
Abstract
Androgen deprivation therapy can induce oxidative stress by increasing reactive oxygen species levels and/or decreasing cellular antioxidant capacity, which in turn cause genetic and epigenetic effects in prostate cancer. Oxidative stress increases androgen receptor (AR) activation through several possible mechanisms, including AR overexpression, AR activation by co-regulators and intracellular signal transduction pathways, mutation of AR and AR-related proteins, expression of AR splice variants, de novo androgen synthesis, and changes in non-AR signaling. Alterations in AR and non-AR signaling appear to have pro-survival and anti-apoptotic effects on prostate cancer cells, resulting in the development of castration-resistant prostate cancer. Thus, antioxidant therapy could be a promising strategy for the treatment of prostate cancer. Oxidative stress also influences the activity of several prostate cancer therapies, such as taxanes, radiotherapy, and AR-targeting agents. Taken together, these observations suggest that oxidative stress-induced AR signaling is a critical resistance factor and a crucial target for prostate cancer treatment.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Androgen deprivation therapy
- Androgen receptor
- Castration-resistant prostate cancer
- Oxidative stress
- Reactive oxygen species
21.1 Introduction
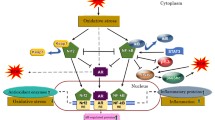
Reactive oxygen species (ROS), which include superoxide (O2 −), hydrogen peroxide (H2O2), and hydroxyl radicals (HO•), are produced by the partial reduction of oxygen and are generated endogenously mainly during mitochondrial oxidative phosphorylation and exogenously predominantly from xenobiotic compounds. ROS levels are controlled through the activity of endogenous antioxidant defense systems such as superoxide dismutase (SOD), catalase, and peroxiredoxin, as well as through exogenous antioxidants such as isoflavones, catechins, carotenes, vitamins, and selenium [1]. Oxidative stress occurs when the cellular antioxidant defense systems are overwhelmed by an increase in ROS levels or a decrease in the antioxidant capacity. Excessive ROS levels lead to damage of macromolecules such as DNA, RNA, proteins, and lipids, which is in part rescued by the DNA repair system and the thioredoxin and glutathione detoxification systems [2]. Damage to DNA can cause genetic aberrations, such as mutations and chromosomal rearrangements, while damage to other molecules can affect epigenetic processes, largely through dysregulation of proteins containing redox-reactive cysteine residues. Oxidation of cysteine produces reactive sulfenic acid (–SOH), which forms disulfide bonds with nearby cysteine residues (–S–S–) or undergoes further oxidation to sulfinic (–SO2H) or sulfonic (–SO3H) acids. With the exception of –SO3H formation, each of these redox modifications can be reversed by reducing systems [3]. These oxidative modifications of cysteines alter the protein structure and function, thereby directly or indirectly affecting a range of events, including intracellular signal transduction and gene expression pathways that modulate various cellular processes (Fig. 21.1) [4].

Relationship between treatment resistance and oxidative stress
Oxidative stress not only plays an important role in prostate carcinogenesis and progression of prostate cancer [5,6,7] but also is involved in the resistance of prostate cancer to therapy, especially androgen deprivation therapy (ADT) [1, 7, 8]. ADT, which consists of surgical or pharmacological castration or anti-androgen therapy, has been commonly used for the treatment of advanced or recurrent prostate cancer since 1941 [9]. Although ADT is initially effective for most prostate cancer patients, therapy resistance invariably develops and the disease becomes lethal castration-resistant prostate cancer (CRPC). Increasing evidence suggests the existence of functional cross talk between oxidative stress and CRPC. Here, we summarize our current knowledge in this area.
21.2 Oxidative Stress Induced by ADT in Prostate Cancer
Several experiments in vitro and in vivo have indicated that castration leads to oxidative stress by promoting increased ROS production and decreased ROS-detoxifying enzyme activity [10,11,12,13]. However, there are also several conflicting studies showing that androgens can induce oxidative stress [14, 15]. This discrepancy may be due to differences in the physiological and nonphysiological conditions in the various studies (Fig. 21.2). For example, Ripple et al. demonstrated that oxidative stress was decreased or increased by physiological or excessive androgen levels, respectively, suggesting that stress can be induced nonspecifically under nonphysiological conditions [16]. Several molecular mechanisms may be responsible for castration-induced oxidative stress, as shown by a reduction in the antioxidant molecules thioredoxin 1, peroxiredoxin 5, and SOD2 in rats after castration [11], a reduction in SOD2 in human prostate cancer tissue after ADT [10], and upregulation of pro-oxidant nicotinamide adenine dinucleotide phosphate oxidases (Noxs) in rat prostate after castration [13]. Collectively, these studies suggest that epigenetic alterations in gene expression and protein function lead to a redox imbalance and induction of oxidative stress in prostate cancer; this is supported by the finding of elevated oxidative stress levels in prostate cancer cells and surgically resected prostate cancer tissues [8, 17]. Thus, ADT-induced oxidative stress can lead to wide-ranging genetic and epigenetic alterations in prostate cancer, as described in more depth in the following sections.

Dose-response relationship between androgen levels and oxidative stress
21.3 Effects of Oxidative Stress on AR and Non-AR Signaling
21.3.1 Effects of Oxidative Stress on AR Signaling
In CRPC, AR signaling is aberrantly augmented by the low androgen milieu via a number of mechanisms, including AR overexpression, AR activation by co-regulators and intracellular signal transduction pathways, mutation of AR and AR-related proteins, expression of AR splice variants, and de novo androgen synthesis. Over the last decade, ADT-induced oxidative stress has been shown to influence AR signaling in prostate cancer. Sharifi et al. showed that suppression of the antioxidant enzyme SOD2 and increased ROS production activated AR signaling through changes in the expression of genes related to steroid metabolism, nuclear receptor co-regulators, and interleukin-6 receptor [7]. We also independently found that ROS play a crucial role in AR signaling and the development of CRPC [1, 8]. Thus, oxidative stress could contribute to castration resistance through AR reactivation by several mechanisms.
