
Abstract
Microglia are the immunocompetent resident macrophages of the central nervous system and constitutes 15–20 % of the glial population. They provides the first line of defence against any disease or insult and display enormous structural and functional plasticity. Microglial cells are also well establised to play a very important role in the pathogenesis of various neurological disorders. Microglial activation not only protect and repair the damaged tissue by eliminating the dying cell and assisting the restorative process but are also implicated in inducing neurodegeneration. This review provides a comprehensive account of development and various physiological states of microglia and their role in healthy and disease brain.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
2.1 Introduction
Our understanding of microglia has moved from being a ‘silent’ cell in healthy brain to an actively involved component in brain physiology, neurogenesis, cognition and behavioural functions. They are the surveyors of the healthy brain with actively retracting and extending their processes and thus maintaining the pre- and post-synaptic elements and fine tuning of the neuronal circuits. Thus, a disruption of this homeostatic act of microglia becomes the prime cause of neuronal disorders. Microglia are nomadic cells of the brain that continuously survey the central nervous system (CNS) with their highly motile extensions for any kind of brain insult (Gehrmann et al. 1995) and constitute 15–20 % of the total glial population in the central nervous system (Carson et al. 2006). They are the resident macrophages of CNS and are immunocompetent phagocytic cells and constitute the first line of defence against any disease or insult and exhibit structural and functional plasticity. Microglia are considered responsible to maintain the homeostasis within the brain and undergo appropriate structural transformations to perform various immunological functions. First recognized by Nissl in 1880, later Pio-del Rio Hortega, a Spanish neuroanatomist, described microglia as resting ramified cells using silver staining methods (Del Rio-Hortega 1932).
Microglial cells are now well recognized as an elementary contributor in the pathogenesis of various neurological diseases and disorders (Heneka et al. 2010; Parpura et al. 2012; Verkhratsky et al. 2014). As affiliate of brain defence system, on any immune breaching or insult, microglia become activated (Saxena et al. 2007; Patro et al. 2010a, b, 2013; Nagayach et al. 2014a, b, 2015; Sharma et al. 2015). On activation, these immune cells get rapidly transformed into the reactive phenotype and slack their highly ramified morphology not only to protect but also to repair the damaged tissue by removing the dying cell debris and facilitating the healing process (Hanisch and Kettenmann 2007; Kettenmann et al. 2011). On the contrary, microglial activation is also responsible in aggravating the neurodegeneration (Block and Hong 2005; Venero et al. 2011). Understanding of the imperative and multitasking attribute of microglial cells, like its stature and response following neuroinflammation deserves pertinent investigation.
2.2 Physiological States of Microglia
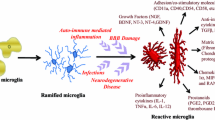
Morphologically, microglia have three major transitional stages that can be distinguished as: amoeboid, ramified or resting and reactive or activated (Fig. 2.1) and these states perform varied functions in the brain.

Microglial transformations both in terms of phenotype and secretory molecules with the advancing age: In developing and adult brain, amoeboid and ramified microglia supports the survival of healthy neurons. During state of insult microglia get activated and attain either of the two phases of activation, i.e. M1 and M2 on the basis of severity and generation of secretory molecules. M1 phase exacerbates microglial activation directed neuronal damage via releasing plethora of pro-inflammatory molecules while M2 phase mitigates the neuroinflammation and promotes tissue repair and neuron survival by secreting growth factors and anti-inflammatory molecules. On disease progression and neuronal death, microglia turn deramified and phagocytic. Gitter cells are the microglia crammed with the phagocytic debris
Amoeboid microglia are round or irregular in shape. They are more prevalent during development, originate from the yolk sac and populate the developing brain early. Association of developmental neuronal cell death and microglia has been reported in most parts of the CNS (Pont-Lezica et al. 2011). Because of phagocytosis as well as their ability to induce apoptosis in unwanted neurons in developing brain, microglia are important participant in the process of CNS development. They also interact with the synapses and modulate synaptic plasticity via pruning of excessive unwanted synapses and this is mediated by the complement pathway (Schafer et al. 2012; Ginhoux et al. 2013; Neiva et al. 2014). Morphologically, they closely resemble the macrophages. Amoeboid microglia are generated from primitive myeloid/ haematopoietic progenitor cells during the embryonic and perinatal stage and sustain up to the early postnatal stages in rats (Prinz and Mildner 2011; Gomez et al. 2013) and finally transform into ramified microglia (Kaur et al. 1985). The development of microglia and role of microglia in brain development have been reviewed by Pont-Lezica et al. (2011) and Nayak et al. (2014).
Ramified or ‘resting surveillent microglia’ of the adult CNS consist of small cell body with short, wispy and fine processes. These processes extend into the brain microenvironment creating a matrix-like structure that helps to better perceive the CNS milieu. Yamasaki et al. (2014) have reviewed the available information on the differentiation of the resident microglia and the monocytes in neuroinflammatory states. Microglia are considered to be the critical effectors and regulators of changes in CNS homeostasis in health and disease (Prinz and Priller 2014) as well as during CNS development. Microglia even in healthy brain continuously survey the CNS for any damage or insult as shown in in vivo time-lapse video microscopy and hence they are never in a state of rest (Nayak et al. 2014). The studies of Hellwig et al. (2013) have established the active role of such cells as depletion of ramified microglia prior to experimental stroke exacerbated the damage, establishing the active and protective role of the so called ‘resting microglia’.
Reactive microglia: Following any unfavourable stimuli ramified microglia get transformed into a reactive or activated state. Such cells have thick and retracted processes with a large and irregular shaped cell body. Reactive microglia even start proliferating to ascertain better screening and support as a hallmark of microglial activation (Niquet et al. 1994). Depending upon the stimulus and progression of the diseased state, microglial activation acts in two ways; either help in efficacious restoration of the injured brain cells or generate a threatening environment that results in exaggerated brain damage. Recent studies with mouse models of neurodegenerative disorders have helped us in better understanding to an extent the role of microglia in health and disease (Hellwig et al. 2013). To overcome the confusion, the activity-dependent microglial activation spectrum (Tang and Le 2015) was developed on the basis of cytoactive factors released by the reactive microglia (Fig. 2.1). The ‘classical activation (popularly known as M1 phase)’ represents the initial innate immune response induced by Toll-like receptor (TLR) ligands and interferon-γ (IFN-γ) followed by the generation of pro-inflammatory cytokines. The reactive release of a plethora of pro-inflammatory molecules like tumour necrosis factor-α (TNF-α), interleukin-6 (IL-6), interleukin-1β (IL-1β), interleukin-12 (IL-12), superoxide anions, nitric oxide synthase, redox molecules like nitrogen dioxide 2 (NOX2), nitrogen dioxide1 (NOX1), a member of Rho family of GTPases (RAC1), inducible nitric oxide synthase (iNOS), nitric oxide synthase 2 (NOS2) and excitotoxic molecules like group II metabotropic glutamate receptor (MGluR2), glutamate transporter-1 (GLT-1), purinergic P2X7 receptor (P2X7-R), etc. (Benoit et al. 2008). The next alternate phase of microglial activation is ‘M2 or alternate activation’ phase, that dampen inflammation by switching over to the anti-inflammatory state by secreting molecules like interleukin-4 (IL-4), interleukin-3 (IL-13), interleukin-1 receptor antagonist (IL-1RA), scavenging receptors and extracellular matrix molecules (Luo and Chen 2012). The cytoactive molecules thus released mitigate the generation of pro-inflammatory molecules. This accelerates the process of wound healing and damaged tissue repair (Martinez et al. 2008). The third or subtype of M2 phase is ‘acquired deactivation’ associated with deactivation of glial inflammation and uptake of apoptotic cells or oxidized lipids via release of anti-inflammatory cytokines like transforming growth factor-β (TGF-β) and interleukin-10 (IL-10; Gregory and Devitt 2004; Colton 2009).
2.3 Development of Microglia
The origin of microglia and its cell lineage still remains highly controversial and debatable. Microglia arise early during development from precursor cells in the embryonic yolk sac that seed the brain rudiment and appear to persist throughout the life. Microglia are the only cell population in the CNS that originate outside the brain. The differentiation of yolk sac macrophages into typical microglia is dependent on transcription factors like IFM regulatory factor-8 (IRF-8; Ginhoux et al. 2013; Prinz and Priller 2014). Bone marrow-derived progenitors or monocytes are also considered to be recruited for supplementing the microglial population (Saijo et al. 2013; Ginhoux et al. 2013; Prinz and Priller 2014).
