
Abstract
We were the first group to successfully sequence the 100%-complete entire eukaryotic genome in 2007, mainly using automated Sanger sequencing of the unicellular, ultrasmall red algal species Cyanidioschyzon merolae 10D. This world record was principally based on the ultrasmall size of the C. merolae genome (ca. 16 megabase pairs) as well as on three excellent previous studies: the 100%-complete mitochondrial genome in 1998, the 100%-complete plastid genome in 2003, and the first algal cell nuclear genome in 2004. The 100%-complete nuclear sequences demonstrated that this ultrasmall red alga contains unusually simple sets of genes and genetic sequences. For example, because introns are lacking in almost all of the protein-coding nuclear genes of C. merolae, the 100%-complete sequence can be used to directly deduce the sequences of all C. merolae proteins, which will be extremely valuable in further proteomics research. Thus, this small red alga represents an ideal model organism for studying the fundamental relationships among the plastid, mitochondrial, and nuclear genomes. The 100%-complete nuclear genome sequence has greatly improved the precision and value of biological analyses of C. merolae.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Algal genome
- Cyanidioschyzon merolae
- Eukaryotic cell
- Mitochondrial genome
- Nuclear genome
- 100%-complete genome
- Plastid genome
- Red alga
1 Introduction
The eukaryotic cell has three types of nuclei that contain nuclear, mitochondrial , and plastid genomes (Kuroiwa 1982). Thus, biological attributes and/or functions in each species of photosynthetic eukaryotes are fundamentally based on consortia of these three genomic information sources, except for some nucleomorph-containing secondary phototrophs such as cryptophytes and chlorarachniophytes (Curtis et al. 2012). Therefore, complete determination of the information from all of the three genomes in a single eukaryotic species was considered tantamount to revealing the complete molecular blueprint of the eukaryote. Thanks to the skillful and patient work of Japanese researchers, the first complete plastid genome was successfully determined through manual DNA sequencing based on the radiolabeling method (Ohyama et al. 1986; Shinozaki et al. 1986) and the first cyanobacterial genome through the automated Sanger DNA sequencing method (Kaneko et al. 1996).
Even in the current era of next-generation sequencing, the 100%-complete sequencing of all three eukaryotic genomes may seem as crazy an endeavor as “eating a bicycle,” which was once recorded in the Guinness Book of World Records (although this disturbing record has since been prohibited from nomination to the book). However, we indeed succeeded in the first 100%-complete eukaryotic genome in 2007, using automated Sanger sequencing of the unicellular red algal species Cyanidioschyzon merolae 10D (Nozaki et al. 2007). This world record was principally based on the ultrasmall genome size (ca. 16 megabase pairs) of C. merolae, as well as on three excellent previous studies: the 100%-complete mitochondrial genome sequence (Ohta et al. 1998), the 100%-complete plastid genome sequence (Ohta et al. 2003), and the first algal cell nuclear genome sequence (Matsuzaki et al. 2004).
Although our original nuclear genomic sequence of C. merolae 10D revealed some unique features such as very few introns, only three copies of ribosomal DNA (rDNA), and a relatively small number of total genes (Matsuzaki et al. 2004), the exact features of certain important repeated elements such as histone gene clusters and telomeres remained uncertain. Due to the functional significance of these elements, it was extremely desirable to complete the sequence and resolve all ambiguities to summarize all of the unique genomic features of C. merolae. Thus, we painstakingly elucidated all of the nucleotide sequences remaining as gaps and telomere-lacking chromosomal ends, to construct the first 100%-complete nuclear genome sequence (Nozaki et al. 2007).
We believed that the establishment of the first 100%-complete eukaryotic genome would be of great widespread interest to the biological sciences as a whole and that demonstration of the simplest set of genomic features from the hot spring red alga C. merolae would represent an important advancement in the fields of genomics and evolutionary biology. More than 10 years have passed since we determined the 100%-complete genome sequences of the mitochondrial, plastid, and nuclear genomes in C. merolae (Ohta et al. 1998, 2003; Nozaki et al. 2007). Recent advancements in next-generation sequencing methods have contributed much to the establishment of the organellar and nuclear whole genomes of numerous eukaryotes (e.g., Hanschen et al. 2016; Session et al. 2016). However, no other 100%-complete eukaryotic genomes have been determined. Thus, our studies (Ohta et al. 1998, 2003; Nozaki et al. 2007) remain the first and only achievement of a 100%-complete eukaryotic genome sequence.