21.3.1.1 AR Overexpression
AR overexpression is thought to be a major cause of CRPC [18]. Indeed, many studies have shown that CRPC progression is associated with increased AR expression [19,20,21,22], which may be attributed to gene amplification, increased transcription and translation, and decreased degradation. Among these, transcriptional upregulation is a particularly important mechanism of increased AR expression. As we summarized previously [1, 18], several transcription factors activated by oxidative stress, including Twist1 [8], YB-1 [23], NF-κB [24], Sp1 [25, 26], Myc [27, 28], CREB [29], and Foxo3a [30], are also known to regulate AR expression, suggesting that ADT-induced oxidative stress may act through these factors to upregulate AR transcription [8].
Many other molecules have also been reported to be involved in regulating AR expression. For example, a pathway linked to 12-hydroxyeicosatetraenoic acid and leukotriene B4 receptor 2 was shown to increase ROS production and upregulate AR expression via the Nox4 pathway [31]. Conversely, treatment with diphenyleneiodonium chloride, an antioxidant that inhibits Nox-mediated ROS production, reduced AR expression via SREBP-1 [32]. The oxidative stress inducers cadmium and zinc chloride increase AR expression in dysplastic prostate glands of rats [33], while the synthetic antimicrobial chemical mequindox induces oxidative stress and AR overexpression in rat testes [34]. Paradoxically, other inducers of oxidative stress, such as a curcumin analog [35] and thymoquinone [36], were reported to suppress AR expression. However, these agents may act through non-redox signaling since the effects were poorly suppressed by the antioxidant N-acetyl cysteine (NAC), an electrophile that supports the production of a major intracellular antioxidant, glutathione.
Collectively, these data suggest that oxidative stress induced by internal and external stimuli induces AR overexpression through stress-induced transcription factors and other pathways.
21.3.1.2 AR Activation by Co-regulators and Intracellular Signal Transduction Pathways
The transcriptional activity of AR is modulated by co-regulators [37], several of which, including peroxiredoxin, Hsp27, and EGR-1, are activated by oxidative stress [1]. We previously showed that cysteine residues in peroxiredoxin are critical for its AR co-regulatory function [1], supporting the possibility that ROS-mediated modification of AR co-regulators affects AR signaling.
In addition, several intracellular signaling pathways play a role in AR transactivation. AR function can be augmented by growth factors and cytokines such as insulin-like growth factor, fibroblast growth factor, epidermal growth factor, and IL-6, as well as key components of their downstream signaling pathways, such as mitogen-activated protein kinase (MAPK), JAK/STAT, protein kinase A, phosphatidylinositol-3-kinase (PI3K)/Akt, and protein kinase C, which may itself be activated by oxidative stress [1]. In fact, we have shown that the ε isoform of protein kinase C increases AR expression through NF-κB signaling and contributes to cellular resistance to castration [38, 39]. Thus, oxidative stress also influences intracellular signaling pathways that interact with transcription factors and co-regulators to modulate AR activity.
21.3.1.3 Mutation of AR-Related Proteins and Generation of AR Splice Variants
Mutations in the AR gene have been shown to change the protein’s ligand-binding affinity, permitting activation by non-cognate steroids and even by anti-androgen agents [6, 40, 41]. Although oxidative stress induces mutations in DNA, it is not yet known whether the AR gene is affected [6]. However, mutations in genes related to AR signaling, including FASN, CYP11B1, HSD17B4 (androgen metabolism), NCOR1, and FOXOA1 (AR cofactors), have been detected in CRPC tissues [42, 43]. Such mutations, probably induced by oxidative stress, may contribute to the development of CRPC through aberrant activation of AR signaling.
Several splice variants of AR exhibit transcriptional activity in the absence of androgen and play a key role in promoting CRPC [44,45,46,47,48]. Although possible, a relationship between expression of the AR splice variants and oxidative stress has not yet been documented. However, we recently reported that the redox-sensitive nuclear factor YB-1 [49] and its upstream kinase RSK [50] regulate the expression of an AR variant [51], supporting a direct link. Based on these intriguing observations, further studies of the effects of oxidative stress on mutation of the AR gene and expression of AR splice variants are warranted.
21.3.1.4 De Novo Androgen Synthesis
De novo synthesis of androgens in the adrenal glands and prostate tumors has been recognized as a potential cause of CRPC [52,53,54], and this was confirmed by clinical trials of abiraterone acetate, an inhibitor of a critical enzyme in androgen biosynthesis, cytochrome P17 (CYP17) [55, 56]. H2O2 regulates androgen synthesis in rat Leydig cells in a biphasic manner, indicating that physiological levels of oxidative stress promote steroidogenesis [57]. Nevertheless, there is no direct evidence at present for the existence of a relationship between oxidative stress and de novo androgen synthesis.
21.3.2 Effects of Oxidative Stress on Non-AR Signaling
In addition to AR signaling, numerous non-AR signaling pathways are activated by oxidative stress and many have been reported to be involved in the development to CRPC through genetic and epigenetic mechanisms. The genotoxic effects of oxidative stress include aberrations such as DNA point mutations and chromosomal rearrangements. In fact, genetic alterations in non-AR signaling molecules, such as PIK3CA, SPOP, RET, RICTOR, and CTNNB1, have been identified in tissues from patients with CRPC [42, 43, 58, 59].
