
Abstract
Extracellular vesicles are now widely recognized as key players in the prevention, repair or progression of cardiovascular disease. Here we first focus on the functional roles of extracellular vesicles in the cross-talk between cardiomyocytes and endothelial cells, important for maintaining normal development and function of the heart. Second, we discuss the role of extracellular vesicles secreted by embryonic and non-embryonic stem cells in repairing cardiomyocyte function and in restoring angiogenic potential after myocardial ischemia-reperfusion injury. Third, we focus on the role of extracellular vesicles in Endothelial to Mesenchymal Transition (EndMT), leading to conversion of endothelial cells to fibroblasts, secretion of extracellular proteins collagen and fibronectin, and fibrosis. Finally, we discuss the role of extracellular vesicles secreted under stress by endothelial cells, macrophages and vascular smooth muscle cells in atherosclerosis. On aggregate, the reviewed preclinical studies present evidence that extracellular vesicles secreted by cardiomyocytes, fibroblasts, endothelial cells, immune-system-related cells, vascular smooth muscle cells and stem cells play an important role in the pathogenesis of cardiovascular disease. However, further studies are needed to gain better insight into the mechanisms used to select specific content to transfer to specific target cells, and to induce or repress signaling pathways in their target cells.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Exosomes are bi-lipid-membranous vesicles containing protein, lipid and nucleic acid material secreted from cells. They are smaller than shedding microvesicles and apoptotic bodies (approximately 40–100 nm for exosomes compared to 100 nm–1 μm for shedding microvesicles and 1–4 μm for apoptotic bodies) and differ by their intracellular origin [19, 42]. Exosomes are identified by their components including integrins and tetraspanins (CD63, CD89, CD81, CD9, and CD82), by maturation-related proteins (flotillin and annexin), and by heat shock proteins (HSP). Together, exosomes and microvesicles are often referred to as extracellular vesicles (EVs). They are not a result of random sampling; instead they contain selective cargo assembled through dedicated packing mechanisms [40, 44], deliver these loads to targeted cells and contain unique trafficking properties. All these mechanisms depend on the individual cell, its cellular state and different physiological, pathological and stress conditions [19, 20]. EVs mediate horizontal, paracrine transfer, delivering microRNAs (miRs), mRNA and proteins between cells of different origin, resulting in silencing or activation of signaling pathways [32]. However, the underlying mechanisms of transfer and the amount of content delivered to the cells remain controversial or unclear.
This chapter will discuss the functional roles of EVs in the prevention, repair or progression of cardiovascular disease, through the communication between several cell types in the heart and vasculature, with an emphasis on signaling pathways.
2 Role of Extracellular Vesicles in Intracardial Communication
2.1 Cardiomyocytes, Myofibroblasts and Cardiac Injury
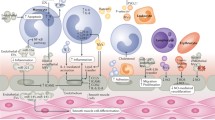
The heart contains cardiomyocytes (CMs), which represent only a third of all cells in the heart, endothelial cells (ECs), immune-system-related cells (macrophages), fibroblasts (FBs), smooth muscle cells (SMCs), sympathetic and parasympathetic neuronal cells, and stem cells. A tight balance between these cell types is needed to maintain the integrity of the heart [4]. Myocardial injury disrupts this integrity by inducing CM death and destroying the vasculature, triggering several effects to repair and maintain cardiac integrity, including cardiac fibrosis by activation of myofibroblasts. This activation involves a complex signaling network containing transforming growth factor (TGF)-β, endothelin-1 (EDN-1), angiotensin II (AGTII), connective tissue growth factor (CCN2), and platelet-derived growth factor (PDGF) (Fig. 3.1a). Myofibroblasts originate from several sources including quiescent tissue FBs, circulating CD34+ fibrocytes, and the phenotypic conversion of various cell types including epithelial cells and ECs. To date, little is known about the inter-organ transfer mechanisms of cardioprotection but recent reports suggest that extracellular vesicles (EVs) release may be involved [13]. In this section we will focus on EVs released from CMs, ECs, macrophages and FBs in cardiac injury and remodeling (Fig. 3.1).