2 Mitochondrial Genome
The 100%-complete nucleotide sequence of the mitochondrial genome of C. merolae 10D is on circular DNA containing 34 or 35 genes encoding proteins (including unidentified open reading frames [ORFs]) and 28 RNA genes (3 rRNAs and 25 tRNAs) (Table 5.1, Fig. 5.1.; Ohta et al. 1998; Yang et al. 2015). The genes are encoded on both strands. This is the largest number of protein-coding genes within the current 37 sequenced red algal mitochondrial genomes (Yang et al. 2015). The G + C content of the C. merolae mitochondrial genome is 27.2%. The genome size is 32,211 base pairs (bp), exceeding the average size (26,866 bp) of the 37 red algal mitochondrial genomes (Yang et al. 2015). The genome size and number of protein-coding genes of another cyanidialean species, Galdieria sulphuraria , are 21,428 bp and 19 genes, respectively. Thus, the large size and gene richness of the C. merolae mitochondrial genome do not represent Cyanidiales -specific characteristics.

Genetic map of the C. merolae mitochondrial genome. Note the C. merolae mtDNA is a circular-mapping molecule . The map was drawn based on the 100%-complete sequence (NC_000887; Ohta et al. 1998) using OrganellarGenomeDRAW http://ogdraw.mpimp-golm.mpg.de/index.shtml
3 Plastid Genome
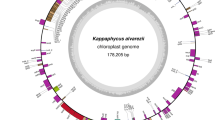
The 100%-complete nucleotide sequence of the plastid genome of C. merolae 10D is on circular DNA composed of 149,987 bp lacking inverted repeats (Table 5.1, Fig. 5.2). The G + C content is 37.6% (Ohta et al. 2003). This organelle genome contains 243 genes on both strands and consists of 207 protein-coding genes (including unidentified ORFs) and 36 RNA genes (a ribonuclease P RNA component, 3 rRNAs, 31 tRNAs, and tmRNA). Approximately 40% of the protein-coding genes overlap, and none of the genes in this plastid genome have introns (Ohta et al. 2003).

Genetic map of the C. merolae chloroplast genome. The map was drawn based on the 100%-complete sequence (NC_004799; Ohta et al. 2003) using OrganellarGenomeDRAW http://ogdraw.mpimp-golm.mpg.de/index.shtml
Recently, the plastid genomes of various red algal species have been sequenced (Janouškovec et al. 2013; Tajima et al. 2014). The C. merolae plastid genome is similar to other red algal plastid genomes in that it is gene rich: 223–250 unique genes are found in plastid genomes of red algae (Janouškovec et al. 2013). The striking feature of the C. merolae genome within the red algae is the high degree of gene compaction: it has the smallest genome size and the shortest intergenic region (Janouškovec et al. 2013). The median intergenic distance of the C. merolae plastid genome is extremely small (only 10 bp), whereas the intergenic distances of other red algal plastid genomes (including that of another cyanidialean species, Cyanidium caldarium) range from 61 to 85 bp (Janouškovec et al. 2013). Thus, the extraordinary compaction of the plastid genome of C. merolae has likely resulted from species-specific factors following the divergence of Cyanidioschyzon from other cyanidialean algae, such as Cyanidium (Janouškovec et al. 2013). The plastid genome of C. merolae encodes several genes that are rarely present in other plastid genomes (Ohta et al. 2003). Recently, 16 cyanobacterial genes were resolved to be present in the plastid genomes of only two Cyanidiales (C. merolae and Cyanidium caldarium) out of all known red algal plastid genomes (Janouškovec et al. 2013). Because plastid gene loss (endosymbiotic gene transfer) is slow in red algal plastid genomes (Nozaki et al. 2003; Janouškovec et al. 2013) and these two cyanidialean species are distantly related to other red algae with published plastid genomes, the 16 genes may be ancestral cyanobacterial genes that were lost in the latter red algae.