The neuroectodermal matrix cells and yolk sac cells are the two distinct sources of microglial precursors (Saijo and Glass 2011). Prenatally, these cells invade the brain through meninges, choroid plexus and ventricles (Boya et al. 1991; Ginhoux et al. 2010). This primeval microglial population was reported in human gestation week 5.5 near the di-telencephalic fissure (Monier et al. 2006). First, the neuroectodermal and yolk sac cells populate the brain during first two trimesters in humans and between embryonic days 10/9.5–10.5 in rodents, and grow as amoeboid microglia (Ginhoux et al. 2010). Subsequently in early days of postnatal development, the circulating monocytes developed from blood borne precursors later give rise to amoeboid microglia (Rezaie and Male 2002). The hematopoietic stem cells in developing and adult brain have also been reported to transform into microglia (Alliot et al. 1991). This has been supported by chimeric animal study following irradiation (Hickey et al. 1992) and in experimental model of allergic encephalomyelitis (EAE; Lassmann and Hickey 1993). However, as a contrast, it has also been reported that microglia also existed before brain vascularization and production of monocytes in hematopoietic tissues indicating thereby that all microglia are not hematopoietic in origin (Shepard and Zon 2000; Takahashi 2001). The perivascular microglia are the only cell population that are continuously replaced in the adulthood by bone marrow-derived haematopoietic precursors (Hickey and Kimura 1988). While we continue debating the microglial lineage and origin, interestingly two independent reports claim that microglia can themselves act like pluripotent stem cells and can also transform into astrocytes, neurons and oligodendrocytes (Yokoyama et al. 2004; Matsuda et al. 2008) although the lineage of microglia is different than the astrocytes and neurons. This is being actively investigated and remains to be established and explored.
2.4 Microglia in Healthy Brain
2.4.1 In Developing Brain
Brain development and maturation involves a continuous refinement of synapses involving pruning of inappropriate synapses and strengthening of the established ones. Microglia have been implicated as a major player for the developmental synaptic pruning (Rakic and Zecevic 2000). The activated microglia surround the regions undergoing developmental synapse turnover, and remove the unnecessary synapses (Paolicelli et al. 2011). This happens in a complement-dependent manner. During the embryonic and early postnatal life amoeboid microglia expressing DNAX associated protein 12 (DAP12), complement and fractalkine receptors are directed towards the developing synaptic sites. Such microglia engulf the complement proteins (C1q and C3) and tagged synapses (Paolicelli et al. 2011; Schafer et al. 2013). Thus, any kind of deviation in microglial involvement leads to deficits in synaptic remodelling and maintenance, resulting in developmental disorders. Microglia are also believed to be involved in regulation of neuronal differentiation (Farinas et al. 2002) and apoptosis (Miller and Kaplan 2001) by producing neurotrophins (Nakajima et al. 2001) and the presence of microglia secreted basic fibroblast growth factors (bFG; Bansal 2002) and cytokine IL-1β have been reported to enhance proliferation and differentiation of oligodendrocytes and astrocytes. Microglia undoubtedly play an authoritatively supportive and directive role in both neurogenesis and gliogenesis in developing brain (Thored et al. 2009).
2.4.2 In Adult Brain
‘Ramified/(resting?) surveillent’ microglia reside at strategic locations throughout the mammalian brain and spinal cord. Such microglia are unremittingly surveying the healthy brain for any disparaging condition at a speed of 1.47 µm s−1 with their long thin processes (Nimmerjahn et al. 2005). Recent in vivo studies have recorded the region specific speed of process motility to be between 0.2 and 6.5 µm/min (Tremblay et al. 2010). During such scrutiny microglial processes constantly establish a direct contact with neuronal synapses (Wake et al. 2009). Such microglia release various neurotrophic growth factors to promote the neuronal survival and also to enhance neurogenesis (Ekdahl et al. 2009). In neurodegenerative diseases and following brain insults the resident microglia get stimulated and transform into activated or reactive state. In such circumstances microglia release numerous inflammatory molecules, growth factors, matrix proteins, chemokines, prostanoids and reactive free radicals (Fig. 2.2) either contributing to neuronal dysfunction and cell death or to provide support in the healing process (Gomes-Leal 2012). The detrimental or beneficial role of microglia depends upon the type and intensity of the insult and associated microglia activation stature. This may even call for microgliosis. Microglia in adult brain are not evidenced to have the ability of restoring their normal density, if depleted experimentally from the pool of precursor cells dispersed all over the brain (Parkhurst et al. 2013; Elmore et al. 2014), rather than depending upon the influx from the peripheral bloodstream as reported previously (Hughes and Bergles 2014). This may be one of the mechanisms how the old and/or damaged microglia are replaced with new healthy microglia during progression of a disease and ageing conditions.

Microglial pathology following neuroinflammation: in response of immune breaching and neuronal damage, a state of neuroinflammation developed inside the brain foremostly activates the microglia. Ramified microglia get transformed into activated microglia and release various neuroinflammatory molecules that leads to blood–brain barrier damage. Such damage promotes the macrophagic infiltration that later on exaggerates the influx of inflammatory cytokines and aggravate the existing neuroinflammatory state in CNS by causing secondary neuronal damage and subsequent microglial activation
It is now clear that microglia are also important in both learning and synaptic remodelling (Parkhurst et al. 2013) and take part in activity-dependent structural remodelling both driven by sensory input and age-related factors (Wake et al. 2009; Tremblay et al. 2012). Microglia in adult brain help in regulation of long-term potentiation (LTP) and tuning of synaptic strength, which is responsible for consistent long-term neural networks (Ben Achour and Pascual 2010; Kettenmann et al. 2011). Microglia also maintain the synaptic plasticity by releasing various soluble molecules responsible for regulating learning and memory and augmentation of N-methyl-d-aspartate (NMDA)-mediated LTP responses. It has been predicted that the absence of microglia-mediated fractalkine receptor CX3CR1 signalling and secretion of glycine and L-serine by experimental intervention results in diminished learning and memory (Hayashi et al. 2006; Rogers et al. 2011). Moreover, microglia also mediate the modulation of GABAergic transmission and basal glutamatergic signalling via brain-derived neurotrophic factor (BDNF) and adenosine triphosphate (ATP; Coull et al. 2005; Pascual et al. 2012). BDNF is required for tyrosine kinase B (TrkB) phosphorylation responsible for synaptic plasticity. This has now been established in mice models depleted of microglia that have impaired ability in multiple learning tasks. Such mice also presented a reduction in motor learning-associated synaptic formation.
2.4.3 In Aged Brain
The immune components in the ageing brain are equally affected with age-associated challenges. The immune system in aged brain is also more susceptible towards age-associated damage and dysfunction (Yung and Julius 2008). Microglial dystrophy has been noted as an indication of microglial senescence in brain (Streit et al. 2004; Kushwaha 2009; Patro et al. 2010c). With advancing age, microglia become more reactive (Streit 2006; Godbout and Johnson 2009), exhibit an amoeboid-like morphology and present an upregulation of major histocompatibility complex class II antigens, toll-like receptors 4 (TLR4) and cluster of differentiation 14 (CD14) receptors on their surface. Concomitantly, microglia also express an elevated pro- (TNF-α, IL-1β, IL-6) and anti-(TGF-β, IL-10) inflammatory cytokines in the healthy senile brain of aged mice (Sierra et al. 2007; Godbout and Johnson 2009). However, it still remains to be established either such primed state is associated with the ageing changes of the brain or ageing of the microglial cells themselves.
Ageing, age-associated exposure to stress and neurodegenerative diseases, all induce a ‘priming’ stimulus to microglia. Microglial priming and impaired microglial response is suggestive of age-related changes in microglial regulation (Wynne et al. 2009). Amplified cytokine response by primed microglia has been related to the behavioural distortions like maladaptive sickness response studied in aged subjects exposed to peripheral stimulation (Dilger and Johnson 2008). Increased cytokine secretion by such ‘primed’ microglia following altered immune reaction also cause cognitive impairment in aged brain (Chen et al. 2008).
