
Abstract
Liver cancer is one of the major causes of cancer-related deaths in the United States, accounting for 4.5% of the total estimated cancer deaths in 2016 and standing as the second leading cause of cancer-related deaths in men worldwide in 2012. There are two major types of primary liver cancers, including hepatocellular carcinoma (HCC) and cholangiocarcinoma (CCA). The most common type of primary liver cancer is HCC, which begins in hepatocytes and accounts for approximately 75% of all liver cancers. Hypoxia, a condition of oxygen deprivation in the tissue, is a common feature of the cancer microenvironment due to increased cell proliferation and limited blood supply. Hypoxia-inducible factor-1 (HIF-1) was the first transcription factor discovered to regulate a wide range of target genes involved in many cellular processes in response to low oxygen levels. HIF-1 is a heterodimeric protein complex composed of two different subunits, α and β. During a condition of hypoxia, HIF-1 heterodimer activates target genes that contain a hypoxia response element (HRE) in the promoter region. The overexpression of HIF-1 is frequently observed in many human solid tumors, including liver cancer, and is associated with tumor development, poor prognosis, and resistance to chemotherapy, suggesting that HIF-1 is a new therapeutic target in liver cancer treatment. In this chapter, we define the molecular mechanism that controls HIF-1 and how it maintains a variation of biological processes in hypoxic environments.
The original version of this chapter was revised. The book was inadvertently published without Abstracts and Keywords, which are now included in all the chapters. An erratum to this chapter can be found at https://doi.org/10.1007/978-981-10-6728-0_39
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
35.1 Introduction
The liver is a central organ that has many functions, including the metabolic change of excessive nutrients, the detoxification of toxic metabolites, the supply of energy-producing substrates, etc., all divided into eight functionally independent segments according to French surgeon Claude Couinaud’s classification system [1]. These eight segments are numbered in Roman numeral fashion (segment I to VIII) in a clockwise manner. Segment I is located posteriorly and is not visible anteriorly. Furthermore, according to Bismuth [2, 3], segment IV can diverge into segment IVa and IVb.
Liver cancer is one of the major causes of cancer-related deaths in the United States, accounting for 4.5% of the total estimated cancer deaths in 2016 and standing as the second leading cause of cancer deaths in men globally during 2012 [4, 5]. In cases of males aged over 55 years in the United States, the incidence rate of liver cancer has increased in contrast to the stable trends for incidences driven by the three major cancers such as lung, prostate, and colon cancer [4]. There are two major types of primary liver cancers, including hepatocellular carcinoma (HCC) and cholangiocarcinoma (CCA). The most common type of primary liver cancer is HCC, also known as hepatoma, which begins in the hepatocytes and accounts for approximately 75% of all liver cancers. Fortunately, well-recognized indicators in HCC give notification to populations that need screen testing and monitoring [6]. The risk factors associated with HCC development are chronic hepatitis B virus (HBV) or the hepatitis C virus (HCV) along with aflatoxin B1 exposure, chronic alcohol consumption, cirrhosis, or diabetes [7,8,9,10]. HBV and HCV infect approximately 2 billion and 170 million individuals worldwide, respectively [11, 12]. Bile duct cancer (CCA) accounts for 10–20% of all liver cancers [13]. Located in the small bile ducts of the liver, CCAs are best classified according to their anatomical location as intrahepatic (iCCA), perihilar (pCCA), or distal (dCCA) [14]. Overall survival rate for liver and intrahepatic bile duct in the United States was 18% from 2005 to 2011 [4]. Other types of liver cancer such as hepatoblastoma originated from immature liver precursor cells and are much less common [15]. Common liver cancer symptoms include weight loss, decrease in appetite, nausea, vomiting, fatigue, enlarged liver (hepatomegaly), enlarged spleen, abdominal swelling (ascites), or jaundice.
Hypoxic regions in HCC are marked by the indicators of increased cell proliferation and limited blood supply. Furthermore, hypoxic conditions can promote HCC tumorigenesis by promoting angiogenesis [6]. Hypoxia-inducible factor-1 (HIF-1), a transcriptional factor responsible for regulating a wide range of target genes involved in many cellular processes such as cell proliferation, cell cycle, metastasis, angiogenesis, apoptosis, cell survival, energy metabolism, and more stands as an essential component in a hypoxic state [16,17,18]. At the current point in research, approximately 60 genes have been identified to be transcriptionally activated by HIF-1 in which the promoter regions of its target genes consist of a cis-acting transcriptional regulatory sequence known as a hypoxia response element (HRE) [19]. The regulation of various genes and their transcription occurs through the binding of HIF-1 to the DNA regions of HRE [20].
HIF-1, first discovered as a transcription factor, regulates erythropoietin expression in response to low oxygen (O2) levels in the blood via de novo protein synthesis [21]. Erythropoietin stimulates bone marrow to produce more red blood cells (i.e., erythrocyte), resulting in the increase of the oxygen-carrying capacity in blood.