4 Nuclear Genome
To construct a 100%-complete nuclear genome sequence of C. merolae 10D, unresolved gaps between contigs and undetermined chromosome ends were filled using our previously constructed C. merolae bacterial artificial chromosome (BAC) clones (Matsuzaki et al. 2004). Polymerase chain reactions (PCRs) of BAC clones containing these gaps were performed using specific primers complementary to sequences flanking the gaps (Nozaki et al. 2007). We used DNA walking annealing control primer technology in order to directly amplify unknown sequences adjacent to known sequences within a contig. PCR products were sequenced by using the cycle sequencing methodology, except for a single gap with extremely high G + C content on chromosome 10, which was filled by an in vitro transcription sequencing reaction (Nozaki et al. 2007). Chromosomal ends were sequenced by the inverse PCR method, polyC-tailing and the anchor primer method, and the asymmetric PCR method (Nozaki et al. 2007). To completely determine the sequences of the histone cluster area in this red algal genome, we performed NotI and ApaI subcloning of the BAC clone GESZ2-b20, which includes possible histone clusters on chromosome 14 (Matsuzaki et al. 2004). We performed Southern blot analysis with histone-related probes, restriction enzyme analysis, and end sequencing of the subclones to reveal the relative positions of the subclones on this chromosome. We completely filled six gaps between contigs/fragments in the area of histone cluster (Matsuzaki et al. 2004) using primer walking of the subclones (Nozaki et al. 2007). Our complete nuclear genome sequence of C. merolae consisting of 16,546,747 nucleotides covers 100% of the 20 linear chromosomes from telomere to telomere (Table 5.1, Fig. 5.3). These 20 unambiguous DNA molecules represent the simple and unique structures of eukaryotic chromosomes (Nozaki et al. 2007): a histone gene cluster of the smallest known size, all chromosomal ends with a unique telomeric repeat, and an extremely low number of transposons. Based on these genomic features and others that were discovered previously (Matsuzaki et al. 2004), C. merolae appears to contain the simplest nuclear genome of the nonsymbiotic species of eukaryotes. These unusually simple genomic features found in this 100%-complete genome sequence were considered very useful for further biological studies of cells of eukaryotes.

Bird’s-eye view of the 100%-complete structures of 20 chromosomes of C. merolae showing regions that were filled by Nozaki et al. (2007; “gap” and “chromosomal end”), the histone cluster area, telomere repeats, putative centromeric regions, and transposable elements (“class I” and “class II”) (From Nozaki et al. (2007))
5 Centromere Regions and Their Dynamics
The centromere is a conserved chromosomal site responsible for attachment of spindles and accurate segregation of chromosomes during mitosis and meiosis . Each chromosome in a eukaryotic cell has a unique centromere where the kinetochore complex assembles and captures the spindle (Cleveland et al. 2003). In addition, the centromere core contains the histone H3 variant, centromere protein A (CENP-A), which replaces canonical H3 at the centromere. The 100%-complete genome sequence of C. merolae clarified that each chromosome also possesses a single A + T-rich region, which is predicted to be the putative centromere region (Matsuzaki et al. 2004; Maruyama et al. 2008). By ChIP-on-chip analysis using an anti-CENP-A antibody and a whole-genome tiling array, core centromere sequences were determined at the predicted region (Kanesaki et al. 2015). The identified centromeres were of the regional type, 1–3 kb in length, and with no consensus sequences or repeat elements. The expression of the CENP-A protein rapidly increased during the S phase of the cell cycle ; subsequently a drastic reconstitution into two discrete foci adjacent to the spindle poles occurred at metaphase (Maruyama et al. 2007). The dynamics of condensins I and II during the cell cycle were also well analyzed (Fujiwara et al. 2013a, b). In C. merolae, condensin II is enriched at centromere regions and is absent along chromosome arms during metaphase of the M phase, whereas condensin I is not enriched at centromere regions. Furthermore, C. merolae possesses the centromere proteins CENP-A, CENP-E, and the hypothetical CENP-C, but does not possess CENP-B or CENP-T. The characteristics of the centromere/kinetochore in lower plants and algae remain largely unknown. Thus, C. merolae is the most well-studied model system for clarifying the detailed mechanisms of centromere dynamics in primitive eukaryotic plant cells.