Extracellular vesicles in cardiac remodeling: Myocardial injury induces CM death and triggers several repair mechanisms, including cardiac fibrosis. The effector cells of fibrosis are activated FBs called myofibroblasts and involves a complex signaling network containing TGF-β, EDN-1, AGTII, CCN2 and PDGF leading to fibrosis (a). EVs secreted from CMs were shown to be enriched in HSP20, pAKT, survivin and SOD1, and were cardioprotective and induced angiogenesis (b). In contrast, diabetes induces CMs to secrete EVs enriched in miR-320 impairing the expression of IGF-1, HSP20 and ETS2 in ECs resulting in impaired angiogenesis (b). CM-derived EVs containing A1TR, β-arrestin and miR-1 and miR133a increase TLR4/HSP27 signaling, resulting in cardioprotection (c). During myocardial ischemia, TNF-α is mainly released in macrophage-derived exosomes, but with persistent ischemia it can also originate from exosomes released by CMs, inducing CM death (d). miR-21-3p (miR-21*) in FB-derived exosomes induced CM hypertrophy by inhibiting SORBS2 and PDLIM5 (e). In addition, these FB-derived exosomes induced MAPK and AKT signaling resulting in intensified AGTII-induced cardiac hypertrophy. Abbreviations: AGT angiotensin, AKT AKT serine/threonine kinase 1, A1TR angiotensin II type I receptors, CM cardiomyocyte, CCN2 connective tissue growth factor, EC endothelial cell, EDN-1 endothelin-1, FB (myo)fibroblast, ETS2 ETS proto-oncogene 2, transcription factor, EV extracellular vesicle, HIF-1α hypoxia inducible factor-1α, HSP heat shock protein, IGF-1 insulin growth factor 1, MAPK mitogen-activated protein kinases, miR microRNA, MQ macrophage, PDGF platelet-derived growth factor, PDLIM5 PDZ and LIM domain 5, SOD superoxide dismutase, SORBS2 sorbin and SH3 domain-containing protein 2, TGF-β transforming growth factor, TLR toll like receptor, TNF tumor necrosis factor
2.2 Extracellular Vesicles from Cardiomyocytes, Endothelial Cells and Myofibroblasts
CMs and ECs have an intimate anatomical relationship that is essential for maintaining normal development and function in the heart. Regulation mechanisms of cardiac and endothelial crosstalk in situations of acute stress remain elusive. This cardiac and endothelial crosstalk may involve EVs, among them exosomes. Recently, HSP20-enriched EVs secreted by CMs acted as a novel cardiokine in regulating myocardial angiogenesis through activation of the vascular endothelial growth factor receptor (VEGFR) signaling cascade [54] (Fig. 3.1b). Indeed, overexpression of HSP20 in streptozotocin (STZ)-induced diabetic mice significantly decreased cardiac dysfunction, hypertrophy, apoptosis, fibrosis, and restored angiogenesis. This protective action against adverse cardiac remodeling involved p-Akt, survivin, and superoxide dismutase 1. In addition, HSP20 exosomes interacted with tumor susceptibility gene 101, an important regulator of cell cycle arrest and p-53 independent cell death [47]. Diabetes also significantly impaired angiogenesis by inhibiting proliferation and migration of mouse cardiac endothelial cells (CECs). Mechanistically, higher levels of microRNA (miR)-320 in exosomes of diabetic animals functionally down-regulated its target genes such as insulin growth factor (IGF-)1, HSP20 and ETS proto-oncogene 2, transcription factor (ETS2) in recipient CECs [48] (Fig. 3.1b).
In myocardial ischemia-reperfusion injury, both miR-1 and miR-133a in the exosome-rich fraction in plasma protected CMs by inhibiting cardiac hypertrophy [7] (Fig. 3.1c). Therefore, they may be utilized to suppress maladapted hypertrophy when blood flow and energy supply is limited [26]. A pro-survival signaling pathway was activated in CMs involving toll-like receptor (TLR)-4 and various kinases, leading to activation of the cardioprotective HSP27.
In addition to secreting microvesicles and exosomes, CMs were found to secrete cytokines, chemokines, and factors like ANP, BNP, TGF-β, and tumor necrosis factor-α (TNF-α) [15, 46]. Excessive TNF-α is thought to be harmful to CMs in acute myocardial infarction (MI). During myocardial ischemia, TNF-α is mainly released in macrophage-derived exosomes, but with persistent ischemia it can also originate from exosomes released by CMs, induced directly by hypoxia and activation of hypoxia inducible factor-1α (HIF-1α) [53] (Fig. 3.1d).
Finally, FBs also have an important function in the pathophysiology of CM death, fibrosis and hypertrophy. MiR-21-3p in cardiac FB-derived exosomes induced CM hypertrophy (Fig. 3.1e). Sorbin and SH3 domain-containing protein 2 (SORBS2) and PDZ and LIM domain 5 (PDLIM5) were identified as miR-21-3p targets by proteome profiling. Silencing SORBS2 or PDLIM5 in CM and inhibition of miR-21-3p induced hypertrophy [5]. In addition, FB-derived exosomes induced mitogen-activated protein kinases (MAPKs) and Akt resulting in increased expression of the Renin Angiotensin System, thereby intensifying AGTII-induced pathological cardiac hypertrophy [29].
3 Vesicles from Stem Cells and Progenitor Cells
3.1 Cardiac Stem Cells
By definition, a stem cell is capable of self-renewal and can differentiate into at least one cell type. Embryonic stem cells (ESCs) are pluripotent stem cells which were first isolated as a small cluster of cells within mouse blastocysts [11], later from human blastocysts [38]. Hematopoietic stem cells (HSCs) differentiate into different blood cells and are CD34+. Mesenchymal stem cells (MSCs) are multipotent stromal cells that can differentiate into a variety of cell types like myocytes and adipocytes [39]. In contrast to HSCs, standardization of MSCs was hampered by the lack of molecular markers to discern MSCs from FBs. However, recently, specific DNA methylation patterns were used to discriminate between MSCs and FBs, and to distinguish between MSCs from bone marrow and adipose tissue [1]. In addition to ESCs, HSCs and MSCs, a population of resident cardiac stem cells (CPCs) have been identified in the heart; but they comprise less than 1% of the cells in the heart. As yet it is not known whether the CPCs actually migrate from bone marrow to the heart, or originate from remnants of embryonic cell populations in the right atrium and right ventricle. CPCs have been sub-classified into c-kit and Scal-1. C-kit cardiogenic stem cells can differentiate into myogenic, vascular endothelial and smooth muscle lineages. Sca-1 are involved in cell signaling and cell adhesion [16]. Finally, induced pluripotent stem cells (iPSC) can generate an abundance of cells without the risk of immune rejection for cell therapy.