As a heterodimeric protein complex, HIF-1 is comprised of both subunits α and β. Both HIF-1α and HIF-1β subunits comprise of the basic helix-loop-helix (bHLH) and PER-ARNT-SIM (PAS) domains [22,23,24], which allow the heterodimerization to occur. DNA binding on HRE with the according sequence (5′-G/ACGTG-3′) in the promoter regions of target genes occurs due to the presence of the bHLH domain [19]. Aryl hydrocarbon receptor nuclear translocator (ARNT), also known as HIF-1β, is constitutively expressed and insensitive to O2; under normoxic conditions, HIF-1α is sensitive to O2 and is degraded through the ubiquitin-proteasome pathway [25, 26].
HIF-1α in normoxic conditions gets hydroxylated on proline residues at the oxygen-dependent degradation domain (ODDD) by prolyl hydroxylase domain enzymes (PHDs), which promote the binding to von Hippel-Lindau (VLH) proteins in the E3 ubiquitin ligase complex for ubiquitination and proteasome-mediated degradation [27, 28]. During hypoxia, PHD activity is a frequent characteristic of the cancer microenvironment (generally at pO2 levels <2%) and leads to the unhydroxylation of HIF-1α even under limitations of O2 availability. HIF-1α then translocates into the nucleus and binds with subunit HIF-1β. The HIF-1 heterodimer forms a complex with CREB-binding protein (CBP)/p300. Hypoxia prevents the hydroxylation of Asn803 and promotes the activity of CBP/p300 in the carboxyl-terminal transactivation domain of HIF-1α [29].
Many human cancers—liver, breast, pancreatic, etc.—contain the elevated expression levels of HIF-1 [30,31,32,33,34]. This elevation in HIF-1 expression in HCC cell lines is correlated with the advancement, poor diagnosis, and resistance of HCC cell lines, indicating that HIF-1 stands as a molecular target in HCC treatment [35,36,37]. Researchers also found HIF-1 as an indicator of HCC progression as higher levels of HIF-1 were found in the sera of HCC patients as compared to the lower levels in cases of benign disease [38, 39].
35.2 Role of HIF-1α in Cell Proliferation and Apoptosis
Hypoxia induces expression of several growth factors including vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), platelet-derived growth factor B (PDGFB), transforming growth factor (TGF)-α or TGF-β, insulin-like growth factor-2 (IGF-2), endothelin-1 (ET-1), and erythropoietin (EPO), which are known to promote cell proliferation in different cancer cells [40,41,42,43]. Cell proliferation stands as the primary concern in maintaining cell survival in the condition of hypoxia. An upregulation of cell survival genes including NK-ĸB, Mcl-1, Bcl-XL, and Bcl2 and the downregulation of pro-apoptotic genes—Bax and Bid—occur through the activation of HIF-1α in hypoxic conditions [44, 45]. Furthermore, the upregulation of Bcl2/adenovirus E1B 19 kDa interacting protein 3 (BNIP3) and BNIP3-like (BNIP3L) proteins in hypoxic conditions has been known to prevent malignant cells from cell death [46].
After cell proliferation, the malignant cells get involved in metastatic processes such as cell migration, adhesion, and invasion under low oxygen conditions. Extracellular growth factors prompt the p42/p44 mitogen-activated protein kinase pathway to phosphorylate HIF-1α, activate HIF-1α target genes transcription, and regulate cell proliferation. A strong correlation exists between elevated levels of HIF-1 and HCC proliferation and apoptosis; the role of HIF-1, however, presents a controversy even though several investigations indicate HIF-1 as an anti-apoptotic agent.
Xia et al. [47] reported that the transcription factor, Forkhead box M1 (FoxM1), induces HCC cell proliferation and resistance to apoptosis and its promoter binds to HIF-1α by the initiation of tumor necrosis factor-α (TNF-α). This observation revealed that TNF-α/HIF-1α stimulated the expression of FoxM1 [48]. Xu et al. [49] reported that HIF-1 stimulated cell cycle progression and cell proliferation by enhancing the expression of cyclins A and D in HCC. HIF-1 also decreases the apoptosis of HCC by increasing the levels of survivin and Bcl2 to prevent mitochondrial Omi/HtrA2 expression [49, 50]. Through several independent and HIF-1-dependent pathways, hypoxia induces the expression of downstream molecule VEGF, resulting in the increase of Bax/Bcl2 levels that leads to the survival of HCC [51]. Furthermore, hypoxia stimulates the levels of cAMP-responsive element-binding protein and is activated by extracellular signals [52].
Apoptosis, coined by Currie and colleagues in 1972, originated from Greek and means “to fall away from.” Stimulated by signaling pathways that result in the activation of caspase cascades and cell death, the apoptosis of a cell depolymerizes the cytoskeleton, condenses chromatin, and translocates nuclear fragments and phosphatidylserine to the surface of the cell. The increase in apoptotic activity stands as an indicator of several diseases, including AIDS, neurodegenerative disorders, insulin-dependent diabetes, myocardial infarction, and atherosclerosis. Two major pathways of apoptosis are responsible for processing stress signals and executing cellular demolition. Because of the importance and lethal nature of apoptosis, it is highly regulated by B-cell CLL/Lymphoma 2 (Bcl2) family proteins. Bcl2 proteins share one or more of four conserved Bcl2 homology (BH) domains (i.e., BH1, BH2, BH3, and BH4). Of these four domains, BH3 domain is found in all Bcl2 proteins, which interact with each other through the BH3 domain.