Oxidative stress also causes epigenetic alterations that activate signaling independently of the AR [60]. For example, in prostate cancer, oxidative stress activates PI3K/Akt [61] and MAPK [62] and elevates the transcriptional activity of NF-κB [63], which promotes survival and inhibits apoptosis. However, many components of these pathways are also involved in AR signaling and show elevated activity in CRPC cells and tissues, as is the case for PI3K/Akt [64], MAPK [65], and NF-κB [66]. Additional non-AR-related mechanisms that contribute to the development of CRPC include inflammation, epithelial–mesenchymal transition, and cancer stem-like characteristics of prostate cancer cells [67]. Intriguingly, these phenomena are also affected by oxidative stress, further supporting the multiple mechanisms through which oxidative stress is involved in CRPC development.
21.4 Oxidative Stress and the Development of CRPC
As described above, the mutual link between oxidative stress and AR signaling supports a role for oxidative stress in CRPC development; indeed, there is direct evidence of such a relationship. We chronically exposed LNCaP, an androgen-dependent prostate cancer cell line, to oxidative stress to generate H2O2-resistant sublines, and found that they expressed increased levels of AR mRNA and protein and exhibited a castration-resistant phenotype [8]. Whereas castration-resistant cells normally exhibit elevated antioxidant protein levels [70, 71] and ROS-scavenging activity [72], overexpression of AR in such cells increases oxidative stress, as indicated by higher intracellular ROS levels [68, 69].
A connection between oxidative stress and CRPC is also supported by clinical findings. Compared with prostate specimens from patients who had undergone radical prostatectomy without ADT, prostate cancer tissues obtained from patients post-ADT show increased 4-hydroxy-2-nonenal levels, indicative of elevated oxidative stress [17]. In addition, a genetic polymorphism in the GSTM3 gene, which encodes an antioxidant enzyme glutathione S-transferase, was recently reported to be associated with increased risk of progression of metastatic prostate cancer to CRPC, which was validated in nonmetastatic prostate cancer [69].
Collectively, these experimental and clinical data are consistent with a close link between oxidative stress and progression to CRPC.
21.5 Clinical Implications of Antioxidant Therapy in CRPC
Given the accumulating evidence that oxidative stress contributes to CRPC, it has been speculated that antioxidant therapy could have therapeutic effects in prostate cancer patients receiving ADT.
Various naturally occurring antioxidative compounds, including isoflavones, catechins, carotenes, vitamins, and selenium, have been investigated as possible prophylactic agents for prostate carcinogenesis and as therapeutic agents for prostate cancer [73, 74]. Among these compounds, the carotenoid lycopene was shown to prevent oxidative damage to proteins, lipids, and DNA. In a preclinical study, lycopene suppressed AR activity and had antitumor effects [75]. In clinical studies, lycopene augmented the therapeutic effects of orchiectomy in advanced prostate cancer patients [76]. A phase II study showed that administration of lycopene at 10 mg per day suppressed elevation of prostate-specific antigen (PSA) in 41 men with prostate cancer [75]. In addition, a case report of a CRPC patient described a reduction in serum PSA levels and disease-associated symptoms after intake of saw palmetto and lycopene supplements [77]. Although the number of patients in these clinical studies was small, the findings support the potential use of lycopene combined with castration in the treatment of prostate cancer, including CRPC. The antioxidants vitamin E and α-tocopherol have also been reported to decrease the risk of prostate cancer mortality, suggesting that they may prevent disease progression [78].
In addition to naturally occurring compounds, synthetic antioxidants might also be useful for the treatment of prostate cancer. In the TRAMP mouse model of prostate cancer, NAC administration reduced 8-hydroxy-2′-deoxyguanosine, nitrotyrosine, and 4-hydroxy-2-nonenal levels in the prostate [79]. In addition, we previously showed that NAC reduced AR expression and that NAC plus ADT successfully suppressed tumor growth in a mouse xenograft model of prostate cancer [17]. SOD mimetics have been shown to reduce oxidative stress, reduce the expression of AR and AR splice variants, and have a therapeutic effect in prostate cancer cells [80]. Finally, the anti-angiogenic agent endostatin inhibits CRPC growth by augmenting antioxidant enzyme activity and suppressing ROS levels [81].
An alternative therapeutic strategy to counter oxidative stress in CRPC is inhibition of ROS production. In support of this, the Nox inhibitor diphenyleneiodonium decreases the viability of prostate cancer cells, including LNCaP cells [82] and another Nox inhibitor, apocynin, suppresses prostate cancer cell invasion [83].
These observations highlight several options for antioxidant therapy, including natural and synthetic antioxidants and ROS inhibitors. However, a critical obstacle for the clinical use of antioxidants is their rapid oxidative degradation under physiological conditions, resulting in poor stability and bioavailability. One potential solution to this problem might be to encapsulate the antioxidant compounds in nanoparticles that also act as oxygen radical scavengers. For example, it was recently reported that curcumin-loaded pH-sensitive redox nanoparticles exert excellent antitumor activity in prostate cancer [84].
21.6 Novel Agents for CRPC and Oxidative Stress
Taxanes such as docetaxel and cabazitaxel, AR-targeting agents such as abiraterone acetate and enzalutamide, and the radiopharmaceutical radium-223 all show benefit in prolonging progression-free and overall survival and have been approved globally for use in CRPC [37, 85].
Similar to other cytotoxic anticancer agents, taxanes have been shown to cause oxidative stress in cancer cells [86]. Moreover, many molecules implicated in oxidative stress signaling, including PI3K/Akt [87], MAPK [88], and NF-κB [89], and their downstream effectors such as Twist1 [90] and YB-1 [91, 92], are all involved in the resistance of prostate cancer to taxanes. In addition, the status of TMPRSS2-ERG fusion gene caused by inflammation-induced oxidative stress through DNA breaks [93] was reported to be associated with the therapeutic effect of taxanes [94, 95]. Thus, oxidative stress appears to contribute to taxane resistance in prostate cancer through various mechanisms.
