroendocrine prostate cancer cells. EZH2 inhibition may be a novel approach for NEPC treatment.
Another study showed that in primary human prostate basal epithelium, overexpression of MYCN and AKT1 was sufficient to transform the cells to grow tumors in mice, and the tumor that developed showed mixed NEPC and adenocarcinoma, which also supports the concept that MYCN facilitates epithelial plasticity [18].
39.5.2 p53, Rb Pathway, and NEPC
p53 mutation and RB1 inactivation have been known to be one of the most common genomic aberrations in lung SCC [40, 41]. In prostate SCC, strongly positive p53 staining by IHC was observed in 56% of SCC with 60% of the cases showing TP53 mutation. Rb protein loss was seen in 90% of SCC with RB1 allelic loss in 85% of the cases [42]. In addition, RB1 copy number loss was identified to be the strongest discriminator between “aggressive variant prostate cancer” and unselected CRPC [43]. However, in routine clinical practice, it is difficult to examine RB1 copy number. Therefore, the usefulness of p16 and cyclin D1 expression by IHC as surrogates for Rb pathway activity was tested [44]. As a result, expression of Cyclin D1 paralleled with loss of Rb signature, and overall, 88% of SCC showed Cyclin D1 loss by IHC compared with less than 10% in high grade PCa, confirming the usefulness of Cyclin D1 IHC as a marker of Rb pathway aberration.
Functionally, p53 and Rb inactivation collaborate to enhance epithelial plasticity, which eventually lead to development of NEPC. In vitro, dual knockdown or knockout of TP53 and RB1 in LNCaP resulted in increase of basal cell and NE markers and reduction of luminal cell markers [20]. Dual knockdown of TP53 and RB1 was sufficient to confer resistance to Enzalutamide. The study also identified that dual knockdown of TP53 and RB1 results in SOX2 elevation, and that the increased expression of NE and basal markers as well as Enzalutamide resistance in these cells can be rescued with SOX2 knockdown. These results indicate that SOX2 overexpression upon p53 and Rb inactivation is one of the major mechanisms of enhanced lineage plasticity. Another study, using the previously discussed in vivo model of conditional knockout mouse, similarly showed that increased lineage plasticity observed upon RB1 and TP53 loss is conferred by increased expression of SOX2 and EZH2 [19]. Even though direct relationship between MYCN/AURKA amplification and p53/ Rb inactivation has not been clarified yet, both pathways seem to drive NEPC by upregulating genes implicated in maintenance of pluripotency and facilitating lineage plasticity.
39.5.3 AR Inhibition and NEPC
Another area of intensive research is how AR inhibition drives NEPC. A recent study identified a neural transcription factor BRN2 to be one of the major genes that link AR inhibition to NEPC development [45]. The gene was identified using a unique panel of Enzalutamide resistant cell lines derived from serial in vivo selection of LNCaP xenografts. The panel consisted of heterogenous clones with different AR and PSA expression levels. One of the clones, 42DENZR represented NEPC, and by comparing the whole transcriptome of the panel of cells, BRN2 was identified to be specifically upregulated in 42DENZR. BRN2 was directly repressed by AR, and BRN2 expression induced NE marker expression and promoted cell growth. Furthermore, BRN2 regulated expression and activity of SOX2, again showing association between NEPC and increased lineage plasticity.
Paternally Expressed 10 (PEG10) is another gene directly repressed by AR that is implicated development of NEPC [10]. PEG10 is a unique retrotransposon derived gene that retains gag and pol domain [46]. Structurally, PEG10 resembles HIV virus, and has a unique −1 ribosomal frameshift sequence which enables balanced expression of gag (RF1) and pol (RF1/2) protein [47]. PEG10 integrated into the therian mammalian genome after the split with prototherians and is indispensable for placental development [48]. PEG10 RF1 promotes cell invasion through TGF-β pathway, and PEG10 RF1/2 promotes cell cycle progression in the absence of TP53 and RB1 [10]. The expression and function of PEG10 is tightly regulated by p53, Rb, and N-myc. Since PEG10 is a testicular antigen whose expression in normal cells is restricted to embryonal organs and neurons [49], and it has domains similar to HIV, PEG10 is potentially targetable [50].