3.2 Stem Cell Derived Vesicles
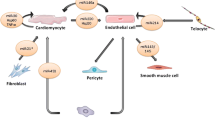
MSCs have been considered to be one of the most promising candidates for regeneration of cardiac cells lost upon injury. But with age, dysregulated MSCs differentiate into dysfunctional inflammatory FBs leading to pathologic fibrosis. The phenotypic change is specific to the heart since MSCs originating from bone or FBs derived from MSCs were free of these defects [43]. In contrast with the original hypothesis that the regenerative capacity of MSCs was due to their potential to engraft, differentiate and replace damaged cardiac cells, recent studies suggested that this was primarily due to paracrine factors released from MSCs in exosomes [35]. For example, GATA binding protein 4 (GATA-4) enriched in exosomes released by MSCs at the border of an ischemic region significantly restored cardiac contractile function and reduced infarct size (Fig. 3.2). Mechanistically, these exosomes increased miR-19a in CMs, resulting in decreased expression of Phosphatase and Tensin Homolog (PTEN), a predicted target of miR-19a, and in the activation of the Akt and ERK signaling pathways [52] (Fig. 3.2). In addition to miR-19, miR profiling analysis revealed that cardiac stem cells exposed to MSC-derived exosomes secreted more miR-147, let-7i-3p, miR-503-5p, and miR-362-3p, and less miR-326-5p, miR-328a-5p, miR-207, miR-760-3pn, and miR-702-5p, associated with activation of target genes involved in angiogenesis and positive regulation of cell proliferation, cell migration, cell differentiation, and response to hypoxia [55]. They activated several signaling pathways important in wound healing (Akt, ERK, and STAT3) and induced the expression of a number of growth factors [hepatocyte growth factor (HGF), IGF1, nerve growth factor (NGF), and stromal-derived growth factor-1 (SDF1)] [37], ultimately leading to preserved cardiac performance after MI [6] (Fig. 3.2). Levels of ATP, NADH and phosphorylated-Akt and phosphorylated- inosine/guanosine kinase (GSK)-3β were increased, while phosphorylated-c-JNK was reduced, thereby decreasing oxidative stress and inflammation in ischemic/reperfused hearts [3]. In addition, the proteome of MSCs and MSC-derived exosomes, from cells cultured under expansion conditions and under ischemic tissue simulated conditions, was shown to contain key angiogenic paracrine effectors and, potentially, differentially expressed in these conditions. In total, 6342 proteins were identified in MSCs and 1927 proteins in MSC-derived exosomes. They included PDGF, epidermal growth factor (EGF), fibroblast growth factor (FGF), and most notably nuclear factor-kappaB (NFkB) signaling pathway proteins. The latter was identified as a key mediator of MSC exosome induced angiogenesis in ECs [2] (Fig. 3.2).

Stem and progenitor cells secrete EVs which protect CMs. EVs released by MSCs overexpressing miR-19 and GATA-4 at the border of an ischemic region following ligation of the left anterior descending coronary artery significantly reduced infarct size and restored cardiac contractile function. Exosomes from iPSC protected against myocardial ischemia/reperfusion injury most likely by delivery of cardioprotective miRs, including nanog-regulated miR-21 and HIF-1α-regulated miR-210. CPCs release EVs containing GATA-4 and MEF2, which are important in the development of pre-cardiac cells. ESC-derived exosomes deliver ESC-specific miR-290-295 cluster and more particular miR-294 to CPCs and CMs, promoting increased survival, cell cycle progression, and proliferation in ECs and CMs. On aggregate, these EVs reduced infarct size and fibrosis and increased cell survival and cardiac function. ASC-derived exosomes carry c-kit and stem cell factor, which play a role in angiogenesis. Exosomes secreted by ASCs also promote EC angiogenesis by transferring miR-125a repressing the NOTCH ligand delta-like 4. HSCs induce angiogenesis by secreting exosomes with increased expression of Shh. CPCs also secrete exosomes with pro-angiogenic properties mediated via ERK/AKT-signaling. They contain high levels of EMMPRIN. MSCs exposed to ischemia secrete exosomes enriched in PDGF, EGF and FGF, and most notably, NFkB signaling pathway proteins inducing angiogenesis in ECs. In addition, MSC-derived exosomes activated several signaling pathways important in wound healing (AKT, ERK, and STAT3) and induced the expression of a number of growth factors (HGF, IGF1, NGF, and SDF1). On aggregate, these EVs induced angiogenesis, cell renewal and wound healing in endothelium. Abbreviations: AKT AKT serine/threonine kinase 1, ASC adipose-derived mesenchymal cell, CPC cardiac progenitor cell, EGF epidermal growth factor, EMMPRIN extracellular matrix metalloproteinase inducer, ERK extracellular signal–regulated kinase, ESC embryonic stem cell, FGF fibroblast growth factor, GATA-4 GATA binding protein 4, GSK inosine/guanosine kinase, HGF hepatocyte growth factor, HIF-1α hypoxia inducible factor-1α, HSCs hematopoietic stem cell, IGF-1 insulin growth factor 1, MEF2 myocyte enhancer factor 2, MSC mesenchymal stem cell, NFkB nuclear factor-kappaB, NGF nerve growth factor, PDGF platelet-derived growth factor, SCF stem cell factor, Shh sonic hedgehog, SDF1 stromal-derived growth factor-1, STAT3 signal transducer and activator of transcription 3
Exosomes derived from iPSC-derived mesenchymal stem cells (iMSC), expressing CD63, CD81, and CD9 at their surface, enhanced microvessel density and blood perfusion in mouse ischemic limbs, consistent with an attenuation of ischemic injury [18], possibly by delivery of cardioprotective miRs, including nanog-regulated miR-21 and HIF-1α-regulated miR-210 [49] (Fig. 3.2).