The Bcl2 protein family has either pro-apoptotic or anti-apoptotic activities. For example, Bcl2, B-cell lymphoma-extra large (Bcl-XL), and myeloid cell leukemia sequence 1 (Mcl-1) have anti-apoptotic activities, whereas Bcl2-associated x protein (Bax), Bcl2 antagonist/killer 1 (Bak), Bcl2-related ovarian killer protein (Bok), and BH3-only proteins have pro-apoptotic functions. Among the BH3-only proteins, activators such as Bax- and Bcl2-interacting mediator of cell death known as Bim interact with Bax and Bak to induce the mitochondrial outer membrane permeabilization (MOMP), whereas depressors neutralize the functions of anti-apoptotic proteins in response to various apoptotic stimuli such as hypoxia, ionizing radiation, cytotoxic agent, DNA damage, and growth factor withdrawal.
35.3 Role of HIF-1α in Cell Cycle
The inactivation of enzymes responsible for nucleotide synthesis causes hypoxically induced cell cycle arrest, and this arrest inhibits DNA replication [53, 54]. The inactivation of the nucleotide synthesis, however, only occurs under the condition of severe hypoxia known as anoxia, which is 0.01% oxygen [55]. On the other hand, relative changes to normoxia occur under moderate hypoxia, which is associated with the hypophosphorylation of retinoblastoma protein (Rb) and the inhibition of the cell cycle [55, 56]. While hypoxia may induce angiogenesis and glycolysis necessary for cell growth, it can also lead to cell cycle arrest and apoptotic activity. Furthermore, HIF-1α upregulates genes under low oxygen tension and has been indicated as a required mechanism for hypoxia-induced growth arrest and for p21cip1, a key cyclin-dependent kinase inhibitor in control of the cell cycle; the specificity of the exact mechanism, however, still remains unclear. HIF-1α acts against the activation of Myc through the downregulation of Myc-activated genes—hTERT and BR—and, thus, induces cell cycle arrest, which leads to the suppression of p21cip1 activity. Thus, Myc is an essential part of the HIF-1α pathway, which regulates Myc genes in hypoxic conditions. Hence, researchers have uncovered a divergent role for HIF-1α as it is not required for cell cycle arrest.
Several investigations have revealed that hypoxia-stimulated cell cycle arrest is associated with the downregulation of cyclin-dependent kinase (CDK) activity and the expression of retinoblastoma (Rb). Although the induction of cyclin G2, a negative regulator of cell cycle progression, was reported under hypoxia via HIF-1 activation, very little information is available to clarify the role of HIF-1 in the regulation of cell cycle machinery [57]. Recent studies have demonstrated the critical role of HIF-1 in the regulation of cell cycle progression under hypoxia by showing that the activation of HIF-1 impedes G1/S transition via two diverse mechanisms [58]. The expression of two CDK inhibitors (CKIs), p21Cip1 and p27Kip1, was increased in a HIF-1-dependent manner. Conserved expression of these CKIs was not observed in HIF-1α null cells and suppressed cyclin/CDK2 activity, leading to the reduction of the ratio of phosphorylated/dephosphorylated Rb protein, resulting in cell cycle arrest at G1/S [58].
35.4 Role of HIF-1α in Metastasis
Tumor metastasis is the multistage process that includes the dissociation, arrest, adhesion, and extravasation. It is also one of the primary causes of poor HCC diagnosis [59]. Adhesive molecules on tumor cells including intercellular adhesive molecule-1 (ICAM-1) and vascular cellular adhesive molecule-1 (VCAM-1) consequently rest the cells entirely and then extravasate from the blood vessels into other tissues or organs where metastatic tumor is formed. Throughout the metastatic signaling cascade, cancer cells communicate with endothelial cells, platelets, lymphocytes, and the homotypic as well as the heterotypic cell clusters from a multicellular emboliform nucleus [60, 61]. The potential of metastasis is associated with the activation of surface adhesive fragments including selectin, ICAM-1, or VCAM-1 [61]. Recently, it was reported that a key role is played by ICAM-1 in the metastatic process. Intra- and extrahepatic metastasis can cause poor prognosis and HCC, and the invasion of such metastases can also consist of epithelial-mesenchymal transition (EMT), which refers to the attainment of motility by tumor cells. Further, EMT requires the loss of E-cadherin, which is a major factor in the maintenance of epithelial polarity [62].
Hypoxia is clinically related to metastasis and poor prognosis [63]. Hypoxic stress speeds up the invasion of liver cancer cells by upregulating ETS-1 as well as the family of matrix metalloproteinases through HIF-1α-independent pathway [64]. The activity of HIF-1α is associated with VEGF [65]. The metastasis of HCC is inhibited by rapamycin and vitexin by downregulating the expression levels of VEGF and HIF-1α [66, 67]. The VEGF expression level in the plasma is elevated after transcatheter arterial chemoembolization (TACE) in patients with varied uptake of venous thrombosis. A 6-month follow-up revealed metastatic foci in almost 70% of the patients with elevated levels of plasma VEGF although patients with reduced plasma VEGF levels did not develop metastasis at all [66]. Consequently, in HCC, improved plasma VEGF level could be correlated with the advancement of metastasis after TACE. Furthermore, amplified plasma insulin-like growth factor II (IGF II) expression levels after TACE appear to be related to metastasis [66, 67].