39.5.4 Clonal Evolution of NEPC
The mode of clonal evolution of NEPC has been recently studied by whole-exome sequencing of sequential biopsies from the same patients during treatment [13]. Divergent clonal evolution, in which CRPC and NEPC cells could arise from the same CRPC clone in a divergent manner, was the most compatible mode of evolution. A recent report from SU2C/PCF/AACR West Coast Prostate Cancer Dream Team reported another distinct subtype of CRPC which histologically shows intermediate pattern between SCC and adenocarcinoma. These results are consistent with in vivo and in vitro data which supports the concept that t-NEPC arises as a result of enhanced epithelial plasticity upon potent AR pathway inhibition and additional aberrations in major oncogenes and tumor suppressor genes.
39.6 Future Perspective
Due to the rarity of NEPC and lack of suitable in vitro and in vivo models that represents clinical NEPC, NEPC was understudied until quite recently. However, next generation sequencing of NEPC samples have opened the door to understanding the genetic hallmarks of NEPC, and novel in vivo models are now at hand to study molecular mechanisms underlying its disease biology. With increasing threat of NEPC, further efforts are required to identify novel therapeutic targets and biomarkers that would lead to effective treatment of this lethal variant.
References
Aparicio AM, Harzstark AL, Corn PG, Wen S, Araujo JC, SM T, et al. Platinum-based chemotherapy for variant castrate-resistant prostate cancer. Clin Cancer Res. 2013;19(13):3621–30.
Beltran H, Rickman DS, Park K, Chae SS, Sboner A, MacDonald TY, et al. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011;1(6):487–95.
Hirano D, Okada Y, Minei S, Takimoto Y, Nemoto N. Neuroendocrine differentiation in hormone refractory prostate cancer following androgen deprivation therapy. Eur Urol. 2004;45(5):586–92. discussion 92
Beltran H, Tagawa ST, Park K, MacDonald T, Milowsky MI, Mosquera JM, et al. Challenges in recognizing treatment-related neuroendocrine prostate cancer. J Clin Oncol. 2012;30(36):e386–9.
Aparicio A, Logothetis CJ, Maity SN. Understanding the lethal variant of prostate cancer: power of examining extremes. Cancer Discov. 2011;1(6):466–8.
Beltran H, Tomlins S, Aparicio A, Arora V, Rickman D, Ayala G, et al. Aggressive variants of castration-resistant prostate cancer. Clin Cancer Res. 2014;20(11):2846–50.
Epstein JI, Amin MB, Beltran H, Lotan TL, Mosquera JM, Reuter VE, et al. Proposed morphologic classification of prostate cancer with neuroendocrine differentiation. Am J Surg Pathol. 2014;38(6):756–67.
Sargos P, Ferretti L, Gross-Goupil M, Orre M, Cornelis F, Henriques de Figueiredo B, et al. Characterization of prostate neuroendocrine cancers and therapeutic management: a literature review. Prostate Cancer Prostatic Dis. 2014;17(3):220–6.
Jeetle SS, Fisher G, Yang ZH, Stankiewicz E, Møller H, Cooper CS, et al. Neuroendocrine differentiation does not have independent prognostic value in conservatively treated prostate cancer. Virchows Arch. 2012;461(2):103–7.
Akamatsu S, Wyatt AW, Lin D, Lysakowski S, Zhang F, Kim S, et al. The placental gene PEG10 promotes progression of neuroendocrine prostate cancer. Cell Rep. 2015;12(6):922–36.
Lin D, Wyatt AW, Xue H, Wang Y, Dong X, Haegert A, et al. High fidelity patient-derived xenografts for accelerating prostate cancer discovery and drug development. Cancer Res. 2014;74(4):1272–83.
Zou M, Toivanen R, Mitrofanova A, Floch N, Hayati S, Sun Y, et al. Transdifferentiation as a mechanism of treatment resistance in a mouse model of castration-resistant prostate cancer. Cancer Discov. 2017;7:736.
Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med. 2016;22:298.
Mosquera JM, Mehra R, Regan MM, Perner S, Genega EM, Bueti G, et al. Prevalence of TMPRSS2-ERG fusion prostate cancer among men undergoing prostate biopsy in the United States. Clin Cancer Res. 2009;15(14):4706–11.
Network CGAR. The molecular taxonomy of primary prostate cancer. Cell. 2015;163(4):1011–25.
Lotan TL, Gupta NS, Wang W, Toubaji A, Haffner MC, Chaux A, et al. ERG gene rearrangements are common in prostatic small cell carcinomas. Mod Pathol. 2011;24(6):820–8.
Bishop JL, Davies A, Ketola K, Zoubeidi A. Regulation of tumor cell plasticity by the androgen receptor in prostate cancer. Endocr Relat Cancer. 2015;22(3):R165–82.