Radiation is known to induce oxidative stress in prostate cancer [96]; however, this is not necessarily beneficial because irradiation-induced oxidative stress can activate pro-survival and anti-apoptotic signaling through molecules such as PI3K/Akt [97], MAPK [98], and NF-κB [99], resulting in resistance to irradiation. Radium-223 is an α particle-emitting isotope [100] and appears to induce oxidative stress in prostate cancer cells. Although the therapeutic effect of this isotope may be affected by oxidative stress-induced signaling, there is currently no direct evidence for this.
Little is known about the interaction between oxidative stress and AR-targeting agents, including abiraterone acetate and enzalutamide. However, oxidative stress levels are increased in enzalutamide-resistant prostate cancer cells established in vitro [69], warranting further investigation. Clinical trials have been initiated for several additional promising agents, including immune checkpoint inhibitors and the poly (ADP-ribose) polymerase inhibitor olaparib. To those agents, biomarkers such as the presence of somatic mutations in DNA repair genes and the number of missense somatic mutations which may be caused by oxidative stress are postulated, and then oxidative stress may commit to the sensitivity to those emerging agents.
21.7 Conclusions and Future Directions
Oxidative stress induced by ADT can activate both AR and non-AR signaling, resulting in the acquisition of castration resistance. Treatment-induced oxidative stress also appears to be involved in the resistance of prostate cancer to therapy. Thus, oxidative stress is a critical resistance factor and a crucial target for prostate cancer treatment. Suppression of oxidative stress signaling by antioxidants or inhibitors of ROS production may thus be a promising strategy to overcome treatment resistance in prostate cancer. However, the relationship between oxidative stress and CRPC is a vast and underexplored area of research, and further investigation is warranted. Such studies will undoubtedly lead to some remarkable discoveries.
References
Shiota M, Yokomizo A, Naito S. Oxidative stress and androgen receptor signaling in the development and progression of castration-resistant prostate cancer. Free Radic Biol Med. 2011;51:1320–8. https://doi.org/10.1016/j.freeradbiomed.2011.07.011.
Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev. Drug Discov. 2009;8:579–91. https://doi.org/10.1038/nrd2803.
Roos G, Messens J. Protein sulfenic acid formation: from cellular damage to redox regulation. Free Radic Biol Med. 2011;51:314–26. https://doi.org/10.1016/j.freeradbiomed.2011.04.031.
Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012;24:981–90. https://doi.org/10.1016/j.cellsig.2012.01.008.
Bostwick DG, Alexander EE, Singh R, Shan A, Qian J, Santella RM, et al. Antioxidant enzyme expression and reactive oxygen species damage in prostatic intraepithelial neoplasia and cancer. Cancer. 2000;89:123–34.
Khandrika L, Kumar B, Koul S, Maroni P, Koul HK. Oxidative stress in prostate cancer. Cancer Lett. 2009;282:125–36. https://doi.org/10.1016/j.canlet.2008.12.011.
Sharifi N, Hurt EM, Thomas SB, Farrar WL. Effects of manganese superoxide dismutase silencing on androgen receptor function and gene regulation: implications for castration-resistant prostate cancer. Clin Cancer Res. 2008;14:6073–80. https://doi.org/10.1158/1078-0432.CCR-08-0591.
Shiota M, Yokomizo A, Tada Y, Inokuchi J, Kashiwagi E, Masubuchi D, et al. Castration resistance of prostate cancer cells caused by castration-induced oxidative stress through Twist1 and androgen receptor overexpression. Oncogene. 2010;29:237–50. https://doi.org/10.1038/onc.2009.322.
Miyamoto H, Messing EM, Chang C. Androgen deprivation therapy for prostate cancer: current status and future prospects. Prostate. 2004;61:332–53. https://doi.org/10.1002/pros.20115.
Best CJ, Gillespie JW, Yi Y, Chandramouli GV, Perlmutter MA, Gathright Y, et al. Molecular alterations in primary prostate cancer after androgen ablation therapy. Clin Cancer Res. 2005;11:6823–34. https://doi.org/10.1158/1078-0432.CCR-05-0585.
Pang ST, Dillner K, Wu X, Pousette A, Norstedt G, Flores-Morales A. Gene expression profiling of androgen deficiency predicts a pathway of prostate apoptosis that involves genes related to oxidative stress. Endocrinology. 2002;143:4897–906. https://doi.org/10.1210/en.2002-220327.
Shan W, Zhong W, Zhao R, Oberley TD. Thioredoxin 1 as a subcellular biomarker of redox imbalance in human prostate cancer progression. Free Radic Biol Med. 2010;49:2078–87. https://doi.org/10.1016/j.freeradbiomed.2010.10.691.
Tam NN, Gao Y, Leung YK, Ho SM. Androgenic regulation of oxidative stress in the rat prostate: involvement of NAD(P)H oxidases and antioxidant defense machinery during prostatic involution and regrowth. Am J Pathol. 2003;163:2513–22. https://doi.org/10.1016/S0002-9440(10)63606-1.
Pathak S, Singh R, Verschoyle RD, Greaves P, Farmer PB, Steward WP, et al. Androgen manipulation alters oxidative DNA adduct levels in androgen-sensitive prostate cancer cells grown in vitro and in vivo. Cancer Lett. 2008;261:74–83. https://doi.org/10.1016/j.canlet.2007.11.015.
