
Abstract
The transient receptor potential canonical (TRPC) family consists of seven isoforms that have been proposed as molecular components essential for nonselective calcium (Ca2+) entry. TRPC1 proteins are expressed in the sarcolemma of skeletal muscles, and TRPC1 is important and necessary for stretch-activated and store-operated channels for Ca2+ entry. Studies have established the essential role of TRPC1 in maintaining Ca2+ homeostasis, regulating myoblast migration and differentiation, regenerating muscle, and contributing to the pathogenesis of muscular dystrophy. This chapter summarizes the evidence for the regulation of TRPC1 to fulfill specific physiological functions in skeletal muscles.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Transient receptor potential canonical type 1
- Calcium
- Mechanosensation
- Myogenesis
- Muscle regeneration
- Duchenne muscular dystrophy
10.1 Introduction
Skeletal muscles exhibit high plasticity in response to mechanical stimuli. Calcium (Ca2+) signaling plays a crucial role in translating mechanical signals into intracellular signaling cascades that control transcription and translation of phenotype-specific genes that affect muscle growth and development [1]. Of particular interest, the transient receptor potential canonical (TRPC) proteins are a family of Ca2+-permeable nonselective cation channels that participate in mechanotransduction in different cell types [2]. TRPC1 is widely expressed in skeletal muscle [3], myoblasts, and myotubes [4, 5]. Even though the physiological function of TRPC1 channels is not yet fully understood, they may contribute to or form stretch-activated (mechanosensitive) channels (SACs) as well as store-operated channels (SOCs) for Ca2+ entry [3, 6, 7]. In addition to interacting with other members of the TRPC family to form heteromeric channels [8], TRPC1 also associates with a range of cytoskeletal and scaffolding proteins to form a macromolecular complex based at the costamere [4, 9]. These associations are critical for the localization, activation, and function of the channel and, subsequently, TRPC1-mediated Ca2+ influx. Moreover, TRPC1 regulates muscle development and regeneration [10]. In this chapter, we will review the biological functions of TRPC1 and its regulatory roles in skeletal muscle physiopathological processes.
10.2 Involvement of TRPC1 in Skeletal Myogenesis
Skeletal muscle stem cells, or satellite cells, were first identified in 1961 and are located underneath the basal lamina between the myofiber sarcolemmas [11]. Satellite cells are the predominant source of myoblasts for replenishing muscle fibers upon muscle injury and regrowth during postnatal life [12]. Satellite cells exist in heterogeneous populations that are able to differentiate and give rise to new myonuclei, as well as to undergo self-renewal to repopulate themselves. In normal, uninjured muscle, satellite cells remain in a quiescent state and are mitotically inactive, expressing the transcription factor Pax7. Upon mechanical or biochemical stimulation, a majority of the satellite cells become activated, express MyoD, and then undergo active proliferation to increase their population. After several rounds of cell division, the satellite cell progeny, which are now called myoblasts, exit from the cell cycle and undergo myogenic differentiation to become post-mitotic myonuclei. The newly differentiated myocytes then fuse with the nearby existing myofibers to form new muscle fibers [13, 14]. Unlike the majority of satellite cells (80%), which commit to myogenic differentiation, a subpopulation of satellite cells (20%) undergo asymmetric cell division, downregulate MyoD expression to replenish the satellite cell niche, and eventually become quiescent again [15, 16]. Adult postnatal myogenesis, sequentially comprising satellite cell activation, migration, and differentiation, as well as the self-renewal processes, is a tightly coordinated biological event that ensures successful skeletal muscle regeneration in response to injury. Extensive research has focused on the signaling cascades that regulate different stages of myogenesis, among which Ca2+ signaling has been a hotspot. This section presents an overview of the involvement of TRPC1-mediated Ca2+ entry in the myogenic program.
10.2.1 Activation of Satellite Cells
In intact muscle, satellite cells are mitotically inactive, expressing Pax7 but not MyoD or myogenin [17]. In the absence of muscle injury, satellite cells are tightly associated with the juxtaposed muscle fibers. In response to muscle injury, biochemical factors released from the damaged tissue trigger the activation of satellite cells, causing the satellite cells to exit their quiescent state [18]. The satellite cells then begin to express the transcription factor MyoD, which is essential for progression from quiescence into S phase of the cell cycle, and thereby become proliferating myoblasts [19]. One soluble factor, fibroblast growth factor 2 (FGF2), is a strong activator that binds to FGF receptors on the satellite cells and triggers a downstream signaling pathway that ultimately leads to active proliferation [20]. Although the FGF-associated downstream signaling events are not fully understood, Ca2+ signaling mediated by TRPC channels has a role in regulating satellite cell activation. FGF2 induces an increase in [Ca2+]i in satellite cells, and the influx of Ca2+ activates nuclear translocation of the members of the nuclear factor of activated T cell (NFAT) family, namely, NFATc2 and NFATc3. NFATc2-null mice display impaired muscle regeneration, while NFATc3 −/− mice exhibit altered primary myogenesis [21, 22] resulting in a more than twofold increase in MyoD immunopositive cells and a concomitant increase in satellite cell proliferation [23]. FGF2-induced intracellular Ca2+ influx in satellite cells was suppressed by SKF 96365, a TRPC channel blocker. Furthermore, SKF 96365 attenuates the nuclear translocation of NFATc3 in satellite cells and the number of MyoD immunoreactive cells [23]. These data suggest that the TRPC channel plays a central role in FGF-2-mediated satellite cell activation. SKF 96365 is, however, a nonselective TRPC channel blocker; it is possible that other TRPC members are involved. We previously reported physical interaction of TRPC1-TRPC3 in C2C12 myotubes, but the absence of TRPC3 in myoblasts and in ex vivo satellite cells makes it unlikely to be directly related in satellite cell activation [23, 24]. Expression of TRPC4 or TRPC6 channel has not been demonstrated in satellite cells. TRPC1 channel is strongly expressed in satellite cells on freshly isolated adult FDB myofibers [23] raising the possibility that TRPC1 channel for responding to the effects of FGF during muscle regeneration.
In embryonic rat neural stem cells, the contribution of TRPC1 in stem cell activation has clearly been determined [25]. TRPC1 and FGF receptor 1 (FGFR-1) are co-expressed in a subpopulation of actively proliferating neuroepithelial cells in the lateral ventricle of the developing rat telencephalon, one of the known domains of neural stem cells. Co-immunoprecipitation experiments show that TRPC1 and FGFR-1 interact either directly or indirectly in a complex present in the telencephalic membrane fraction, where the active functional forms of FGF receptors are located. Basic FGF induces Ca2+ entry into embryonic neural stem cells and promotes stem cell proliferation. The influx of Ca2+ and the associated cell proliferation are antagonized by SKF 96365 and, more importantly, by antisense oligos specifically targeting TRPC1, suggesting that FGF-triggered stem cell proliferation occurs via TRPC1-mediated Ca2+ entry. Thus it is possible, and perhaps likely, that the activation of muscle satellite cells in response to FGF2-induced Ca2+ entry also occurs through TRPC1 channel.
10.2.2 Migration of Satellite Cells
During early stage of myogenesis, satellite cells activate from the quiescent state and commit to active proliferation to generate myoblasts that express MyoD. Although skeletal muscle damage usually occurs in a localized region, satellite cell activation is not restricted to only the site of muscle injury. In fact, satellite cells are capable of migrating not only from adjacent non-injured regions to damage sites on the same muscle fiber but also across the basal lamina between myofibers [26, 27]. Therefore, activated satellite cells, also termed MyoD-positive myoblasts, possess migratory features. In order to migrate, myoblasts must reorganize their contractile and cytoskeletal components to attain structural asymmetry or, in other words, polarity. Moreover, directional cell movement depends on tight spatiotemporal coordination of protrusion and retraction in different parts of the cell as well as the presence of mechanosensitive machinery, which is required to sense mechanical stimuli in the nearby environment. Cells experiencing different substrate rigidities exhibit fluctuations in [Ca2+]i, which is regulated by mechanosensitive channels, and Ca2+ gradients confer the cellular polarity necessary for the directional migration process [28, 29]. Repetitive Ca2+ transients, particularly mediated via mechanosensitive TRPC1 channels, are involved in myoblast migration.