35.4.1 Adhesion
Hypoxic conditions include the regulation of the transcription factor known as hypoxia-inducible factor 1α (HIF-1α), which is a master regulator that has pro-angiogenic activities such as the regulation of vascular endothelial growth factor (VEGF). Hypoxia-induced angiogenesis prompts researchers to investigate the multistep process by analyzing aortic and coronary artery smooth muscle cells with the additional treatment of cobalt chloride. Results show that HIF-1α activation reduced migration of smooth muscle cells and adhesion to the extracellular matrix. The presence of HIF-1α and cobalt chloride reduces the expression levels of tyrosine phosphorylation of focal adhesion kinase (FAK). Through FAK activation, HIF-1α acts as a suppressor of adhesion and migration of these smooth muscle cells by, but HIF-1α expression can also expand vessel growth by allowing smooth muscle cells to migrate and detach from the basement membrane and endothelial cells [68].
35.4.2 Invasion
Invasion consists of multiple steps including initiation. Epithelial-mesenchymal transition (EMT) offers tumor cells with motility to initiate the invasion. EMT includes the loss of E-cadherin, which influences on the adhesion junctions, which aids in maintaining the epithelial polarity [69]. HIF-1 may act as a leading modulator of EMT through upregulating the expression of various transcription repressors including transcription factor 3 (TCF3), E-cadherin, Snail, Twist1, Zfhx1a, and Zfhx1b [70]. It has been elucidated that HIF-1 enhances invasion as well as metastasis of HCC by inducing EMT during hypoxic conditions. HIF-1 possibly communicates with two HREs in the promoter region of Snail and also upregulates its activities in order to indirectly influence the expression levels of vimentin, E-cadherin, and N-cadherin [71].
35.4.3 Migration
Caused potentially by hypoxic conditions, pathological characteristics such as invasion and vascular proliferation can stand as indicators of malignant gliomas. Hypoxia-inducible factor-1 (HIF-1), which is a transcription factor derived from the HIF-1α subunit, forms the cellular response in hypoxic conditions. Though hypoxia has been correlated to the progression of angiogenesis, HIF-1 does not have a clear role in malignant gliomas. Therefore, investigations of the role of HIF-1α in the migration and invasion of human glioma cells in hypoxic conditions have been undertaken. Cell migration plays an essential role in expanding the breadth and growth of various cellular responses such as embryogenesis, inflammatory responses, and tumor metastasis [72].
35.4.4 Angiogenesis
Cells tend to undergo many biological and physiological responses to low oxygen levels known as hypoxia. One of the most studied responses to hypoxia is the expression of pro-angiogenic growth factors that activate their receptors and result in new blood vessel formation known as angiogenesis [73, 74]. Angiogenesis is an essential component of tumor cell migration and formation [75, 76]. HIF-1α upregulates the expression level of VEGF, an important pro-angiogenic factor [40, 77, 78]. Growth factors activated by HIF-1α regulate endothelial cell proliferation and blood vessel formation. HIF-1α activates the transcription of VEGF, VEGF receptor 1, adrenomedullin, COX-2, angiopoietin-2, angiopoietin receptor Tie-2, endothelin-1, endothelin-2, monocyte chemotactic protein-1, fibroblast growth factor-3, osteopontin, hepatocyte growth factor, transforming growth factor (TFG)-α, histone deacetylase, placental endothelial factor, nitric oxide synthase, TGF-β1, and TFG-β2. VEGF is one of the most critical angiogenic factors, which is secreted by normal as well as oncogenic cells in response to hypoxic conditions, and the receptors are mainly expressed on the endothelial cells. Angiogenesis that is induced via hypoxia is inhibited by agents blocking RAS, EGFR, and the receptor tyrosine kinase ERBB2 indicating that carcinogenic and hypoxia response signaling pathways are overlapped. HIF-1 stimulation can also reduce the expression levels of anti-angiogenic genes such as thrombospondin-1 and thrombospondin-2.
Angiogenesis is essential for tumor growth and progression by supplying oxygen and nutrients through the newly created blood vessels. It is already proven that the effective way to treat HCC is through anti-angiogenic therapy [79]. Under hypoxic conditions, HIF-1 acts as a direct transcriptional activator of the VEGF pathway, which promotes the migration and proliferation of vascular endothelial cells. HIF-1 directly activates the BEGF transcriptional pathway during hypoxic conditions as well. Sorafenib, an approved multi-kinase inhibitor and approved drug for advanced HCC patients, contains mechanisms that inhibit the expression levels of VEGF and HIF-1 proteins in order to result in the reduction of the expression of HCC vascularization [80]. A study done by Wang et al. [81] reports that a rat model experienced elevated levels of HIF-1 and VEGF after 20 weeks of hepatocarcinogenesis induction, revealing pro-angiogenic roles. Other pro-angiogenic markers include angiopoietin-2 (ANGPT2), stromal-derived factor 1 (SDF1), platelet-derived growth factor-B (PDGF-B), placental growth factor (PGF), and stem cell factor (SCF) [82]. Further research is required in order to clarify the pro-angiogenic role of HIF-1 in hypoxic conditions and possibly placing HIF-1 as a therapeutic target in HCC treatment.