Pinthus JH, Bryskin I, Trachtenberg J, Lu JP, Singh G, Fridman E, et al. Androgen induces adaptation to oxidative stress in prostate cancer: implications for treatment with radiation therapy. Neoplasia. 2007;9:68–80.
Ripple MO, Henry WF, Rago RP, Wilding G. Prooxidant-antioxidant shift induced by androgen treatment of human prostate carcinoma cells. J Natl Cancer Inst. 1997;89:40–8.
Shiota M, Song Y, Takeuchi A, Yokomizo A, Kashiwagi E, Kuroiwa K, et al. Antioxidant therapy alleviates oxidative stress by androgen deprivation and prevents conversion from androgen dependent to castration resistant prostate cancer. J Urol. 2012;187:707–14. https://doi.org/10.1016/j.juro.2011.09.147.
Shiota M, Yokomizo A, Naito S. Increased androgen receptor transcription: a cause of castration-resistant prostate cancer and a possible therapeutic target. J Mol Endocrinol. 2011;47:R25–41. https://doi.org/10.1530/JME-11-0018.
Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–9. https://doi.org/10.1038/nm972.
Gregory CW, Hamil KG, Kim D, Hall SH, Pretlow TG, Mohler JL, et al. Androgen receptor expression in androgen-independent prostate cancer is associated with increased expression of androgen-regulated genes. Cancer Res. 1998;58:5718–24.
Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol. 2005;23:8253–61. https://doi.org/10.1200/JCO.2005.03.4777.
Zegarra-Moro OL, Schmidt LJ, Huang H, Tindall DJ. Disruption of androgen receptor function inhibits proliferation of androgen-refractory prostate cancer cells. Cancer Res. 2002;62:1008–13.
Shiota M, Takeuchi A, Song Y, Yokomizo A, Kashiwagi E, Uchiumi T, et al. Y-box binding protein-1 promotes castration-resistant prostate cancer growth via androgen receptor expression. Endocr Relat Cancer. 2011;18:505–17. https://doi.org/10.1530/ERC-11-0017.
Zhang L, Altuwaijri S, Deng F, Chen L, Lal P, Bhanot UK, et al. NF-kappaB regulates androgen receptor expression and prostate cancer growth. Am J Pathol. 2009;175:489–99. https://doi.org/10.2353/ajpath.2009.080727.
Faber PW, van Rooij HC, Schipper HJ, Brinkmann AO, Trapman J. Two different, overlapping pathways of transcription initiation are active on the TATA-less human androgen receptor promoter. The role of Sp1. J Biol Chem. 1993;268:9296–301.
Yuan H, Gong A, Young CY. Involvement of transcription factor Sp1 in quercetin-mediated inhibitory effect on the androgen receptor in human prostate cancer cells. Carcinogenesis. 2005;26:793–801. https://doi.org/10.1093/carcin/bgi021.
Grad JM, Dai JL, Wu S, Burnstein KL. Multiple androgen response elements and a Myc consensus site in the androgen receptor (AR) coding region are involved in androgen-mediated up-regulation of AR messenger RNA. Mol Endocrinol. 1999;13:1896–911.
Lee JG, Zheng R, McCafferty-Cepero JM, Burnstein KL, Nanus DM, Shen R. Endothelin-1 enhances the expression of the androgen receptor via activation of the c-myc pathway in prostate cancer cells. Mol Carcinog. 2009;48:141–9. https://doi.org/10.1002/mc.20462.
Mizokami A, Yeh SY, Chang C. Identification of 3′,5′-cyclic adenosine monophosphate response element and other cis-acting elements in the human androgen receptor gene promoter. Mol Endocrinol. 1994;8:77–88.
Yang L, Xie S, Jamaluddin MS, Altuwaijri S, Ni J, Kim E, et al. Induction of androgen receptor expression by phosphatidylinositol 3-kinase/Akt downstream substrate, FOXO3a, and their roles in apoptosis of LNCaP prostate cancer cells. J Biol Chem. 2005;280:33558–65. https://doi.org/10.1074/jbc.M504461200.
Lee JW, Kim GY, Kim JH. Androgen receptor is up-regulated by a BLT2-linked pathway to contribute to prostate cancer progression. Biochem Biophys Res Commun. 2012;420:428–33. https://doi.org/10.1016/j.bbrc.2012.03.012.
Huang WC, Li X, Liu J, Lin J, Chung LW. Activation of androgen receptor, lipogenesis, and oxidative stress converged by SREBP-1 is responsible for regulating growth and progression of prostate cancer cells. Mol Cancer Res. 2012;10:133–42. https://doi.org/10.1158/1541-7786.MCR-11-0206.
Arriazu R, Pozuelo JM, Martín R, Rodríguez R, Santamaría L. Quantitative and immunohistochemical evaluation of PCNA, androgen receptors, apoptosis, and Glutathione-S-Transferase P1 on preneoplastic changes induced by cadmium and zinc chloride in the rat ventral prostate. Prostate. 2005;63:347–57. https://doi.org/10.1002/pros.20192.
Ihsan A, Wang X, Liu Z, Wang Y, Huang X, Liu Y, et al. Long-term mequindox treatment induced endocrine and reproductive toxicity via oxidative stress in male Wistar rats. Toxicol Appl Pharmacol. 2011;252:281–8. https://doi.org/10.1016/j.taap.2011.02.020.
Fajardo AM, MacKenzie DA, Ji M, Deck LM, Vander Jagt DL, Thompson TA, et al. The curcumin analog ca27 down-regulates androgen receptor through an oxidative stress mediated mechanism in human prostate cancer cells. Prostate. 2012;72:612–25. https://doi.org/10.1002/pros.21464.