Furthermore, CPC-derived EVs had the same beneficial effects as their parent cells in the treatment of chronic heart failure in mice [23]. Like MSCs, CPCs secreted exosomes with pro-angiogenic properties mediated via ERK/Akt-signaling (Fig. 3.2). Analysis of pro-angiogenic factors revealed high levels of extracellular matrix metalloproteinase inducer (EMMPRIN) [45]. Also mouse ESC-derived exosomes augmented function in infarcted hearts by enhancing neovascularization and CM survival and by reducing fibrosis, most likely by delivery of ESC-specific miR-294 to CPCs, promoting increased survival, cell cycle progression, and proliferation [24] (Fig. 3.2).
Finally, adipose mesenchymal stem cell (ASC)-derived EVs induced in vitro vessel-like structure formation by human microvascular endothelial cells (MECs) (Fig. 3.2). Treatment of ASCs with PDGF stimulated secretion of EVs, carrying c-kit and stem cell factor, SCF, regulated by HIF-1α, inducing angiogenesis [28]. Exosomes secreted by ASCs also promoted EC angiogenesis by transferring miR-125a, which repressed the NOTCH ligand delta-like 4 [27]. Similarly, neovascularization was induced by transplantation of human HSCs to ischemic tissues in preclinical models (Fig. 3.2). These cells secreted exosomes with increased expression of the angiogenic factor sonic hedgehog (Shh), to offset age- and health-related angiogenic declines [36]. They also reduced infarct size and increased border zone capillary density compared with unmodified CD34+ cells [30].
4 Role of Extracellular Vesicles in Endothelial to Mesenchymal Transition
Recent studies demonstrated that the phenotypic transition of ECs into MSCs, called Endothelial to Mesenchymal Transition or EndMT, plays an important role in the pathogenesis of fibrotic disorders. During EndMT, resident ECs acquire a mesenchymal phenotype characterized by an increased ability to migrate and invade, thereby contributing to tissue remodeling and fibrosis [25, 31].
Hypoxia was found to efficiently induce human coronary artery endothelial cells (CAECs) to undergo EndMT, resulting in EndMT-derived FBs (Fig. 3.3). This process was mediated through a HIF-1α-dependent pathway, TGF/SMAD signaling pathways and DNA (cytosine-5)-methyltransferase 3A (DNMT3a)-mediated hypermethylation of Ras-Gap-like protein 1 (RASAL1) promoter and direct zinc-finger transcription factor Snail (SNAIL) induction [50]. Ultimately, this resulted in increased expression of extracellular matrix proteins such as collagen COLI and COLIII [51]. Focal myocardial fibrosis is also a structural hallmark of diabetic cardiomyopathy resulting from hyperglycemia-induced endothelial injury leading to EndMT associated with reduced expression of EC markers, such as CD31 and CD34, and increased expression of multiple mesenchymal markers, such as COLI and COLIV, and vimentin. Mir-200b reverted diabetes-associated EndMT by directly interacting with VEGF, SMAD2/3 and regulating p300-dependant histone acetylation and expression of for example extracellular matrix proteins [12].

Endothelial to Mesenchymal Transition, fibrosis and atherosclerosis. Hypoxia and diabetes induce secretion of EVs by FBs enriched in HIF-1α and TGF-β, respectively. They induce endothelial cells to undergo endothelial to mesenchymal transition (EndMT) through direct induction of SNAIL, a target of HIF-1α, and through TGF/SMAD signaling pathways and DNMT3a-mediated hypermethylation of RASAL1 promoter. EndMT results in the generation of EndMT-derived FBs with increased expression extracellular matrix proteins such as COLI, COLIII and FN. Hypoxia and diabetes cause endothelial dysfunction with increased secretion of leukocyte adhesion molecules leading to increased macrophage accumulation and oxidative stress. Macrophages secrete EVs enriched in CD36 and TNF-α which induce VSMC migration. Stimulated VSMCs secrete extracellular vesicles inducing matrix accumulation and calcification, resulting in complex atherosclerotic plaques. Collagen and fibronectin in extracellular vesicles secreted by EndMT-derived FBs exacerbate the growth of atherosclerotic plaques by inducing macrophage infiltration and making the atherosclerotic plaques more complex due to extracellular matrix deposition. Abbreviations: COL collagen, DNMT3a DNA (cytosine-5)-methyltransferase 3A, EC endothelial cell, EV extracellular vesicle, FB fibroblast, FN fibronectin, HIF-1α hypoxia inducible factor-1α, ICAM-1 intercellular adhesion molecule 1, RASAL1 Ras-Gap-like protein 1, ROS reactive oxygen species, SMC smooth muscle cells, SNAIL zinc-finger transcription factor Snail, TGF-β transforming growth factor, TNF tumor necrosis factor, VCAM-1 vascular cell adhesion molecule-1
Cardiac fibrosis does, however, not only result from a decrease in microvessels and oxygen supply but also from increased oxygen consumption by activated immune and inflammatory cells and fibroblasts leading to localized tissue hypoxia predominantly within inflammatory lesions (“inflammatory hypoxia”). Herein, HIF-1α, an oxygen-sensitive transcription factor that allows adaptation to hypoxia environments, plays an important role. Recent data suggest that the HIF-1 α-mediated metabolic shift and fibrosis is not only related to cardiovascular diseases but also to immune-related disorders [10] (Fig. 3.3). A common direct target of HIF-1α in hypoxia-induced EndMT is SNAIL [51] (Fig. 3.3).