2.5 General Microglial Physiology
2.5.1 Ion Channels
Myriad of microglial patch-clamp studies in tissue slices and in cell culture showed that microglia possess various ion channels, comprising K+, Ca2+ and Na+ channels. These ion channels undoubtedly play a potential role in both regulation and maintenance of microglial functions (Färber and Kettenmann 2005, 2006a, b; Eder 2005; Schilling and Eder 2007; Black et al. 2009). In general, ion channels in all living cells may influence several cellular processes like, proliferation, migration, apoptosis, secretion and excitability, etc. via movement of cations or anions across the membrane through hydrophilic pores. The functional stature of the microglia evidently states the expression patterns of the ion channels. Expression of various cytokines or immune molecules fluctuate the pH along the gradient and/or activation of the G proteins or protein kinase C, that in turn could modulate the microglial ion channels. The functional coherence and transforming ability of microglial ion channels during various conditions make them a suitable target to study under pathophysiological process like neuroinflammation that further contributes to the onset or progression of neurological disorders.
2.5.2 Sodium Channels
In the healthy CNS (in vivo) the evidential data regarding the functional activity of voltage-operated Na+ channels in microglial cells is scanty (Black and Waxman 2012). However, in vitro study in rat microglia (Black et al. 2009) depicted the three major isoforms of sodium channels enlisted as, Nav1.1, Nav1.6 (tetrodotoxin-sensitive) and Nav1.5 (tetrodotoxin-insensitive). Reportedly, Nav1.6 is the most abundant isoform that also participate in the modification of microglial functions. In an experiment of primary cultures, mice lacking Nav1.6 express decrement in the LPS-exposed phagocytosis (Craner et al. 2005).
2.5.3 Calcium-Permeable Channels
Expression of classical voltage-operated Ca2+ channels is considered to be absent in microglia in the CNS (both in vivo and in vitro). Store-operated channels and channels of TRP family are the two main types of Ca2+ permeable channels in microglia. Similar to all other non-excitable cells, microglial cells also possess store-operated Ca2+ entry that was mediated by the Ca2+ release activated channels, i.e. TRP channels.
2.5.4 Calcium Signalling in Microglia
Calcium signalling is a homeostatic mechanism controlled by an evolutionary conserved cascade of molecules, that directs both intracellular calcium buffering and calcium transportation across the cellular membrane (Petersen et al. 2005). In resting microglial cells, calcium signalling is triggered by the calcium entry through ligand-gated plasmalemma and store-operated calcium permeable channels that further direct the release of intracellular stored calcium (Färber and Kettenmann 2006a). Microglia constitute both type of intracellular calcium channels, i.e. ryanodine receptors (RyRs) and inositol 1,4,5-trisphosphate (InsP3)-gated Ca2+ (InsP3Rs). Microglial calcium signalling is mainly initiated by InsP3Rs that further activates the G-protein-coupled metabotropic receptors connected to phospholipase C (PLC; Kettenmann et al. 2011).
2.5.5 Potassium Channels
Potassium channels (Kv) were credited as one of the first ion channels characterized in the microglia (Kettenmann et al. 1990). Precisely, the inward rectifier Kv (KIR), is the first marker channel identified as the marker of activated microglia. Potassium channels in microglia have largely been studied in cultures and/or in tissue slices. The inward rectifier K+ currents are the main source of membrane permeability in invading amoeboid microglia during perinatal brain development. Such currents become almost undetectable as the cells get transferred to their ramified surveillance (or so called resting) states (Boucsein et al. 2000).
2.5.6 Anion Channels
Microglial proliferation, phagocytic activity, the control of ramified morphology mainly of cell volume and microglial resting potential are all considered to be regulated by the volume-regulated Cl− channels. These channels are activated by a hypo-osmotic state. Such channels have been largely studied in microglia cultures and such cells also express the chloride intracellular channel-1 (CLIC-1). CLIC-1 play a major role in release of pro-inflammatory factors from microglia and have been considered to play an important role in progression of brain disorders.
2.5.7 Proton Channels
Functionally H+ channels are considered to be associated with the regulation of respiratory bursts in phagocytic states of microglia. These are voltage-operated proton channels with single channel conductance having high selectivity to H+. Extracellular pH is supposed to be regulating these channel expression in microglia in culture. Activated states of microglia decrease the H+ current in cell culture experiments. In respiratory bursts, activation of NADPH oxidase generates protons and superoxide anions. These ions are effluxed by H+ channels and protect the cytosol by regulating the intracellular pH.
2.6 Potent Immune Response During Brain Insult: Neuroinflammation
Neuroinflammation is an essential biological progression that stands as the foreground of various acute and chronic neuropathological conditions. Any alteration in brain’s cellular and functional integrity grounds the incidence of neuroinflammation. Neuroinflammation as a defending responder aims to refurbish the tissue homeostasis via inducing several repair processes (Goldszmid and Trinchieri 2012). However, if the regulation of this mechanism remains uncontrolled then the initial inflammatory response amplifies exceedingly and the protective mode shifts towards the collateral destruction that would further result in severe disease progression.
Neuroinflammation is a dynamic process in which both microglia and astroglia may migrate, proliferate, release potentially harmful factors (i.e. cytokines and reactive oxygen species), display different surface proteins (i.e. MHC-I/II, etc.) and blend in functions such as antigen presentation and phagocytosis in response to signals like protein aggregates, neuronal degeneration and glial products (i.e. colony stimulating factor and cytokines, etc.). Cytokine signalling following neuroinflammation is actively involved in the regulation of various brain functions like synaptic signalling modulation, neurotransmission, neuroendocrine functions and neural circuitry of behaviour and cognition (Camacho-Arroyo et al. 2009; del Rey et al. 2013; Aprile-Garcia et al. 2013; Cuartas and Jorge 2014). Therefore, it is relatively apparent to presume an altered behavioural and cognitive outcome as a consequence of a dysregulation in cytokine signalling which might result in depression, anxiety, behavioural deficits and cognitive dysfunction as observed previously (Lynch 2002; Bains and Oliet 2007; Baune et al. 2008; McAfoose and Baune 2009). Additionally in various cross-sectional and prospective population studies, it was shown that any alteration in the level of these cytokines in hippocampus lead to Alzheimer’s disease (AD), dementia and cognitive impairment (Dik et al. 2005; Magaki et al. 2007; Holmes et al. 2009; Brosseron et al. 2014).
2.7 Microglia in Immune Regulation
Following injury (Patro et al. 2005, 2010a), inflammation (Patro and Patro 2004; Patro et al. 2010b), blood–brain barrier (BBB) damage (Davies et al. 1998) or metabolic disorder (Nagayach et al. 2014a, b) associated stimuli, microglia get activated. Microglia have also been activated exogenously by various inflammatory stimuli such as lipopolysaccharide (Rivest 2003; Sharma et al. 2015), β-amyloid (Sondag et al. 2009), interferon-γ (Chao et al. 1993), thrombin (Möller et al. 2006), Poly I:C (Patro and Patro 2004), etc. for screening the stature and pathological role of activated microglia. Such activated microglia release an array of immunocompetent molecules comprising of numerous chemokines like KC, MIP-1α (Macrophage-Inflammatory Protein-1α), MIP-1β, MIP-2, MCP-1 (Monocyte chemoattractant protein-1), RANTES (regulated on activation, normal T cell expressed and secreted), IP-10 (IFN-γ-inducible protein-10), and interleukins like IL-1α/β, IL-3, IL-6, IL-10, IL-12, IL-15, IL-18, tumour necrosis factor α (TNF-α), interferon gamma inducing factor (IGIF), inflammatory proteins, TGF-β, etc. Collectively, these molecules not only control the inflammatory processes, but also regulate immune response of the brain and even contribute to neuropathogenesis in CNS inflammation. Activated microglia also promote neuroprotection by releasing anti-inflammatory molecules and growth factors like nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3), basic fibroblast growth factor, etc. following immunological stimuli (Hanisch and Kettenmann 2007). In conclusion, microglia as immune regulators of the nervous system can secrete several types of molecules or express various receptors that facilitate the integration of microglial response towards the changing microenvironment (Saxena et al. 2007; Neumann et al. 2008; Patro et al. 2014).
2.7.1 Phagocytic Behaviour of Microglial Cells
Microglia that maintain homeostasis in normal cells that include phagocytic clearance of neuronal damage products or debris. During early development they are innate immune cells and clear the supernumerary synaptic processes and apoptotic neurons. In the adult CNS this phagocytic ability becomes a boon following injury and associated frequent loss of neurons and microglial recruitment at the site of injury. Such microglial presence does more than just debris clearance, which includes axonal and myelin debris in spinal cord injury or multiple sclerosis, amyloid-β deposits in AD, etc. Earlier in this review, we have explained how inefficiency of microglia not only affects clearing up the injury site but also fail reorganization of the neuronal circuits. With age, such inefficiency also enhances the prevalence of neurodegenerative disease and inadequate regeneration. However, the mechanism, action and consequence of microglial phagocytosis have not been deciphered. Thus, there is now a call for considering new therapeutic avenues involving the mechanisms of microglia-mediated tissue repair.