TRPC1 gene silencing by gene-specific siRNA in the transformed renal epithelial cell line MDCK alters the morphology and the polarity of MDCK cells in a dose-dependent manner [30]. While mock-transfected or TRPC1-overexpressing MDCK cells exhibit clear polarity in the form of persistent directional protrusion, cells transfected with siRNAs targeting TRPC1 show a random orientation and a significant reduction in their distance displaced from original position. TRPC1-knockdown endothelial progenitor cells [31] and synovial fibroblasts from TRPC1 −/− mice [32] give similar results, suggesting that TRPC1-associated Ca2+ transients have a regulatory role in cell migration across cell types.
TRPC1 complexed with STIM1 and Orai1 constitutes the core component of store-operated Ca2+ entry (SOCE), the Ca2+ entry mechanism that is activated by depletion of Ca2+ stores [33]. It has been demonstrated that SOCE-mediated Ca2+ signaling regulates cell polarity [28]. A study using multiple fluorescence labeling and siRNA strategy have demonstrated that TRPC1, but not Orai1, displays a high degree of co-localization with STIM1 and is responsible for regulating cell polarity. The crucial role of TRPC1 in regulating myoblast migration was revealed in a study showing that stimulation of C2C12 myoblasts with IGF-1 triggers Ca2+ influx, activates calpain, and accelerates migration [34]. The presence of a calpain inhibitor significantly impairs the effect of IGF-1 on myoblast migration. GsMTx-4, a specific inhibitor of SACs, completely abolishes IGF-1-induced Ca2+ entry. Similarly, when myoblasts are treated with TRPC1-specific siRNA and shRNA, the effect of IGF-1 on calpain activity is completely lost and the percentage of migrating cells diminishes significantly, confirming that Ca2+ influx through TRPC1 is responsible for calpain activity and myoblast migration [34].
In addition, calpain-dependent myristoylated alanine-rich C kinase substrate (MARCKS), an actin-binding protein expressed at the focal adhesion sites of myoblasts, is important in regulating cell adhesion and migration processes [35]. Treatment with calpain blocks MARCKS accumulation resulting in enhanced myoblast migration. Conversely, inhibition of calpain activity with siRNA leads to MARCKS accumulation, accompanied with retardation of myoblast migration [36]. TRPC1-knockdown myoblasts exhibit accumulated MARCKS expression in the cytosol, indicating that IGF-1 promotes myoblast migration by regulating calpain activity via TRPC1-mediated Ca2+ influx and possibly through inhibition of MARCKS accumulation [34].
10.2.3 Differentiation of Satellite Cells
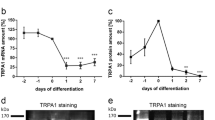
TRPC1 has been implicated in satellite cell differentiation because of its upregulation during C2C12 myogenesis [24, 37]. During C2C12 differentiation, proliferative MyoD-expressing myoblasts switch on myogenin, a basic helix-loop-helix transcription factor that regulates myogenic differentiation through transactivation of muscle-specific genes such as myosin heavy chains (MHCs). Soon after expressing myogenin, the myoblasts exit from the cell cycle and commit to differentiating into myocytes. The myocytes become elongated, align with adjacent myocytes, and fuse to form multinucleated myotubes. We previously reported the dynamic expression of TRPC1 in C2C12 cells in which TRPC1 is initially expressed in all proliferating myoblasts but is soon restricted to multinucleated myotubes upon terminal differentiation and is totally absent from the undifferentiated mononucleated myocytes, suggesting its differential roles in myogenesis [24]. Indeed, a significant increase in Ca2+ influx is observed 24 h post-differentiation in both C2C12 and primary myoblasts [38]; however, this Ca2+ influx is abolished in TRPC1-knockdown C2C12 and in primary myoblasts derived from TRPC1 −/− mice. Unlike TRPC1 +/+ myoblasts, TRPC1 −/− myoblasts do not line up with each other [38]. In addition, other studies show that myocyte fusion in TRPC1-knockdown cells occurs much more slowly than in control cells [34, 37]. A possible explanation for these findings is that loss of TRPC1-mediated Ca2+ influx impairs the fusion process. In vitro myogenic cell fusion can be characterized as undergoing two phases: the first phase is the fusion between individual myoblasts to generate nascent myotubes, followed by the second phase, which is the incorporation of the newly differentiated myocytes into nascent myotubes [39]. In TRPC1-knockdown myoblasts, siTRPC1 treatment does not alter the fusion index but significantly reduces the number of nuclei per myotube, implying that the second fusion phase requires the regulation of TRPC1 [40]. Another possibility is a defect in cell migration or downregulation of MyoD and myogenin. The transcriptional activity of MyoD is known to be inhibited by a myogenic repressor prohibitin 2 [41]. It has been demonstrated that phosphorylated Akt interacts specifically with prohibitin 2 and partially decreases the binding of prohibitin 2 to MyoD, thereby increasing MyoD transcriptional activity and subsequently increasing expression of myogenin and the MHC [38, 41]. In addition the phosphoinositide 3-kinase (PI3K)/Akt pathway plays a major role in the transcriptional activity of MyoD in muscle hypertrophy and regeneration. Inhibition of TRPC1 expression attenuates subsequent activation of Akt and the p85 unit of PI3K during early myogenic differentiation in myoblasts [38]. TRPC1 −/− mice present delays in skeletal muscle regeneration in response to cardiotoxin-induced injury, with smaller fiber size and reduced myofibrillar protein content. Therefore, impairment of the Akt/PI3K pathway may account for the poor regenerative capacity in TRPC1 −/− muscles.
TRPC1- and TRPC4-mediated SOCE are necessary for the expression of a crucial player in myogenesis, myocyte enhancer factor 2 (MEF2), in human postnatal myoblasts [40]. Overexpression of STIM1 with TRPC1 or TRPC4 increases SOCE and enhances myoblast fusion, leading to hypertrophic myotubes. Nevertheless, in cells that are deficient in TRPC1 or TRPC4 expression, overexpression of STIM1 or Orai1 to normalize Ca2+ concentration for SOCE is insufficient to restore the normal size of myotubes unless TRPC channels are reexpressed. This evidence further confirms the importance of TRPC1-mediated SOCE in myocyte fusion.
In yeast two-hybrid screening, TRPC1 is an interacting partner of the a-isoform of the inhibitor of the MyoD family (I-mfa) [42]. I-mfa directly represses myogenic regulator factors such as MyoD and myogenin. I-mfa binds MyoD and myogenin, allowing MyoD and myogenin to retain their subcellular localization in the cytosol, and inhibits the transactivation activity of the MyoD family, thus repressing myogenesis [43]. TRPC1 interacts with I-mfa both in vitro and in vivo [42]. Ectopic expression of I-mfa in CHO-K1 cells suppresses endogenous TRPC1-mediated SOCE. Conversely, inactivating endogenous I-mfa expression through RNAi enhances SOCE in A431 human epidermoid carcinoma cells. Although TRPC1 is present in A431 cells and the protein level appears unaltered after I-mfa knockdown, thapsigargin (TG)-induced SOCE is dramatically enhanced in I-mfa-knockdown cells, and such enhancement can be reduced substantially in the presence of TRPC1-neutralizing antibody. This suggests that I-mfa inhibits TRPC1-mediated SOCE not by modulating the transcription or translation of TRPC1 but instead most likely through physical interaction. Importantly, myogenin transfection reduces the association between TRPC1 and I-mfa in a concentration-dependent manner, suggesting that I-mfa may not bind to myogenin and TRPC1 simultaneously. Instead, myogenin appears to compete with TRPC1 for I-mfa. As such, we believe that during early differentiation, TRPC1 may physically interact with I-mfa, compete with myogenin for binding to I-mfa, and thus release myogenin for the subsequent myogenic differentiation program. We share this view with other investigators [38].