5 Role of Extracellular Vesicles in Atherosclerosis
Hypoxia and diabetes are known to cause endothelial dysfunction, with increased secretion of leukocyte adhesion molecules leading to increased macrophage accumulation, reactive oxygen species (ROS) and oxidative stress, ultimately leading to atherosclerosis (Fig. 3.3). The circulation of atherosclerotic patients has been shown to contain more leukocyte-derived EVs promoting vascular SMC adhesion and migration, than those of healthy participants. In addition, macrophage-derived foam cells secrete EVs which promote increased levels of vascular SMC migration and adhesion, regulating the actin cytoskeleton and focal adhesion pathways to a greater extent than macrophage-derived EVs. Western blotting revealed that foam cell-derived EVs could also promote the phosphorylation of ERK and Akt in SMCs in a time-dependent manner. Foam cell-derived EVs could enter the SMCs and transfer integrins to the surface of these cells [33].
The calcification of SMC is also mediated by regulated exosome secretion. Comparative proteomics showed that vascular SMC-derived exosomes shared components with osteoblast-derived EVs including calcium-binding and extracellular matrix proteins (Fig. 3.3). Elevated extracellular calcium was found to induce sphingomyelin phosphodiesterase 3 and the secretion of calcifying exosomes from vascular SMCs in vitro. Chemical inhibition of sphingomyelin phosphodiesterase 3 prevented SMC calcification. In vivo, EVs containing exosomes were observed in vessels from chronic kidney disease patients on dialysis, and CD63 was located where there was calcification. Importantly, factors such as TNF-α and PDGF-BB were also found to increase exosome production, leading to increased calcification of SMC [22]. Comparison between exosomes from quiescent and activated SMCs showed evidence of 29 differentially expressed proteins which are involved in cytoskeleton organization, chaperones, cell adhesion, cell signaling, metabolic pathways, vesicle trafficking and extracellular matrix production [9] (Fig. 3.3). Foam cell formation and enhanced VSMC and extracellular matrix accumulation with calcification resulted in the generation of complex atherosclerotic plaques (Fig. 3.3). The extracellular matrix proteins COL and fibronectin, induced in EndMT, were associated with increased luminal endothelial expression of intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1). Mechanistically, the relation between EndMT and atherosclerosis also depends on loss of endothelial fibroblast growth factor receptor 1 (FGFR1) expression and activation of endothelial TGF-β signaling [8].
6 Research Limitations and Future Outlook
Facing a shortage of human data we focused our review to studies containing in vivo data obtained in relevant preclinical models. Even with these restrictions, it proved to be difficult to compare the outcome of selected in vivo studies because of differences in isolation procedures of EVs (for example ultracentrifugation, size exclusion chromatography, or immunoadsorption) which may yield EVs with different sizes (not necessarily purified exosomes), use of different sets of surface markers which may yield information about cell origin of EVs and differences in extent of evaluation of cargo (miRs, proteins or none of them). Therefore standardization of separation procedures and protocols to analyze biogenesis, composition and function are needed to improve our insight in the mechanistic role of EVs [34]. Novel isolation procedures may involve microfluidics devices for on-chip isolation and quantification of circulating micro-particles [21], and microchip-based RNA extraction, amplification and RT-PCR analysis [41].
A major shortcoming of many of the previous studies is that the full content of EVs was not all analyzed or that the selection of compounds to which functional roles were attributed was biased. Indeed, many of the reviewed studies focused on one of a few numbers of compounds without displaying information about the complete cargo or specifying the reason why these compounds were selected. Furthermore, data on the signaling pathways involved in the mechanisms of action of EVs are rare and again incomplete. Therefore, an unbiased systems biology approach is needed to generate and test hypotheses about the effect of context (e.g. spatial organization of endothelial cells in relation to other cell-types like macrophages or myofibroblasts and interaction through paracrine factors) dependent on state of the disease on the functional role of specific EVs. To this end Gray and colleagues [14] proposed cue-signal-response studies using partial least square regression (PLSR) methods that study how signals (exosome content or cargo) translate cues from the secreting cell (its gene/protein expression state) to elicit a specific response in the recipient cells.
In reviewing the role of EVs in cardiovascular diseases it became obvious that, although it is generally accepted that macrophages play a crucial role in the development of cardiac fibrosis and atherosclerosis, information about the role of their precursor cells, monocytes, and exosomes derived from monocytes is limited. Recently, we found that low mitochondrial cytochrome oxidase-1, a marker of mitochondrial dysfunction, in monocyte-derived exosomes predicted the risk of future cardiovascular events in the same way as low mitochondrial cytochrome oxidase-1 in monocytes. Therefore, the role monocyte-derived exosomes should be investigated further [17].
7 Conclusions
The studies reviewed present evidence that extracellular vesicles secreted by CMs, ECs, immune-system-related cells (macrophages), fibroblasts (FBs) and stem cells play an important role in the regulation of endothelial cardiomyocyte and endothelial function in relation to cardiovascular diseases. However, knowledge of the underlying signaling pathways is still too sparse to identify targets for EV-mediated treatment of these diseases.