Koka PS, Mondal D, Schultz M, Abdel-Mageed AB, Agrawal KC. Studies on molecular mechanisms of growth inhibitory effects of thymoquinone against prostate cancer cells: role of reactive oxygen species. Exp Biol Med. 2010;235:751–60. https://doi.org/10.1258/ebm.2010.009369.
Shiota M, Yokomizo A, Fujimoto N, Naito S. Androgen receptor cofactors in prostate cancer: potential therapeutic targets of castration-resistant prostate cancer. Curr Cancer Drug Targets. 2011;11:870–81. https://doi.org/10.2174/156800911796798904.
Shiota M, Yokomizo A, Takeuchi A, Imada K, Kashiwagi E, Song Y, et al. Inhibition of protein kinase C/Twist1 signaling augments anticancer effects of androgen deprivation and enzalutamide in prostate cancer. Clin Cancer Res. 2014;20:951–61. https://doi.org/10.1158/1078-0432.CCR-13-1809.
Shiota M, Yokomizo A, Takeuchi A, Kashiwagi E, Dejima T, Inokuchi J, et al. Protein kinase C regulates Twist1 expression via NF-κB in prostate cancer. Endocr Relat Cancer. 2017. https://doi.org/10.1530/ERC-16-0384.
Brooke GN, Bevan CL. The role of androgen receptor mutations in prostate cancer progression. Curr Genomics. 2009;10:18–25. https://doi.org/10.2174/138920209787581307.
Azad AA, Volik SV, Wyatt AW, Haegert A, Le Bihan S, Bell RH, et al. Androgen receptor gene aberrations in circulating cell-free DNA: biomarkers of therapeutic resistance in castration-resistant prostate cancer. Clin Cancer Res. 2015;21:2315–24. https://doi.org/10.1158/1078-0432.CCR-14-2666.
Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–43. https://doi.org/10.1038/nature11125.
Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215–28. https://doi.org/10.1016/j.cell.2015.05.001.
Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008;68:5469–77. https://doi.org/10.1158/0008-5472.CAN-08-0594.
Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009;69:16–22. https://doi.org/10.1158/0008-5472.CAN-08-2764.
Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H, et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer Res. 2009;69:2305–13. https://doi.org/10.1158/0008-5472.CAN-08-3795.
Sun S, Sprenger CC, Vessella RL, Haugk K, Soriano K, Mostaghel EA, et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J Clin Invest. 2010;120:2715–30. https://doi.org/10.1172/JCI41824.
Watson PA, Chen YF, Balbas MD, Wongvipat J, Socci ND, Viale A, et al. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc Natl Acad Sci U S A. 2010;107:16759–65. https://doi.org/10.1073/pnas.1012443107.
Hayakawa H, Uchiumi T, Fukuda T, Ashizuka M, Kohno K, Kuwano M, et al. Binding capacity of human YB-1 protein for RNA containing 8-oxoguanine. Biochemistry. 2002;41:12739–44.
Siebel A, Cubillos-Rojas M, Santos RC, Schneider T, Bonan CD, Bartrons R, et al. Contribution of S6 K1/MAPK signaling pathways in the response to oxidative stress: activation of RSK and MSK by hydrogen peroxide. PLoS One. 2013;8:e75523. https://doi.org/10.1371/journal.pone.0075523.
Shiota M, Fujimoto N, Imada K, Yokomizo A, Itsumi M, Takeuchi A, et al. Potential role for YB-1 in castration-resistant prostate cancer and resistance to enzalutamide through the androgen receptor V7. J Natl Cancer Inst. 2016;108:djw005. https://doi.org/10.1093/jnci/djw005.
Locke JA, Guns ES, Lubik AA, Adomat HH, Hendy SC, Wood CA, et al. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Res. 2008;68(15):6407. https://doi.org/10.1158/0008-5472.CAN-07-5997.
Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, et al. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. 2008;68:4447–54. https://doi.org/10.1158/0008-5472.CAN-08-0249.
Stanbrough M, Bubley GJ, Ross K, Golub TR, Rubin MA, Penning TM, et al. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66:2815–25. https://doi.org/10.1158/0008-5472.CAN-05-4000.
de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995–2005. https://doi.org/10.1056/NEJMoa1014618.
Ryan CJ, Smith MR, de Bono JS, Molina A, Logothetis CJ, de Souza P, et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N Engl J Med. 2013;368:138–48. https://doi.org/10.1056/NEJMoa1209096.
Zhao Y, Ao H, Chen L, Sottas CM, Ge RS, Li L, Zhang Y. Mono-(2-ethylhexyl) phthalate affects the steroidogenesis in rat Leydig cells through provoking ROS perturbation. Toxicol In Vitro. 2012;26:950–5. https://doi.org/10.1016/j.tiv.2012.04.003.
Beltran H, Yelensky R, Frampton GM, Park K, Downing SR, MacDonald TY, et al. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. Eur Urol. 2013;63:920–6. https://doi.org/10.1016/j.eururo.2012.08.053.
Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med. 2016;22:298–305. https://doi.org/10.1038/nm.4045.
Zhang Z, Hou X, Shao C, Li J, Cheng JX, Kuang S, et al. Plk1 inhibition enhances the efficacy of androgen signaling blockade in castration-resistant prostate cancer. Cancer Res. 2014;74:6635–47. https://doi.org/10.1158/0008-5472.CAN-14-1916.
Ning P, Zhong JG, Jiang F, Zhang Y, Zhao J, Tian F, et al. Role of protein S in castration-resistant prostate cancer-like cells. Endocr Relat Cancer. 2016;23:595–607. https://doi.org/10.1530/ERC-16-0126.
Kumar B, Koul S, Khandrika L, Meacham RB, Koul HK. Oxidative stress is inherent in prostate cancer cells and is required for aggressive phenotype. Cancer Res. 2008;68:1777–85. https://doi.org/10.1158/0008-5472.CAN-07-5259.