10.2.4 Regulation of Quiescent Satellite Cells
In an in vitro mass myoblast culture, whether it is a primary myoblast culture or a well-established myogenic cell line (e.g., C2C12), myoblasts are highly proliferative upon serum stimulation and express MyoD just like activated satellite cells in vivo [44, 45]. Nevertheless, the generation of differentiation-resistant, mononucleated cells that may be analogous to quiescent satellite cells in vivo always accompanies the formation of multinucleated myotubes [44]. This undifferentiated cell population, the reserve cells, is characterized by the absence of MyoD expression. When isolated, reserve cells retain the capacity to reexpress MyoD and reinitiate the myogenic program [44]. Therefore, generation of reserve cells is considered essential to maintaining a consistent, self-sustained population in the face of repeated injury [46].
We have previously revealed that, although TRPC1 expression in C2C12 increases upon differentiation, the channel is present only in multinucleated myotubes. The mononucleated cells that represent the majority (if not all) of the reserve cells in a differentiated C2C12 culture do not express the TRPC1 channel [24], raising the possibility that downregulation of TRPC1 may play a role in maintaining the reserve-cell population. In fact, as discussed previously, persistent expression of MyoD requires TRPC1 expression. Downregulation of TRPC1 may be required to suppress expression of MyoD in the reserve-cell population and prevent the cells from progressing into S phase, instead arresting in G0 phase. A cyclin-dependent kinase inhibitor, p27, is expressed at a high level in the quiescent reserve cells, whereas its expression decreases following mitogen stimulation [47, 48]. In a thyroid cancer cell model, p27 expression is significantly elevated in TRPC1-knockdown cells [49], raising the question of whether TRPC1 downregulation in reserve cells would also enhance p27 and downregulate MyoD. The underlying mechanism that governs the selection of either maintenance or downregulation of TRPC1 expression is entirely unknown, but asymmetric cell division that regulates differential cell fates in satellite cells may be involved [50]. Since TRPC1 channel activity and expression is responsive to growth factors such as IGF-1 and FGF2, the presence or absence of receptors for growth factors or even of the TRPC1 channel itself, as a result of asymmetric cell division, may alter the ultimate daughter cell fates, determining whether a cell fuses to myotubes or remains an undifferentiated reserve cell. Another possibility is that the absence of TRPC1 channels releases l-mfa that may then bind to MyoD and prevent transcriptional activity. The role of TRPC1-mediated Ca2+ signaling in potentially regulating cell fate, and its possible involvement in asymmetric cell division, remained to be explored.
10.3 Involvement of TRPC1 in Muscle Regeneration
Skeletal muscle demonstrates high adaptability to altered mechanical loading and also possesses a remarkable capacity for regeneration [51, 52]. Muscle disuse (or unloading) leads to muscle atrophy, fiber-type transition, and compromised contractile function, as seen in astronauts and patients on bedrest. Subjecting disused muscles to mechanical reloading can restore muscle mass, cross-sectional area, fiber-type transition, and contractile function. However, disused muscles are also prone to reloading-induced muscle injury, and improper rehabilitation may lead to further functional deterioration and impaired muscle regrowth [53].
Skeletal muscle regeneration is a coordinated process that involves an early phase of tissue inflammation to scavenge necrotic cell debris and establish a favorable environment for myogenic cell activities and a late phase of myofiber regeneration. Infiltrating inflammatory cells triggers the recruitment and activation of myogenic cells [54]. Activated myogenic cells proliferate and migrate to the injured site in order to repair injured myofibers. During the late stage, anti-inflammatory responses resolve inflammation and confer a favorable microenvironment for myoblasts to commit to myogenic differentiation and subsequent fusion [54, 55]. Regeneration is considered completed upon resolution of inflammation, absence of central nucleated myofibers at the injured area, and recovery of contractile functions. This section focuses on the role of TRPC1 in muscle regeneration following disuse and injury.
10.3.1 Muscle Regeneration Following Reduced Mechanical Load
Skeletal muscle is well known for its mechanosensitive responsiveness; sensors recognize mechanical stimuli and translate the inputs into biochemical signals that undergo complicated molecular cascades and ultimately elicit an appropriate cellular response. The mechanosensitive nature of skeletal muscle is best demonstrated by the response during gravitational unloading and reloading. In response to gravitational unloading, postural muscles, such as the soleus that is composed predominantly of slow-twitch fibers, display the most prominent phenotypic changes, including an increase in fast MHC fibers due to slow-to-fast fiber-type shifting, accompanied by a reduction in muscle mass and fiber cross-sectional area due to atrophy [56]. Such remodeling occurs only very mildly in fast-twitch muscles [57].
TRPC1 channel-mediated Ca2+ entry is mechanosensitive [28, 58–60]. Involvement of TRPC1 signaling in unloading-induced atrophy is evident in a C2C12 model exposed to simulated microgravity [59]. This condition retards myoblast proliferation and inhibits terminal differentiation concomitant with repressed TRPC1 expression. The TRPC blocker SKF 96365 retards proliferation and retains myoblast at the G2/M phase of the cell cycle in a dose-dependent manner, similar to microgravity conditions. Yet the role of TRPC1 in mechanical unloading was unclear until our group reported dynamic TRPC1 expression in the soleus muscle when mice were subjected to hind limb unloading [61]. Despite a rather stable expression of TRPC1 mRNA transcript, the TRPC1 protein level significantly decreases after 14 days of hind limb unloading; we confirmed the reduction by immunohistochemistry, in which TRPC1 immunoreactivity was significantly diminished in the sarcolemma of the soleus muscle. These in vivo data echo the in vitro observation [59] that C2C12 cells under microgravity downregulate TRPC1, indicating that TRPC1 expression in skeletal muscle is mechanosensitive. This notion is further supported by the findings that TRPC1 transcripts in the soleus muscle were seven times higher than those in fast-twitch EDL muscle (unpublished data), corroborated by immunohistochemical staining. In contrast, the TRPC3 channel, which is the predominant TRPC isoform in EDL muscle [62], displays rather stable expression during hind limb unloading [61]. The preferential expression of TRPC1 in slow muscles rather than in fast muscles fits well with the response of the channel expression upon mechanical unloading.
Given the evidence that TRPC1 is mechanosensitive, we initially expected that when the unloaded mouse was allowed to resume normal weight bearing, TRPC1 expression would immediately revert. Surprisingly, TRPC1 expression decreases further in the first few days of reloading [61]. Restoration of TRPC1 expression commences at around 7 days of reloading; however, we are unsure by which day the expression of TRPC1 reaches its lowest point, because we have not examined the time points between day 3 and day 7 of reloading. The expression does not fully return to control level until day 28 of reloading. The time window of TRPC1 protein restoration correlates strongly with the recovery of muscle mass and fiber cross-sectional area and the fiber-type shifting that unloading alters [63]. This correlation prompted us to hypothesize that TRPC1 expression is related to reloading-induced muscle regrowth. The hypothesis led us to further experiments on in vivo TRPC1 silencing in soleus muscles immediately following reloading [63]. By employing focal electroporation of siRNA, we achieved specific downregulation of TRPC1 only within the target muscle, while the TRPC1 channel expression in other regions and even in adjacent muscles remained unaltered. Inhibition of TRPC1 expression lasted for at least 7 days post-electroporation, which allowed sufficient time to observe the effects of TRPC1 suppression in reloaded muscle regrowth. Our hypothesis was confirmed by the observation that silencing of TRPC1 in soleus muscle impairs the overall muscle regrowth process. We demonstrate significant reductions in muscle size, mass, and fiber cross-sectional area in TRPC1-knockdown soleus muscles, compared to control oligo-electroporated reloaded soleus muscles [63]. Moreover, TRPC1 silencing blocks the typical fast-to-slow fiber remodeling in the reloaded soleus, thus establishing the physiological relevance of TRPC1 in reloading-induced muscle regrowth. We subsequently correlated the expression of TRPC1 during the unloading-reloading process with expression of calcineurin (CaN) and NFATc1, which are well known for their roles in fiber-type conversion.