References
de Almeida DC, Ferreira MRP, Franzen J, Weidner CI, Frobel J, Zenke M, Costa IG, Wagner W (2016) Epigenetic classification of human mesenchymal stromal cells. Stem Cell Rep 6:168–175
Anderson JD, Johansson HJ, Graham CS, Vesterlund M, Pham MT, Bramlett CS, Montgomery EN, Mellema MS, Bardini RL, Contreras Z, Hoon M, Bauer G, Fink KD, Fury B, Hendrix KJ, Chedin F, EL-Andaloussi S, Hwang B, Mulligan MS, Lehtiö J, Nolta JA (2016) Comprehensive proteomic analysis of mesenchymal stem cell exosomes reveals modulation of angiogenesis via nuclear factor-KappaB signaling: MSC exosomes induce angiogenesis via NFkB pathway. Stem Cells 34:601–613
Arslan F, Lai RC, Smeets MB, Akeroyd L, Choo A, Aguor ENE, Timmers L, van Rijen HV, Doevendans PA, Pasterkamp G, Lim SK, de Kleijn DP (2013) Mesenchymal stem cell-derived exosomes increase ATP levels, decrease oxidative stress and activate PI3K/Akt pathway to enhance myocardial viability and prevent adverse remodeling after myocardial ischemia/reperfusion injury. Stem Cell Res 10:301–312
Banerjee I, Fuseler JW, Price RL, Borg TK, Baudino TA (2007) Determination of cell types and numbers during cardiac development in the neonatal and adult rat and mouse. Am J Phys Heart Circ Phys 293:H1883–H1891
Bang C, Batkai S, Dangwal S, Gupta SK, Foinquinos A, Holzmann A, Just A, Remke J, Zimmer K, Zeug A, Ponimaskin E, Schmiedl A, Yin X, Mayr M, Halder R, Fischer A, Engelhardt S, Wei Y, Schober A, Fiedler J, Thum T (2014) Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J Clin Investig 124:2136–2146
Bian S, Zhang L, Duan L, Wang X, Min Y, Yu H (2014) Extracellular vesicles derived from human bone marrow mesenchymal stem cells promote angiogenesis in a rat myocardial infarction model. J Mol Med 92:387–397
Carè A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, Bang M-L, Segnalini P, Gu Y, Dalton ND, Elia L, Latronico MVG, Høydal M, Autore C, Russo MA, Dorn GW, Ellingsen O, Ruiz-Lozano P, Peterson KL, Croce CM, Peschle C, Condorelli G (2007) MicroRNA-133 controls cardiac hypertrophy. Nat Med 13:613–618
Chen P-Y, Qin L, Baeyens N, Li G, Afolabi T, Budatha M, Tellides G, Schwartz MA, Simons M (2015) Endothelial-to-mesenchymal transition drives atherosclerosis progression. J Clin Investig 125:4514–4528
Comelli L, Rocchiccioli S, Smirni S, Salvetti A, Signore G, Citti L, Trivella MG, Cecchettini A (2014) Characterization of secreted vesicles from vascular smooth muscle cells. Mol Biosyst 10:1146–1152
Deng W, Feng X, Li X, Wang D, Sun L (2016) Hypoxia-inducible factor 1 in autoimmune diseases. Cell Immunol 303:7–15
Evans MJ, Kaufman MH (1981) Establishment in culture of pluripotential cells from mouse embryos. Nature 292:154–156
Feng B, Cao Y, Chen S, Chu X, Chu Y, Chakrabarti S (2016) miR-200b mediates endothelial-to-mesenchymal transition in diabetic cardiomyopathy. Diabetes 65:768–779
Giricz Z, Varga ZV, Baranyai T, Sipos P, Pálóczi K, Kittel Á, Buzás EI, Ferdinandy P (2014) Cardioprotection by remote ischemic preconditioning of the rat heart is mediated by extracellular vesicles. J Mol Cell Cardiol 68:75–78
Gray WD, French KM, Ghosh-Choudhary S, Maxwell JT, Brown ME, Platt MO, Searles CD, Davis ME (2015) Identification of therapeutic covariant microRNA clusters in hypoxia-treated cardiac progenitor cell exosomes using systems biology. Circ Res 116:255–263
Gupta S, Knowlton AA (2007) HSP60 trafficking in adult cardiac myocytes: role of the exosomal pathway. Am J Phys Heart Circ Phys 292:H3052–H3056
Henning RJ (2011) Stem cells in cardiac repair. Futur Cardiol 7:99–117
Holvoet P, Vanhaverbeke M, Bloch K, Baatsen P, Sinnaeve P, Janssens S (2016) Low MT-CO1 in monocytes and microvesicles is associated with outcome in patients with coronary artery disease. J Am Heart Assoc 5:e004207
Hu G, Li Q, Niu X, Hu B, Liu J, Zhou S, Guo S, Lang H, Zhang C, Wang Y, Deng Z (2015) Exosomes secreted by human-induced pluripotent stem cell-derived mesenchymal stem cells attenuate limb ischemia by promoting angiogenesis in mice. Stem Cell Res Ther 6:10
Huber HJ, Holvoet P (2015) Exosomes: emerging roles in communication between blood cells and vascular tissues during atherosclerosis. Curr Opin Lipidol 26:412–419
Hulsmans M, Holvoet P (2013) MicroRNA-containing microvesicles regulating inflammation in association with atherosclerotic disease. Cardiovasc Res 100:7–18