35.5 Molecular Mechanism of HIF-1α
Cells respond to decreased oxygen levels through HIF-1 that is a heterodimer composed of the hypoxia response factor known as HIF-1α and the aryl hydrocarbon receptor nuclear translocator (ARNT) also identified as the HIF-1β. During the absence of oxygen, HIF-1 and hypoxia response elements (HREs) bind to each other in the promoter regions of the hypoxia response genes and as a result activate the expression of a number of genes such as VEGF, which is a pro-angiogenic growth factor. The redox active apurinic/apyrimidinic endonuclease-1 (APE1) has been shown to allow HIF-1α to function transcriptionally in a reduced state.
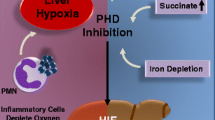
Under normoxic conditions, posttranslational HIF-1α is rapidly degraded by the proteasome. Prolyl hydroxylases (PHDs) hydroxylate the proline residues (402 and 564) at the ODDD of HIF-1α (Fig. 35.1). Hydroxylation of these residues facilitates the binding of the von Hippel-Lindau tumor suppressor gene (pVHL), which is a key component recognized by the E3 ubiquitin ligase complex in targeting HIF-1α for ubiquitination and degradation by the 26S proteasome [83]. In this degradation process, three PHDs (PHD1, PHD2, and PHD3) are the oxygen sensors controlling HIF-1α. These homologs are recently identified in mammals that have an immense potential to hydroxylate HIF-1α in normoxia [22]. The expression of PHD2 is controlled through the concentration of oxygen in the cell, and the conversion of hydroxyproline from proline involves ascorbate, iron, and 2-oxoglutarate. Under hypoxic environment, the inactivation of PHDs releases HIF-1α from hydroxylation by inhibiting pVHL from binding to proline residues and leading to the stabilization of HIF-1α in the cytoplasm.

Molecular mechanism of HIF-1α. HIF-1 consists of HIF-1α and HIF-1β. In hypoxia, HIF-1, a heterodimer, binds to DNA promoter regions of HRE and thereby activates the expression of numerous hypoxia response genes involved in cell proliferation, cell cycle, metastasis, angiogenesis, and cell survival. In contrast, HIF-1α is hydroxylated by PHDs and rapidly degraded by the proteasome under normoxic conditions. HIF indicates hypoxia-inducible factor; HRE indicates hypoxia response element; PHD indicates prolyl hydroxylase domain enzyme
In reference to the oxygen-dependent pVHL pathway, arrest defective 1 (ARD1) acetylates HIF-1α in order to induce HIF-1α degradation by acetylation of Lys532, resulting in an enhanced interaction of HIF-1α with pVHL [84].
Due to hypoxic conditions, certain miRNAs become responsible for the regulation of the HIF-α expression [85,86,87]. The miRNAs miR-424, miR-200b, and miR-429 stabilize HIF-α expression, while other miRNAs such as miR-199a and miR-22 reduce HIF-α activity. Endothelial cells in hypoxic conditions experience HIF-1α accumulation in the nucleus. Hypoxic conditions drive the upregulation of miR-424, miR-200b, and miR-429 expression, which stabilizes HIF-1α isoforms through the inhibition of the pVHL scaffold protein Cullin 2 (CUL2) or PHD activation [86]. Further, miR-199a downregulates the HIF-1α activity of cardiomyocytes [87]. Hypoxic conditions decrease miR-199a activity but enhance HIF-1α expression.
EMT is characterized by the loss of cell-cell adhesion and apical-basal polarity and important to tumor development as it promotes invasion and metastasis. During EMT, epithelial cells lose E-cadherin expression, a hallmark of EMT, and obtain mesenchymal markers such as vimentin and fibronectin. There are three types of EMT: type 1 appears in embryogenesis and organ development, type 2 is essential for tissue regeneration and organ fibrosis, and type 3 is associated with cancer stem cell properties. The EMT process involves master regulators, including SNAIL, TWIST, and ZEB transcription factors. By being activated earlier in the EMT process, these transcription factors play a central role in the development, fibrosis, and cancer and depend on the type of cell or tissue involved in the initiation of the EMT process.
Hypoxia in the tumor environment can promote EMT through HIF-1α expression, which activates TWIST, SNAIL, and other EMT inducers. In epithelial cells undergoing EMT, the mammalian TOR complex 1 (mTORC1) and mTORC2 are activated due to the presence of the AKT activity. TWIST is a direct transcriptional target of HIF-1α, whereas SNAIL is regulated by hypoxia at the posttranscriptional level. The upregulation of TWIST activity, stemness of cancer cells through the TWIST-BMI1 axis, members of the LOX/LOXL2 family, and other EMT inducers such as ZEB1/2 are crucial for the progression of the metastasis in a hypoxic environment. The hypoxic environment allows for the stability of HIF-1α, which leads to increased stemness and EMT of cancer cells and the activation of the following pathways: Wnt, Notch, and TGF-β. Furthermore, the activity of HIF-2α induces more stemness by increasing the expression levels of Oct-4. Under severe hypoxic conditions (0.1% O2), PERK, ATF4, and ATF6 potentiate the EMT of cancer cells.