A temporal increase in CaN expression and NFAT nuclear translocation precedes the onset of TRPC1 upregulation during the reloading process [63]. CaN inhibitors (FK506 and cyclosporine A) suppress CaN activity and block the recovery of the percentage of MHC I fiber and fiber cross-sectional area in reloaded mice, which is consistent with results from previous reports [64–66]. We considered the possibility that CaN/NFAT activity modulates TRPC1 expression in reloading muscle. Inhibition of CaN activity results in repression of TRPC1 compared with the vehicle control. This novel finding helps establish the possible role of CaN/NFAT in regulating TRPC1 expression in reloaded muscle, suggesting that the TRPC1 channel participates in CaN/NFAT-mediated muscle regrowth. SOCE regulates gene expression via TRPC1 when Ca2+ entry triggers the activation of CaN followed by dephosphorylation of NFAT, NFAT nuclear translocation, and transactivation of gene expression [67–70]. The possibility that TRPC1 expression being regulated by CaN/NFAT may represent a novel pathway deserves further investigation. The detailed pathway of CaN/NFAT leading to TRPC1-mediated SOCE and the involvement of SOCE in fiber-type conversion have yet to be explored, though it is possible that the PI3K/Akt pathway could be involved in the muscle mass and fiber cross-sectional area recovery mediated by TRPC1. Nevertheless, the research thus far strongly indicates that TRPC1 is a mechanosensitive channel and that Ca2+ entry via TRPC1 plays an important role in muscle remodeling during mechanical reloading [38].
10.3.2 Muscle Regeneration Following Injury
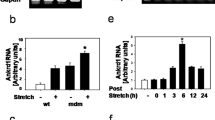
Under normal circumstances, muscle repair after injury follows a finely coordinated process that involves inflammation, removal of necrotic cellular debris, and regeneration. Intracellular signaling cascades that control transcription and protein translation of phenotype-specific genes regulate this process. Reloading from disuse induces muscle damage similar to that as in stretch-induced or eccentric muscle contractions [71]. Eccentric contraction-induced muscle damage is characterized by immediate muscle weakness with prolonged stiffness. Eccentric contractions cause leakage of soluble cytosolic proteins from the muscle into the plasma due to increased membrane permeability [72]. They also trigger a rise in resting [Ca2+]i, possibly caused by SACs [73, 74]. We previously demonstrated that the eccentric contraction-induced proteolysis of cytoskeletal proteins coincides with a reduction in muscle force, while the absence of extracellular Ca2+ or treatment with a calpain inhibitor attenuates the damage of cytoskeletal proteins and improves muscle tetanic force [75]. These data indicate that muscle cytoskeletal damage following eccentric contraction is due to an increase in Ca2+ influx that activates calpain and its associated proteolytic activity. In addition, the loss of immunogenicity of cytoskeletal proteins was reduced in muscles treated with SAC blocker streptomycin as well as in TRPC1 knockout muscles [7], suggesting that the channel responsible for Ca2+ entry after eccentric contraction is possibly the SAC-TRPC1 channel. All these evidence support the hypothesis that TRPC1 functions as SAC to increase Ca2+ influx and activate calpain-mediated proteolysis of cytoskeletal proteins.
Apart from the loss of cytoskeletal proteins by eccentric contractions, inflammation, such as infiltration of neutrophils into injured fibers, contributes a significant proportion of muscle damage during eccentric contractions or reloading following disuse atrophy [76]. Inflammation is a critical biological event and a prerequisite to muscle regeneration; blocking the inflammatory response results in poor or suboptimal muscle regeneration [77, 78]. Proinflammatory cytokines provide an important stimulus in the early phase of reloading that modulates satellite cell activation and the eventual recovery of muscle mass. There are two classes of macrophages that are implicated in muscle regeneration, M1 and M2 macrophages. The invasion of phagocytic M1 macrophages (in addition to neutrophils) regulates satellite cell activation, proliferation, and migration, while non-phagocytic M2 macrophages in the second phase of infiltration may modulate muscle regrowth during regeneration [79]. Tumor necrosis factor-α (TNF-α), which is primarily synthesized by macrophages, has been described as a “master regulator” of inflammatory cytokine production. TNF-α receptor −/− mice exhibit a reduction in MyoD expression and a slower recovery of muscle force [80], as well as suppression of transactivation of early myogenic markers and a consequent decrease in expression of myosin heavy chain [81]. TNF-α is important in promoting satellite cell proliferation and avoiding premature myogenic differentiation following injury [82]. In spite of the critical role of inflammation in muscle regeneration, prolonged inflammatory responses (e.g., responses to chronic injury) impair muscle regeneration by delaying myogenesis [54], suggesting that the cross talk between inflammatory cells and skeletal muscle helps tightly regulate the timing of muscle regeneration.
Despite the established involvement of TRPC1 in muscle regeneration upon injury, there is a lack of data supporting the participation of TRPC1-induced Ca2+ entry during the initial inflammatory response following skeletal muscle injury. Indirect evidence, however, can shed light on the potential interactions between TRPC1 and inflammatory responses during the regenerative process. For example, treatment of cultured rat hippocampal astrocytes with proinflammatory cytokines such as TNF-α, IL-1β, and LPS significantly suppresses endogenous TRPC1 expression [83]. Similarly, in gene manipulation studies, there is a negative association between TRPC1 and TNF-α, where inhibition of TRPC1 expression using siRNA impairs TRPC1-associated Ca2+ influx. This results in the enhancement of TNF-α-induced COX2 and prostaglandin E2 production, whereas overexpression of TRPC1 suppressed PGE2 release and inhibits COX-2 expression [84]. Blocking TRPC1-associated SOCE produces identical results [84]. Interestingly, TNF-α treatment drives TRPC1-mediated SOCE, suggesting that either the two processes are not entirely mutually exclusive or there might be a negative feedback mechanism in the immune response. In an induced inflammatory setting, antigen-induced elevation of systemic TNF-α levels is more profound in TRPC1 −/−-knockdown mice than in wild-type mice [85]. During the initial stage of muscle regeneration, the inflammatory response is mediated by proinflammatory cytokines such as TNF-α and IL-1β. Considering that TNF-α and IL-1β promote satellite cell activation but inhibit myogenic differentiation [55, 86, 87] and are dominant during the initial phase of muscle injury, TNF-α may possibly act on TRPC1-associated satellite cell activation and proliferation. Following inflammation, TNF-α may trigger the activation of TRPC1 expression and its associated SOCE, and TRPC1-mediated SOCE, in turn, may suppress the activity of TNF-α and take charge of the subsequent regenerative muscle regrowth program. Another possibility is that activation of TRPC1 leads to inhibition of the TNF-α-mediated inflammatory response and thus initiates the onset of the regenerative process.