Kanwar SS, Dunlay CJ, Simeone DM, Nagrath S (2014) Microfluidic device (ExoChip) for on-chip isolation, quantification and characterization of circulating exosomes. Lab Chip 14:1891
Kapustin AN, Chatrou MLL, Drozdov I, Zheng Y, Davidson SM, Soong D, Furmanik M, Sanchis P, De Rosales RTM, Alvarez-Hernandez D, Shroff R, Yin X, Muller K, Skepper JN, Mayr M, Reutelingsperger CP, Chester A, Bertazzo S, Schurgers LJ, Shanahan CM (2015) Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ Res 116:1312–1323
Kervadec A, Bellamy V, El Harane N, Arakélian L, Vanneaux V, Cacciapuoti I, Nemetalla H, Périer M-C, Toeg HD, Richart A, Lemitre M, Yin M, Loyer X, Larghero J, Hagège A, Ruel M, Boulanger CM, Silvestre J-S, Menasché P, Renault NKE (2016) Cardiovascular progenitor–derived extracellular vesicles recapitulate the beneficial effects of their parent cells in the treatment of chronic heart failure. J Heart Lung Transplant 35:795–807
Khan M, Nickoloff E, Abramova T, Johnson J, Verma SK, Krishnamurthy P, Mackie AR, Vaughan E, Garikipati VNS, Benedict C, Ramirez V, Lambers E, Ito A, Gao E, Misener S, Luongo T, Elrod J, Qin G, Houser SR, Koch WJ, Kishore R (2015) Embryonic stem cell-derived exosomes promote endogenous repair mechanisms and enhance cardiac function following myocardial infarction. Circ Res 117:52–64
Kovacic JC, Mercader N, Torres M, Boehm M, Fuster V (2012) Epithelial-to-mesenchymal and endothelial-to-mesenchymal transition: from cardiovascular development to disease. Circulation 125:1795–1808
Kuwabara Y, Ono K, Horie T, Nishi H, Nagao K, Kinoshita M, Watanabe S, Baba O, Kojima Y, Shizuta S, Imai M, Tamura T, Kita T, Kimura T (2011) Increased microRNA-1 and microRNA-133a levels in serum of patients with cardiovascular disease indicate myocardial damage. Circ Cardiovasc Genet 4:446–454
Liang X, Zhang L, Wang S, Han Q, Zhao RC (2016) Exosomes secreted by mesenchymal stem cells promote endothelial cell angiogenesis by transferring miR-125a. J Cell Sci 129:2182–2189
Lopatina T, Bruno S, Tetta C, Kalinina N, Porta M, Camussi G (2014) Platelet-derived growth factor regulates the secretion of extracellular vesicles by adipose mesenchymal stem cells and enhances their angiogenic potential. Cell Commun Signal 12:26
Lyu L, Wang H, Li B, Qin Q, Qi L, Nagarkatti M, Nagarkatti P, Janicki JS, Wang XL, Cui T (2015) A critical role of cardiac fibroblast-derived exosomes in activating renin angiotensin system in cardiomyocytes. J Mol Cell Cardiol 89:268–279
Mackie AR, Klyachko E, Thorne T, Schultz KM, Millay M, Ito A, Kamide CE, Liu T, Gupta R, Sahoo S, Misener S, Kishore R, Losordo DW (2012) Sonic hedgehog-modified human CD34+ cells preserve cardiac function after acute myocardial infarction. Circ Res 111:312–321
Markwald RR, Fitzharris TP, Manasek FJ (1977) Structural development of endocardial cushions. Am J Anat 148:85–119
Melo SA, Sugimoto H, O’Connell JT, Kato N, Villanueva A, Vidal A, Qiu L, Vitkin E, Perelman LT, Melo CA, Lucci A, Ivan C, Calin GA, Kalluri R (2014) Cancer exosomes perform cell-independent microRNA biogenesis and promote tumorigenesis. Cancer Cell 26:707–721
Niu C, Wang X, Zhao M, Cai T, Liu P, Li J, Willard B, Zu L, Zhou E, Li Y, Pan B, Yang F, Zheng L (2016) Macrophage foam cell–derived extracellular vesicles promote vascular smooth muscle cell migration and adhesion. J Am Heart Assoc 5:e004099
Peterson MF, Otoc N, Sethi JK, Gupta A, Antes TJ (2015) Integrated systems for exosome investigation. Methods 87:31–45
Safari S, Malekvandfard F, Babashah S, Alizadehasl A, Sadeghizadeh M, Motavaf M (2016) Mesenchymal stem cell-derived exosomes: a novel potential therapeutic avenue for cardiac regeneration. Cell Mol Biol (Noisy-le-grand) 62:66–73
Sahoo S, Klychko E, Thorne T, Misener S, Schultz KM, Millay M, Ito A, Liu T, Kamide C, Agrawal H, Perlman H, Qin G, Kishore R, Losordo DW (2011) Exosomes from human CD34+ stem cells mediate their proangiogenic paracrine activity. Circ Res 109:724–728
Shabbir A, Cox A, Rodriguez-Menocal L, Salgado M, Badiavas EV (2015) Mesenchymal stem cell exosomes induce proliferation and migration of normal and chronic wound fibroblasts, and enhance angiogenesis in vitro. Stem Cells Dev 24:1635–1647