Lee JK, Phillips JW, Smith BA, Park JW, Stoyanova T, McCaffrey EF, et al. N-Myc drives neuroendocrine prostate cancer initiated from human prostate epithelial cells. Cancer Cell. 2016;29(4):536–47.
SY K, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science. 2017;355(6320):78–83.
Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, Chen CC, et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science. 2017;355(6320):84–8.
Shen R, Dorai T, Szaboles M, Katz AE, Olsson CA, Buttyan R. Transdifferentiation of cultured human prostate cancer cells to a neuroendocrine cell phenotype in a hormone-depleted medium. Urol Oncol. 1997;3(2):67–75.
Zhang XQ, Kondrikov D, Yuan TC, Lin FF, Hansen J, Lin MF. Receptor protein tyrosine phosphatase alpha signaling is involved in androgen depletion-induced neuroendocrine differentiation of androgen-sensitive LNCaP human prostate cancer cells. Oncogene. 2003;22(43):6704–16.
Bang YJ, Pirnia F, Fang WG, Kang WK, Sartor O, Whitesell L, et al. Terminal neuroendocrine differentiation of human prostate carcinoma cells in response to increased intracellular cyclic AMP. Proc Natl Acad Sci U S A. 1994;91(12):5330–4.
Cox ME, Deeble PD, Lakhani S, Parsons SJ. Acquisition of neuroendocrine characteristics by prostate tumor cells is reversible: implications for prostate cancer progression. Cancer Res. 1999;59(15):3821–30.
Cox ME, Deeble PD, Bissonette EA, Parsons SJ. Activated 3′,5′-cyclic AMP-dependent protein kinase is sufficient to induce neuroendocrine-like differentiation of the LNCaP prostate tumor cell line. J Biol Chem. 2000;275(18):13812–8.
Qiu Y, Robinson D, Pretlow TG, Kung HJ. Etk/Bmx, a tyrosine kinase with a pleckstrin-homology domain, is an effector of phosphatidylinositol 3′-kinase and is involved in interleukin 6-induced neuroendocrine differentiation of prostate cancer cells. Proc Natl Acad Sci U S A. 1998;95(7):3644–9.
Deeble PD, Murphy DJ, Parsons SJ, Cox ME. Interleukin-6- and cyclic AMP-mediated signaling potentiates neuroendocrine differentiation of LNCaP prostate tumor cells. Mol Cell Biol. 2001;21(24):8471–82.
Kim J, Adam RM, Freeman MR. Activation of the Erk mitogen-activated protein kinase pathway stimulates neuroendocrine differentiation in LNCaP cells independently of cell cycle withdrawal and STAT3 phosphorylation. Cancer Res. 2002;62(5):1549–54.
Mori S, Murakami-Mori K, Bonavida B. Interleukin-6 induces G1 arrest through induction of p27(Kip1), a cyclin-dependent kinase inhibitor, and neuron-like morphology in LNCaP prostate tumor cells. Biochem Biophys Res Commun. 1999;257(2):609–14.
Deeble PD, Cox ME, Frierson HF, Sikes RA, Palmer JB, Davidson RJ, et al. Androgen-independent growth and tumorigenesis of prostate cancer cells are enhanced by the presence of PKA-differentiated neuroendocrine cells. Cancer Res. 2007;67(8):3663–72.
Ohsaki Y, Yang HK, Le PT, Jensen RT, Johnson BE. Human small cell lung cancer cell lines express functional atrial natriuretic peptide receptors. Cancer Res. 1993;53(13):3165–71.
Mertz KD, Setlur SR, Dhanasekaran SM, Demichelis F, Perner S, Tomlins S, et al. Molecular characterization of TMPRSS2-ERG gene fusion in the NCI-H660 prostate cancer cell line: a new perspective for an old model. Neoplasia. 2007;9(3):200–6.
Aparicio A, Tzelepi V, Araujo JC, Guo CC, Liang S, Troncoso P, et al. Neuroendocrine prostate cancer xenografts with large-cell and small-cell features derived from a single patient's tumor: morphological, immunohistochemical, and gene expression profiles. Prostate. 2011;71(8):846–56.
Lapuk AV, Wu C, Wyatt AW, McPherson A, McConeghy BJ, Brahmbhatt S, et al. From sequence to molecular pathology, and a mechanism driving the neuroendocrine phenotype in prostate cancer. J Pathol. 2012;227(3):286–97.