Zhang J, Johnston G, Stebler B, Keller ET. Hydrogen peroxide activates NFkappaB and the interleukin-6 promoter through NFκB-inducing kinase. Antioxid Redox Signal. 2001;3:493–504.
Chung S, Furihata M, Tamura K, Uemura M, Daigo Y, Nasu Y, et al. Overexpressing PKIB in prostate cancer promotes its aggressiveness by linking between PKA and Akt pathways. Oncogene. 2009;28:2849–59. https://doi.org/10.1038/onc.2009.144.
Mukherjee R, McGuinness DH, McCall P, Underwood MA, Seywright M, Orange C, et al. Upregulation of MAPK pathway is associated with survival in castrate-resistant prostate cancer. Br J Cancer. 2011;104:1920–8. https://doi.org/10.1038/bjc.2011.163.
McCall P, Bennett L, Ahmad I, Mackenzie LM, Forbes IW, Leung HY, et al. NFκB signalling is upregulated in a subset of castrate-resistant prostate cancer patients and correlates with disease progression. Br J Cancer. 2012;107:1554–63. https://doi.org/10.1038/bjc.2012.372.
Zong Y, Goldstein AS. Adaptation or selection--mechanisms of castration-resistant prostate cancer. Nat Rev. Urol. 2013;10:90–8. https://doi.org/10.1038/nrurol.2012.237.
Shigemura K, Sung SY, Kubo H, Arnold RS, Fujisawa M, Gotoh A, et al. Reactive oxygen species mediate androgen receptor- and serum starvation-elicited downstream signaling of ADAM9 expression in human prostate cancer cells. Prostate. 2007;67:722–31. https://doi.org/10.1002/pros.20565.
Shiota M, Fujimoto N, Itsumi M, Takeuchi A, Inokuchi J, Tatsugami K, et al. Gene polymorphisms in antioxidant enzymes correlate with the efficacy of androgen-deprivation therapy for prostate cancer with implications of oxidative stress. Ann Oncol. 2016. https://doi.org/10.1093/annonc/mdw646.
Kuruma H, Egawa S, Oh-Ishi M, Kodera Y, Satoh M, Chen W, et al. High molecular mass proteome of androgen-independent prostate cancer. Proteomics. 2005;5:1097–112. https://doi.org/10.1002/pmic.200401115.
Shiota M, Yokomizo A, Kashiwagi E, Takeuchi A, Fujimoto N, Uchiumi T, et al. Peroxiredoxin 2 in the nucleus and cytoplasm distinctly regulates androgen receptor activity in prostate cancer cells. Free Radic Biol Med. 2011;51:78–87. https://doi.org/10.1016/j.freeradbiomed.2011.04.001.
Wu CT, Chen WC, Liao SK, Hsu CL, Lee KD, Chen MF. The radiation response of hormone-resistant prostate cancer induced by long-term hormone therapy. Endocr Relat Cancer. 2007;14:633–43. https://doi.org/10.1677/ERC-07-0073.
Hori S, Butler E, McLoughlin J. Prostate cancer and diet: food for thought? BJU Int. 2011;107:1348–59. https://doi.org/10.1111/j.1464-410X.2010.09897.x.
Itsumi M, Shiota M, Takeuchi A, Kashiwagi E, Inokuchi J, Tatsugami K, et al. Equol inhibits prostate cancer growth through degradation of androgen receptor by S-phase kinase-associated protein 2. Cancer Sci. 2016;107:1022–8. https://doi.org/10.1111/cas.12948.
Zhang X, Wang Q, Neil B, Chen X. Effect of lycopene on androgen receptor and prostate-specific antigen velocity. Chin Med J. 2010;123:2231–6.
Ansari MS, Gupta NP. A comparison of lycopene and orchidectomy vs orchidectomy alone in the management of advanced prostate cancer. BJU Int. 2003;92:375–8.
Matlaga BR, Hall MC, Stindt D, Torti FM. Response of hormone refractory prostate cancer to lycopene. J Urol. 2001;166:613.
Watters JL, Gail MH, Weinstein SJ, Virtamo J, Albanes D. Associations between alpha-tocopherol, beta-carotene, and retinol and prostate cancer survival. Cancer Res. 2009;69:3833–41. https://doi.org/10.1158/0008-5472.CAN-08-4640.
Tam NN, Nyska A, Maronpot RR, Kissling G, Lomnitski L, Suttie A, et al. Differential attenuation of oxidative/nitrosative injuries in early prostatic neoplastic lesions in TRAMP mice by dietary antioxidants. Prostate. 2006;66:57–69. https://doi.org/10.1002/pros.20313.
Thomas R, Sharifi N. SOD mimetics: a novel class of androgen receptor inhibitors that suppresses castration-resistant growth of prostate cancer. Mol Cancer Ther. 2012;11:87–97. https://doi.org/10.1158/1535-7163.MCT-11-0540.
Lee JH, Kang M, Wang H, Naik G, Mobley JA, Sonpavde G, et al. Endostatin inhibits androgen-independent prostate cancer growth by suppressing nuclear receptor-mediated oxidative stress. FASEB J. 2017. https://doi.org/10.1096/fj.201601178R.
Chaiswing L, Bourdeau-Heller JM, Zhong W, Oberley TD. Characterization of redox state of two human prostate carcinoma cell lines with different degrees of aggressiveness. Free Radic Biol Med. 2007;43:202–15. https://doi.org/10.1016/j.freeradbiomed.2007.03.031.
Chaiswing L, Zhong W, Cullen JJ, Oberley LW, Oberley TD. Extracellular redox state regulates features associated with prostate cancer cell invasion. Cancer Res. 2008;68:5820–6. https://doi.org/10.1158/0008-5472.CAN-08-0162.