6 Conclusions
A histone gene cluster of the smallest known size, all chromosomal ends with a unique telomeric repeat, and an extremely low number of transposons in C. merolae (Nozaki et al. 2007), as well as other simple features of the C. merolae nuclear genome (Matsuzaki et al. 2004; Misumi et al. 2005), are extremely distinctive and represent the simplest set of features of genomes recognized in any nonsymbiotic eukaryote yet studied (Fig. 5.4). It is generally considered that such simple genomic features are the consequences of reductive evolution of a eukaryote of ultrasmall size (Derelle et al. 2006). However, none of these features are shared by Ostreococcus, a similarly ultrasmall green alga, in which histone genes are distributed among 6 or more chromosomes, 39% of the genes harbor introns, and 8166 protein genes and 417 transposable elements are dispersed across the chromosomes (Derelle et al. 2006). These characteristics suggest differences in modes of reduction of genomes between the ancestors of Cyanidioschyzon (red algae ) and Ostreococcus (Chloroplastida [green plants and algae]). On the other hand, algae growing in acidic hot springs (pH 1.5, 45 °C) may be candidates for retaining ancient or ancestral attributes of plants, because throughout Earth’s history, volcanic activity is thought to have provided such an extreme environment. According to Cunningham et al. (2006), C. merolae has perhaps the simplest chlorophyll and carotenoid assortment found in any photosynthetic eukaryote. In addition, the C. merolae plastid and mitochondrial genomes contain large numbers of genes, which are thought to be ancestral features, because it is generally considered that reversal of plastid gene loss is impossible (Martin et al. 2002; Nozaki et al. 2003). Thus, we hypothesized that some of the unusual or simple characteristics of the C. merolae genome may represent ancestral features that have been conserved in the Cyanidioschyzon lineage but have become modified extensively during the evolution of other lineages of plants/algae (Nozaki et al. 2007). Alternatively, the unique features of the C. merolae genome may reflect adaptations to their extreme environment. However, many simple features found in the C. merolae genome, such as the rare presence of introns and few rRNA regions in the nuclear genome, are not present in the nuclear genome sequence of another hot spring red alga, Galdieria (Barbier et al. 2005; Schönknecht et al. 2013). Organellar genomic features are also different between Cyanidioschyzon and Galdieria, as discussed above. Based on the 100%-complete nuclear, mitochondrial, and plastid genome sequences (Ohta et al. 1998, 2003; Matsuzaki et al. 2004; Nozaki et al. 2007), all of the major types of genetic information in eukaryotes are present in C. merolae. Furthermore, C. merolae contains unusually simple sets of genes and sequences as revealed by the 100%-complete genome. Because introns are lacking in almost all protein-coding nuclear genes of this ultrasmall red alga, the 100%-complete genome can be used to directly deduce the sequences of all red algal proteins, which will be very valuable in future research of proteomics . Thus, C. merolae represents an ideal model for studying the fundamental relationships among the chloroplast, mitochondrial, and nuclear genomes. The 100%-complete nuclear genome sequence (Nozaki et al. 2007) has greatly improved the precision of biological analyses of C. merolae, including studies of the dividing machineries of plastids (Yoshida et al. 2010) and peroxisomes (Imoto et al. 2013). The 100%-complete genome sequence also enables us to design various tools for genome-wide analyses, such as an organellar DNA microarray (Minoda et al. 2005; Kanesaki et al. 2012), nuclear DNA microarray (Fujiwara et al. 2009; Kanesaki et al. 2009; Imamura et al. 2009), and high-density whole-genome tiling array (Kanesaki et al. 2015). Furthermore, the 100%-complete genome sequence greatly contributed to the establishment of homologous recombination techniques (Minoda et al. 2004; Fujiwara et al. 2013a, b; Taki et al. 2015) and conditional gene expression systems (Sumiya et al. 2014; Fujiwara et al. 2015). The establishment of these advanced molecular biological techniques has made C. merolae one of the most exceptional model organisms among the eukaryotic algae.