Tzelepi V, Zhang J, JF L, Kleb B, Wu G, Wan X, et al. Modeling a lethal prostate cancer variant with small-cell carcinoma features. Clin Cancer Res. 2012;18(3):666–77.
Dardenne E, Beltran H, Benelli M, Gayvert K, Berger A, Puca L, et al. N-Myc induces an EZH2-mediated transcriptional program driving neuroendocrine prostate cancer. Cancer Cell. 2016;30(4):563–77.
Gingrich JR, Barrios RJ, Kattan MW, Nahm HS, Finegold MJ, Greenberg NM. Androgen-independent prostate cancer progression in the TRAMP model. Cancer Res. 1997;57(21):4687–91.
Qi J, Nakayama K, Cardiff RD, Borowsky AD, Kaul K, Williams R, et al. Siah2-dependent concerted activity of HIF and FoxA2 regulates formation of neuroendocrine phenotype and neuroendocrine prostate tumors. Cancer Cell. 2010;18(1):23–38.
Foster BA, Gingrich JR, Kwon ED, Madias C, Greenberg NM. Characterization of prostatic epithelial cell lines derived from transgenic adenocarcinoma of the mouse prostate (TRAMP) model. Cancer Res. 1997;57(16):3325–30.
Olivier M, Eeles R, Hollstein M, Khan MA, Harris CC, Hainaut P. The IARC TP53 database: new online mutation analysis and recommendations to users. Hum Mutat. 2002;19(6):607–14.
Kaye FJ. RB and cyclin dependent kinase pathways: defining a distinction between RB and p16 loss in lung cancer. Oncogene. 2002;21(45):6908–14.
Tan HL, Sood A, Rahimi HA, Wang W, Gupta N, Hicks J, et al. Rb loss is characteristic of prostatic small cell neuroendocrine carcinoma. Clin Cancer Res. 2014;20(4):890–903.
Aparicio AM, Shen L, Tapia EL, JF L, Chen HC, Zhang J, et al. Combined tumor suppressor defects characterize clinically defined aggressive variant prostate cancers. Clin Cancer Res. 2016;22(6):1520–30.
Tsai H, Morais CL, Alshalalfa M, Tan HL, Haddad Z, Hicks J, et al. Cyclin D1 loss distinguishes prostatic small-cell carcinoma from most prostatic adenocarcinomas. Clin Cancer Res. 2015;21(24):5619–29.
Bishop JL, Thaper D, Vahid S, Davies A, Ketola K, Kuruma H, et al. The master neural transcription factor BRN2 is an androgen receptor-suppressed driver of neuroendocrine differentiation in prostate cancer. Cancer Discov. 2017;7(1):54–71.
Clark MB, Jänicke M, Gottesbühren U, Kleffmann T, Legge M, Poole ES, et al. Mammalian gene PEG10 expresses two reading frames by high efficiency −1 frameshifting in embryonic-associated tissues. J Biol Chem. 2007;282(52):37359–69.
Lux H, Flammann H, Hafner M, Lux A. Genetic and molecular analyses of PEG10 reveal new aspects of genomic organization, transcription and translation. PLoS One. 2010;5(1):e8686.
Ono R, Nakamura K, Inoue K, Naruse M, Usami T, Wakisaka-Saito N, et al. Deletion of Peg10, an imprinted gene acquired from a retrotransposon, causes early embryonic lethality. Nat Genet. 2006;38(1):101–6.
Okabe H, Satoh S, Furukawa Y, Kato T, Hasegawa S, Nakajima Y, et al. Involvement of PEG10 in human hepatocellular carcinogenesis through interaction with SIAH1. Cancer Res. 2003;63(12):3043–8.
Cardno TS, Shimaki Y, Sleebs BE, Lackovic K, Parisot JP, Moss RM, et al. HIV-1 and human PEG10 Frameshift elements are functionally distinct and distinguished by novel small molecule modulators. PLoS One. 2015;10(10):e0139036.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Akamatsu, S. (2018). Molecular Basis of Neuroendocrine Prostate Cancer. In: Arai, Y., Ogawa, O. (eds) Hormone Therapy and Castration Resistance of Prostate Cancer. Springer, Singapore. https://doi.org/10.1007/978-981-10-7013-6_39
Download citation
DOI: https://doi.org/10.1007/978-981-10-7013-6_39
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-7012-9
Online ISBN: 978-981-10-7013-6
eBook Packages: MedicineMedicine (R0)