Though cardiotoxin-injured TRPC1 −/− mice show deficient muscle regeneration, it is also essential to investigate whether they exhibit prolonged inflammatory responses that delay muscle regeneration. We believe that investigating the interaction between proinflammatory cytokines and TRPC1 during skeletal muscle injury will enrich our understanding of muscle plasticity and the role of TRPC1 in muscle regeneration. Nevertheless, this proposed hypothesis may be an oversimplification of the interaction between TRPC1-mediated SOCE and inflammatory responses, considering that the inflammation-muscle regeneration process is a very complicated biological concert involving multiple components (e.g., TRPC1 for migration and chemotaxis of neutrophils [88], macrophage phagocytosis [89], and caspase-dependent cytokine secretion [90]), all of which are important in the regulation of the muscle injury and regeneration process. Furthermore, the unidentified relationships between TRPC1 and other important inflammatory signals such as interferon-gamma (IFNγ), IL-6, and other immune chemokines limit our understanding and await further investigation.
10.4 Involvement of TRPC1 in Muscular Dystrophy
Duchenne muscular dystrophy (DMD), a devastating X-linked recessive disease, is a rapidly progressing form of muscular dystrophy. The pathogenic mechanisms are complex and there is currently no cure. This disease is caused by the lack of the cytoskeletal protein dystrophin [91] and is characterized by unremitting degeneration-regeneration cycles with chronic inflammation, progressive degeneration, and fibrosis. It has long been suggested that excessive intracellular Ca2+ directly initiates a cascade of pathological events causing activation of proteases and muscle degeneration in DMD [92].
The disruption of dystrophin alters the protein and activity levels of TRPC1, causing excessive Ca2+ influx into the dystrophic muscles. Dystrophin and its associated glycoprotein complex (DAGC) link the actin cytoskeleton to laminin in the extracellular matrix [93]. Apart from providing structural stability during muscle contraction, there is clear evidence that dystrophin and the DAGC anchor signaling molecules for various signaling pathways [94]. TRPC1 channels are likely part of the costameric macromolecular complex linked to cytoskeletal dystrophin-DAGC, and they interact with many scaffolding proteins such as Homer 1 and caveolin-3, among others [4]. The channels are regulated by the presence of this complex, and therefore the absence of dystrophin leads to channel dysregulation and increased Ca2+ influx, as observed in mdx (a mouse model of DMD) muscles. TRPC1 is localized on the plasma membrane. TRPC1/TRPC4 channels interact with the α1-syntrophin-dystrophin complex and regulate the activity of SACs and SOCs [95]. α1-Syntrophin-deficient myotubes display abnormal Ca2+ influx dependent on TRPC1 [4]. The scaffolding protein Homer 1 is linked to and stabilizes TRPC1, and Homer 1 −/− mice exhibit myopathy associated with aberrant Ca2+ entry due to TRPC1 overactivity [96]. The scaffolding protein caveolin-3 also regulates TRPC1 expression. Caveolin-3 expression assists in localizing TRPC1 to the plasma membrane and mediates SOCE [97]. Together, these results provide evidence that intact dystrophin-macromolecular complex is required for the expression, localization, and activation of TRPC1 channels to regulate Ca2+ influx.
Increased expression and function of TRPC1 in mdx muscles [97, 98] may contribute to disease onset and progression. TRPC1-mediated Ca2+ entry induces muscle damage in mdx mice. It appears that the channel can be directly or indirectly activated from membrane stretch or Ca2+ store depletion. Even though there is no clear conclusion about whether TRPC1 contributes to SACs in skeletal muscle [99, 100], the activation of SACs following stretch-induced muscle contractions provides evidence that the influx of Ca2+ is likely to be caused by SACs. A substantial increase in intracellular Ca2+ concentration follows eccentric contractions in mdx muscle fibers; more importantly, SAC blockers (Gd3+, streptomycin, and GsMTx-4) prevent muscle damage and the early rise in Ca2+ concentration [6]. In vivo, streptomycin-treated mdx mice show less muscle damage and membrane permeability when subjected to downhill treadmill running [101]. Higher levels of TRPC1 protein in the mdx diaphragm muscles correlate with a more severe dystrophic phenotype, while daily injections of streptomycin significantly reduce Evans Blue uptake and blood CK levels [98]. These studies all support the view that TRPC1 contributes to SAC-Ca2+ entry and is part of the mechanism underlying the muscle damage process in dystrophic muscles.
The association of Orai1 and TRPC1 to form distinct STIM1-gated channels that are activated following store depletion may be a source of Ca2+ overload in mdx muscle. The contribution of TRPC1 channels to SOCE in DMD is supported by studies showing that SOCE is more active in mdx muscles and contributes to elevated intracellular Ca2+ [102, 103]. STIM1 and Orai1 are also upregulated in mdx muscles [103]. Furthermore, RNA silencing of either TRPC1 [3] or Orai1 [104] restores aberrant SOCE in mdx muscles. Other families of TRP channels, such as TRPC3, TRPV2, and TRPV4, are also sources of Ca2+ entry and stretch-induced muscle damage in mdx muscles [62, 105, 106]. An increase in reactive oxygen species (ROS) production could provide another pathway for SOCE activation, and details of the regulatory signaling pathways involved have been reviewed recently [107].
The inflammatory response to muscle injury is of considerable importance in physiological and pathological contexts. Inflammation is a major pathogenic feature that significantly contributes to disease progression in DMD [108–110]. Emerging evidence indicates that the complex roles myeloid cells play can influence DMD pathology. The temporal and spatial patterns of macrophage distribution are disturbed in dystrophic muscle; for instance, M1 macrophages persist in mdx muscles and cause further muscle damage due to the continuous inflammatory response [111]. This disrupts the timing and balance of the M1-M2 transition and leads to aberrant regeneration [112]. Given the possibility that TRPC1 has roles in inflammation and the muscle regenerative process (as described earlier), further studies are needed to elucidate its regulation in DMD in order to attenuate the pathogenic process without affecting muscle regeneration.
10.5 Concluding Remarks
TRPC1 channel is reported to contribute to both store-operated and stretch-activated Ca2+ entry pathways. The channel functions on its own or may interact with other TRP channels and scaffolding proteins for numerous physiological functions in skeletal muscles. In vivo and in vitro studies demonstrate that TRPC1 contributes to muscle cell proliferation, differentiation, and regeneration. TRPC1 has also been implicated in muscle regeneration following disuse atrophy. Moreover, dysregulation of TRPC1 channel causes disturbances in Ca2+ homeostasis and is involved in the pathogenesis and progression of Duchenne muscular dystrophy. There are numerous studies that highlight the interactions between TRPC1 and inflammatory responses in other cell types during the regenerative process. It remains to be elucidated to explore the roles of inflammatory cells in modulating TRPC1 in muscle diseases. Further understanding of the mechanism in regulating the channel may provide therapeutic implications targeting TRPC1 or its upstream or downstream pathways to promote muscle regeneration.