Thangavel S, Yoshitomi T, Sakharkar MK, Nagasaki Y. Redox nanoparticles inhibit curcumin oxidative degradation and enhance its therapeutic effect on prostate cancer. J Control Release. 2015;209:110–9. https://doi.org/10.1016/j.jconrel.2015.04.025.
Fujimoto N. Novel agents for castration-resistant prostate cancer: early experience and beyond. Int J Urol. 2016;23:114–21. https://doi.org/10.1111/iju.12907.
Bellezza I, Grottelli S, Gatticchi L, Mierla AL, Minelli A. α-Tocopheryl succinate pre-treatment attenuates quinone toxicity in prostate cancer PC3 cells. Gene. 2014;539:1–7. https://doi.org/10.1016/j.gene.2014.02.009.
Kosaka T, Miyajima A, Shirotake S, Suzuki E, Kikuchi E, Oya M. Long-term androgen ablation and docetaxel up-regulate phosphorylated Akt in castration resistant prostate cancer. J Urol. 2011;185:2376–81. https://doi.org/10.1016/j.juro.2011.02.016.
Shiota M, Itsumi M, Yokomizo A, Takeuchi A, Imada K, Kashiwagi E, et al. Targeting ribosomal S6 kinases/Y-box binding protein-1 signaling improves cellular sensitivity to taxane in prostate cancer. Prostate. 2014;74:829–38. https://doi.org/10.1002/pros.22799.
Tantivejkul K, Loberg RD, Mawocha SC, Day LL, John LS, Pienta BA, et al. PAR1-mediated NFkappaB activation promotes survival of prostate cancer cells through a Bcl-xL-dependent mechanism. J Cell Biochem. 2005;96:641–52. https://doi.org/10.1002/jcb.20533.
Shiota M, Izumi H, Tanimoto A, Takahashi M, Miyamoto N, Kashiwagi E, et al. Programmed cell death protein 4 down-regulates Y-box binding protein-1 expression via a direct interaction with Twist1 to suppress cancer cell growth. Cancer Res. 2009;69:3148–56. https://doi.org/10.1158/0008-5472.CAN-08-2334.
Shiota M, Zoubeidi A, Kumano M, Beraldi E, Naito S, Nelson CC, et al. Clusterin is a critical downstream mediator of stress-induced YB-1 transactivation in prostate cancer. Mol Cancer Res. 2011;9:1755–66. https://doi.org/10.1158/1541-7786.MCR-11-0379.
Shiota M, Kashiwagi E, Yokomizo A, Takeuchi A, Dejima T, Song Y, et al. Interaction between docetaxel resistance and castration resistance in prostate cancer: implications of Twist1, YB-1, and androgen receptor. Prostate. 2013;73:1336–44. https://doi.org/10.1002/pros.22681.
Mani RS, Amin MA, Li X, Kalyana-Sundaram S, Veeneman BA, Wang L, et al. Inflammation-induced oxidative stress mediates gene fusion formation in prostate cancer. Cell Rep. 2016;17:2620–31. https://doi.org/10.1016/j.celrep.2016.11.019.
Galletti G, Matov A, Beltran H, Fontugne J, Miguel Mosquera J, Cheung C, et al. ERG induces taxane resistance in castration-resistant prostate cancer. Nat Commun. 2014;5:5548. https://doi.org/10.1038/ncomms6548.
Reig Ò, Marín-Aguilera M, Carrera G, Jiménez N, Paré L, García-Recio S, et al. TMPRSS2-ERG in blood and docetaxel resistance in metastatic castration-resistant prostate cancer. Eur Urol. 2016;70:709–13. https://doi.org/10.1016/j.eururo.2016.02.034.
Josson S, Xu Y, Fang F, Dhar SK, St Clair DK, St Clair WH. RelB regulates manganese superoxide dismutase gene and resistance to ionizing radiation of prostate cancer cells. Oncogene. 2006;25:1554–9. https://doi.org/10.1038/sj.onc.1209186.
Goldberg Z, Rocke DM, Schwietert C, Berglund SR, Santana A, Jones A, et al. Human in vivo dose-response to controlled, low-dose low linear energy transfer ionizing radiation exposure. Clin Cancer Res. 2006;12:3723–9. https://doi.org/10.1158/1078-0432.CCR-05-2625.
Yacoub A, McKinstry R, Hinman D, Chung T, Dent P, Hagan MP. Epidermal growth factor and ionizing radiation up-regulate the DNA repair genes XRCC1 and ERCC1 in DU145 and LNCaP prostate carcinoma through MAPK signaling. Radiat Res. 2003;159:439–52.
Kim BY, Kim KA, Kwon O, Kim SO, Kim MS, Kim BS, et al. NF-κB inhibition radiosensitizes Ki-Ras-transformed cells to ionizing radiation. Carcinogenesis. 2005;26:1395–403. https://doi.org/10.1093/carcin/bgi081.
Shore ND. Radium-223 dichloride for metastatic castration-resistant prostate cancer: the urologist’s perspective. Urology. 2015;85:717–24. https://doi.org/10.1016/j.urology.2014.11.031.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Shiota, M. (2018). Oxidative Stress and Castration-Resistant Prostate Cancer. In: Arai, Y., Ogawa, O. (eds) Hormone Therapy and Castration Resistance of Prostate Cancer. Springer, Singapore. https://doi.org/10.1007/978-981-10-7013-6_21
Download citation
DOI: https://doi.org/10.1007/978-981-10-7013-6_21
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-7012-9
Online ISBN: 978-981-10-7013-6
eBook Packages: MedicineMedicine (R0)