35.6 Conclusions and Future Perspective
Hypoxia in the liver regulates gene expression in physiological conditions and diseases such as cirrhosis and cancer. Hypoxic conditions are known to promote the tumorigenic factors of proliferation, angiogenesis, invasion, and even resistance (chemo and radio) in the progression of HCC. Understanding hypoxic conditions in HCC can allow researchers to further investigate the progression of HCC malignancies. However, only HCC patients suited to palliative treatment can benefit from therapeutic methods that induce hypoxic conditions. Both treatment methods TACE and TAE induce hypoxia and, thus, promote to HCC angiogenesis; thus combining the benefits of TACE and TAE along with anti-angiogenic targeted therapy can introduce new approaches for HCC treatment. Unfortunately, hypoxic conditions prevent HCC cells from the combination therapy featuring the benefits from TACE, TAE, and anti-angiogenic factors. As angiogenesis becomes a major factor in the frequency of HCC recurrence, anti-angiogenic therapy can stand as the solution for future prevention and even benefit those who have surgical resections. By favoring a therapeutic approach of agent-inducing hypoxia with agent-targeting hypoxia factors, researchers can provide new treatment approaches for HCC patients.
In conclusion, compelling evidence reveals that HIF-1 activity promotes HCC proliferation, invasion, and metastasis. Along with the promotion of angiogenesis, HIF-1 activity also increases resistance of HCC cells to both chemotherapy and radiotherapy. Furthermore, clinical data supports the correlation between an increase in HIF-1 activity and the HCC’s poor prognosis. Thus, researchers have considered HIF-1 as targeted molecule in targeted therapy in HCC treatment, including inhibition of the HIF-1 pathway via small molecular targets. However, these molecular targets exhibited some disadvantages: detrimental side effects, low specificity, and the lack of evaluation in clinical trials. Therefore, further research is required to understand how to utilize the full efficacy of the therapeutic methods targeting HIF-1 in HCC treatment.
References
van Leeuwen MS et al (1995) Planning of liver surgery using three dimensional imaging techniques. Eur J Cancer 31A(7–8):1212–1215
Fasel JH (2008) Portal venous territories within the human liver: an anatomical reappraisal. Anat Rec (Hoboken) 291(6):636–642
Fasel JH, Majno PE, Peitgen HO (2010) Liver segments: an anatomical rationale for explaining inconsistencies with Couinaud’s eight-segment concept. Surg Radiol Anat 32(8):761–765
Siegel RL, Miller KD, Jemal A (2016) Cancer statistics, 2016. CA Cancer J Clin 66(1):7–30
Torre LA et al (2015) Global cancer statistics, 2012. CA Cancer J Clin 65(2):87–108
Sherman M, Llovet JM (2011) Smoking, hepatitis B virus infection, and development of hepatocellular carcinoma. J Natl Cancer Inst 103(22):1642–1643
Badvie S (2000) Hepatocellular carcinoma. Postgrad Med J 76(891):4–11
Sherman M (2005) Hepatocellular carcinoma: epidemiology, risk factors, and screening. Semin Liver Dis 25(2):143–154
El-Serag HB, Tran T, Everhart JE (2004) Diabetes increases the risk of chronic liver disease and hepatocellular carcinoma. Gastroenterology 126(2):460–468
Bosch FX et al (2005) Epidemiology of hepatocellular carcinoma. Clin Liver Dis 9(2):191–211. v
Lavanchy D (2004) Hepatitis B virus epidemiology, disease burden, treatment, and current and emerging prevention and control measures. J Viral Hepat 11(2):97–107
Chisari FV (2005) Unscrambling hepatitis C virus-host interactions. Nature 436(7053):930–932
Shaib Y, El-Serag HB (2004) The epidemiology of cholangiocarcinoma. Semin Liver Dis 24(2):115–125
Razumilava N, Gores GJ (2013) Classification, diagnosis, and management of cholangiocarcinoma. Clin Gastroenterol Hepatol 11(1):13–21. e1; quiz e3-4
De Ioris M et al (2008) Hepatoblastoma with a low serum alpha-fetoprotein level at diagnosis: the SIOPEL group experience. Eur J Cancer 44(4):545–550
Rankin EB, Giaccia AJ (2008) The role of hypoxia-inducible factors in tumorigenesis. Cell Death Differ 15(4):678–685
Carmeliet P et al (1998) Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 394(6692):485–490
Pennacchietti S et al (2003) Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell 3(4):347–361
Jiang BH et al (1996) Dimerization, DNA binding, and transactivation properties of hypoxia-inducible factor 1. J Biol Chem 271(30):17771–17778
Semenza GL (2000) HIF-1: mediator of physiological and pathophysiological responses to hypoxia. J Appl Physiol (1985) 88(4):1474–1480
Semenza GL, Wang GL (1992) A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol 12(12):5447–5454
Ziello JE, Jovin IS, Huang Y (2007) Hypoxia-inducible factor (HIF)-1 regulatory pathway and its potential for therapeutic intervention in malignancy and ischemia. Yale J Biol Med 80(2):51–60