References
Berchtold MW, Brinkmeier H, Muntener M (2000) Calcium ion in skeletal muscle: its crucial role for muscle function, plasticity, and disease. Physiol Rev 80:1215–1265
Patel A, Sharif-Naeini R, Folgering JR, Bichet D, Duprat F, Honore E (2010) Canonical TRP channels and mechanotransduction: from physiology to disease states. Pflugers Arch 460:571–581
Vandebrouck C, Martin D, Colson-Van Schoor M, Debaix H, Gailly P (2002) Involvement of TRPC in the abnormal calcium influx observed in dystrophic (mdx) mouse skeletal muscle fibers. J Cell Biol 158:1089–1096
Sabourin J, Cognard C, Constantin B (2009) Regulation by scaffolding proteins of canonical transient receptor potential channels in striated muscle. J Muscle Res Cell Motil 30:289–297
Sabourin J, Lamiche C, Vandebrouck A, Magaud C, Rivet J, Cognard C et al (2009) Regulation of TRPC1 and TRPC4 cation channels requires an alpha1-syntrophin-dependent complex in skeletal mouse myotubes. J Biol Chem 284:36248–36261
Yeung EW, Whitehead NP, Suchyna TM, Gottlieb PA, Sachs F, Allen DG (2005) Effects of stretch-activated channel blockers on [Ca2 +]i and muscle damage in the mdx mouse. J Physiol 562:367–380
Zhang BT, Whitehead NP, Gervasio OL, Reardon TF, Vale M, Fatkin D et al (2012) Pathways of Ca2+ entry and cytoskeletal damage following eccentric contractions in mouse skeletal muscle. J Appl Physiol (1985) 112:2077–2086
Dietrich A, Fahlbusch M, Gudermann T (2014) Classical Transient Receptor Potential 1 (TRPC1): channel or channel regulator? Cell 3:939–962
Smani T, Dionisio N, Lopez JJ, Berna-Erro A, Rosado JA (1838) Cytoskeletal and scaffolding proteins as structural and functional determinants of TRP channels. Biochim Biophys Acta 2014:658–664
Brinkmeier H (2011) TRP channels in skeletal muscle: gene expression, function and implications for disease. Adv Exp Med Biol 704:749–758
Mauro A (1961) Satellite cell of skeletal muscle fibers. J Biophys Biochem Cytol 9:493–495
Zammit P, Beauchamp J (2001) The skeletal muscle satellite cell: stem cell or son of stem cell? Differentiation 68:193–204
Bischoff R, Heintz C (1994) Enhancement of skeletal muscle regeneration. Dev Dyn 201:41–54
Schultz E, McCormick KM (1994) Skeletal muscle satellite cells. Rev Physiol Biochem Pharmacol 123:213–257
Collins CA, Olsen I, Zammit PS, Heslop L, Petrie A, Partridge TA et al (2005) Stem cell function, self-renewal, and behavioral heterogeneity of cells from the adult muscle satellite cell niche. Cell 122:289–301
Schultz E (1996) Satellite cell proliferative compartments in growing skeletal muscles. Dev Biol 175:84–94
Cornelison DD, Wold BJ (1997) Single-cell analysis of regulatory gene expression in quiescent and activated mouse skeletal muscle satellite cells. Dev Biol 191:270–283
Cooper RN, Tajbakhsh S, Mouly V, Cossu G, Buckingham M, Butler-Browne GS (1999) In vivo satellite cell activation via Myf5 and MyoD in regenerating mouse skeletal muscle. J Cell Sci 112:2895–2901
Zhang K, Sha J, Harter ML (2010) Activation of Cdc6 by MyoD is associated with the expansion of quiescent myogenic satellite cells. J Cell Biol 188:39–48
Bischoff R (1990) Interaction between satellite cells and skeletal muscle fibers. Development 109:943–952
Horsley V, Friday BB, Matteson S, Kegley KM, Gephart J, Pavlath GK (2001) Regulation of the growth of multinucleated muscle cells by an NFATC2-dependent pathway. J Cell Biol 153:329–338
Kegley KM, Gephart J, Warren GL, Pavlath GK (2001) Altered primary myogenesis in NFATC3(−/−) mice leads to decreased muscle size in the adult. Dev Biol 232:115–126
Liu Y, Schneider MF (2014) FGF2 activates TRPC and Ca2+ signaling leading to satellite cell activation. Front Physiol 5:38
Cheung KK, Yeung SS, Au SW, Lam LS, Dai ZQ, Li YH et al (2011) Expression and association of TRPC1 with TRPC3 during skeletal myogenesis. Muscle Nerve 44:358–365
Fiorio Pla A, Maric D, Brazer SC, Giacobini P, Liu X, Chang YH et al (2005) Canonical transient receptor potential 1 plays a role in basic fibroblast growth factor (bFGF)/FGF receptor-1-induced Ca2 + entry and embryonic rat neural stem cell proliferation. J Neurosci 25:2687–2701
Jockusch H, Voigt S (2003) Migration of adult myogenic precursor cells as revealed by GFP/nLacZ labelling of mouse transplantation chimeras. J Cell Sci 116:1611–1616
Schultz E, Jaryszak DL, Valliere CR (1985) Response of satellite cells to focal skeletal muscle injury. Muscle Nerve 8:217–222
Huang YW, Chang SJ, I-Chen HH, Huang HT, Lin HH, Shen MR et al (2015) Mechanosensitive store-operated calcium entry regulates the formation of cell polarity. J Cell Physiol 230:2086–2097
Kobayashi T, Sokabe M (2010) Sensing substrate rigidity by mechanosensitive ion channels with stress fibers and focal adhesions. Curr Opin Cell Biol 22:669–676
Fabian A, Fortmann T, Dieterich P, Riethmuller C, Schon P, Mally S et al (2008) TRPC1 channels regulate directionality of migrating cells. Pflugers Arch 457:475–484
Kuang CY, Yu Y, Wang K, Qian DH, Den MY, Huang L (2012) Knockdown of transient receptor potential canonical-1 reduces the proliferation and migration of endothelial progenitor cells. Stem Cells Dev 21:487–496
Fabian A, Bertrand J, Lindemann O, Pap T, Schwab A (2012) Transient receptor potential canonical channel 1 impacts on mechanosignaling during cell migration. Pflugers Arch 464:623–630
Cheng KT, Ong HL, Liu X, Ambudkar IS (2013) Contribution and regulation of TRPC channels in store-operated Ca2 + entry. Curr Top Membr 71:149–179
Louis M, Zanou N, Van Schoor M, Gailly P (2008) TRPC1 regulates skeletal myoblast migration and differentiation. J Cell Sci 121:3951–3959
Spizz G, Blackshear PJ (2001) Overexpression of the myristoylated alanine-rich C-kinase substrate inhibits cell adhesion to extracellular matrix components. J Biol Chem 276:32264–32273
Dedieu S, Mazeres G, Poussard S, Brustis JJ, Cottin P (2003) Myoblast migration is prevented by a calpain-dependent accumulation of MARCKS. Biol Cell 95:615–623
Formigli L, Sassoli C, Squecco R, Bini F, Martinesi M, Chellini F et al (2009) Regulation of transient receptor potential canonical channel 1 (TRPC1) by sphingosine 1-phosphate in C2C12 myoblasts and its relevance for a role of mechanotransduction in skeletal muscle differentiation. J Cell Sci 122:1322–1333
Zanou N, Schakman O, Louis P, Ruegg UT, Dietrich A, Birnbaumer L et al (2012) TRPC1 ion channel modulates phosphatidylinositol 3-kinase/Akt pathway during myoblast differentiation and muscle regeneration. J Biol Chem 287:14524–14534
Rochlin K, Yu S, Roy S, Baylies MK (2010) Myoblast fusion: when it takes more to make one. Dev Biol 341:66–83
Antigny F, Koenig S, Bernheim L, Frieden M (2013) During post-natal human myogenesis, normal myotube size requires TRPC1- and TRPC4-mediated Ca(2)(+) entry. J Cell Sci 126:2525–2533
Sun L, Liu L, Yang XJ, Wu Z (2004) Akt binds prohibitin 2 and relieves its repression of MyoD and muscle differentiation. J Cell Sci 117:3021–3029