Comparison of the three nuclear genomes of photosynthetic eukaryotes, Cyanidioschyzon, Ostreococcus (a green alga of ultrasmall size), and Arabidopsis (a seed plant). Telomere repeat sequences are indicated by asterisks above the generic names (Based on Nozaki et al. (2007))
References
Barbier G, Oesterhelt C et al (2005) Comparative genomics of two closely related unicellular thermo-acidophilic red algae, Galdieria sulphuraria and Cyanidioschyzon merolae, reveals the molecular basis of the metabolic flexibility of Galdieria sulphuraria and significant differences in carbohydrate metabolism of both algae. Plant Physiol, 137:460–474. https://doi.org/10.1104/pp.104.051169
Cleveland DW, Mao Y et al (2003) Centromeres and kinetochores: from epigenetics to mitotic checkpoint signaling. Cell 112:407–421. https://doi.org/10.1016/S0092-8674(03)00115-6
Cunningham FX Jr, Lee H et al (2006) Carotenoid biosynthesis in the primitive red alga Cyanidioschyzon merolae. Eukaryot Cell 6:533–545. https://doi.org/10.1128/EC.00265-06
Curtis BA, Tanifuji G et al (2012) Algal genomes reveal evolutionary mosaicism and the fate of nucleomorphs. Nature 492:59–65. https://doi.org/10.1038/nature11681
Derelle E, Ferraz C et al (2006) Genome analysis of the smallest free-living eukaryote Ostreococcus tauri unveils many unique features. Proc Natl Acad Sci U S A 103:11647–11652. https://doi.org/10.1073/pnas.0604795103
Fujiwara T, Misumi O et al (2009) Periodic gene expression patterns during the highly synchronized cell nucleus and organelle division cycles in the unicellular red alga Cyanidioschyzon merolae. DNA Res 16:59–72. https://doi.org/10.1093/dnares/dsn032
Fujiwara T, Ohnuma M et al (2013a) Gene targeting in the red alga Cyanidioschyzon merolae: single- and multi-copy insertion using authentic and chimeric selection markers. PLoS One 8:e73608. https://doi.org/10.1371/journal.pone.0073608
Fujiwara T, Tanaka K et al (2013b) Spatiotemporal dynamics of condensins I and II: evolutionary insights from the primitive red alga Cyanidioschyzon merolae. Mol Biol Cell 24:2515–2527. https://doi.org/10.1091/mbc.E13-04-0208
Fujiwara T, Kanesaki Y et al (2015) A nitrogen source-dependent inducible and repressible gene expression system in the red alga Cyanidioschyzon merolae. Front Plant Sci 6:657. https://doi.org/10.3389/fpls.2015.00657
Hanschen ER, Marriage TN et al (2016) The Gonium pectorale genome demonstrates co-option of cell cycle regulation during the evolution of multicellularity. Nat Commun 7:11370. https://doi.org/10.1038/ncomms11370
Imamura S, Kanesaki Y et al (2009) R2R3-type MYB transcription factor, CmMYB1, is a central nitrogen assimilation regulator in Cyanidioschyzon merolae. Proc Natl Acad Sci U S A 106:12548–12553. https://doi.org/10.1073/pnas.0902790106
Imoto Y, Kuroiwa H et al (2013) Single-membrane-bounded peroxisome division revealed by isolation of dynamin-based machinery. Proc Natl Acad Sci U S A 110:9583–9588. https://doi.org/10.1073/pnas.1303483110
Janouškovec J, Liu S-L et al (2013) Evolution of red algal plastid genomes: ancient architectures, introns, horizontal gene transfer, and taxonomic utility of plastid markers. PLoS One 8:e59001. https://doi.org/10.1371/journal.pone.0059001
Kaneko T, Sato S et al (1996) Sequence analysis of the genome of the unicellular cyanobacterium Synechocystis sp. strain PCC6803. II. Sequence determination of the entire genome and assignment of potential protein-coding regions. DNA Res 3:109–136
Kanesaki Y, Kobayashi Y et al (2009) Mg-protoporphyrin IX signaling in Cyanidioschyzon merolae: multiple pathways may involve the retrograde signaling in plant cells. Plant Signal Behav 4:1190–1192. https://doi.org/10.4161/psb.4.12.10061
Kanesaki Y, Imamura S et al (2012) External light conditions and internal cell cycle phases coordinate accumulation of chloroplast and mitochondrial transcripts in the red alga Cyanidioschyzon merolae. DNA Res 19:289–303. https://doi.org/10.1093/dnares/dss013
Kanesaki Y, Imamura S et al (2015) Identification of centromere regions in chromosomes of a unicellular red alga, Cyanidioschyzon merolae. FEBS Lett 589:1219–1224. https://doi.org/10.1016/j.febslet.2015.04.009
Kuroiwa T (1982) Mitochondrial nuclei. Int Rev Cytol 75:1–59
Martin W, Rujan T et al (2002) Evolutionary analysis of Arabidopsis, cyanobacterial, and chloroplast genomes reveals plastid phylogeny and thousands of cyanobacterial genes in the nucleus. Proc Natl Acad Sci U S A 99:12246–11251. https://doi.org/10.1073/pnas.182432999