While phagocytosis is beneficial as it cleans up CNS and induces anti-inflammatory response but also produces toxic ROS which we have discussed above. For more details, we would refer you to Neumann and Takahashi (2007) and Sierra et al. (2013). In physiological conditions the highly motile ramified processes respond to the chemotactic ‘find me’ signals like fractalkine, ATP, UDP, etc. from apoptotic cells. Subsequently ‘Eat me’ signals, i.e. the ligands for a plethora of microglial receptors are produced by the apoptotic cells that manifest cutting and engulfing of apoptotic debris. Following this the phagocytic microglia with the help of lysosomes and other organelles finally degrade and digest the debris.
2.8 Microglial Pathophysiology Following Neuroinflammation
2.8.1 ATP Signalling
Microglia gets activated through various signalling pathways including chemokine/ chemokine receptors, nucleotides/purinergic receptors (P1, P2X and P2Y) and high-mobility group box (HMGB)/toll-like receptors. Consequently, activated microglia may secrete various soluble factors that act in inflammatory, trophic or protective manner (Suzuki et al. 2004; Di Virgilio et al. 2009). Microglia express purinergic (P2) receptors by means of elevated concentrations of extracellular ATP-induced intracellular Ca2+ elevation in a receptor-dependent manner (Fig. 2.3; Ferrari et al. 1996). Different concentrations of purine mediate ATP- and ADP-induced microglial chemokinesis and chemotaxis (Honda et al. 2001; Davalos et al. 2005). ATP signalling plays a major role not only in normal CNS function but also during the pathological states. Signals triggering microglia activation following any insult are directed by the release of purine nucleotides, comprising ADP, ATP and UTP by damaged neuronal cells (Nimmerjahn et al. 2005; Domercq et al. 2013). These signals are received by the P2Y receptors present in both microglia and astrocytes-mediating chemotactic response of microglia. During neuroinflammatory condition, the extracellular ATP concentration increase is noted that further inhibit activation and overexpression of P2X7 receptors in microglial cells.

Microglial physiology following neuroinflammation: Neuroinflammation mediated by various brain insults result in neuronal damage. Dying neurons and activated microglia themselves increases the level of extracellular ATP that further triggers the purinergic receptors (P2Y, P2X4 and P27) present in the microglial cells. Activation of these receptors trigger an inward cationic current and initiate a cascade of second messenger signalling via G protein-phospholipase C (PLC) signal transduction pathway. This further elevates intracellular calcium. Iontropic receptors, P2X are involved in the expression, posttranslational processing and secretion of several inflammatory molecules and reactive oxidative radicals (ROS, RNS). P2X7 receptors eventually mediate apoptosis by caspase activation that also modulates the secretion of inflammatory molecules. Excessive release of glutamate via dying neurons activates glutamate receptors in microglia and exert inflammatory effect and aggravate the pro-inflammatory action
2.8.2 Sodium Channel Signalling
Microglial voltage-gated sodium channels (Nav) are involved in a wide range of regulatory functions such as proliferation, morphological alterations, migration and phagocytosis (Eder 2005; Black et al. 2009) in response to inflammatory stimulus. Activated sodium channels stimulate a transient and rapid depolarization in microglial cells. Such depolarization of microglial cell membrane triggers the signalling cascades that further activates the microglial cell and subsequent immune activation. Furthermore, activated microglial membrane depolarization is a critical participant in the conformational change of MHC I molecule, which is essentially required for the antigen presentation functions (Bene et al. 1997). Studies on human multiple sclerosis lesions and animal models of experimental autoimmune encephalopathy (EAE), have indicated that expression of Nav 1.6 isoform of sodium channel in activated microglia gets increased (Craner et al. 2005). Consequently, the blocking of these channels was evidently used in developing the antiepileptic therapies and drugs (Black et al. 2007).
2.8.3 Potassium Channel Signalling
Inward rectifier K+ currents are generally recorded in the activated states of microglia in various pathological conditions. Insults to the nervous tissue like ischemia, peripheral nerve damage have been recorded to induce a several fold increase in the amplitudes of inward rectifier K+ channels (Boucsein et al. 2000).
The activated (delayed) receptor K+ channels in microglia are KV1.2, KV1.3 and KV1.5. The microglia in prenatal brain also express KV1.1 and KV1.2 channels. These channels are responsible for the increased delay rectifier currents in the activated state of microglia. An increased expression of delayed rectifier channels is considered as indicatives of functional responses of hyperactive microglia and during active proliferation. Such increased expression have been experimentally evidenced in microglial cells in cultures with LPS or interferon-γ (Norenberg et al. 1992), in situ following axotomy (Boucsein et al. 2000) and as a reference to microglial activation following exposure to experimental condition like in vitro exposure to LPS, interferon-γ, β-amyloid or HIV-1 regulating protein Tat, etc. (Norenberg et al. 1992; Boucsein et al. 2000). High conductance (BK) and small conductance (KCNNG/KCa3.1/SK4/IK1) type Ca2+ dependent potassium (KCa) channels are also considered to be responsible in regulation of activated state of microglia in various pathological conditions (Schlichter et al. 2010). Interestingly, studies have also reported that the stimulation of ATP-sensitive K+ channels (KATP) decreases the probability of microglial activation and are neuroprotective in several models of neurodegeneration involving neuroinflammation (Dolga and Culmsee 2012; Ortega et al. 2012).
2.8.4 Calcium Signalling
Calcium receptor activation generates two intracellular second messengers, the InsP3 and the diacylglycerol (DAG) in activated microglia which further in response activates InsP3Rs of the endoplasmic reticulum, and thus directs calcium release that regulates various cellular functions. In cell culture of rodent microglia, Ca2+ release activates Ca2+ currents (ICRAC). In activated microglia, the amplification of such currents gets decreased. ICRAC occurs after the activation of a complex of ORAI (pore forming) and STIM (Ca2+ sensor) protein (Ohana et al. 2009). Microglial cells also express a range of TRPM, TRPV and TRPC channels considered to produce intracellular Ca2+ signals regulating release of cytokines.
2.8.5 Neurotransmitter Receptors
Microglia express several neurotransmitter receptors (Färber and Kettenmann 2006a, b) such as glutamate receptors (AMPA/Kainate, NMDA receptors, metabotropic glutamate receptors, GABA, adrenergic, dopaminergic and cholintergic receptors. Interestingly, the pathological and physiological role of these receptors in microglial cells is still under investigation (Fig. 2.3). Although studies had depicted that these neurotransmitters could exert inflammatory (both pro- and anti-) effects on microglial cells (Hagino et al. 2004; Pocock and Kettenmann 2007). Like, GABA receptors can modulate the interleukin (IL-6 and IL-12) release and glutamate receptors are capable in controlling the TNF-α release. Microglial activation of metabotropic glutamate receptors induces TNF-α and Fas ligand secretion which further trigger the neuronal caspase-3 activation through Fas receptor and TNFR1 (also known as p55), leading to neuronal damage (Taylor et al. 2005). Via receiving the signals from dying neurons microglia NMDA receptor gets activated and triggers the secretion of neurotoxic factors through microglia directing towards the microglia ability of inducing and aggravating the neurological damage (Kaindl et al. 2012). In the neurodegenerative diseases like hypoxia, AD and multiple sclerosis the altered concentration and expression of glutamate receptors in microglia depict the possibility of glutamate mediated toxicity in the progression of these pathological states (Newcombe et al. 2008; Sivakumar et al. 2010). Furthermore, a lipopolysaccharide (LPS) induced activated microglia culture study shows the releases of pro-inflammatory molecules which was attenuated by the simultaneous activation of the GABA(B) receptors directing towards the role of GABA(B) receptors in the modulation of microglia immune response (Kuhn et al. 2004).
The role and relevance of ATP and calcium signalling and neurotransmitters in microglia during neuroinflammatory conditions of several pathological diseases makes them a valuable target for developing therapeutic strategies for neuroprotection. Aspects of neurotransmitter signalling in the pathophysiology of microglia have been aptly reviewed by Domercq et al. (2013).
2.9 Microglia in Neurological Diseases
Neuroinflammatory mechanism is comprised of an organized set of interaction between varied mediators like cytokines, chemokines and prostaglandins, etc. Rather than pathological conditions, the secretion of inflammatory molecules following neuroinflammation is highly influenced by the microglial activation. In response to any injury, insult or disparaged conditions, the apparent activation of microglia triggers the secretion of inflammatory molecules (Fig. 2.2) that circumstantially become superfluous at chronic glial activation (Block et al. 2007).