Mandl M, Depping R (2014) Hypoxia-inducible aryl hydrocarbon receptor nuclear translocator (ARNT) (HIF-1beta): is it a rare exception? Mol Med 20:215–220
Koh MY, Powis G (2012) Passing the baton: the HIF switch. Trends Biochem Sci 37(9):364–372
Salceda S, Caro J (1997) Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem 272(36):22642–22647
Huang LE et al (1998) Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci U S A 95(14):7987–7992
Ohh M et al (2000) Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol 2(7):423–427
Maxwell PH et al (1999) The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399(6733):271–275
Lando D et al (2002) FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev 16(12):1466–1471
Bangoura G et al (2004) Expression of HIF-2alpha/EPAS1 in hepatocellular carcinoma. World J Gastroenterol 10(4):525–530
Krieg M et al (2000) Up-regulation of hypoxia-inducible factors HIF-1alpha and HIF-2alpha under normoxic conditions in renal carcinoma cells by von Hippel-Lindau tumor suppressor gene loss of function. Oncogene 19(48):5435–5443
Wu XY et al (2010) Identification of differential proteins in colon cancer SW480 cells with HIF1-alpha silence by proteome analysis. Neoplasma 57(4):299–305
Chiavarina B et al (2010) HIF1-alpha functions as a tumor promoter in cancer associated fibroblasts, and as a tumor suppressor in breast cancer cells: autophagy drives compartment-specific oncogenesis. Cell Cycle 9(17):3534–3551
Talks KL et al (2000) The expression and distribution of the hypoxia-inducible factors HIF-1alpha and HIF-2alpha in normal human tissues, cancers, and tumor-associated macrophages. Am J Pathol 157(2):411–421
Wilson WR, Hay MP (2011) Targeting hypoxia in cancer therapy. Nat Rev Cancer 11(6):393–410
Onnis B, Rapisarda A, Melillo G (2009) Development of HIF-1 inhibitors for cancer therapy. J Cell Mol Med 13(9A):2780–2786
Xia Y, Choi HK, Lee K (2012) Recent advances in hypoxia-inducible factor (HIF)-1 inhibitors. Eur J Med Chem 49:24–40
Unruh A et al (2003) The hypoxia-inducible factor-1 alpha is a negative factor for tumor therapy. Oncogene 22(21):3213–3220
Li S et al (2011) Expression characteristics of hypoxia-inducible factor-1alpha and its clinical values in diagnosis and prognosis of hepatocellular carcinoma. Hepat Mon 11(10):821–828
Forsythe JA et al (1996) Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol 16(9):4604–4613
Feldser D et al (1999) Reciprocal positive regulation of hypoxia-inducible factor 1alpha and insulin-like growth factor 2. Cancer Res 59(16):3915–3918
Grimshaw MJ (2007) Endothelins and hypoxia-inducible factor in cancer. Endocr Relat Cancer 14(2):233–244
Chen TM et al (2014) Overexpression of FGF9 in colon cancer cells is mediated by hypoxia-induced translational activation. Nucleic Acids Res 42(5):2932–2944
Chen N et al (2009) BCL-xL is a target gene regulated by hypoxia-inducible factor-1{alpha}. J Biol Chem 284(15):10004–10012
Erler JT et al (2004) Hypoxia-mediated down-regulation of Bid and Bax in tumors occurs via hypoxia-inducible factor 1-dependent and -independent mechanisms and contributes to drug resistance. Mol Cell Biol 24(7):2875–2889
Bellot G et al (2009) Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol 29(10):2570–2581
Xia LM et al (2009) Transcriptional up-regulation of FoxM1 in response to hypoxia is mediated by HIF-1. J Cell Biochem 106(2):247–256
Xia L et al (2012) The TNF-alpha/ROS/HIF-1-induced upregulation of FoxMI expression promotes HCC proliferation and resistance to apoptosis. Carcinogenesis 33(11):2250–2259
Xu Z et al (2012) Role of hypoxia-inducible-1alpha in hepatocellular carcinoma cells using a Tet-on inducible system to regulate its expression in vitro. Oncol Rep 27(2):573–578
Piret JP et al (2005) Hypoxia-inducible factor-1-dependent overexpression of myeloid cell factor-1 protects hypoxic cells against tert-butyl hydroperoxide-induced apoptosis. J Biol Chem 280(10):9336–9344
Baek JH et al (2000) Hypoxia-induced VEGF enhances tumor survivability via suppression of serum deprivation-induced apoptosis. Oncogene 19(40):4621–4631
Abramovitch R et al (2004) A pivotal role of cyclic AMP-responsive element binding protein in tumor progression. Cancer Res 64(4):1338–1346
Thelander L, Graslund A, Thelander M (1983) Continual presence of oxygen and iron required for mammalian ribonucleotide reduction: possible regulation mechanism. Biochem Biophys Res Commun 110(3):859–865
Loffler M (1989) The biosynthetic pathway of pyrimidine (deoxy)nucleotides: a sensor of oxygen tension necessary for maintaining cell proliferation? Exp Cell Res 182(2):673–680
Gardner LB et al (2001) Hypoxia inhibits G1/S transition through regulation of p27 expression. J Biol Chem 276(11):7919–7926
Krtolica A, Krucher NA, Ludlow JW (1998) Hypoxia-induced pRB hypophosphorylation results from downregulation of CDK and upregulation of PP1 activities. Oncogene 17(18):2295–2304