Ma R, Rundle D, Jacks J, Koch M, Downs T, Tsiokas L (2003) Inhibitor of myogenic family, a novel suppressor of store-operated currents through an interaction with TRPC1. J Biol Chem 278:52763–52772
Chen CM, Kraut N, Groudine M, Weintraub H (1996) I-mf, a novel myogenic repressor, interacts with members of the MyoD family. Cell 86:731–741
Yoshida N, Yoshida S, Koishi K, Masuda K, Nabeshima Y (1998) Cell heterogeneity upon myogenic differentiation: down-regulation of MyoD and Myf-5 generates ‘reserve cells’. J Cell Sci 111:769–779
Hawke TJ, Garry DJ (2001) Myogenic satellite cells: physiology to molecular biology. J Appl Physiol (1985) 91:534–551
Collins CA (2006) Satellite cell self-renewal. Curr Opin Pharmacol 6:301–306
Cao Y, Zhao Z, Gruszczynska-Biegala J, Zolkiewska A (2003) Role of metalloprotease disintegrin ADAM12 in determination of quiescent reserve cells during myogenic differentiation in vitro. Mol Cell Biol 23:6725–6738
Ochiai N, Nishizuka M, Osada S, Imagawa M (2016) Fad24, a Positive Regulator of Adipogenesis, Is Required for S Phase Re-entry of C2C12 Myoblasts Arrested in G0 Phase and Involved in p27(Kip1) Expression at the Protein Level. Biol Pharm Bull 39:807–814
Asghar MY, Magnusson M, Kemppainen K, Sukumaran P, Lof C, Pulli I et al (2015) Transient receptor potential canonical 1 (TRPC1) channels as regulators of sphingolipid and VEGF receptor expression: Implications for thyroid cancer cell migration and proliferation. J Biol Chem 290:16116–16131
Shinin V, Gayraud-Morel B, Gomes D, Tajbakhsh S (2006) Asymmetric division and cosegregation of template DNA strands in adult muscle satellite cells. Nat Cell Biol 8:677–687
Favier FB, Benoit H, Freyssenet D (2008) Cellular and molecular events controlling skeletal muscle mass in response to altered use. Pflugers Arch 456:587–600
Rennie MJ, Wackerhage H, Spangenburg EE, Booth FW (2004) Control of the size of the human muscle mass. Annu Rev Physiol 66:799–828
Kortebein P, Symons TB, Ferrando A, Paddon-Jones D, Ronsen O, Protas E et al (2008) Functional impact of 10 days of bed rest in healthy older adults. J Gerontol A Biol Sci Med Sci 63:1076–1081
Tidball JG, Villalta SA (2010) Regulatory interactions between muscle and the immune system during muscle regeneration. Am J Phys Regul Integr Comp Phys 298:R1173–R1187
Arnold L, Henry A, Poron F, Baba-Amer Y, van Rooijen N, Plonquet A et al (2007) Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflammatory macrophages to support myogenesis. J Exp Med 204:1057–1069
Ohira Y, Jiang B, Roy RR, Oganov V, Ilyina-Kakueva E, Marini JF et al (1992) Rat soleus muscle fiber responses to 14 days of spaceflight and hindlimb suspension. J Appl Physiol (1985) 73:51s–57s
Jiang B, Ohira Y, Roy RR, Nguyen Q, Ilyina-Kakueva EI, Oganov V et al (1992) Adaptation of fibers in fast-twitch muscles of rats to spaceflight and hindlimb suspension. J Appl Physiol (1985) 73:58s–65s
Seth M, Zhang ZS, Mao L, Graham V, Burch J, Stiber J et al (2009) TRPC1 channels are critical for hypertrophic signaling in the heart. Circ Res 105:1023–1030
Benavides Damm T, Richard S, Tanner S, Wyss F, Egli M, Franco-Obregon A (2013) Calcium-dependent deceleration of the cell cycle in muscle cells by simulated microgravity. FASEB J 27:2045–2054
Kerstein PC, Jacques-Fricke BT, Rengifo J, Mogen BJ, Williams JC, Gottlieb PA et al (2013) Mechanosensitive TRPC1 channels promote calpain proteolysis of talin to regulate spinal axon outgrowth. J Neurosci 33:273–285
Zhang BT, Yeung SS, Cheung KK, Chai ZY, Yeung EW (2014) Adaptive responses of TRPC1 and TRPC3 during skeletal muscle atrophy and regrowth. Muscle Nerve 49:691–699
Zanou N, Shapovalov G, Louis M, Tajeddine N, Gallo C, Van Schoor M et al (2010) Role of TRPC1 channel in skeletal muscle function. Am J Phys Cell Phys 298:C149–C162
Xia L, Cheung KK, Yeung SS, Yeung EW (2016) The involvement of transient receptor potential canonical type 1 in skeletal muscle regrowth after unloading-induced atrophy. J Physiol 594:3111–3126
Bush EW, Hood DB, Papst PJ, Chapo JA, Minobe W, Bristow MR et al (2006) Canonical transient receptor potential channels promote cardiomyocyte hypertrophy through activation of calcineurin signaling. J Biol Chem 281:33487–33496
Sugiura T, Abe N, Nagano M, Goto K, Sakuma K, Naito H et al (2005) Changes in PKB/Akt and calcineurin signaling during recovery in atrophied soleus muscle induced by unloading. Am J Phys Regul Integr Comp Phys 288:R1273–R1278
Oishi Y, Ogata T, Yamamoto KI, Terada M, Ohira T, Ohira Y et al (2008) Cellular adaptations in soleus muscle during recovery after hindlimb unloading. Acta Physiol (Oxford) 192:381–395
Ohba T, Watanabe H, Murakami M, Takahashi Y, Iino K, Kuromitsu S et al (2007) Upregulation of TRPC1 in the development of cardiac hypertrophy. J Mol Cell Cardiol 42:498–507
Wang C, Li JF, Zhao L, Liu J, Wan J, Wang YX et al (2009) Inhibition of SOC/Ca2 +/NFAT pathway is involved in the anti-proliferative effect of sildenafil on pulmonary artery smooth muscle cells. Respir Res 10:123
Wu X, Eder P, Chang B, Molkentin JD (2010) TRPC channels are necessary mediators of pathologic cardiac hypertrophy. Proc Natl Acad Sci U S A 107:7000–7005
Makarewich CA, Zhang H, Davis J, Correll RN, Trappanese DM, Hoffman NE et al (2014) Transient receptor potential channels contribute to pathological structural and functional remodeling after myocardial infarction. Circ Res 115:567–580
Riley DA, Thompson JL, Krippendorf BB, Slocum GR (1995) Review of spaceflight and hindlimb suspension unloading induced sarcomere damage and repair. Basic Appl Myol 5:139–145
Newham DJ, Jones DA, Edwards RH (1986) Plasma creatine kinase changes after eccentric and concentric contractions. Muscle Nerve 9:59–63
Yeung EW, Ballard HJ, Bourreau JP, Allen DG (2003) Intracellular sodium in mammalian muscle fibers after eccentric contractions. J Appl Physiol (1985) 94:2475–2482
Yeung EW, Allen DG (2004) Stretch-activated channels in stretch-induced muscle damage: role in muscular dystrophy. Clin Exp Pharmacol Physiol 31:551–556
Zhang BT, Yeung SS, Allen DG, Qin L, Yeung EW (2008) Role of the calcium-calpain pathway in cytoskeletal damage after eccentric contractions. J Appl Physiol 105:352–357
Tidball JG, Berchenko E, Frenette J (1999) Macrophage invasion does not contribute to muscle membrane injury during inflammation. J Leukoc Biol 65:492–498
Summan M, Warren GL, Mercer RR, Chapman R, Hulderman T, Van Rooijen N et al (2006) Macrophages and skeletal muscle regeneration: a clodronate-containing liposome depletion study. Am J Phys Regul Integr Comp Phys 290:R1488–R1495
Tidball JG, Wehling-Henricks M (2007) Macrophages promote muscle membrane repair and muscle fibre growth and regeneration during modified muscle loading in mice in vivo. J Physiol 578:327–336
Tidball JG (2011) Mechanisms of muscle injury, repair, and regeneration. Commun Phys 1:2029–2062