Microglia through various pattern–recognition receptors (like Toll-like receptors, NOD-like receptors, receptors of cell wall components or DNA/RNA of pathogens), purinergic receptors, advanced glycation endproducts receptors and scavenger receptors receive signals from injured or dying cells and vascular damage (Block et al. 2007; Brown and Neher 2010). Connections of these receptors initiate a microglial activation cascade with the expression of various proteins, comprising CD-45, COX-II, iNOS, MHC-II and various co-stimulatory molecules amongst others. These molecules facilitate the microglial expression of antigens to T cells, entered through the damaged BBB during neuroinflammation (Aloisi 2001; Carson 2005; Gertig and Hanisch 2014). Activity-dependent microglial morphological heterogenity and population segregation has previously been discussed and exemplified via the proliferative ability and/or release spectrum of the constitutive or inducible mRNAs, proteins (e.g. major histocompatibility complex class II, TNF-α, IL-6/12/1β, integrins, IGF-I, CD4/11c/34/40/86/45, FcγRII, iNOS and molecules of the neurotrophin family), superoxide anions, nitric oxide synthase and proteases, etc. redox (NOX2, NOX1, RAC1, iNOS, NOS2, etc.) and excitotoxic molecules (MGluR2, GLT-1, P2X7-R, etc.).
Furthermore, microglia also shares a bidirectional collaborative relationship with neurons that assumed to be imperative in establishing a pertinent physiological, behavioural and immunological response against any injury or disorder. Neurons via a set of unique ligand-receptor pairs (CX3CL1-CX3CR1 and CD200-CD200R), microRNA-124 (mir-124), neurotransmitters (GABA, glutamate, catecholamines), peptides and/or growth factors, CD22, CCL21, fraktalkine (that act on receptors present on microglial membrane) maintain and regulate the microglial activation (Gomes-Leal 2012; Eyo and Wu 2013).
As described above, the secretion of pro-inflammatory cytokines following microglia activation is the classic theory of neuroinflammation recognized and reviewed widely (Carson et al. 2006; Luo and Chen 2012; Boche et al. 2013). Microglia activation is generally accompanied by the proliferation of cells, mobilization towards the damaged or dying cell and the expression and secretion of pro-inflammatory cytokines, like IL-6, TNFα. IL-1β and chemokines, such as cytokine (C-C motif) ligand (CCL)2, CCL3, CCL4, CCL5, CXCL10 and/or CCL12 (Olson and Miller 2004; Semple et al. 2010). Later on, inflammatory molecules stimulate other astroglia and microglia leading to the exacerbation of glial (microglia and astroglia) activation. Furthermost alterations in cytokines expression are a result of stimulation of the transcription factor NF-κB (nuclear factor kappa enhancer of B cells) via phosphorylation-induced activation of IκB kinase (Brown and Neher 2010). Neurotransmitter signalling in the pathophysiology of microglia has been aptly reviewed by Domercq et al. (2013).
During pathological condition, cellular damage following activation of microglial cells further initiates and perpetuates the state of excitoxicity and oxidative stress within the brain. The oxidative stress and cell death caused by the microglial activation are also contributing in the generation and propagation of pro-inflammatory cytokines as documented widely (Shi et al. 2013; Sandireddy et al. 2014; Muriach et al. 2014). Furthermore, oxidative stress leads to the excessive dicarbonyl glycation which further activates the calpain expression and degrades the brain-derived neurotrophic factor (BDNF). This contributes to retard the process of neurogenesis and synaptic plasticity and stimulates NFkB-dependent inflammation and secretion of inflammatory molecules. Recurrent increment in cytokine levels increases the permeability of BBB to peripheral immune molecules and prolongs the central immune inflammatory response that generates an inordinate environment of oxidative and inflammatory stresses, accelerating CNS damage and elicits adverse structural and functional consequences. Intriguingly, reciprocal relationship between neuroinflammation, cell death and microglial activation is popularly considered as a prerequisite for the onset and pathogenesis of various psychiatric disorders like AD, PD, dementia and bipolar disorder, etc. (Hojo et al. 2004; Enciu and Popescu 2013; Najjar et al. 2013; Watkins et al. 2014).
Interestingly microglia plays a dual role during various neurological insults including neuroinflammation in CNS (Table 2.1). On the basis of secretory molecules released by the activated microglial cells microglial activation is divided into two major phases (Fig. 2.1), i.e. classical activation (M1 phase) and alternate activation (M2 phase; Colton 2009). During classical activation, microglia get triggered by the activated Toll-like receptors (TLRs) through intracellular proteins or pathogen-associated molecular patterns (PAMPs) released from injured neurons and release an array of inflammatory molecules (TNF-α, IL-6, IL-1β, IL-12), redox molecules (NOX2, NOX1, RAC1, iNOS, NOS2) and excitotoxic molecules (MGluR2, GLT-1, P2X7-R), nitric oxide synthase, superoxide anions, etc. (Ransohoff and Brown 2012; Boche et al. 2013) that further resulted in blood–brain barrier (BBB) disruption. The BBB damage promotes the infiltration of macrophages which later on exaggerate the influx of inflammatory cytokines and aggravate the existing neuroinflammatory state in CNS (Fig. 2.2; Zipser et al. 2007; Lassman et al. 2012; Obermeier et al. 2013). While in alternate activation, microglia secrete molecules like trophic growth factors, IL-4, IL-13, IL-1RA, scavenging receptors and extracellular matrix (Luo and Chen 2012; Cherry et al. 2014). Considering the imperious role of microglia in neuroinflammation, a particular attention is warranted on the microglia mediated neuro-immunological aspects of neurodegeneration and neuroregulation.
In conclusion, brain function and dysfunction, without any reservations, has a direct connection with microglial forms and functional states. They contribute to the pathogenesis and progression of various neurological disorders. Microglial activation also acts as a defence mechanism for various insults to the brain including infections. Being the immune cells of the CNS, they protect and repair the damage as also facilitate the healing process. Our understanding even today on microglial functions in normal and diseased brain is limited. Further insights on the physiology and pathophysiology of microglia using in vivo models are likely to contribute to our knowledge on the mechanisms and role of neuroinflammation for prevention or progression of brain disorders.