Shamblott MJ, Axelman J, Wang S, Bugg EM, Littlefield JW, Donovan PJ, Blumenthal PD, Huggins GR, Gearhart JD (1998) Derivation of pluripotent stem cells from cultured human primordial germ cells. Proc Natl Acad Sci U S A 95:13726–13731
Singh A, Singh A, Sen D (2016) Mesenchymal stem cells in cardiac regeneration: a detailed progress report of the last 6 years (2010–2015). Stem Cell Res Ther 7(1):82
Squadrito ML, Baer C, Burdet F, Maderna C, Gilfillan GD, Lyle R, Ibberson M, De Palma M (2014) Endogenous RNAs modulate microRNA sorting to exosomes and transfer to acceptor cells. Cell Rep 8:1432–1446
Takahashi K, Yan IK, Kim C, Kim J, Patel T (2014) Analysis of extracellular RNA by digital PCR. Front Radiat Ther Oncol 4:129
Théry C, Ostrowski M, Segura E (2009) Membrane vesicles as conveyors of immune responses. Nat Rev Immunol 9:581–593
Trial J, Entman ML, Cieslik KA (2016) Mesenchymal stem cell-derived inflammatory fibroblasts mediate interstitial fibrosis in the aging heart. J Mol Cell Cardiol 91:28–34
Villarroya-Beltri C, Baixauli F, Gutiérrez-Vázquez C, Sánchez-Madrid F, Mittelbrunn M (2014) Sorting it out: regulation of exosome loading. Semin Cancer Biol 28:3–13
Vrijsen KR, Maring JA, Chamuleau SAJ, Verhage V, Mol EA, Deddens JC, Metz CHG, Lodder K, van Eeuwijk ECM, van Dommelen SM, Doevendans PA, Smits AM, Goumans M-J, Sluijter JPG (2016) Exosomes from cardiomyocyte progenitor cells and mesenchymal stem cells stimulate angiogenesis via EMMPRIN. Adv Healthc Mater 5:2555–2565
Waldenström A, Gennebäck N, Hellman U, Ronquist G (2012) Cardiomyocyte microvesicles contain DNA/RNA and convey biological messages to target cells. PLoS One 7:e34653
Wang X, Gu H, Huang W, Peng J, Li Y, Yang L, Qin D, Essandoh K, Wang Y, Peng T, Fan G-C (2016) Hsp20-mediated activation of exosome biogenesis in cardiomyocytes improves cardiac function and angiogenesis in diabetic mice. Diabetes 65:3111–3128
Wang X, Huang W, Liu G, Cai W, Millard RW, Wang Y, Chang J, Peng T, Fan G-C (2014) Cardiomyocytes mediate anti-angiogenesis in type 2 diabetic rats through the exosomal transfer of miR-320 into endothelial cells. J Mol Cell Cardiol 74:139–150
Wang Y, Zhang L, Li Y, Chen L, Wang X, Guo W, Zhang X, Qin G, He S, Zimmerman A, Liu Y, Kim I, Weintraub NL, Tang Y (2015) Exosomes/microvesicles from induced pluripotent stem cells deliver cardioprotective miRNAs and prevent cardiomyocyte apoptosis in the ischemic myocardium. Int J Cardiol 192:61–69
Xu X, Tan X, Hulshoff MS, Wilhelmi T, Zeisberg M, Zeisberg EM (2016) Hypoxia-induced endothelial-mesenchymal transition is associated with RASAL1 promoter hypermethylation in human coronary endothelial cells. FEBS Lett 590:1222–1233
Xu X, Tan X, Tampe B, Sanchez E, Zeisberg M, Zeisberg EM (2015) Snail is a direct target of hypoxia-inducible factor 1α (HIF1α) in hypoxia-induced endothelial to mesenchymal transition of human coronary endothelial cells. J Biol Chem 290:16653–16664
Yu B, Kim HW, Gong M, Wang J, Millard RW, Wang Y, Ashraf M, Xu M (2015) Exosomes secreted from GATA-4 overexpressing mesenchymal stem cells serve as a reservoir of anti-apoptotic microRNAs for cardioprotection. Int J Cardiol 182:349–360
Yu X, Deng L, Wang D, Li N, Chen X, Cheng X, Yuan J, Gao X, Liao M, Wang M, Liao Y (2012) Mechanism of TNF-α autocrine effects in hypoxic cardiomyocytes: initiated by hypoxia inducible factor 1α, presented by exosomes. J Mol Cell Cardiol 53:848–857
Zhang X, Wang X, Zhu H, Kranias EG, Tang Y, Peng T, Chang J, Fan G-C (2012) Hsp20 functions as a novel cardiokine in promoting angiogenesis via activation of VEGFR2. PLoS One 7:e32765
Zhang Z, Yang J, Yan W, Li Y, Shen Z, Asahara T (2016) Pretreatment of cardiac stem cells with exosomes derived from mesenchymal stem cells enhances myocardial repair. J Am Heart Assoc 5:e002856
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Nature Singapore Pte Ltd.
About this chapter
Cite this chapter
Vanhaverbeke, M., Gal, D., Holvoet, P. (2017). Functional Role of Cardiovascular Exosomes in Myocardial Injury and Atherosclerosis. In: Xiao, J., Cretoiu, S. (eds) Exosomes in Cardiovascular Diseases. Advances in Experimental Medicine and Biology, vol 998. Springer, Singapore. https://doi.org/10.1007/978-981-10-4397-0_3
Download citation
DOI: https://doi.org/10.1007/978-981-10-4397-0_3
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-4396-3
Online ISBN: 978-981-10-4397-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)