Ortmann B, Druker J, Rocha S (2014) Cell cycle progression in response to oxygen levels. Cell Mol Life Sci 71(18):3569–3582
Goda N et al (2003) Hypoxia-inducible factor 1alpha is essential for cell cycle arrest during hypoxia. Mol Cell Biol 23(1):359–369
Uchino K et al (2011) Hepatocellular carcinoma with extrahepatic metastasis: clinical features and prognostic factors. Cancer 117(19):4475–4483
Jiang WG et al (2015) Tissue invasion and metastasis: molecular, biological and clinical perspectives. Semin Cancer Biol 35(Suppl):S244–S275
Bendas G, Borsig L (2012) Cancer cell adhesion and metastasis: selectins, integrins, and the inhibitory potential of heparins. Int J Cell Biol 2012:676731
van Zijl F et al (2009) Epithelial-mesenchymal transition in hepatocellular carcinoma. Future Oncol 5(8):1169–1179
Vaupel P, Mayer A (2007) Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev 26(2):225–239
Miyoshi A et al (2006) Hypoxia accelerates cancer invasion of hepatoma cells by upregulating MMP expression in an HIF-1alpha-independent manner. Int J Oncol 29(6):1533–1539
Liu K et al (2016) The changes of HIF-1alpha and VEGF expression after TACE in patients with hepatocellular carcinoma. J Clin Med Res 8(4):297–302
Wu XZ, Xie GR, Chen D (2007) Hypoxia and hepatocellular carcinoma: the therapeutic target for hepatocellular carcinoma. J Gastroenterol Hepatol 22(8):1178–1182
Lin D, Wu J (2015) Hypoxia inducible factor in hepatocellular carcinoma: a therapeutic target. World J Gastroenterol 21(42):12171–12178
Corley KM, Taylor CJ, Lilly B (2005) Hypoxia-inducible factor 1alpha modulates adhesion, migration, and FAK phosphorylation in vascular smooth muscle cells. J Cell Biochem 96(5):971–985
Lamouille S, Xu J, Derynck R (2014) Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 15(3):178–196
Haase VH (2009) Oxygen regulates epithelial-to-mesenchymal transition: insights into molecular mechanisms and relevance to disease. Kidney Int 76(5):492–499
Luo D et al (2014) The role of hypoxia inducible factor-1 in hepatocellular carcinoma. Biomed Res Int 2014:409272
Lauffenburger DA, Horwitz AF (1996) Cell migration: a physically integrated molecular process. Cell 84(3):359–369
Carmeliet P (2003) Angiogenesis in health and disease. Nat Med 9(6):653–660
Yancopoulos GD et al (2000) Vascular-specific growth factors and blood vessel formation. Nature 407(6801):242–248
Folkman J (2002) Role of angiogenesis in tumor growth and metastasis. Semin Oncol 29(6 Suppl 16):15–18
Jain RK (2002) Tumor angiogenesis and accessibility: role of vascular endothelial growth factor. Semin Oncol 29(6 Suppl 16):3–9
Jiang BH et al (1997) V-SRC induces expression of hypoxia-inducible factor 1 (HIF-1) and transcription of genes encoding vascular endothelial growth factor and enolase 1: involvement of HIF-1 in tumor progression. Cancer Res 57(23):5328–5335
Ryan HE, Lo J, Johnson RS (1998) HIF-1 alpha is required for solid tumor formation and embryonic vascularization. EMBO J 17(11):3005–3015
Semela D, Dufour JF (2004) Angiogenesis and hepatocellular carcinoma. J Hepatol 41(5):864–880
Zhu YJ et al (2017) New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol Sin 38(5):614–622
Wang W et al (2009) Expression and correlation of hypoxia-inducible factor-1alpha, vascular endothelial growth factor and microvessel density in experimental rat hepatocarcinogenesis. J Int Med Res 37(2):417–425
Rey S, Semenza GL (2010) Hypoxia-inducible factor-1-dependent mechanisms of vascularization and vascular remodelling. Cardiovasc Res 86(2):236–242
Semenza GL (2007) Hypoxia-inducible factor 1 (HIF-1) pathway. Sci STKE 2007(407):cm8
Jeong JW et al (2002) Regulation and destabilization of HIF-1alpha by ARD1-mediated acetylation. Cell 111(5):709–720
Yoshioka Y et al (2012) Micromanaging iron homeostasis: hypoxia-inducible micro-RNA-210 suppresses iron homeostasis-related proteins. J Biol Chem 287(41):34110–34119
Nagaraju GP et al (2015) Hypoxia inducible factor-1alpha: its role in colorectal carcinogenesis and metastasis. Cancer Lett 366(1):11–18
el Azzouzi H et al (2013) The hypoxia-inducible microRNA cluster miR-199a approximately 214 targets myocardial PPARdelta and impairs mitochondrial fatty acid oxidation. Cell Metab 18(3):341–354
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Nature Singapore Pte Ltd
About this chapter

Cite this chapter
Choi, I., Lammata, S., Merchant, N., Park, D. (2017). Role of Hypoxia-Inducible Factor (HIF) in Liver Cancer. In: Nagaraju, G., Bramhachari, P. (eds) Role of Transcription Factors in Gastrointestinal Malignancies. Springer, Singapore. https://doi.org/10.1007/978-981-10-6728-0_35
Download citation
DOI: https://doi.org/10.1007/978-981-10-6728-0_35
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-6727-3
Online ISBN: 978-981-10-6728-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)