References
Alliot F, Lecain E, Grima B, Pessac B (1991) Microglial progenitors with a high proliferative potential in the embryonic and adult mouse brain. Proc Natl Acad Sci USA 88(4):1541–1545
Aloisi F (2001) Immune function of microglia. Glia 36(2):165–179
Aprile-Garcia F, Antunica-Noguerol M, Budziñski ML, Liberman AC, Arzt E (2013) Novel insights into the neuroendocrine control of inflammation: the role of GR and PARP1. Endocr Connect 3(1):R1–R12
Bains JS, Oliet SH (2007) Glia: they make your memories stick! Trends Neurosci 30(8):417–424
Bansal R (2002) Fibroblast growth factors and their receptors in oligodendrocyte development: implications for demyelination and remyelination. Dev Neurosci 24(1):35–46
Baune BT, Ponath G, Rothermundt M, Riess O, Funke H, Berger K (2008) Association between genetic variants of IL-1beta, IL-6 and TNF-alpha cytokines and cognitive performance in the elderly general population of the MEMO-study. Psychoneuroendocrinology 33(1):68–76
Ben Achour S, Pascual O (2010) Glia: the many ways to modulate synaptic plasticity. Neurochem Int 57(4):440–445
Bene L, Szollosi J, Balazs M, Matyus L, Gaspar R, Ameloot M, Dale RE, Damjanovich S (1997) Major histocompatibility complex class I protein conformation altered by transmembrane potential changes. Cytometry 27:353–357
Benoit M, Benoit D, Jean-Louis M (2008) Macrophage polarizaition in bacterial infections. J Immunol 181:3733–3739
Black JA, Waxman SG (2012) Sodium channels and microglial function. Exp Neurol 234(2):302–315
Black JA, Liu S, Carrithers M, Carrithers LM, Waxman SG (2007) Exacerbation of experimental autoimmune encephalomyelitis after withdrawal of phenytoin and carbamazepine. Ann Neurol 62:21–33
Black JA, Liu S, Waxman SG (2009) Sodium channel activity modulates multiple functions in microglia. Glia 57(10):1072–1081
Block ML, Hong JS (2005) Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol 76(2):77–98
Block ML, Zecca L, Hong JS (2007) Microglia–mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci 8(1):57–69
Boche D, Perry VH, Nicoll JA (2013) Review: activation patterns of microglia and their identification in the human brain. Neuropathol Appl Neurobiol 39:3–18
Boucsein C, Kettenmann H, Nolte C (2000) Electrophysiological properties of microglial cells in normal and pathologic rat brain slices. Eur J Neurosci 12(6):2049–2058
Boya J, Calvo JL, Carbonell AL, Borregon A (1991) A lectin histochemistry study on the development of rat microglial cells. J Anat 175:229–236
Brosseron F, Krauthausen M, Kummer M, Heneka MT (2014) Body fluid cytokine levels in mild cognitive impairment and Alzheimer’s disease: a comparative overview. Mol Neurobiol 50(2):534–544
Brown GC, Neher JJ (2010) Inflammatory neurodegeneration and mechanisms of microglial killing of neurons. Mol Neurobiol 41(2–3):242–247
Camacho-Arroyo I, López-Griego L, Morales-Montor J (2009) The role of cytokines in the regulation of neurotransmission. NeuroImmunomodulation 16(1):1–12
Carson MJ (2005) Microglia-the professional antigen-presenting cells of the CNS? In: Kropshofer H, Vogt AB (eds) Antigen presenting cells: from mechanisms to drug development. Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, FRG. doi:10.1002/3527607021.ch12
Carson MJ, Doose JM, Melchior B, Schmid CD, Ploix CC (2006) CNS immune privilege: hiding in plain sight. Immunol Rev 213:48–65
Chao CC, Hu S, Gekker G, Novick WJ Jr, Remington JS, Peterson PK (1993) Effects of cytokines on multiplication of Toxoplasma gondii in microglial cells. J Immunol 150(8 Pt 1):3404–3410
Chen J, Buchanan JB, Sparkman NL, Godbout JP, Freund GG, Johnson RW (2008) Neuroinflammation and disruption in working memory in aged mice after acute stimulation of the peripheral innate immune system. Brain Behav Immun 22(3):301–311
Cherry JD, Olschowka JA, O’Banion MK (2014) Are “resting microglia more M2”? Front Immunol. doi:10.3389/fimmu.2014.00594
Colton CA (2009) Heterogeneity of microglial activation in the innate immune response in the brain. J Neuroimmune Pharmacol 4:399–418
Coull JA, Beggs S, Boudreau D, Boivin D, Tsuda M, Inoue K et al (2005) BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 438(7070):1017–1021
Craner MJ, Damarjian TG, Liu S et al (2005) Sodium channels contribute to microglia/macrophage activation and function in EAE and MS. Glia 49:220–229
Cuartas A, Jorge M (2014) Cognition and inflammation: the role of cytokines in cognitive performance. Int J Psychol Res 7(2):8–10
Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S et al (2005) ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 8:752–758
Davies CA, Loddick SA, Stroemer RP, Hunt J, Rothwell NJ (1998) An integrated analysis of the progression of cell responses induced by permanent focal middle cerebral artery occlusion in the rat. Exp Neurol 154(1):199–212
del Rey A, Balschun D, Wetzel W, Randolf A, Besedovsky HO (2013) A cytokine network involving brain-borne IL-1β, IL-1ra, IL-18, IL-6, and TNFα operates during long-term potentiation and learning. Brain Behav Immun 33:15–23
Del Rio-Hortega P (1932) Microglia. In: Penfield W (ed) Cytology and cellular pathology of the nervous system. Paul B Hoeber, New York, pp 482–534
Di Virgilio F, Ceruti S, Bramanti P, Abbracchio MP (2009) Purinergic signalling in inflammation of the central nervous system. Trends Neurosci 32:79–87
Dik MG, Jonker C, Hack CE, Smit JH, Comijs HC, Eikelenboom P (2005) Serum inflammatory proteins and cognitive decline in older persons. Neurology 64(8):1371–1377
Dilger RN, Johnson RW (2008) Aging, microglial cell priming, and the discordant central inflammatory response to signals from the peripheral immune system. J Leukoc Biol 84(4):932–939
Dolga AM, Culmsee C (2012) Protective roles for potassium SK/KCa2 channels in microglia and neurons. Front Pharmacol 3:196
Domercq M, Vázquez-Villoldo N, Matute (2013) Neurotransmitter signaling in the pathophysiology of microglia. Front Cell Neurosci 7:49
Eder C (2005) Regulation of microglial behavior by ion channel activity. J Neurosci Res 81:314–321
Ekdahl CT, Kokaia Z, Lindvall O (2009) Brain inflammation and adult neurogenesis: the dual role of microglia. Neuroscience 158(3):1021–1029
Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA et al (2014) Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 82(2):380–397
Enciu AM, Popescu BO (2013) Is there a causal link between inflammation and dementia? Biomed Res Int 2013:316495
Eyo UB, Wu LJ (2013) Bidirectional microglia-neuron communication in the healthy brain. Neural Plast. doi:10.1155/2013/456857
Färber K, Kettenmann H (2005) Physiology of microglial cells. Brain Res Rev 48(2):133–143
Färber K, Kettenmann H (2006a) Functional role of calcium signals for microglial function. Glia 54(7):656–665
Färber K, Kettenmann H (2006b) Purinergic signaling and microglia. Pflugers Arch 452(5):615–621
Farinas I, Cano-Jaimez M, Bellmunt E, Soriano M (2002) Regulation of neurogenesis by neurotrophins in developing spinal sensory ganglia. Brain Res Bull 57(6):809–816
Ferrari D, Villalba M, Chiozzi P, Falzoni S, Ricciardi-Castagnoli P, Di Virgilio F (1996) Mouse microglial cells express a plasma membrane pore gated by extracellular ATP. J Immunol 156:1531–1539
Gehrmann J, Matsumoto Y, Kreutzberg JW (1995) Microglia: intrinsic immuneffector cell of the brain. Brain Res Brain Res Rev 20(3):269–287
Gertig U, Hanisch UK (2014) Microglial diversity by responses and responders. Front Cell Neurosci 8:101
Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S et al (2010) Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330(6005):841–845
Ginhoux F, Lim S, Hoeffel G, Low D, Huber T (2013) Origin and differentiation of microglia. Front Cell Neurosci 7:45
Godbout JP, Johnson RW (2009) Age and neuroinflammation: a lifetime of psychoneuroimmune consequences. Immunol Allergy Clin North Am 29(2):3213–3237
Goldszmid RS, Trinchieri G (2012) The price of immunity. Nat Immunol 13(10):932–938
Gomes-Leal W (2012) Microglial physiopathology: how to explain the dual role of microglia after acute neural disorders? Brain Behav 2(3):345–356
Gomez PE, Schulz C, Geissmann F (2013) Development and homeostasis of “resident” myeloid cells: the case of the microglia. Glia 61(1):112–120
Gregory CD, Devitt A (2004) The macrophage and the apoptotic cell: an innate immune interaction viewed simplistically? Immunology 113:1–14