Maruyama S, Kuroiwa H et al (2007) Centromere dynamics in the primitive red alga Cyanidioschyzon merolae. Plant J 49:1122–1129. https://doi.org/10.1111/j.1365-313X.2006.03024.x
Maruyama S, Matsuzaki M et al (2008) Centromere structures highlighted by the 100%-complete Cyanidioschyzon merolae genome. Plant Signal Behav 3:140–141. https://doi.org/10.4161/psb.3.2.5066
Matsuzaki M, Misumi O et al (2004) Genome sequence of the ultrasmall unicellular red alga Cyanidioschyzon merolae 10D. Nature 428:653–657. https://doi.org/10.1038/nature02398
Minoda A, Sakagami R et al (2004) Improvement of culture conditions and evidence for nuclear transformation by homologous recombination in a red alga, Cyanidioschyzon merolae 10D. Plant Cell Physiol 45:667–671. https://doi.org/10.1093/pcp/pch087
Minoda A, Nagasawa K et al (2005) Microarray profiling of plastid gene expression in a unicellular red alga, Cyanidioschyzon merolae. Plant Mol Biol 59:375–385. https://doi.org/10.1007/s11103-005-0182-1
Misumi O, Matsuzaki M et al (2005) Cyanidioschyzon merolae genome. A tool for facilitating comparable studies on organelle biogenesis in photosynthetic eukaryotes. Plant Physiol 137:567–585. https://doi.org/10.1104/pp.104.053991
Nozaki H, Ohta N et al (2003) Phylogeny of plastids based on cladistic analysis of gene loss inferred from complete plastid genome sequences. J Mol Evol 57:377–382. https://doi.org/10.1007/s00239-003-2486-6
Nozaki H, Takano H et al (2007) A 100%-complete sequence reveals unusually simple genomic features in the hot-spring red alga Cyanidioschyzon merolae. BMC Biol 5:28. https://doi.org/10.1186/1741-7007-5-28
Ohta N, Sato N et al (1998) Structure and organization of the mitochondrial genome of the unicellular red alga Cyanidioschyzon merolae deduced from the complete nucleotide sequence. Nucleic Acids Res 26:5190–5198
Ohta N, Matsuzaki M et al (2003) Complete sequence and analysis of the plastid genome of the unicellular red alga Cyanidioschyzon merolae. DNA Res 10:67–77
Ohyama K, Fukuzawa H et al (1986) Chloroplast gene organization deduced from complete sequence of liverwort Marchantia polymorpha chloroplast DNA. Nature 322:572–574
Schönknecht G, Chen WH et al (2013) Gene transfer from bacteria and archaea facilitated evolution of an extremophilic eukaryote. Science 339:1207–1210. https://doi.org/10.1126/science.1231707
Session AM, Uno Y et al (2016) Genome evolution in the allotetraploid frog Xenopus laevis. Nature 538:336–343. https://doi.org/10.1038/nature19840
Shinozaki K, Ohme M et al (1986) The complete nucleotide sequence of the tobacco chloroplast genome: its gene organization and expression. EMBO J 5:2043–2049
Sumiya N, Fujiwara T et al (2014) Development of a heat-shock inducible gene expression system in the red alga Cyanidioschyzon merolae. PLoS One 9:e111261. https://doi.org/10.1371/journal.pone.0111261.
Tajima N, Sato S et al (2014) Analysis of the complete plastid genome of the unicellular red alga Porphyridium purpureum. J Plant Res 127:389–397. https://doi.org/10.1007/s10265-014-0627-1
Taki K, Sone T, Kobayashi Y, Watanabe S, Imamura S, Tanaka K (2015) Construction of a URA5.3 deletion strain of the unicellular red alga Cyanidioschyzon merolae: a background less host strain for transformation experiments. J Gen Appl Microbiol 61:211–214. https://doi.org/10.2323/jgam.61.211
Yang EC, Kim KM et al (2015) Highly conserved mitochondrial genomes among multicellular red algae of the Florideophyceae. Genome Biol Evol 7:2394–23406. https://doi.org/10.1093/gbe/evv147
Yoshida Y, Kuroiwa H et al (2010) Chloroplasts divide by contraction of a bundle of nanofilaments consisting of polyglucan. Science 329:949–953. https://doi.org/10.1126/science.1190791
Acknowledgments
HN was supported by a Grant-in-Aid for Scientific Research (grant number 16H02518) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT)/Japan Society for the Promotion of Science (JSPS) KAKENHI.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Nozaki, H., Kanesaki, Y., Matsuzaki, M., Hirooka, S. (2017). The 100%-Complete Nuclear and Organellar Genome Sequences of the Ultrasmall Red Algal Species Cyanidioschyzon merolae 10D. In: Kuroiwa, T., et al. Cyanidioschyzon merolae. Springer, Singapore. https://doi.org/10.1007/978-981-10-6101-1_5
Download citation
DOI: https://doi.org/10.1007/978-981-10-6101-1_5
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-6100-4
Online ISBN: 978-981-10-6101-1
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)