Warren GL, Hulderman T, Jensen N, McKinstry M, Mishra M, Luster MI et al (2002) Physiological role of tumor necrosis factor alpha in traumatic muscle injury. FASEB J 16:1630–1632
Chen SE, Jin B, Li YP (2007) TNF-alpha regulates myogenesis and muscle regeneration by activating p38 MAPK. Am J Phys Cell Phys 292:C1660–C1671
Fu X, Xiao J, Wei Y, Li S, Liu Y, Yin J et al (2015) Combination of inflammation-related cytokines promotes long-term muscle stem cell expansion. Cell Res 25:655–673
Ronco V, Grolla AA, Glasnov TN, Canonico PL, Verkhratsky A, Genazzani AA et al (2014) Differential deregulation of astrocytic calcium signalling by amyloid-beta, TNFalpha, IL-1beta and LPS. Cell Calcium 55:219–229
Hai L, Kawarabayashi Y, Imai Y, Honda A, Inoue R (2011) Counteracting effect of TRPC1-associated Ca2 + influx on TNF-alpha-induced COX-2-dependent prostaglandin E2 production in human colonic myofibroblasts. Am J Physiol Gastrointest Liver Physiol 301:G356–G367
Medic N, Desai A, Olivera A, Abramowitz J, Birnbaumer L, Beaven MA et al (2013) Knockout of the Trpc1 gene reveals that TRPC1 can promote recovery from anaphylaxis by negatively regulating mast cell TNF-alpha production. Cell Calcium 53:315–326
Li YP (2003) TNF-alpha is a mitogen in skeletal muscle. Am J Phys Cell Phys 285:C370–C376
Broussard SR, McCusker RH, Novakofski JE, Strle K, Shen WH, Johnson RW et al (2004) IL-1beta impairs insulin-like growth factor i-induced differentiation and downstream activation signals of the insulin-like growth factor i receptor in myoblasts. J Immunol 172:7713–7720
Lindemann O, Strodthoff C, Horstmann M, Nielsen N, Jung F, Schimmelpfennig S et al (2015) TRPC1 regulates fMLP-stimulated migration and chemotaxis of neutrophil granulocytes. Biochim Biophys Acta 1853:2122–2130
Zhou X, Ye Y, Sun Y, Li X, Wang W, Privratsky B et al (2015) Transient receptor potential channel 1 deficiency impairs host defense and proinflammatory responses to bacterial infection by regulating protein kinase c alpha signaling. Mol Cell Biol 35:2729–2739
Py BF, Jin M, Desai BN, Penumaka A, Zhu H, Kober M et al (2014) Caspase-11 controls interleukin-1beta release through degradation of TRPC1. Cell Rep 6:1122–1128
Hoffman EP, Brown RH Jr, Kunkel LM (1987) Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell 51:919–928
Bodensteiner JB, Engel AG (1978) Intracellular calcium accumulation in Duchenne dystrophy and other myopathies: a study of 567,000 muscle fibers in 114 biopsies. Neurology 28:439–446
Blake DJ, Weir A, Newey SE, Davies KE (2002) Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev 82:291–329
Constantin B (1838) Dystrophin complex functions as a scaffold for signalling proteins. Biochim Biophys Acta 2014:635–642
Vandebrouck A, Sabourin J, Rivet J, Balghi H, Sebille S, Kitzis A et al (2007) Regulation of capacitative calcium entries by alpha1-syntrophin: association of TRPC1 with dystrophin complex and the PDZ domain of alpha1-syntrophin. FASEB J 21:608–617
Stiber JA, Zhang ZS, Burch J, Eu JP, Zhang S, Truskey GA et al (2008) Mice lacking Homer 1 exhibit a skeletal myopathy characterized by abnormal transient receptor potential channel activity. Mol Cell Biol 28:2637–2647
Gervasio OL, Whitehead NP, Yeung EW, Phillips WD, Allen DG (2008) TRPC1 binds to caveolin-3 and is regulated by Src kinase - role in Duchenne muscular dystrophy. J Cell Sci 121:2246–2255
Matsumura CY, Taniguti AP, Pertille A, Santo Neto H, Marques MJ (2011) Stretch-activated calcium channel protein TRPC1 is correlated with the different degrees of the dystrophic phenotype in mdx mice. Am J Phys Cell Phys 301:C1344–C1350
Maroto R, Raso A, Wood TG, Kurosky A, Martinac B, Hamill OP (2005) TRPC1 forms the stretch-activated cation channel in vertebrate cells. Nat Cell Biol 7:179–185
Gottlieb P, Folgering J, Maroto R, Raso A, Wood TG, Kurosky A et al (2008) Revisiting TRPC1 and TRPC6 mechanosensitivity. Pflugers Arch 455:1097–1103
Whitehead NP, Streamer M, Lusambili LI, Sachs F, Allen DG (2006) Streptomycin reduces stretch-induced membrane permeability in muscles from mdx mice. Neuromuscul Disord 16:845–854
Ducret T, Vandebrouck C, Cao ML, Lebacq J, Gailly P (2006) Functional role of store-operated and stretch-activated channels in murine adult skeletal muscle fibres. J Physiol 575:913–924
Edwards JN, Friedrich O, Cully TR, von Wegner F, Murphy RM, Launikonis BS (2010) Upregulation of store-operated Ca2+ entry in dystrophic mdx mouse muscle. Am J Phys Cell Phys 299:C42–C50
Zhao X, Moloughney JG, Zhang S, Komazaki S, Weisleder N (2012) Orai1 mediates exacerbated Ca(2+) entry in dystrophic skeletal muscle. PLoS One 7:e49862
Millay DP, Goonasekera SA, Sargent MA, Maillet M, Aronow BJ, Molkentin JD (2009) Calcium influx is sufficient to induce muscular dystrophy through a TRPC-dependent mechanism. Proc Natl Acad Sci U S A 106:19023–19028
Pritschow BW, Lange T, Kasch J, Kunert-Keil C, Liedtke W, Brinkmeier H (2011) Functional TRPV4 channels are expressed in mouse skeletal muscle and can modulate resting Ca2+ influx and muscle fatigue. Pflugers Arch 461:115–122
Allen DG, Whitehead NP, Froehner SC (2016) Absence of dystrophin disrupts skeletal muscle signaling: roles of Ca2+, reactive oxygen species, and nitric oxide in the development of muscular dystrophy. Physiol Rev 96:253–305
Tidball JG, Wehling-Henricks M (2005) Damage and inflammation in muscular dystrophy: potential implications and relationships with autoimmune myositis. Curr Opin Rheumatol 17:707–713
Evans NP, Misyak SA, Robertson JL, Bassaganya-Riera J, Grange RW (2009) Dysregulated intracellular signaling and inflammatory gene expression during initial disease onset in Duchenne muscular dystrophy. Am J Phys Med Rehabil 88:502–522
De Paepe B, De Bleecker JL (2013) Cytokines and chemokines as regulators of skeletal muscle inflammation: presenting the case of Duchenne muscular dystrophy. Mediat Inflamm 2013:540370
Villalta SA, Nguyen HX, Deng B, Gotoh T, Tidball JG (2009) Shifts in macrophage phenotypes and macrophage competition for arginine metabolism affect the severity of muscle pathology in muscular dystrophy. Hum Mol Genet 18:482–496
Tidball JG, Dorshkind K, Wehling-Henricks M (2014) Shared signaling systems in myeloid cell-mediated muscle regeneration. Development 141:1184–1196
Acknowledgments
This work was supported by the Hong Kong Research Grants Council General Research Funds (PolyU 5636/12M, 5636/13M) and PolyU Central Research Grant (G-Y88P).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Nature Singapore Pte Ltd.
About this chapter
Cite this chapter
Yeung, E.W., Cheung, KK., Sun, KT. (2017). Biological Role of TRPC1 in Myogenesis, Regeneration, and Disease. In: Sakuma, K. (eds) The Plasticity of Skeletal Muscle. Springer, Singapore. https://doi.org/10.1007/978-981-10-3292-9_10
Download citation
DOI: https://doi.org/10.1007/978-981-10-3292-9_10
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-3291-2
Online ISBN: 978-981-10-3292-9
eBook Packages: MedicineMedicine (R0)