Hagino Y, Kariura Y, Manago Y, Amano T, Wang B et al (2004) Heterogeneity and potentiation of AMPA type of glutamate receptors in rat cultured microglia. Glia 47:68–77
Hanisch UK, Kettenmann H (2007) Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci 10(11):1387–1394
Hayashi Y, Ishibashi H, Hashimoto K, Nakanishi H (2006) Potentiation of the NMDA receptor-mediated responses through the activation of the glycine site by microglia secreting soluble factors. Glia 53(6):660–668
Hellwig S, Heinrich A, Biber K (2013) The brain’s best friend: microglial neurotoxicity revisited. Front Cell Neurosci 7:71
Heneka MT, Rodriguez JJ, Verkhratsky A (2010) Neuroglia in neurodegeneration. Brain Res Rev 63(1–2):189–211
Hickey WF, Kimura H (1988) Perivascular microglial cells of the CNS are bone marrow-derived and present antigen in vivo. Science 239:290–292
Hickey WF, Vass K, Lassmann H (1992) Bone marrow-derived elements in the central nervous system: an immunohistochemical and ultrastructural survey of rat chimeras. J Neuropathol Exp Neurol 51(3):246–256
Hojo K, Kawamata T, Tanaka C, Maeda K (2004) Inflammatory glial activation in the brain of a patient with hereditary sensory neuropathy type 1 with deafness and dementia. Neurosci Lett 367(3):340–343
Holmes C, Cunningham C, Zotova E, Woolford J, Dean C, Kerr S, Culliford D, Perry VH (2009) Systemic inflammation and disease progression in Alzheimer disease. Neurology 73(10):768–774
Honda S, Sasaki Y, Ohsawa K, Imai Y, Nakamura Y, Inoue K et al (2001) Extracellular ATP or ADP induce chemotaxis of cultured microglia through Gi/o-coupled P2Y receptors. J Neurosci 21:1975–1982
Hughes EG, Bergles DE (2014) Hidden progenitors replace microglia in the adult brain. Neuron 82(2):253–255
Kaindl AM, Degos V, Peineau S, Gouadon E, Chhor V et al (2012) Activation of microglial N-methyl-d-aspartate receptors triggers inflammation and neuronal cell death in the developing and mature brain. Ann Neurol 72:536–549
Kaur C, Ling EA, Wong WC (1985) Transformation of amoeboid microglial cells into microglia in the corpus callosum of the postnatal rat brain. An electron microscopical study. Arch Histol Jpn 48(1):17–25
Kettenmann H, Hoppe D, Gottmann K, Banati R, Kreutzberg G (1990) Cultured microglial cells have a distinct pattern of membrane channels different from peritoneal macrophages. J Neurosci Res 26(3):278–287
Kettenmann H, Hanisch UK, Noda M, Verkhratsky A (2011) Physiology of microglia. Physiol Rev 91(2):461–553
Kuhn SA, van Landeghem FK, Zacharias R, Färber K, Rappert A, Pavlovic S, Hoffmann A, Nolte C, Kettenmann H (2004) Microglia express GABA(B) receptors to modulate interleukin release. Mol Cell Neurosci 25(2):312–322
Kushwaha S (2009) Age related changes in astrocytes and microglia in hippocampus and striate cortex. PhD thesis, Jiwaji University, Gwalior, India, p 93
Lassman ME, McLaughlin TM, Somers EP, Stefanni AC, Chen Z, Murphy BA et al (2012) A rapid method for cross-species quantitation of apolipoproteins A1, B48 and B100 in plasma by ultra-performance liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Spectrom 26(2):101–108
Lassmann H, Hickey WF (1993) Radiation bone marrow chimeras as a tool to study microglia turnover in normal brain and inflammation. Clin Neuropathol 12(5):284–285
Luo XG, Chen SD (2012) The changing phenotype of microglia from homeostasis to disease. Transl Neurodegeneration 1:9
Lynch MA (2002) Interleukin-1 beta exerts a myriad of effects in the brain and in particular in the hippocampus: analysis of some of these actions. Vitam Horm 64:185–219
Magaki S, Mueller C, Dickson C, Kirsch W (2007) Increased production of inflammatory cytokines in mild cognitive impairment. Exp Geron 42(3):233–240
Martinez FO, Sica A, Mantovani A, Locati M (2008) Macrophage activation and polarization. Front Biosci 13:453–461
Matsuda S, Niidome T, Nonaka H, Goto Y, Fujimura K, Kato M et al (2008) Microtubule–associated protein 2–positive cells derived from microglia possess properties of functional neurons. Biochem Biophys Res Commun 368(4):971–976
McAfoose J, Baune BT (2009) Evidence for a cytokine model of cognitive function. Neurosci Biobehav Rev 33(3):355–366
Miller FD, Kaplan DR (2001) Neurotrophin signalling pathways regulating neuronal apoptosis. Cell Mol Life Sci 58(8):1045–1053
Möller T, Weinstein JR, Hanisch UK (2006) Activation of microglial cells by thrombin: past, present and future. Semin Thromb Hemost 32(Suppl 1):69–76
Monier A, Evrard P, Gressens P et al (2006) Distribution and differentiation of microglia in the human encephalon during the first two trimesters of gestation. J Comp Neurol 499(4):565–582
Muriach M, Flores-Bellver M, Romero FJ, Barcia JM (2014) Diabetes and the brain: oxidative stress, inflammation, and autophagy. Oxid Med Cell Longev 2014:102158. doi:10.1155/2014/102158
Nagayach A, Patro N, Patro I (2014a) Astrocytic and microglial response in experimentally induced diabetic rat brain. Metab Brain Dis 29(3):747–761
Nagayach A, Patro N, Patro I (2014b) Experimentally induced diabetes causes glial activation, glutamate toxicity and cellular damage leading to changes in motor function. Front Cell Neurosci 8:355. doi:10.3389/fncel.2014.00355
Nagayach A, Patro N, Patro I (2015) Microglia in physiology and pathology of brain. Proc Natl Acad Sci, India, Sect-B. doi:10.1007/s40011-
Najjar S, Pearlman DM, Alper K, Najjar A, Devinsky O (2013) Neuroinflammation and psychiatric illness. J Neuroinflammation 10:43
Nakajima K, Honda S, Tohyama Y, Imai Y, Kohsaka S, Kurihara T (2001) Neurotrophin secretion from cultured microglia. J Neurosci Res 65(4):322–331
Nayak D, Roth TL, McGavern DB (2014) Microglia development and function. Annu Rev Immunol 32:367–402
Neiva I, Malva JO, Valero J (2014) Can we talk about microglia without neurons? A discussion of microglial cell autonomous properties in culture. Front Cell Neurosci 8:202
Neumann H, Takahashi K (2007) Essential role of microglial triggering receptor expressed on myeloid cells-2 (TREM-2) for central nervopus tissue immune homeostasis. J Neuroimmunol 184:92–99
Neumann H, Kotter MR, Franklin JM (2008) Debris clearance by microglia: an essential link between degenereation and regeneration. Brain 132(2):288–295
Newcombe J, Uddin A, Dove R, Patel B, Turski L et al (2008) Glutamate receptor expression in multiple sclerosis lesions. Brain Pathol 18:52–61
Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308:1314–1318
Niquet J, Ben-Ari Y, Repressa A (1994) Glial reaction after seizure induced hippocampal lesion: immunohistochemical characterization of proliferating glial cells. J Neurocytol 23(10):641–656
Norenberg W, Gebicke-Haerter PJ, Illes P (1992) Inflammatory stimuli induce a new K+ outward current in cultured rat microglia. Neurosci Lett 147:171–174
Obermeier B, Daneman R, Ransohoff RM (2013) Development, maintenance and disruption of the blood-brain barrier. Nat Med 19(12):1584–1596
Ohana L, Newell EW, Stanley EF, Schlichter LC (2009) The Ca2+ release-activated Ca2+ current (I(CRAC)) mediates store-operated Ca2+ entry in rat microglia. Channels (Austin) 3(2):129–139
Olson JK, Miller SD (2004) Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol 173:3916–3924
Ortega FJ, Gimeno-Bayon J, Espinosa-Parrilla JF et al (2012) ATP-dependent potassium channel blockade strengthens microglial neuroprotection after hypoxia-ischemia in rats. Exp Neurol 235:282–296
Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P et al (2011) Synaptic pruning by microglia is necessary for normal brain development. Science 333(6048):1456–1458
Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR 3rd, Lafaille JJ, Hempstead BL, Littman DR, Gan WB (2013) Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155(7):1596–1609
Parpura V, Heneka MT, Montana V, Oliet SH, Schousboe A, Haydon PG et al (2012) Glial cells in (patho)physiology. J Neurochem 121(1):4–27
Pascual O, Ben Achour S, Rostaing P, Triller A, Bessis A (2012) Microglia activation triggers astrocyte-mediated modulation of excitatory neurotransmission. Proc Natl Acad Sci USA 109(4):E197–E205
Patro N, Patro IK (2004) Effect of immunactivator (poly I:C) on the rat cerebral cortex. J Tissue Res 4(1):71–74
Patro IK, Pathak S, Patro N (2005) Central responses to peripheral nerve injury: role of non–neuronal cells. In: Thakur MK, Prasad S (eds) Molecular and cellular neurobiology. Narosa Publishing House, New Delhi, p 217
Patro IK, Amit Shrivastava M, Bhumika S, Patro N (2010a) Poly I: C induced microglial activation impairs motor activity in adult rats. Indian J Exp Biol 48(2):104–109
Patro N, Kushwah S, Patro IK (2010b) Aging of microglia: does it influence neuroprotection? Proc Natl Acad Sci India Sect B 80(1):14–23
Patro N, Nagayach A, Patro IK (2010c) Iba1 expressing microglia in the dorsal root ganglia become activated following peripheral nerve injury in rats. Indian J Exp Biol 48(2):110–116
Patro N, Singh K, Patro IK (2013) Differential microglial and astrocytic response to bacterial and viral infection in the developing hippocampus of neonatal rats. Indian J Exp Biol 51(8):606–614
Patro IK, Saxena K, Nagayach A, Patro N (2014) Involvement of glilal cells in pathophysiology of peripheral nerve injury. In: Mathur PP (ed) Contemporary topics in life sciences. Narendra Publishing House, New Delhi, pp 153–173
Petersen OH, Michalak M, Verkhratsky A (2005) Calcium signalling: past, present and future. Cell Calcium 38:161–169
Pocock JM, Kettenmann H (2007) Neurotransmitter receptors on microglia. Trends Neurosci 30(10):527–535
Pont-Lezica L, Béchade C, Belarif-Cantaut Y, Pascual O, Bessis A (2011) Physiological roles of microglia during development. J Neurochem 119(5):901–908
Prinz M, Mildner A (2011) Microglia in the CNS: immigrants from another world. Glia 59(2):177–187
Prinz M, Priller J (2014) Microglia and brain macrophages in the molecular age: from origin to neuropsychiatric disease. Nat Rev Neurosci 15(5):300–312
Rakic S, Zecevic N (2000) Programmed cell death in the developing human telencephalon. Eur J Neurosci 12(8):2721–2734
Ransohoff RM, Brown MA (2012) Innate immunity in the central nervous system. J Clin Invest 122:1164–1171
Rezaie P, Male D (2002) Mesoglia and microglia—A historical review of the concept of mononuclear phagocytes within the central nervous system. J Hist Neurosci 11(4):325–374
Rivest S (2003) Molecular insights on the cerebral innate immune system. Brain Behav Immun 17(1):13–19
Rogers JT, Morganti JM, Bachstetter AD, Hudson CE, Peters MM, Grimmig BA et al (2011) CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. J Neurosci 31(45):16241–16250
Saijo K, Glass CK (2011) Microglial cell origin and phenotypes in health and disease. Nat Rev 11(11):775–787
Saijo K, Crotti A, Glass CK (2013) Regulation of microglia activation and deactivation by nuclear receptors. Glia. 61:104–111
Sandireddy R, Yerra VG, Areti A, Komirishetty P, Kumar A (2014) Neuroinflammation and oxidative stress in diabetic neuropathy: futuristic strategies based on these targets. Int J Endocrinol 2014:674987
Saxena K, Patro N, Patro I (2007) FK506 protects neurons following peripheral nerve injury via immunosuppression. Cell Mol Neurobiol 27:1049–1057
Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, Stevens B (2012) Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74(4):691–705
Schafer DP, Lehrman EK, Stevens B (2013) The “Quad-Partite” synapse: microglia-synapse interactions in the developing and mature CNS. Glia 61(1):24–36
Schilling T, Eder C (2007) Ion channel expression in resting and activated microglia of hippocampal slices from juvenile mice. Brain Res 1186(1):21–28
Schlichter LC, Kaushal V, MoxonEmre I, Sivagnanam V, Vincent C (2010) The Ca2+ activated SK3 channel is expressed in microglia in the rat striatum and contributes to microglia-mediated neurotoxicity in vitro. J Neuroinflammation 7:4
Semple BD, Kossmann T, Morganti-Kossmann MC (2010) Role of chemokines in CNS health and pathology: a focus on the CCL2/CCR2 and CXCL8/CXCR2 networks. J Cereb Blood Flow Metab 30:459–473
Sharma A, Patro N, Patro IK (2015) Lipopolysaccharide-Induced apoptosis of astrocytes: therapeutic intervention by minocycline. Cell Mol Neurobiol. 07/2015. doi:10.1007/s10571-015-0238-y
Shepard JL, Zon LI (2000) Developmental derivation of embryonic and adult macrophages. Curr Opin Hematol 7(1):3–8
Shi X, Chen Y, Nadeem L, Xu G (2013) Beneficial effect of TNF-α inhibition on diabetic peripheral neuropathy. J Neuroinflammation 10:69
Sierra A, Gottfried-Blackmore AC, McEwen BS, Bulloch K (2007) Microglia derived from aging mice exhibit an altered inflammatory profile. Glia 55:412–424
Sierra A, Abiega O, Shahraz A, Neumann H (2013) Janus-faced microglis: beneficial and detrimental consequences of microglial phagocytosis. Front Cell Neurosci 7:355. doi:10.3389/fncel.2013.00006
Sivakumar V, Ling EA, Lu J, Kaur C (2010) Role of glutamate and its receptors and insulin-like growth factors in hypoxia induced periventricular white matter injury. Glia 58:507–523
Sondag CM, Dhawan G, Combs CK (2009) Beta amyloid oligomers and fibrils stimulate differential activation of primary microglia. J Neuroinflammation 6:1
Streit WJ (2006) Microglial senescence: does the brain’s immune system have an expiration date? Trends Neurosci 29(9):506–510
Streit WJ, Sammons NW, Kuhns AJ, Sparks DL (2004) Dystrophic microglia in the aging human brain. Glia 45(2):208–212
Suzuki T, Hide I, Ido K, Kohsaka S, Inoue K, Nakata Y (2004) Production and release of neuroprotective tumor necrosis factor by P2X7 receptor-activated microglia. J Neurosci 24:1–7
Takahashi K (2001) Development and differentiation of macrophages and related cells: historical review and current concepts. J Clin Exp Hematopathol 41:1–33
Tang Yu, Le W (2015) Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol Neurobiol. doi:10.1007/s12035-014-9070-5
Taylor DL, Jones F, Kubota ES, Pocock JM (2005) Stimulation of microglial metabotropic glutamate receptor mGlu2 triggers tumor necrosis factor-alpha induced neurotoxicity in concert with microglial-derived Fas ligand. J Neurosci 25:2952–2964
Thored P, Heldmann U, Gomes-Leal W, Gisler R, Darsalia V, Taneera J et al (2009) Long-term accumulation of microglia with proneurogenic phenotype concomitant with persistent neurogenesis in adult subventricular zone after stroke. Glia 57(8):835–849
Tremblay MÈ, Lowery RL, Majewska AK (2010) Microglial interactions with synapses are modulated by visual experience. PLoS Biol 8:e1000527
Tremblay MÈ, Zettel ML, Ison JR, Allen PD, Majewska AK (2012) Effects of aging and sensory loss on glial cells in mouse visual and auditory cortices. Glia 60(4):541–558
Venero JL, Burguillos MA, Brundin P, Joseph B (2011) The executioners sing a new song: killer caspases activate microglia. Cell Death Differ 18(11):1679–1691
Verkhratsky A, Parpura V, Pekna M, Pekny M, Sofroniew M (2014) Glia in the pathogenesis of neurodegenerative diseases. Biochem Soc Trans 42(5):1291–1301
Wake H, Moorhouse AJ, Jinno S, Kohsaka S, Nabekura J (2009) Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci 29(13):3974–3980
Watkins CC, Sawa A, Pomper MG (2014) Glia and immune cell signaling in bipolar disorder: insights from neuropharmacology and molecular imaging to clinical application. Transl Psychiatry 4:e350
Wynne AM, Henry CJ, Godbout JP (2009) Immune and behavioral consequences of microglial reactivity in the aged brain. Integr Comp Biol 49(3):254–266
Yamasaki R, Lu H, Butovsky O, Ohno N, Rietsch AM, Cialic R (2014) Differential roles of microglia and monocytes in the inflamed central nervous system. J Exp Med 211(8):1533–1549
Yokoyama A, Yang L, Itoh S, Mori K, Tanaka J (2004) Microglia, a potential source of neurons, astrocytes and oligodendrocytes. Glia 45(1):96–104
Yung RL, Julius A (2008) Epigenetics, aging and autoimmunity. Autoimmunity 41(4):329–335
Zipser BD, Johanson CE, Gonzalez L, Berzin TM, Tavares R, Hulette CM, Vitek MP, Hovanesian V, Stopa EG (2007) Microvascular injury and blood-brain barrier leakage in Alzheimer’s disease. Neurobiol Aging 28(7):977–986
Acknowledgments
The work of the authors is variously supported by the Department of Biotechnology and the Indian Council of Medical Research, Govt. of India, New Delhi.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media Singapore
About this chapter
Cite this chapter
Patro, I., Nagayach, A., Sinha, S., Patro, N. (2016). General Physiology and Pathophysiology of Microglia During Neuroinflammation. In: Jana, N., Basu, A., Tandon, P. (eds) Inflammation: the Common Link in Brain Pathologies. Springer, Singapore. https://doi.org/10.1007/978-981-10-1711-7_2
Download citation
DOI: https://doi.org/10.1007/978-981-10-1711-7_2
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-1710-0
Online ISBN: 978-981-10-1711-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)